
Abstract
Background
Malaria transmission in Tanzania is driven by mosquitoes of the Anopheles gambiae complex and Anopheles funestus group. The latter includes An. funestus s.s., an anthropophilic vector, which is now strongly resistant to public health insecticides, and several sibling species, which remain largely understudied despite their potential as secondary vectors. This paper provides the initial results of a cross-country study of the species composition, distribution and malaria transmission potential of members of the Anopheles funestus group in Tanzania.
Methods
Mosquitoes were collected inside homes in 12 regions across Tanzania between 2018 and 2022 using Centres for Disease Control and Prevention (CDC) light traps and Prokopack aspirators. Polymerase chain reaction (PCR) assays targeting the noncoding internal transcribed spacer 2 (ITS2) and 18S ribosomal DNA (18S rDNA) were used to identify sibling species in the An. funestus group and presence of Plasmodium infections, respectively. Where DNA fragments failed to amplify during PCR, we sequenced the ITS2 region to identify any polymorphisms.
Results
The following sibling species of the An. funestus group were found across Tanzania: An. funestus s.s. (50.3%), An. parensis (11.4%), An. rivulorum (1.1%), An. leesoni (0.3%). Sequencing of the ITS2 region in the nonamplified samples showed that polymorphisms at the priming sites of standard species-specific primers obstructed PCR amplification, although the ITS2 sequences closely matched those of An. funestus s.s., barring these polymorphisms. Of the 914 samples tested for Plasmodium infections, 11 An. funestus s.s. (1.2%), and 2 An. parensis (0.2%) individuals were confirmed positive for P. falciparum. The highest malaria transmission intensities [entomological inoculation rate (EIR)] contributed by the Funestus group were in the north-western region [108.3 infectious bites/person/year (ib/p/y)] and the south-eastern region (72.2 ib/p/y).
Conclusions
Whereas An. funestus s.s. is the dominant malaria vector in the Funestus group in Tanzania, this survey confirms the occurrence of Plasmodium-infected An. parensis, an observation previously made in at least two other occasions in the country. The findings indicate the need to better understand the ecology and vectorial capacity of this and other secondary malaria vectors in the region to improve malaria control.
Graphical Abstract

Similar content being viewed by others

Background
Africa has witnessed significant progress in the fight against malaria from 2000, thanks to extensive vector control using insecticide-treated nets and indoor spraying, alongside improved diagnosis and treatment [1]. However, this progress began to stagnate around 2015, and currently, malaria causes ≤ 249 million cases and 608,000 deaths annually, predominantly in sub-Saharan Africa [2].
Other than the weak health systems and socio-economic conditions [3, 4], the persistent malaria burden in the region is exacerbated by several biological challenges, notably insecticide resistance [5, 6], anti-malarial drug resistance [7,8,9], failing diagnostics [10,11,12,13] and an invasive vector species, Anopheles stephensi [14,15,16]. The problem is compounded by human behaviour and lifestyles leading to inadequate protection during peak transmission periods, and insufficient community and stakeholder engagement in malaria prevention efforts [17,18,19,20]. Evidence also suggests that the traditional methods used to disrupt malaria transmission, notably insecticide treated bed nets (ITNs) and indoor residual spraying (IRS) are insufficient against certain vector species with atypical behaviours, such as those that do not bite or rest primarily indoors [21,22,23].
In east and southern Africa, the major malaria vectors are Anopheles gambiae sensu stricto, Anopheles arabiensis and Anopheles funestus mosquitoes [24, 25]. In most settings, An. gambiae s.s. and An. funestus mosquitoes have been historically dominating the malaria transmission [26,27,28,29]. However, recently, in some localities, such as in parts of western Kenya and south-eastern Tanzania, the wide coverage of ITNs likely in concert with environmental changes, appears to have significantly suppressed An. gambiae s.s. leaving An. arabiensis and An. funestus s.s. as the main drivers of transmission [30,31,32,33]. In Tanzania, An. funestus is now the dominant malaria vector across the country [34]. More detailed studies have revealed that even when outnumbered by An. arabiensis, An. funestus s.s. mediates over 90% of the ongoing malaria transmission in south-eastern Tanzania [31]. With An. funestus being highly anthropophilic, and in some settings having stronger resistance to public health insecticides compared with the other major malaria vectors [35], this vector species poses a significant challenge to the existing vector control interventions. It is noteworthy, that most studies have so far focused on only one member of the An. funestus group, i.e. An. funestus s.s. despite this species being one member of a large species complex [36].
The An. funestus group is thought to comprise 13 sibling species: An. funestus s.s.[25], An. funestus-like, An. vaneedeni [37], An. parensis [38,39,40], An. rivulorum[41], An. rivulorum-like [42], An. leesoni, An. aruni, An. confusus, An. brucei, An. fuscivenosus and An. longipalpis types A and C [43]. Of these, An. funestus s.s. is the most competent malaria vector in the group, though other sibling species, such as An. rivulorum, An. leesoni and An. parensis have also been reported to carry Plasmodium falciparum to lesser extent [37,38,39, 41, 43]. Despite these important observations, the species composition, distribution, and role in malaria transmission of the An. funestus group remains understudied, and several members of this group are likely to be misidentified. For instance, Ogola et al. [44] reported an unidentified sibling species within the An. funestus group in Kenya, and existing polymerase chain reaction (PCR) assays commonly return unamplified samples, including those morphologically confirmed as belonging to the group [45, 46]. More importantly, our understanding of the ecological dynamics and potential roles of these in perpetuating persistent malaria transmission remains limited.
