
Abstract
As one of most common and severe mental disorders, major depressive disorder (MDD) significantly increases the risks of premature death and other medical conditions for patients. Neuroinflammation is the abnormal immune response in the brain, and its correlation with MDD is receiving increasing attention. Neuroinflammation has been reported to be involved in MDD through distinct neurobiological mechanisms, among which the dysregulation of neurogenesis in the dentate gyrus (DG) of the hippocampus (HPC) is receiving increasing attention. The DG of the hippocampus is one of two niches for neurogenesis in the adult mammalian brain, and neurotrophic factors are fundamental regulators of this neurogenesis process. The reported cell types involved in mediating neuroinflammation include microglia, astrocytes, oligodendrocytes, meningeal leukocytes, and peripheral immune cells which selectively penetrate the blood–brain barrier and infiltrate into inflammatory regions. This review summarizes the functions of the hippocampus affected by neuroinflammation during MDD progression and the corresponding influences on the memory of MDD patients and model animals.
Similar content being viewed by others


Introduction
Characterized by depressed mood, persistent anxiety, loss of interest in daily activities, and even suicidal thoughts, major depressive disorder (MDD) has been ranked as the third leading cause of the disease burden worldwide, especially for the elderly population, with a reported average incidence of 13.3% in 2021 [1]. MDD stands out among psychiatric disorders for that patients are at significantly increased risk of premature death by suicide and other medical conditions, such as sleep disorders or cardiovascular diseases [2, 3]. The neurobiology of MDD can be categorized into three major aspects: the reduced serotonin neurotransmission, the dysfunctional hypothalamus–pituitary–adrenal (HPA) axis, and the impaired neurogenesis in the dentate gyrus of the adult hippocampus [4,5,6]. To further explore the molecular and cellular mechanisms of MDD development and potential treatment, several types of animal models have been established and are generally recognized as sufficient to mimic the behavioral characteristics of MDD patients, including learned helplessness (LH), chronic unpredictable mild stress (CUMS), and repeated social defeat (RSD) [7,8,9,10].
In both MDD animal models and patients, various types of cognitive deficits have been widely observed, such as attenuated memory performance [11, 12]. Short-term and long-term memory have been manifested to involve different neural systems [13]. The well-studied case of Henry Molaison, in which the patient showed memory impairment after the bilateral removal of the hippocampus, pioneered the exploration of memory formation by providing the first clear correlation between memory and the hippocampus [14]. To date, the structures of the medial temporal lobe, including the hippocampus, have been verified to be responsible for episodic long-term memory processing [15] and defined as the medial temporal lobe memory system [16]. Therefore, the conditions of the hippocampus during MDD progression require our attention.
Initially referred to as the inflammatory functions of microglia under pathological conditions, the definition “neuroinflammation” is now used ubiquitously to describe all immunological activities within the central nervous system (CNS) toward acute and chronic diseases, including traumatic, infectious, ischemic, autoimmune, and degenerative ones [17]. The previous theory of “brain immune privilege”, which postulates that the CNS could tolerate the introduction of antigens and thus limit any excessive inflammatory damage, was based on the absence of any lymphatic drainage and the blood–brain barrier (BBB) [18]. However, recent discoveries have improved the general understanding of immune surveillance of the CNS and remotivated discussions about the soundness of “brain immune privilege” [19]. In healthy brains, various types of lymphocytes and myeloid cells have been found to be located in the meninges, cerebrospinal fluid (CSF), and parenchyma [20, 21]. The current perception of neuroinflammation includes three major categories: microglia and resident macrophages in the CNS mediate innate immune responses [22], more peripheral immune cells migrate and infiltrate into the CNS during neuroinflammation [23], and infiltrated T cells may play an important role in adaptive immunity [24,25,26].
