
Abstract
Background
Most malaria vectors belong to species complexes. Sibling species often exhibit divergent behaviors dictating the measures that can be deployed effectively in their control. Despite the importance of the Anopheles funestus complex in malaria transmission in sub-Saharan Africa, sibling species have rarely been identified in the past and their vectoring potential remains understudied.
Methods
We analyzed 1149 wild-caught An. funestus (senso lato) specimens from 21 sites in Kenya, covering the major malaria endemic areas including western, central and coastal areas. Indoor and outdoor collection tools were used targeting host-seeking and resting mosquitoes. The identity of sibling species, infection with malaria Plasmodium parasites, and the host blood meal sources of engorged specimens were analyzed using PCR-based and sequencing methods.
Results
The most abundant sibling species collected in all study sites were Anopheles funestus (59.8%) and Anopheles rivulorum (32.4%) among the 1062 successfully amplified specimens of the An. funestus complex. Proportionally, An. funestus dominated in indoor collections whilst An. rivulorum dominated in outdoor collections. Other species identified were Anopheles leesoni (4.6%), Anopheles parensis (2.4%), Anopheles vaneedeni (0.1%) and for the first time in Kenya, Anopheles longipalpis C (0.7%). Anopheles funestus had an overall Plasmodium infection rate of 9.7% (62/636), predominantly Plasmodium falciparum (59), with two infected with Plasmodium ovale and one with Plasmodium malariae. There was no difference in the infection rate between indoor and outdoor collections. Out of 344 An. rivulorum, only one carried P. falciparum. We also detected P. falciparum infection in two non-blood-fed An. longipalpis C (2/7) which is the first record for this species in Kenya. The mean human blood indices for An. funestus and An. rivulorum were 68% (93/136) and 64% (45/70), respectively, with feeding tendencies on a broad host range including humans and domestic animals such as cow, goat, sheep, chicken and pig.
Conclusions
Our findings underscore the importance of active surveillance through application of molecular approaches to unravel novel parasite-vector associations possibly contributed by cryptic species with important implications for effective malaria control and elimination.
Similar content being viewed by others

Background
Malaria remains the most important vector-borne disease in tropical countries of the world, especially in sub-Saharan Africa (SSA). The scale-up of long lasting insecticidal net (LLIN) distribution has contributed to a decline in malaria transmission, number of cases and mortality within the last decade [1], prompting efforts toward a target of 2030 for elimination. Sustained malaria reduction has proved challenging in many disease-endemic countries of Africa. Whilst in many settings LLIN coverage is high, malaria transmission persists [2] and there is need for a better understanding of the vector species composition. Surveillance of vector populations becomes increasingly important to inform fine-tuning of control strategies in the event of changing local conditions [3] or to discern cryptic species contributing to stealth transmission.
The major African malaria vectors belong to two species complexes: Anopheles gambiae and Anopheles funestus [4, 5]. Few studies in Kenya have examined the contribution of Anopheles funestus sibling species to malaria transmission, despite its well-established role in malaria transmission [6]. It is well known that An. funestus (sensu stricto) (hereafter referred to as An. funestus) is the primary vector species in the An. funestus group [5]. Recent reports of novel Plasmodium falciparum sporozoite associations in An. vaneedeni [7], as well as yet-to-be-identified species [8, 9], suggest the possibility of adaptive changes in vector systems among the group. This underscores the importance of active surveillance of vector populations for potential emerging threats as malaria is being controlled.
Bionomic traits and susceptibility to Plasmodium infection vary among the 13 sibling species known to occur throughout the Afrotropical region in the An. funestus group: An. funestus, An. funestus-like, An. aruni, An. confusus, An. parensis, An. vaneedeni, An. longipalpis types A and C, An. leesoni, An. rivulorum, An. rivulorum-like, An. brucei and An. fuscivenosus [5]. Five species have been documented to occur in Kenya, namely An. funestus, An. rivulorum, An. leesoni, An. parensis and An. vaneedeni [6, 10]. Of these, P. falciparum infection has been found in An. funestus as the main vector [11, 12] and to a minor extent in An. rivulorum [13, 14] and An. leesoni [9]. The often small sample sizes of An. funestus (s.l.) mosquitoes analyzed by molecular means in most of these studies preclude a detailed understanding of the occurrence and ecological distribution of member species in this complex and corollary, their vectoring potential of malaria parasites.