To bridge these knowledge gaps, our research team initiated and implemented a cross-country survey of malaria vectors aimed at determining the species composition, spatial distribution, and the relative contribution of different Anopheles spp. to malaria transmission in mainland Tanzania. This paper presents the results from the initial phase of these surveys, covering 14 districts in 12 regions across Tanzania.
Methods
Study area
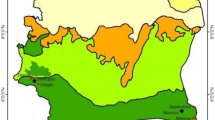
Mosquitoes were collected from 14 districts in 12 administrative regions across Tanzania mainland (Fig. 1). Tanzania has a broadly tropical climate, with four primary climatic zones: the hot and humid coastal plain (i.e. Pwani, Tanga, Lindi and Mtwara), the semi-arid central plateau (i.e. Dodoma, Kigoma, Katavi and Rukwa), the high rainfall lake regions (i.e. Kagera and Mwanza) and the cooler highlands (i.e. Morogoro and Ruvuma). On the Tanzanian coast and offshore islands, temperatures typically fluctuate between 27 °C and 29 °C. In the central, northern, and western regions, temperatures vary between 20 °C and 30 °C. The extended rainy season spans from March to May, while the shorter rainy season extends from October to early December, with the dry season lasting from June to September. Overall, annual rainfall ranges from 550 mm in the central areas to 3690 mm in certain parts of the southwestern highlands [47]. In most of these districts, the majority of the rural households are subsistence farmers [48, 49]. Malaria prevalence in children under the age of 5 years differs significantly in the study area, with the highest in the north-western (i.e. Kagera and Kigoma) and south-eastern regions (i.e. Mtwara and Lindi) to less than 1% in the central region (i.e. Dodoma)[50].

Map of Tanzania showing the regions where Anopheles funestus mosquitoes were collected
The data collection sites are shown in Fig. 1. Specific districts were: Misenyi in Kagera, Kakonko and Kibondo in Kigoma, Chamwino in Dodoma, Ulanga and Kilombero in Morogoro, Tunduru in Ruvuma, Bagamoyo in Pwani, Nkasi in Rukwa, Tanganyika in Katavi, Misungwi in Mwanza, Mtama in Lindi, Mahurunga in Mtwara and Muheza in Tanga. These collection sites represent diverse geographical regions, including the hot and humid coastal plain (i.e. Bagamoyo, Muheza, Mtama and Mahurunga), the semi-arid central plateau (i.e. Chamwino, Kakonko, Kibondo, Tanganyika and Nkasi), the high rainfall lake regions (i.e. Misenyi and Misungwi) and the cooler highlands (i.e. Kilombero, Ulanga and Tunduru).
Mosquito collection and processing
Mosquito collections, conducted as part of a larger project on the population genetics of An. funestus sensu lato, were sporadic and completed between December 2018 and December 2022. These collections spanned both dry and wet seasons. Whereas multiple mosquito species were collected, only An. funestus s.l. are used in this analysis. In each of the districts, at least two houses were selected upon consent from the household heads and used for the collection of adult mosquitoes. Centres for Disease Control and Prevention (CDC) light traps [51] and Prokopack aspirators [52] were used to sample indoor host-seeking and resting mosquitoes, respectively. The overall sampling approach had been specifically designed to collect An. funestus s.l for population genetics studies, and was specifically focused on indoor collections, with no outdoor trapping in this phase of the study. Thus, mosquitoes were morphologically sorted to the species level and females of An. funestus s.l individually packed in an Eppendorf tube with 80% ethanol. In addition, in some locations, such as Dodoma, Tanga and Morogoro regions, where sampling of adult An. funestus s.l was insufficient, larval collections were conducted using standard larval dippers [53]. The collected larvae were reared to adults as previously described [54], then also sorted by taxa as above.
Extraction of genomic DNA
Genomic DNA was extracted from the heads and thoraxes of collected mosquitoes using DNAzol method [55]. Bead Ruptor 96 well plate homogenizer (OMNI international, Kennesaw, GA, USA) was used for homogenization and the resultant DNA pellets were eluted in 50 µl of Tris–ethylenediaminetetraacetic acid (EDTA) buffer.
Identification of the sibling species in the An. funestus group and detection of Plasmodium spp. infections
A cocktail of species-specific primers for the identification of the sibling species in the An. funestus group was used, as previously described by Koekemoer et al. [56]; with a slight adaptation to include a primer for An. rivulorum-like (Table 1) in the cocktail [42]. A nested PCR assay was used for the detection of the Plasmodium spp., of which the first round of the PCR included universal forward and reverse primers for 18S rDNA Plasmodium spp. (Table 2) regardless of species; followed by a second round using the amplicon from the first round as DNA template. Species-specific primers for Plasmodium falciparum, Plasmodium ovale, Plasmodium vivax and Plasmodium malariae were used in the second round (Table 2).