The relationships between neuroinflammation and neurodegenerative disorders have been gradually elucidated [27, 28]. In specific, multiple psychosocial stressors have been demonstrated to accelerate the development of neuroinflammation and mental illness according to the clinical evidence [29]. Ongoing neuroinflammation could in turn induce depressive-like behaviors [30, 31] or promote MDD progression [32]. Currently available antidepressants eliminate depressive symptoms for only 37% of MDD patients [33], together with an overall cumulative remission rate of 67% [33]. These facts suggest the potential advantages of considering immune-regulatory mechanisms in the development of MDD treatment, compared to the current first-line antidepressants and their focus on inhibiting neurotransmitter reuptake or sensitizing their receptors. Moreover, the abilities of these antidepressants to ameliorate cognitive deficits in patients suffering from MDD, especially memory impairments, have not been evaluated sufficiently during their development [34]. Since the neuroinflammatory dynamics observed in MDD animal models have been reported to regulate memory impairments [35, 36] and hippocampal neurogenesis [37, 38], this review intended to summarize the links and interactions between neuroinflammation and cognitive deficits which are correlated with the proliferation and functions of neurons in the hippocampus.
The effects of neuroinflammation on memory impairment via the modulation of hippocampal neurogenesis
Neurogenesis in the hippocampus
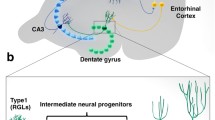
Among the different regions in the cerebral parenchyma, the hippocampus is predominantly associated with memory functions. Anatomically, the mammalian hippocampus consists of the dentate gyrus (DG), subiculum, and cornu ammonis (CA) areas 1–3 [39]. Neurogenesis is the process by which neural stem cells (NSCs) gradually differentiate into mature neurons [40]. Neurotrophins (NTs) serve as the essential and irreplaceable mediators during this process [41]. The DG is one of two recognized microenvironments/niches for neurogenesis in adult mammalian brains, which refers to the sub-granular zone (SGZ) of the DG and the border of the lateral ventricles of the brain (sub-ventricular zone, SVZ) [42]. The DG can be divided into three distinct layers: the molecular layer (ML) containing mainly the dendritic projections of granule cells (GCs), the granular cell layer (GCL) containing the cell bodies of GCs, and the polymorphic layer named the hilus [43]. The thin layer of cells forming the border of both the GCL and hilus is defined as the SGZ, where NSCs are located together with other cell types, such as neural progenitor cells (NPCs), GCs, interneurons, mossy cells, astrocytes, oligodendrocytes, and microglia [44, 45]. Activated NSCs in the DG differentiate into oligodendrocytes, astrocytes, and mature neurons through distinct differentiation pathways [46]. The proliferation, differentiation, and survival of these NSCs are mediated by various factors within the SGZ microenvironment [47], such as neurotransmitters, growth factors, cytokines, and hormones secreted from other cell types [48, 49].
The prolonged capacity of NSCs in the SGZ to self-renew, retain cellular identity, and react to specific signals is fundamental for mammals to ensure life-long neurogenesis in the DG [50]. The differentiation from NSCs into mature neurons includes a series of sequential and time-consuming processes [51]. Type 1 NSCs, also named radial glial cells for their radial morphology, are mainly located in the SGZ [52]. Newborn type 1 NSCs first transdifferentiate into transient amplifying progenitor cells (TAPCs), which exhibit limited self-renewal ability and a higher proliferation rate than NSCs and can be further divided into subtypes 2a and 2b [53]. Type 2b TAPCs continue to differentiate to generate neuroblasts (NBs)/type 3 cells and migrate to the inner half of the GCL [54]. The process from type 1 NSCs to type 3 NBs normally lasts 1–4 days, during which 60% of newborn NPCs undergo cell death by apoptosis [55]. Type 3 NBs go through another 5–27 days of differentiation into immature neurons [56] and subsequently 28–60 days into mature neurons with integrated synaptic structure [57]. That is, the entire process of neurogenesis in the adult mammalian brains takes 5–11 weeks after the birth of NSCs, which ensures the persistent maturation of fully functional neurons throughout the life circle [58]. Therefore, developments in techniques and equipment have been continually focusing on evaluating neurogenesis more precisely for both animal models [59] and human patients [60].
Neuroinflammation inhibits hippocampal neurogenesis during depression
In both animal models and human patients, the participation of neuroinflammation during MDD progression has been increasingly recognized. For example, Haapakoski et al. [61] discovered strong correlations between depression and the upregulation of peripheral markers of inflammation in CSF samples, such as interleukin-6 (IL-6) and C-reactive protein (CRP). In addition, interferon-α (IFN-α) in the brain parenchyma has been reported by Su et al. to be interrelated with the pathogenesis of MDD [62]. In a meta-analysis, the clinical use of various anti-inflammatory drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs), cytokine inhibitors, statins, and minocycline, significantly ameliorated depressive symptoms in some MDD patients [63]. In mice with depressive-like behaviors, elevated transcription of proinflammatory and injury-repair genes was observed in hippocampal tissues [64].