This study focused on the An. funestus group in relation to possible ongoing variations in vector-parasite associations in Kenya. Specifically, we examined the identity and occurrence in the An. funestus group sibling species, Plasmodium infection rates and blood-feeding patterns from specimens sampled from varied malaria-endemic locations in coastal, western and Rift Valley areas of Kenya.
Methods
Anopheles samples
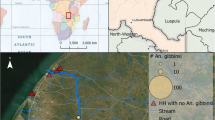
We analysed female specimens of the An. funestus complex collected from randomly selected consenting households in diverse sites in the different epidemiological risk zones for malaria in Kenya [15]. These included Kwale County (Kidomaya, Marigiza, Fihoni), Kilifi County (Jaribuni, Malindi), Taita-Taveta County (Kiwalwa, Njoro), Tana River County (Tana Delta) in the coastal region and Busia County (Bunyala, East Bukusu, Samia, Lwanya-Bumala), Kisumu County (Ahero) and Siaya County (Usenge, West Alego, Mageta) in the Lake Victoria basin in western Kenya and more seasonal malaria transmission areas including the arid and semi-arid areas of northern and central Kenya such as Baringo County (Kapkuikui, Kamnarok) (Fig. 1). Samples from the study sites were mainly collected in 2017, except for those from Ahero, Usenge and Kamnarok (2015), and Mageta (2014).

Map of Kenya showing the sampling locations
Mosquito collections were undertaken both indoors and outdoors with CDC light traps targeting host-seeking mosquitoes for most sites (Bunyala, East Bukusu, Samia, Lwanya-Bumala, West Alego, Kidomaya, Marigiza, Fihoni, Jaribuni). This was achieved by deploying 10 traps per site (5 indoors and 5 outdoors) in separate randomly selected households. At the sites in Ahero, Tana Delta, Kapkuikui, collections were limited to outdoors only using CDC light traps (10) while in Kamnarok, Usenge, Mageta, Njoro and Kiwalwa, collections were limited to indoors using pyrethrum spray catches (PSC) or backpack aspiration (ASP) targeting resting mosquitoes. CDC light traps were set at 18:00 h and retrieved at 06:00 h the following day with PSC and ASP collections conducted between 06:00 and 08:00 h. Indoor and outdoor catches were not undertaken systematically to compare trapping efficiency indoors and outdoors at a particular site, but different tools and locations were used in an attempt to identify the mosquito species richness.
DNA extraction and identification of Anopheles funestus sibling species
We extracted genomic DNA from individual whole specimens using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. The DNA was used for identification of the sibling species as well as probing for Plasmodium infection and host blood meal sources.
Molecular identification of member species of the An. funestus complex involved polymerase chain reaction (PCR) amplification of the polymorphic ITS2 region of ribosomal DNA using established primers [16, 17]. The PCR in a 15 μl reaction involved 0.5 μM each of the primers, 3 μl of 5× Hot Firepol Blend Master Mix Ready to Load (Solis BioDyne, Tartu, Estonia) and 2 μl of DNA template. PCR cycling conditions were as follows: initial denaturation at 95 °C for 15 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 46 °C for 30 s and extension at 72°C for 40 s, and a final extension at 72 °C for 10 min. Thermal reactions were conducted on SimpliAmp Thermal Cycler (Applied Biosystems, Loughborough, UK). Size fragments characteristic of each species were distinguished after separation in agarose gel electrophoresis (1.5%) stained with ethidium bromide against a 100 bp DNA ladder (O’ Gene Ruler, Fermentas, Fisher Scientific, Loughborough, UK).