Further analysis of the internal transcribed spacer 2 (ITS2) region in non-amplified An. funestus samples to investigate polymorphisms
A total of ten samples underwent cloning and sequencing, employing the following primers: ITS2A: 5′ TGT GAA CTG CAG GAC ACA T 3′ (forward) and ITS2B: 5′ TAT GCT TAA ATT CAG GGG GT 3′ (reverse). The PCR reaction mixture, conditions and procedures for the thermal cycling and electrophoresis were similar to those described earlier. The amplicons (approximately 840 base pairs) were excised from the gel and cleaned using Wizard® SV Gel and PCR Clean-Up System (Catalogue number: #A9281, Promega). The purified product was cloned using a plasmid vector pJET1.2/blunt (CloneJET PCR Cloning Kit, Catalogue number: #K1231, Thermo Scientific). The resulting recombinant plasmid DNA was isolated and purified (QIAprep Spin Miniprep Kit, Catalogue number #27106, Qiagen) and sent for sequencing. Sequencing of the recombinant plasmid DNA was carried out using the reverse PJET1.2 primer (5′-AAGAACATCGATTTTCCATGGCAG-3′). Plasmid primer regions trimming, sequence alignment and analysis were performed using SeaView software [57].
Data analysis
The data collected from the field included the number of traps used, the number of collection days, and the mosquitoes captured per trap, facilitating the calculation of trap nights (defined as the product of the number of traps and collection days). The annual entomological inoculation rate (EIR) was determined by multiplying human biting rates and Plasmodium sporozoite prevalence, then adjusted for 365 days [31]. A coefficient of 0.68 was used for conversion of the sampling efficiency of the CDC light trap relative to human landing catch (HLC) [31, 46]. Mosquitoes collected using Prokopack aspirators and from larval collections were excluded from the EIR calculation, as these methods do not accurately reflect host-seeking behaviour or the potential for infectivity. Basic Local Alignment Search Tool for nucleotides (BLASTn) analysis [58] was used to identify and characterize nucleotide sequences by finding homologous sequences in the National Center for Biotechnology Information nucleotide (NCBI nt) database. The top hits were retrieved and analysed to determine sequence similarity, alignment scores and query coverage. For the alignment and analysis of ITS2 sequences cloned from PCR-negative samples, SeaView software [57] was utilized.
Results
Species composition and distribution
A total of 1092 An. funestus s.l were analysed, of which 549 (50.3%) were An. funestus s.s., 124 were An. parensis (11.4%) and 12 were An. rivulorum (1.1%; Table 3). No An. vaneedeni or An. rivulorum-like were found during this study. While the other species, An. funestus s.s. and An. rivulorum were more widespread in the study sites, the An. parensis samples were found most abundantly in the central and northern regions (Table 3; Dodoma and Mwanza). There were 404 samples (37%) for which the DNA fragments did not amplify during the PCR (Table 3). Subsequent cloning and sequencing of the ITS2 region in these non-amplified samples revealed multiple polymorphisms within the reverse primer’s priming region specific to An. funestus s.s. (examples are shown in Additional file 1). The ITS2 sequences were found to be similar to those of An. funestus s.s., with the exception of polymorphisms within the priming site. BLASTn analysis revealed that the highest identity was 99.5%, considering sequences with 100% query coverage. The analysis confirmed that these sequences closely matched An. funestus s.s. sequences from the NCBI nt database.
Prevalence of Plasmodium sporozoite infections in the mosquitoes
Of 914 An. funestus s.l tested for Plasmodium spp. infection, 13 were found positive for P. falciparum (Table 4). The majority of the infections were in An. funestus s.s. (n = 11). In addition, there were two An. parensis mosquitoes infected with P. falciparum (n = 2). No other Plasmodium species were detected, nor were any sibling species of An. funestus (besides An. funestus s.s. and An. parensis) found to be infected with Plasmodium spp.
Transmission intensities mediated by An. funestus group
The annualized EIR estimates varied significantly across the regions. The highest EIR estimates were recorded in Kagera [108.3 infectious bites/person/year (ib/p/y)], Ruvuma (72.2 ib/p/y) and Morogoro regions (65.6 ib/p/y). Since no infected mosquitoes were collected in Dodoma, Rukwa, Lindi, Katavi and Mtwara, it was not possible to estimate EIRs from the An. funestus s.l collected in these regions (Table 5).
Discussion
Anopheles funestus mosquitoes are among the most widespread, and yet also among the least studied species of malaria vectors. However, in recent years, there has been an increasing awareness that populations of An. funestus s.s., known for their high degree of anthropophily and now marked by significant pesticide resistance [35, 46], are becoming predominant in many malaria transmission areas, particularly in East and Southern Africa [59, 60]. In areas, such as south-eastern Tanzania, this species now mediates 85–98% of new malaria infections, even in villages where it is outnumbered by other species, such as An. arabienesis [31, 46, 61]. Today, the species composition and distribution of the An. funestus group, particularly in Tanzania, are well described. However, despite field collections regularly capturing several other members of the complex in many locations, the ecology and vectorial importance of these potential secondary vectors are poorly understood. This current study was therefore aimed at expanding on the existing knowledge towards understanding the vectorial role of An. funestus species across Tanzania mainland.
We found four known and previously reported sibling species of the An. funestus group (i.e. An. funestus s.s., An. parensis, An. rivulorum and An. leesoni), with An. funestus s.s. dominating malaria transmission across all the 12 regions surveyed. Moreover, 37% of the collected mosquitoes were not amplified by the available species-specific PCR assay [56] designed for the An. funestus group, despite being morphologically identified as An. funestus s.l. While this is a significantly high failure rate of the recommended PCR assays, similar non-amplification problems have been reported in previous studies, albeit at lower rates, including in south-eastern Tanzania [35, 45, 46]. Nonetheless, upon cloning and sequencing, it was confirmed that the ITS2 sequences were similar to that of An. funestus s.s. with the exception of polymorphisms present within the priming site of the common and widely used species-specific reverse primer. This PCR mis-priming is hereby considered the main reason for the high rates of non-amplification observed in this study; and may also have affected the aforementioned past studies. It is noteworthy, that all technologies based on PCR amplification, including higher throughput species identification multilocus amplicon panel approaches [62], will at times face similar issues because of the highly polymorphic genomes of Anopheline vector species. This suggests the need to continue improving the methods for identifying members of such species groups and complexes.
The incrimination of An. parensis with transmission of P. falciparum in this study provides only the third such report in Tanzania in the past 15 years [39, 63]. The two previous reports [39, 63] utilized CDC light traps, pyrethrum spray catch and aspirators for indoor sampling of host-seeking An. parensis mosquitoes, with nested PCR and enzyme linked immunosorbent assay (ELISA) as methods of Plasmodium sporozoite detection. Furthermore, the first report which was based on four specimens reported a 25% sporozoite rate [39], whilst the one conducted within the similar geographical area surrounding Lake Victoria as our present study and based on hundreds of samples reported 1.1% rate [63]. Collectively, these repeated observations suggest that An. parensis may be playing a modest but considerable role as a secondary malaria vector in Tanzania and should be further investigated to optimize the control of malaria transmission.
In this study, the two Plasmodium-infected An. parensis mosquitoes were found in the village of Ngaya in the Misungwi district in north-western Tanzania where long-lasting insecticidal nets (LLINs) were already widely implemented [63]. A 2018 study [64] focusing on An. funestus group species composition in several villages of the same district reported over 90% An. funestus s.s. and 6.5% An. parensis. While the aquatic ecology of An. parensis was not within the scope of this paper, studies in rural south-eastern Tanzania noted that An. parensis generally shared aquatic habitats with An. funestus s.s. and An. rivulorum (Kahamba et al. Unpublished data). Further studies are required to understand how vector control interventions might have been associated with the apparently higher importance of An. parensis in this location.
On the basis of our present findings there is a possibility that An. parensis may be contributing to residual malaria transmission, particularly in localities where An. funestus s.s. and other major vector species have been significantly impacted by chemical control interventions. This has previously been observed in the north-eastern part of South Africa, where An. parensis was reported to minimally contribute to residual malaria transmission, following an almost complete suppression of An. funestus s.s. following large-scale IRS implementation [38].
In previous studies, various sibling species within the Anopheles funestus group have been implicated as malaria vectors [37,38,39, 41, 43], resulting in multiple questions regarding the factors influencing their prevalence and roles in disease transmission. For instance, a study conducted in central Kenya reported significant densities of An. parensis inside human dwellings, though with a low human blood index [40]. In our present study, we also collected a significant number of resting An. parensis inside houses in the northern region. Additionally, our current findings, coupled with a previous study [65] conducted in the Muheza district of north-eastern Tanzania, which reported that over 60% of An. parensis caught inside houses had fed on humans despite the availability of cattle, signify the potential role of An. parensis as a contributor to the residual malaria transmission. Consequently, it will be necessary to extend our control efforts beyond the current indoor vector control interventions, to address not just An. parensis, but also other important species, such as An. arabiensis, which is also widespread in Tanzania [34] and tends to bite outdoors [66]. Additionally, there is a need for a thorough understanding of the ecology of An. parensis and other sibling species within the An. funestus group; as well as their responsiveness to current vector control interventions.
Annual entomological inoculation rates (EIR) were computed for different regions and were found to be the highest in areas where An. funestus s.s. dominate as the member of the An. funestus group, such as north-western and southern regions of Kagera and Ruvuma. Notably, Kigoma exhibited the lowest measurable EIR at 18.2 infectious bites per person per year (ib/p/yr) among all regions where infected mosquitoes were found. Among infected mosquitoes, Plasmodium falciparum was the only malarial parasite detected. However, it is essential to note that other Plasmodium species, such as P. ovale and P. malariae, have been previously reported in other country-wide surveys [67,68,69]. One limitation of this study was that the mosquito sampling, primarily designed for species identification and genomic analysis, was insufficient to definitively rule out Plasmodium infections in regions where none of the tested mosquitoes were found to be infected. Consequently, areas reporting zero EIR estimates are simply categorized as having non-estimable EIRs, rather than being considered as having no risk of malaria transmission. It is expected that expanded surveys would reveal non-zero prevalence rates within either the An. funestus group or the An. gambiae complex. Additionally, another limitation of the present study was the inability of the available species-specific PCR assay [56] designed for the An. funestus group, to identify 37% of the collected mosquitoes that had otherwise been morphologically identified as An. funestus.