PCR detection of Plasmodium malaria parasites
We screened for Plasmodium infection in individual specimens by analyzing high resolution melting (HRM) profiles generated from real time-PCR (RT-PCR-HRM) products of non-coding mitochondrial sequence (ncMS) as described before [18] and/or amplification of the cytochrome c oxidase 1 (cox1) gene [19]. Plasmodium falciparum DNA obtained from the National Institute for Biological Standards and Control (NIBSC; London, UK) was used as a standard reference positive control. RT-PCR-HRM was performed in RotorGene Q thermocycler (Qiagen) with RotorGene Q software v.2.1 (Qiagen) with Plasmodium infection determined by comparing melt curves to those obtained using a positive control. Conventional PCR for Plasmodium parasite detection targeting the cox1 gene was carried out in a 15 μl reaction containing 0.5 μM of each primer, 9 μl of PCR water, 3 μl of 5× Hot Firepol® Blend Master Mix (Solis BioDyne) and 2 μl of DNA template. Amplification was carried out with the following cycling parameters: 95 °C for 15 min, 40 cycles of 95 °C for 30 s, 59 °C for 30 s and 72 °C for 40 s, followed by 72 °C for 10 min. For further confirmation, the PCR product of all positive samples by RT-PCR-HRM and a subset of cox1 gene amplicons were purified using ExoSAP-IT (USB Corporation, Cleveland, OH, USA) and outsourced for sequencing (Macrogen, Seoul, Republic of Korea or University of Notre Dame, Notre Dame, Indiana, USA).
Sequences were viewed and edited in Chromas, embedded in MEGA v.6 software [20] prior to querying GenBank using BLAST. Multiple sequence alignments of the resulting contiguous sequences were performed using ClustalW with default parameters. Maximum likelihood phylogeny was computed using the best fit model of sequence evolution with nodal support for the different groupings evaluated through 1000 bootstrap resampling.
We further confirmed the species identity of Plasmodium-positive mosquito specimens by amplifying and sequencing of ribosomal DNA internal transcribed spacer region 2 (rDNA ITS2) gene [21] and/or the mitochondrial cytochrome c oxidase subunit 1 (cox1) gene [22]. PCR volumes for rDNA ITS2 and cox1 were: 15 μl containing 0.5 μM final concentrations for each primer, 9 μl of PCR water, 3 μl of 5× Hot Firepol Blend Master Mix (Solis BioDyne) and 2 μl of DNA template. Amplification was undertaken with the following cycling parameters: initial denaturation at 95 °C for 15 min, followed by 40 cycles of denaturation at 95 °C for 30 s, 30 s annealing at 60 and 50 °C for rDNA ITS2 and cox1, respectively, and extension at 72 °C for 40 s, and a final extension at 72 °C for 10 min. Thermal reactions were conducted on SimpliAmp Thermal Cycler (Applied Biosystems). The samples were purified as previously described and outsourced for sequencing (DNA Sequencing Facility, University of Illinois at Urbana-Champaign, Champaign, IL, USA). Sequences were similarly cleaned and compared to GenBank database and consensus sequences aligned using ClustalW or MUSCLE in MEGA v.6 for cox1 and ITS, respectively. Maximum likelihood (ML) trees were constructed (1000 bootstraps) with inclusion of reference sequences as outgroups utilizing the GTR+G and Kimura-2 parameter model of sequence evolution for cox1 and ITS gene fragments, respectively, in MEGA v.6.
Blood-meal analyses
To detect blood-meal host sources, cytochrome b (cyt b), 16S ribosomal rRNA and cytochrome c oxidase subunit 1 (cox1) fragments were amplified from genomic DNA extracted from engorged Anopheles mosquitoes using RT-PCR-HRM (Rotor Gene Q thermocycler; Qiagen) and compared to profiles of known positive controls as previously described [23]. Extracted DNA from sugar-fed insectary-reared Anopheles mosquitoes served as negative controls. Known vertebrate DNA used in a previous study, namely cow, Swiss mouse, pig, goat, chicken and human, were included as standard reference positive controls. PCR volumes of 10 μl contained 0.5 μM concentrations of each primer, 6 μl of water, 2 μl of 5× Hot Firepol Evagreen HRM Mix (Solis BioDyne) and 1 μl of DNA template. Amplification for cyt b and 16S rRNA primers was carried out as previously described [23]. High resolution melting profiles generated were analyzed using HRM analysis tools present in the RGQ software. Vertebrate hosts were determined through comparison of the Anopheles mosquitoes’ blood meal HRM melt profiles to those of the standard reference control species.
Statistical analysis
We used Pearson’s Chi-square tests in R v.3.3.1 [24] to evaluate the difference in proportions at an α = 0.05 level of significance. We established the Plasmodium infection rate (IR) as the percentage of the number positive out of the total number examined. Overall proportions of mosquitoes and IR were compared indoors and outdoors using Pearson’s Chi-square test.
Results
We analyzed 1149 specimens of which 87 (7.6%) did not amplify in PCR. Of the 1062 that successfully amplified, 627 specimens originated from indoor sampling and 435 from outdoor sampling. Anopheles funestus was predominant (59.9%) followed by An. rivulorum (32.4%), An. leesoni (4.6%), An. parensis (2.4%), An. longipalpis C (0.7%) and An. vaneedeni (0.1%). Species composition of the An. funestus complex, however, varied between sampling areas, with higher proportions of An. rivulorum collected in coastal sites (Fig. 1). Proportionally, An. funestus dominated indoor collections (476/627; 75.9%), while specimens of An. rivulorum dominated in the outdoor collections, mainly from the coastal area (253/435; 58.2%) (Fig. 2, Table 1).

Relative frequency of species in the Anopheles funestus group, in the different malaria-endemic areas, Kenya. The numbers in parentheses indicate the total sample size of collected mosquitoes per site
Overall, we observed a significantly higher proportion of An. funestus indoors than outdoors (476/636 vs 160/636; χ2 = 156.0, df = 1, P < 0.001), confirming its highly endophilic nature. Likewise, captures of An. leesoni were significantly more frequent indoors than outdoors (40/49 vs 9/49; χ2 = 18.4, df = 1, P < 0.001). In contrast, a much higher proportion of An. rivulorum was trapped outdoors than indoors (253/344 vs 91/344; χ2 = 75.4, df = 1, P < 0.001) (Table 1) suggesting an exophilic tendency for this species. Comparable numbers of An. parensis were encountered indoors (12/25) and outdoors (13/25) (Fig 2, Table 1).
Sequencing of ncMS and/or cox1 gene amplicons indicated Plasmodium infections in 65 specimens, of which 62 shared 100% identity to Plasmodium falciparum [GenBank: CP017005 (ncMS); NC037526 (cox1)], two shared 100% identity to P. ovale [GenBank: AB354571 (ncMS); KP050417 (cox1)] and one shared 100% identity with Plasmodium malariae [MF693428, GQ355486 (cox1)]. These Plasmodium species identifications are supported by the sequence alignment (Additional file 1: Figure S1).
Among cox1 gene (615–658 nt) and/or ITS2 rDNA (267–763 nt) PCR product sequences of specimens found positive for Plasmodium infection, 62 shared 100% identity with An. funestus [GenBank: KJ522832 (cox1); KJ522816 (ITS2)], one shared 100% identity with An. rivulorum [GenBank: KR014839 (cox1)] and two shared 100% identity with An. longipalpis C [GenBank: KR014831, EF095767 (ITS); KR014848 (cox1)]. The mosquito specimen identifications are further supported by ML phylogeny (Fig. 3a, b).

Phylogenetic trees for Plasmodium positive mosquito specimens in the An. funestus group inferred by applying maximum likelihood analysis in MEGA v.6 based on a ITS (267–763 bp), using Kimura-2 model and b cox1 barcode region (615–658 bp) using GTR+G model. Bootstrap support values from 1000 replications are indicated above nodes. Anopheles dthali was included for outgroup purposes. The branch length scale represents substitutions per site. Furthermore, selected sequences of Plasmodium-positive specimens were deposited in GenBank [accession numbers: An. funestus, MH299885-MH299890 (cox1), MH298707-MH298752 (ITS2); An. rivulorum, MH547425 (cox1); An. longipalpis, C MH547426-MH547427 (cox1), MH536653-MH536654 (ITS2)]
Selected sequences of Plasmodium isolates detected by the cox1 gene (369–477 nt) were deposited in the GenBank database under the accession numbers (MH547428-MH547442 (P. falciparum), MH547443 (P. ovale wallickeri) and MH547444 (P. malariae). Furthermore, selected sequences of Plasmodium-positive specimens were deposited in GenBank [accession numbers: An. funestus MH299885-MH299890 (cox1), MH298707-MH298752 (ITS2); An. rivulorum MH547425 (cox1), An. longipalpis C MH547426-MH547427 (cox1), MH536653-MH536654 (ITS2)].