Conclusions
This study underscores the pivotal role of the An. funestus group in malaria transmission with a particular focus on the prominent An. funestus s.s. Additionally, the study sheds light on the lesser-studied sibling species, An. parensis, which is identified here, for the third time, as playing a role in the transmission of Plasmodium falciparum. Challenges in PCR amplification owing to ITS2 region polymorphisms highlight the limitations of current molecular tools for distinguishing species within the Funestus group. This study contributes to the body of knowledge on malaria vector composition and distribution in Tanzania and emphasizes the critical need for the adaptation of vector control interventions to regional specificities in malaria transmission dynamics. More importantly, the findings call for a deeper investigation into the ecology and vectorial capacity of secondary vectors to enhance malaria control strategies.
Availability of data and materials
All data supporting the conclusions of this article are provided within the text, including the GenBank accession numbers PP853609, PP853610 and PP853611.
Abbreviations
- ITN:
-
Insecticide treated bed net
- IRS:
-
Indoor residual spraying
- PCR:
-
Polymerase chain reaction
- CDC:
-
Centres for Disease Control and Prevention
- EDTA:
-
Ethylene diamine tetra acetic acid
- ITS:
-
Internal transcribed spacer
- HLC:
-
Human landing catch
- EIR:
-
Entomological inoculation rate
- BLASTn:
-
Basic local alignment search tool for nucleotides
- NCBI nt:
-
National centre for biotechnology information nucleotide
References
Bhatt S, Weiss DJ, Cameron E, Bisanzio D, Mappin B, Dalrymple U, et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature. 2015;526:207–11.
WHO. World malaria report 2023 [Internet]. 2023. Available from: https://www.who.int/publications/i/item/9789240086173.
The malERA Consultative Group on Health Systems. A research agenda for malaria eradication: health systems and operational research. PLoS Med. 2011;8:e1000397.
Okumu F, Gyapong M, Casamitjana N, Castro MC, Itoe MA, Okonofua F, et al. What Africa can do to accelerate and sustain progress against malaria. PLOS Glob Public Health. 2022;2:e0000262.
Hancock PA, Hendriks CJM, Tangena JA, Gibson H, Hemingway J, Coleman M, et al. Mapping trends in insecticide resistance phenotypes in African malaria vectors. PLoS Biol. 2020;18:e3000633.
Moyes CL, Athinya DK, Seethaler T, Battle KE, Sinka M, Hadi MP, et al. Evaluating insecticide resistance across African districts to aid malaria control decisions. Proc Natl Acad Sci USA. 2020;117:28063–70.
Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. 2019;19:e338–51.
Ikeda M, Kaneko M, Tachibana SI, Balikagala B, Sakurai-Yatsushiro M, Yatsushiro S, et al. Artemisinin-resistant Plasmodium falciparum with high survival rates, Uganda, 2014–2016. Emerg Infect Dis. 2018;24:718–26.
Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana SI, Yamauchi M, et al. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med. 2021;385:1163–71.
Feleke SM, Reichert EN, Mohammed H, Brhane BG, Mekete K, Mamo H, et al. Plasmodium falciparum is evolving to escape malaria rapid diagnostic tests in Ethiopia. Nat Microbiol. 2021;6:1289–99.
Mwangonela ZE, Ye Y, Quaye R, Msuya HM, Mwamlima TG, Mswata SS, et al. Field evaluation of the novel one step malaria Pf and Pf/Pv rapid diagnostic tests and the proportion of HRP-2 gene deletion identified on samples collected in the Pwani region, Tanzania. Bull Natl Res Cent. 2023;47:29.
Thomson R, Beshir KB, Cunningham J, Baiden F, Bharmal J, Bruxvoort KJ, et al. pfhrp2 and pfhrp3 gene deletions that affect malaria rapid diagnostic tests for Plasmodium falciparum: analysis of archived blood samples from 3 African countries. J Infect Dis. 2019;220:144–52.
Berhane A, Anderson K, Mihreteab S, Gresty K, Rogier E, Mohamed S, et al. Major threat to malaria control programs by Plasmodium falciparum lacking histidine-rich protein 2, Eritrea. Emerg Infect Dis. 2018;24:462–70.
Mnzava A, Monroe AC, Okumu F. Anopheles stephensi in Africa requires a more integrated response. Malar J. 2022;21:17.
Al-Eryani SM, Irish SR, Carter TE, Lenhart A, Aljasari A, Montoya LF, et al. Public health impact of the spread of Anopheles stephensi in the WHO Eastern Mediterranean Region countries in Horn of Africa and Yemen: need for integrated vector surveillance and control. Malar J. 2023;22:56.
Sinka ME, Pironon S, Massey NC, Longbottom J, Hemingway J, Moyes CL, et al. A new malaria vector in Africa: predicting the expansion range of Anopheles stephensi and identifying the urban populations at risk. Proc Natl Acad Sci USA. 2020;117:24900–8.
Finda MF, Moshi IR, Monroe A, Limwagu AJ, Nyoni AP, Swai JK, et al. Linking human behaviours and malaria vector biting risk in south-eastern Tanzania. PLoS ONE. 2019;14:e0217414.