Fifty-nine, two, and one specimens found infected with P. falciparum, P. ovale and P. malariae, respectively, were associated with An. funestus. This translated to a Plasmodium infection rate (IR) of 9.7% (62/636). One P. falciprum infection was associated with An. rivulorum (IR = 0.3%, 1/344) and two with An. longipalpis C (IR = 28.6%, 2/7). The two Plasmodium infected An. longipalpis C were collected indoors (2/7) from Kamnarok (Baringo County), while the infective An. rivulorum was recorded outdoors (1/253) in Jaribuni from coastal Kenya. Plasmodium falciparum infections, especially in An. funestus, occurred in diverse sites: P. ovale from West Alego and Bunyala, Plasmodium malariae from Samia, western Kenya (Fig. 1). Site differences were observed with Plasmodium rates ranging from 3.5% in Bunyala to 50% in Lwanya-Bumala; however, these figures need to be interpreted with caution since sampling dates and seasons were variable, hence direct comparisons could not be made. Notably, Plasmodium infections were more frequently detected in the malaria endemic western Kenya sites than other sites (Table 1).
We analyzed 231 engorged specimens for blood-host sources, comprising 48 collected indoors by PSC/aspiration and 183 outdoors using CDC-light trap. Overall, the human blood index for An. funestus was 68.4% (93/136) and 64.3% (45/70) for An. rivulorum. Human feeds were also encountered for An. leesoni and An. parensis. The two blood-fed An. longipalpis C specimens had each fed on cow and goat. Mixed feeding on humans and other animal hosts and was observed for An. funestus and An. rivulorum, both of which displayed a feeding tendency on a broad host range (Fig. 4).

Host blood meal sources for the sibling species in the Anopheles funestus group, Kenya. The numbers in parentheses indicates total sample size for each species analyzed
Discussion
Most malaria endemic countries including Kenya have embraced the global plan to eliminate the disease through upscaling of vector control measures, particularly with LLINs [25]. However, these measures have impacted vector behaviors, bionomics and vectorial systems [26, 27]. Our data indicate high Plasmodium infection rates in An. funestus, affirming its role as the main vector relative to the other sibling species in the group (reviewed in [5]). Furthermore, we provide the first report of An. longipalpis C for Kenya. This species, known for its zoophagic but endophilic tendencies has never been associated with Plasmodium transmission [28]. However, we found two out of seven specimens P. falciparum-positive, indicating that feeding on humans had taken place. Although recorded in low numbers, its potential role as secondary vector in the more arid areas of Kenya should be further investigated.
We uncovered a higher species richness among the An. funestus group than previously documented in Kenya. Furthermore, sympatric occurrence of more than three sibling species in a locality as observed in this study, highlights the importance of integrating molecular approaches in surveillance efforts for adequate assessment of the impact of interventions on vector populations [29]. A lack of amplification of some specimens calls for further improvement of existing protocols to adequately discriminate and identify An. funestus group members.
We found considerable site differences in Plasmodium infection rates in An. funestus ranging from 3.5% in Bunyala to 50% in Lwanya-Bumala (Table 1). However, these figures should be interpreted with caution since sampling dates and seasons were variable, hence direct comparisons could not be made. Notably, Plasmodium infections were more frequently detected in this species in the malaria endemic western Kenya sites compared to coastal sites. Such site-to-site variation in infection rates is well recognized and may result from differences among populations and their impact on malaria transmission in different ecologies. Differences have been observed at the genetic level between coastal and western populations of An. funestus [30] although whether this pattern potentially impacts differentially on transmission has not been assessed. Nonetheless, the high infection rates observed in western Kenya where malaria persists despite indoor vector control [2] warrants further research.
Residual transmission has emerged as a major threat to effective malaria control, as key vectors alter their phenotypes to render current proven vector control interventions such as LLINs less effective. Examples include exhibition of behavioral plasticity in resting and feeding patterns. We found a rather low human blood index (64.8%) for An. funestus compared to the near exclusive human feeding habit previously noted for this species [6, 12, 31]. Although variation in this pattern by site or ecology cannot be discounted, the data indicates a certain degree of zoophily and an unusual behavioral divergence in its feeding habits supported by feeding on a broad host-range. Its outdoor behavior might reduce the impact of LLINs on the species and hence contribute to residual transmission. This assertion is supported by its growing prominence in malaria transmission in recent times in Kenya [32] following the dwindling importance of Anopheles gambiae (s.s.) at large in East Africa [32], which calls for further research into its adaptive biology and role in the disease persistence and even resurgence.
Species records are based on sampling efforts, which in our study was limited in scope (i.e. intensity and duration of trapping in each location) and targeted the rainy seasons only. Seasonal occurrence including species succession has been shown to influence changes in sibling species composition [33]. Studies over longer periods covering seasonal scales need to be encouraged to establish the full profile of sibling species in this group in the context of persistent malaria transmission in Kenya and elsewhere.
Our findings underscore the need for improved surveillance of vector populations employing molecular techniques including sequencing to improve malaria vector control and working towards malaria elimination. This is essential for fine-tuning control strategies in the event of changing local conditions [3] and for discerning cryptic vectors that may contribute to stealth transmission.
Conclusions
Our findings have revealed a higher species richness among the An. funestus group than previously documented in Kenya. Notably is the occurrence of An. longipalpis C and for the first time, naturally infected with Plasmodium falciparum. Anopheles funestus remains the main vector species in the Funestus group in Kenya which was found to exhibit a rather low human blood index. Our findings underscore the importance of active surveillance through application of molecular approaches to unravel novel parasite-vector associations possibly contributed by cryptic species with important implications for effective malaria control and elimination.
Abbreviations
- ASP:
-
Aspiration
- CDC:
-
Centers for Disease Control and Prevention
- COX1:
-
Cytochrome c oxidase subunit 1
- HRM:
-
High resolution melting
- LLIN:
-
Long-lasting insecticidal net
- PCR:
-
Polymerase chain reaction
- PSC:
-
Pyrethrum spray catches
- s.l:
-
sensu lato
- s.s:
-
sensu stricto
- SSA:
-
sub-Saharan Africa
References
Bhatt S, Weiss D, Cameron E, Bisanzio D, Mappin B, Dalrymple U, et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature. 2015;526:207.
Obala AA, Mangeni JN, Platt A, Aswa D, Abel L, Namae J, et al. What is threatening the effectiveness of insecticide-treated bednets? A case-control study of environmental, behavioral, and physical factors associated with prevention failure. PLoS One. 2015;10:e0132778.
Castro M. Malaria transmission and prospects for malaria eradication: the role of the environment. Col Spring Harb Perspect Med. 2017;7:1–12.
Tchouassi DP, Quakyi IA, Addison EA, Bosompem KM, Wilson MD, Appawu MA, et al. Characterization of malaria transmission by vector populations for improved interventions during the dry season in the Kpone-on-Sea area of coastal Ghana. Parasit Vectors. 2012;5:212.
Dia I, Guelbeogo MW, Ayala D. Advances and perspectives in the study of the malaria mosquito Anopheles funestus. In: Manguin S, editor. Anopheles Mosquitoes. New Insights into Malaria Vectors. London: Intech; 2013. p. 828.
Muturi EJ, Kamau L, Jacob BG, Muriu S, Mbogo CM, Shililu J, et al. Spatial distribution, blood feeding pattern, and role of Anopheles funestus complex in malaria transmission in central Kenya. Parasitol Res. 2009;105:1041–6.
Burke A, Dandalo L, Munhenga G, Dahan-Moss Y, Mbokazi F, Ngxongo S, et al. A new malaria vector mosquito in South Africa. Sci Rep. 2017;7:43779.
Stevenson J, Laurent BS, Lobo NF, Cooke MK, Kahindi SC, Oriango RM, et al. Novel vectors of malaria parasite in the western highlands of Kenya. Emerg Infect Dis. 2012;18:1547.