Monroe A, Asamoah O, Lam Y, Koenker H, Psychas P, Lynch M, et al. Outdoor-sleeping and other night-time activities in northern Ghana: implications for residual transmission and malaria prevention. Malar J. 2015;14:35.
Adhikari B, Pell C, Cheah PY. Community engagement and ethical global health research. Glob Bioeth. 2020;31:1–12.
Asale A, Kussa D, Girma M, Mbogo C, Mutero CM. Community-based integrated vector management for malaria control: lessons from three years’ experience (2016–2018) in Botor-Tolay district, southwestern Ethiopia. BMC Public Health. 2019;19:1208.
Okumu F, Finda M. Key characteristics of residual malaria transmission in two districts in south-eastern Tanzania—implications for improved control. J Infect Dis. 2021;223:S143–54.
Wilkes TJ, Matola YG, Charlwood JD. Anopheles rivulorum, a vector of human malaria in Africa. Med Vet Entomol. 1996;10:108–10.
Durnez L, Coosemans M. Residual transmission of malaria: an old issue for new approaches. In: Manguin S, editor. Anopheles mosquitoes—new insights into malaria vectors. InTech: Rijeka; 2013. p. 671–704.
Mwangangi JM, Muturi EJ, Muriu SM, Nzovu J, Midega JT, Mbogo CM. The role of Anopheles arabiensis and Anopheles coustani in indoor and outdoor malaria transmission in Taveta District, Kenya. Parasit Vectors. 2013;6:114.
Ogola EO, Fillinger U, Ondiba IM, Villinger J, Masiga DK, Torto B, et al. Insights into malaria transmission among Anopheles funestus mosquitoes, Kenya. Parasit Vectors. 2018;11:577.
Coetzee M, Craig M, Le Sueur D. Distribution of African malaria mosquitoes belonging to the Anopheles gambiae complex. Parasitol Today. 2000;16:74–7.
Gillies MT, Coetzee M. A supplement to the Anophelinae of Africa south of the Sahara (Afrotropical region). Johannesburg: South African Institute for Medical Research; 1987;55:1–43.
Sinka ME, Bangs MJ, Manguin S, Rubio-Palis Y, Chareonviriyaphap T, Coetzee M, et al. A global map of dominant malaria vectors. Parasit Vectors. 2012;5:69.
Coetzee M, Fontenille D. Advances in the study of Anopheles funestus, a major vector of malaria in Africa. Insect Biochem Mol Biol. 2004;34:599–605.
Lwetoijera DW, Harris C, Kiware SS, Dongus S, Devine GJ, McCall PJ, et al. Increasing role of Anopheles funestus and Anopheles arabiensis in malaria transmission in the Kilombero Valley, Tanzania. Malar J. 2014;13:331.
Mapua SA, Hape EE, Kihonda J, Bwanary H, Kifungo K, Kilalangongono M, et al. Persistently high proportions of Plasmodium-infected Anopheles funestus mosquitoes in two villages in the Kilombero valley, south-eastern Tanzania. Parasite Epidemiol Control. 2022;18:e00264.
McCann RS, Ochomo E, Bayoh MN, Vulule JM, Hamel MJ, Gimnig JE, et al. Reemergence of Anopheles funestus as a vector of Plasmodium falciparum in western Kenya after long-term implementation of insecticide-treated bed nets. Am J Trop Med Hyg. 2014;90:597–604.
Bayoh MN, Mathias DK, Odiere MR, Mutuku FM, Kamau L, Gimnig JE, et al. Anopheles gambiae: historical population decline associated with regional distribution of insecticide-treated bed nets in western Nyanza Province, Kenya. Malar J. 2010;9:62.
Mwalimu CD, Kiware S, Nshama R, Derua Y, Machafuko P, Gitanya A, et al. The changing composition of malaria vector populations in the Lower Moshi area of north-eastern Tanzania following deployment of mosquito-proofed housing. Malar J. 2023;22:91.
Pinda PG, Eichenberger C, Ngowo HS, Msaky DS, Abbasi S, Kihonda J, et al. Comparative assessment of insecticide resistance phenotypes in two major malaria vectors, Anopheles funestus and Anopheles arabiensis in south-eastern Tanzania. Malar J. 2020;19:354.
Dia I, Guelbeogo MW, Ayala D. Advances and perspectives in the study of the malaria mosquito Anopheles funestus. Parasit Vectors. 2013;6:5.
Burke A, Dandalo L, Munhenga G, Dahan-Moss Y, Mbokazi F, Ngxongo S, et al. A new malaria vector mosquito in South Africa. Sci Rep. 2017;7:15488.
Burke A, Dahan-Moss Y, Duncan F, Qwabe B, Coetzee M, Koekemoer L, et al. Anopheles parensis contributes to residual malaria transmission in South Africa. Malar J. 2019;18:221.
Temu EA, Minjas JN, Tuno N, Kawada H, Takagi M. Identification of four members of the Anopheles funestus (Diptera: Culicidae) group and their role in Plasmodium falciparum transmission in Bagamoyo coastal Tanzania. Acta Trop. 2007;104:38–46.
Kamau L, Koekemoer LL, Coetzee M, Hunt RH. Anopheles parensis: the main member of the Anopheles funestus species group found resting inside human dwellings in Mwea area of central Kenya toward the end of the rainy season. J Am Mosq Control Assoc. 2003;19:130–3.
Kawada H, Dida GO, Sonye G, Njenga SM, Mwandawiro C, Minakawa N. Reconsideration of Anopheles rivulorum as a vector of Plasmodium falciparum in western Kenya: some evidence from biting time, blood preference, sporozoite positive rate, and pyrethroid resistance. Parasit Vectors. 2012;5:230.
Cohuet A, Simard F, Toto JC, Kengne P, Coetzee M, Fontenille D. Species identification within the Anopheles funestus group of malaria vectors in Cameroon and evidence for a new species. Am J Trop Med Hyg. 2003;69:200–5.
Kinya F, Mutero CM, Sang R, Owino EA, Rotich G, Ogola EO, et al. Outdoor malaria vector species profile in dryland ecosystems of Kenya. Sci Rep. 2022;12:7131.
Ogola EO, Chepkorir E, Sang R, Tchouassi DP. A previously unreported potential malaria vector in a dry ecology of Kenya. Parasit Vectors. 2019;12:314.
Matowo NS, Koekemoer LL, Moore SJ, Mmbando AS, Mapua SA, Coetzee M, et al. Combining synthetic human odours and low-cost electrocuting grids to attract and kill outdoor-biting mosquitoes: field and semi-field evaluation of an improved mosquito landing box. PLoS ONE. 2016;11:e0145653.
Kaindoa EW, Matowo NS, Ngowo HS, Mkandawile G, Mmbando A, Finda M, et al. Interventions that effectively target Anopheles funestus mosquitoes could significantly improve control of persistent malaria transmission in south-eastern Tanzania. PLoS ONE. 2017;12:e0177807.
World Bank Group. Climate Change Knowledge Portal. 2021. Available from: https://climateknowledgeportal.worldbank.org/country/tanzania/climate-data-historical.
Derua YA, Alifrangis M, Hosea KM, Meyrowitsch DW, Magesa SM, Pedersen EM, et al. Change in composition of the Anopheles gambiae complex and its possible implications for the transmission of malaria and lymphatic filariasis in north-eastern Tanzania. Malar J. 2012;11:188.
Swai JK, Finda MF, Madumla EP, Lingamba GF, Moshi IR, Rafiq MY, et al. Studies on mosquito biting risk among migratory rice farmers in rural south-eastern Tanzania and development of a portable mosquito-proof hut. Malar J. 2016;15:564.
National Bureau of Statistics. Tanzania Demographic and Health Survey and Malaria Indicator Survey 2022 Key Indicators.
Mboera LEG, Kihonda J, Braks MAH, Knols BGJ. Influence of Centers for Disease Control light trap position, relative to a human-baited bed net, on catches of Anopheles gambiae and Culex quinquefasciatus in Tanzania. Am J Trop Med Hyg. 1998;59:595–6.
Maia MF, Robinson A, John A, Mgando J, Simfukwe E, Moore SJ. Comparison of the CDC Backpack aspirator and the Prokopack aspirator for sampling indoor- and outdoor-resting mosquitoes in southern Tanzania. Parasit Vectors. 2011;4:124.
Vanek MJ, Shoo B, Mtasiwa D, Kiama M, Lindsay SW, Fillinger U, et al. Community-based surveillance of malaria vector larval habitats: a baseline study in urban Dar es Salaam, Tanzania. BMC Public Health. 2006;6:154.
Batista EPA, Mapua SA, Ngowo H, Matowo NS, Melo EF, Paixão KS, et al. Videographic analysis of flight behaviours of host-seeking Anopheles arabiensis towards BG-Malaria trap. PLoS ONE. 2019;14:e0220563.
Sangba MLO, Deketramete T, Wango SP, Kazanji M, Akogbeto M, Ndiath MO. Insecticide resistance status of the Anopheles funestus population in Central African Republic: a challenge in the war. Parasit Vectors. 2016;9:230.
Koekemoer LL, Kamau L, Hunt RH, Coetzee M. A cocktail polymerase chain reaction assay to identify members of the Anopheles funestus (Diptera: Culicidae) group. Am J Trop Med Hyg. 2002;66:804–11.
Gouy M, Guindon S, Gascuel O. Sea view version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Msugupakulya BJ, Urio NH, Jumanne M, Ngowo HS, Selvaraj P, Okumu FO, et al. Changes in contributions of different Anopheles vector species to malaria transmission in east and southern Africa from 2000 to 2022. Parasit Vectors. 2023;16:408.
Kahamba NF, Finda M, Ngowo HS, Msugupakulya BJ, Baldini F, Koekemoer LL, et al. Using ecological observations to improve malaria control in areas where Anopheles funestus is the dominant vector. Malar J. 2022;21:183.
Swai JK, Mmbando AS, Ngowo HS, Odufuwa OG, Finda MF, Mponzi W, et al. Protecting migratory farmers in rural Tanzania using eave ribbons treated with the spatial mosquito repellent, transfluthrin. Malar J. 2019;18:364.