Laurent BS, Cooke M, Krishnankutty SM, Asih P, Mueller JD, Kahindi S, et al. Molecular characterization reveals diverse and unknown malaria vectors in the western Kenyan highlands. Am J Trop Med Hyg. 2016;94:327–35.
Kamau LMG, Koekemoer LL, Hunt RH. Coetzee M. A survey of the Anopheles funestus (Diptera: Culicidae) group of mosquitoes from 10 sites in Kenya with special emphasis on population genetic structure based on chromosomal inversion karyotypes. J Med Entomol. 2003;40:664–71.
Shililu JI, Mbogo CM, Mutero CM, Gunter JT, Swalm C, Regens JL, et al. Spatial distribution of Anopheles gambiae and Anopheles funestus and malaria transmission in Suba District, western Kenya. Int J Trop Insect Sci. 2003;23:187–96.
Mwangangi JM, Mbogo CM, Nzovu JG, Githure JI, Yan G, Beier JC. Blood-meal analysis for anopheline mosquitoes sampled along the Kenyan coast. J Am Mosq Contr Assoc. 2003;19:371–5.
Gillies M, Smith A. The effect of a residual house-spraying campaign in East Africa on species balance in the Anopheles funestus group. The replacement of A. funestus Giles by An. rivulorum Leeson. Bull Entomol Res. 1960;51:243–52.
Kawada H, Dida GO, Sonye G, Njenga SM, Mwandawiro C, Minakawa N. Reconsideration of Anopheles rivulorum as a vector of Plasmodium falciparum in western Kenya: some evidence from biting time, blood preference, sporozoite positive rate, and pyrethroid resistance. Parasit Vectors. 2012;5:230.
Noor AM, Kinyoki DK, Mundia CW, Kabaria CW, Mutua JW, Alegana VA, et al. The changing risk of Plasmodium falciparum malaria infection in Africa: 2000–10: a spatial and temporal analysis of transmission intensity. Lancet. 2014;383:1739–47.
Koekemoer L, Kamau L, Hunt R, Coetzee M. A cocktail polymerase chain reaction assay to identify members of the Anopheles funestus (Diptera: Culicidae) group. Am J Trop Med Hyg. 2002;66:804–11.
Cohuet A, Simard F, Toto J, Kengne P, Coetzee M, Fontenille D. Species identification within the Anopheles funestus group of malaria vectors in Cameroon and evidence for a new species. Am J Trop Med Hyg. 2003;69:200–5.
Chaumeau V, Andolina C, Fustec B, Ndam NT, Brengues C, Herder S, et al. Comparison of the performances of five primer sets for the detection and quantification of Plasmodium in anopheline vectors by real-time PCR. PLoS One. 2016;11:e0159160.
Echeverry DF, Deason NA, Makuru V, Davidson J, Xiao H, Niedbalski J, et al. Fast and robust single PCR for Plasmodium sporozoite detection in mosquitoes using the cytochrome oxidase I gene. Malar J. 2017;16:230.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Beebe NW, Saul A. Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction-restriction fragment length polymorphism analysis. Am J Trop Med Hyg. 1995;53:478–81.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Ogola E, Villinger J, Mabuka D, Omondi D, Orindi B, Mutunga J, et al. Composition of Anopheles mosquitoes, their blood-meal hosts, and Plasmodium falciparum infection rates in three islands with disparate bed net coverage in Lake Victoria, Kenya. Malar J. 2017;16:360.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. http://www.R-project.org/.
President’s Malaria Initiative Kenya. Malaria Operational Plan Fy 2017. USA: US Department of State, US Agency for International Development, Centers for Disease Control and Prevention; 2017.
Mwangangi JM, Mbogo CM, Orindi BO, Muturi EJ, Midega JT, Nzovu J, et al. Shifts in malaria vector species composition and transmission dynamics along the Kenyan coast over the past 20 years. Malar J. 2013;12:13.
Conn JE, Norris DE, Donnelly MJ, Beebe NW, Burkot TR, Coulibaly MB, et al. Entomological monitoring and evaluation: diverse transmission settings of ICEMR projects will require local and regional malaria elimination strategies. Am J Trop Med Hyg. 2015;93(Suppl. 3):28–41.