Makunin A, Korlević P, Park N, Goodwin S, Waterhouse RM, von Wyschetzki K, et al. A targeted amplicon sequencing panel to simultaneously identify mosquito species and Plasmodium presence across the entire Anopheles genus. Mol Ecol Resour. 2022;22:813–28.
Kakilla C, Manjurano A, Nelwin K, Martin J, Mashauri F, Kinung’hi SM, et al. Malaria vector species composition and entomological indices following indoor residual spraying in regions bordering Lake Victoria, Tanzania. Malar J. 2020;19:135.
Matowo NS, Martin J, Kulkarni MA, Mosha JF, Lukole E, Isaya G, et al. An increasing role of pyrethroid-resistant Anopheles funestus in malaria transmission in the Lake Zone, Tanzania. Sci Rep. 2021;11:13457.
Kweka EJ, Mahande AM, Nkya WM, Assenga C, Lyatuu EE, Nyale E, et al. Vector species composition and malaria infectivity rates in Mkuzi, Muheza District, north-eastern Tanzania. Tanzan J Health Res. 2008;10:192–4.
Limwagu AJ, Kaindoa EW, Ngowo HS, Hape E, Finda M, Mkandawile G, et al. Using a miniaturized double-net trap (DN-Mini) to assess relationships between indoor-outdoor biting preferences and physiological ages of two malaria vectors, Anopheles arabiensis and Anopheles funestus. Malar J. 2019;18:327.
Tarimo BB, Nyasembe VO, Ngasala B, Basham C, Rutagi IJ, Muller M, et al. Seasonality and transmissibility of Plasmodium ovale in Bagamoyo District, Tanzania. Parasit Vectors. 2022;15:18.
Yman V, Wandell G, Mutemi DD, Miglar A, Asghar M, Hammar U, et al. Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in Eastern Tanzania. PLoS Negl Trop Dis. 2019;13:e0007414.
Popkin-Hall ZR, Seth MD, Madebe RA, Budodo R, Bakari C, Francis F, et al. Malaria species positivity rates among symptomatic individuals across regions of differing transmission intensities in Mainland Tanzania. medRxiv. 2023;229:959–68.
Acknowledgements
We would like to thank the community members across all districts for their participation in this study. We would also like to extend our sincere gratitude to Mr. Simon Ashall, Dr. Florian Noulin at Keele University and Ms. Prisca A. Kweyamba, Mr. Said Abbas, Mr. Faraji Abilahi, Mr. Augusto Mwambaluka and Mr. Jonael Msangi at Ifakara Health Institute.
Funding
This work was supported in whole or in part by the Bill and Melinda Gates Foundation (Grant No. INV-002138 to Ifakara Health Institute). Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the author accepted manuscript version that might arise from this submission. This work was also supported by Howard Hughes Medical institute (Grant No: OPP1099295) awarded to F.O.O., and UKRI-Medical Research Council and the UK Foreign, Commonwealth and Development office (FCDO) under the MRC/FCDO concordant agreement which is also part of the EDCTP2 programme supported by the European Union (under the African Research Leaders Award number MR/T008873/1 awarded to N.J.G.).
Author information
Authors and Affiliations
Contributions
S.A.M. and B.S. contributed substantially to this report; I.H.N., G.M., H.B., J.O., J.P., E.W.K., N.J.G., N.K. and E.H. facilitated the sample collections; S.A.M. and B.S. carried out all laboratory examinations and wrote the manuscript with help from F.T. and F.O.O.; N.J.G. and F.O.O. contributed to the design of the study. All authors have read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approvals for this project were obtained from Ifakara Health Institute’s Institutional Review Board (Protocol ID: IHI/IRB/No: 19-2017 and IHI/IRB/EXT/No: 33-2022) and the Medical Research Coordinating Committee (MRCC) at the National Institute for Medical Research, in Tanzania (Protocol ID: NIMR/HQ/R.8a/Vol.IX/2697 and NIMR/HQ/R.8a/Vol.IX/3494). Written consents were sought from all participants of this study, after they had understood the purpose and procedure of the discussions.
Consent for publication
Permission to publish this study was obtained from National Institute for Medical Research, in Tanzania NIMR/HQ/R.8c/VOL. I/1185.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13071_2024_6348_MOESM1_ESM.docx
Additional file 1: (A) An example sequence from NCBI (approximately 844 base pairs including primers) with accession number JN994135.1, comprising of partial sequences of 5.8S and 28S ribosomal RNA genes flanking the internal transcribed spacer 2 region. The outer forward and reverse primer sequences for the complete ITS2 region with 5.8S and 28S rRNA genes flanks are highlighted (green and yellow highlights). The reverse primer specific to An. funestus s.s. in the species identification assay is also shown (dark green). (B), (C) and (D) represent the same region cloned and sequenced from non-amplified samples which revealed polymorphisms within different sections of the reverse primer’s priming region (red).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mapua, S.A., Samb, B., Nambunga, I.H. et al. Entomological survey of sibling species in the Anopheles funestus group in Tanzania confirms the role of Anopheles parensis as a secondary malaria vector. Parasites Vectors 17, 261 (2024). https://doi.org/10.1186/s13071-024-06348-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06348-9