Kent R, Coetzee M, Mharakurwa S, Norris D. Feeding and indoor resting behaviour of the mosquito Anopheles longipalpis in an area of hyperendemic malaria transmission in southern Zambia. Med Vet Entomol. 2006;20:459–63.
Coetzee M, Koekemoer LL. Molecular systematics and insecticide resistance in the major African malaria vector Anopheles funestus. Annu Rev Entomol. 2013;58:393–412.
Braginets OP, Minakawa N, Mbogo CM, Yan G. Population genetic structure of the African malaria mosquito Anopheles funestus in Kenya. Am J Trop Med Hyg. 2003;69:303–8.
Dadzie SK, Brenyah R, Appawu MA. Role of species composition in malaria transmission by the Anopheles funestus group (Diptera: Culicidae) in Ghana. J Vector Ecol. 2013;38:105–10.
National Malaria Control Programme (NMCP) Kenya National Bureau of Statistics (KNBS), and ICF International. Kenya Malaria Indicator Survey 2015. Nairobi; Rockville: NMCP, KNBS, and ICF International; 2016. p. 165.
Njoroge MM, Tirados I, Lindsay SW, Vale GA, Torr SJ, Fillinger U. Exploring the potential of using cattle for malaria vector surveillance and control: a pilot study in western Kenya. Parasit Vectors. 2017;10:18.
Acknowledgements
We thank Dr Joseph Mwangangi and Ms Julia Wanjiru for contributing samples from Taita-Taveta and Ahero, and Jackson Kimani for design of the map used in this study. Special thanks to Nixon Onyimbo for support with field mosquito collection and for the community members in the villages who provided access to their homesteads.
Funding
The study was funded by a grant from the Foundation for the National Institutes of Health through the Vector-Based Transmission of Control: Discovery Research (VCTR) programme of the Grand Challenges in Global Health Initiative. Additionally, we gratefully acknowledge the financial support for this research by the following organizations and agencies: UK Department for International Development (DFID), Swedish International Development Cooperation Agency (Sida), Swiss Agency for Development and Cooperation (SDC), and the Kenyan Government. The views expressed herein do not necessarily reflect the official opinion of the donors.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional file. The raw data can be obtained from the corresponding author upon reasonable request. Selected sequences of Plasmodium isolates detected by cox1 gene (369–477 nt) have been deposited in GenBank (accession numbers: P. falciparum, MH547428-MH547442; P. ovale wallickeri, MH547443; and P. malariae, MH547444). Furthermore, selected sequences of Plasmodium-positive specimens were deposited in GenBank [accession numbers: An. funestus, MH299885-MH299890 (cox1), MH298707-MH298752 (ITS2); An. rivulorum, MH547425 (cox1); An. longipalpis, C MH547426-MH547427 (cox1), MH536653-MH536654 (ITS2)].
Author information
Authors and Affiliations
Contributions
UF, DKM, BT and DPT conceived the experiment(s). EOO, IMO and DPT conducted the experiment(s). EOO, JV and DPT analyzed the results. DPT wrote the draft manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We acquired ethical approval for mosquito trapping from the Scientific and Ethical Review Unit (SERU) of the Kenya Medical Research Institute (Non-SSC Protocol #388 and Non-SSC Protocol #310). We sought informed consent to trap mosquitoes in and around homesteads from village elders and household heads.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Sequence alignment of 162 nucleotide (nt) Plasmodium ncMS sequences corresponding to nucleotide positions 5759–5920 of GenBank accession CP017005. Study sequences are highlighted in bold and GenBank accession numbers and species identifications are indicated for each ncMS gene sequence. In sequence alignment, red = A, green = T, yellow = G, blue = C. grey = nt identity with first sequence. (TIF 503 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ogola, E.O., Fillinger, U., Ondiba, I.M. et al. Insights into malaria transmission among Anopheles funestus mosquitoes, Kenya. Parasites Vectors 11, 577 (2018). https://doi.org/10.1186/s13071-018-3171-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3171-3