MDD-induced neuroinflammation has been reported to affect cognitive and memory functions by affecting neurogenesis in the DG (Fig. 1A, B; the corresponding experimental methods were described in Additional file 1), leading to inattention, impaired working memory, and increased negative cognitive bias [65, 66]. Mckim et al. [67] reported that RSD promoted neuroinflammation (mainly through microglial modulation) in the mouse hippocampus, resulting in transient spatial memory impairment. Although the cell number and proliferation rate of NPCs were not affected by RSD, their differentiation into mature neurons was significantly inhibited. Correspondingly, minocycline, an inhibitor of microglial differentiation into proinflammatory phenotype, was found to penetrate the BBB into the CNS and suppress the recruitment and infiltration of peripheral lymphocytes into the hippocampus during RSD [68], thereby attenuating spatial memory impairment after RSD by inhibiting neuroinflammation [36].

Microglial activation and attenuated hippocampal neurogenesis in DG were observed in depression model of mice. A Immunostaining with anti-Dcx and anti-NeuN of hippocampal coronal sections of mice under CUMS treatment. B The statistical analysis of Dcx+ cells located at the SGZ. The mice received CUMS treatment showed significantly decreased newborn, immature neurons as indicated by anti-Dcx staining (***p < 0.001 vs. Ctrl). C Immunostaining with anti-Iba1 of hippocampal coronal sections of mice under CUMS treatment. The mice received CUMS treatment showed increased Iba1+ signals in the DG, which might suggest increased numbers of reactive microglia. The experimental details are described in Additional file 1
Previous studies have shown that exercise serves as a powerful inducer of hippocampal neurogenesis, and the benefits of regular exercise on cognitive function might be attributed to its proneurogenic effects [69]. In addition, in the DG with improved neurogenesis, newborn mature neurons also exhibit enhanced synaptic plasticity [70] and long-term potentiation (LTP) [71]. In 1999, Gage et al. found [72] that voluntary running increased the proliferation and survival of new neurons in the mouse DG. In subsequent studies, it was further demonstrated that long-term voluntary running in mice not only augmented hippocampal neurogenesis, but also enhanced the spatial learning ability of mice [73]. This exercise-induced hippocampal neurogenesis has been shown to be necessary to improve spatial memory functions in rodents [74] and to be limited to the hippocampus, which means that neurogenesis in the SVZ is not affected [75]. In addition, exercise-induced hippocampal neurogenesis is accompanied by increased LTP, dendritic spine density, and reactivity to NTs, together with increased fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF) expression levels [76, 77]. The interplay between exercise and cognitive functions influenced by neuroinflammation, however, was investigated much later. Wu et al. [78] demonstrated that lipopolysaccharide (LPS)-induced neurogenesis deficits and impaired Morris water maze (MWM) test performance in mouse models could be gradually rescued during the subsequent 5 weeks of forced treadmill training. Nevertheless, negative results were also observed in subsequent studies [79], which was explained by the authors’ hypothesis that exercise interventions might miss the time window of the influence of neuroinflammation on neurogenesis. For human subjects, similar effects of exercise on regulating neuroinflammation, promoting cognitive functions, and maintaining the structural integrities of the corresponding brain regions have also been demonstrated, but the existing evidence is not sufficiently informative as research at the animal level and needs to be further studied [80, 81].
Neurotrophins and hippocampal neurogenesis
Neurotrophin definition and functions in neurogenesis
The understanding of NTs started from the discovery of nerve growth factor (NGF) [82]. NTs are growth factors that play irreplaceable roles in neuronal proliferation, survival, development, function, and plasticity [83]. To date, the discovered NTs could be listed as follows: NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3/4 (NT-3/4; they are similar in gene sequences and protein structures due to the same ancestral gene), neurotrophin-5 (NT-5), and neurotrophin-6 (NT-6) [84]. Other cytokines, including leukemia inhibitory factor (LIF), insulin-like growth factor-1 (IGF-1), transforming growth factor (TGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) [85], have also been detected in the CNS and reported to modulate neurogenesis, but they are not defined as NTs. Both neurons and neuroglial cells have been shown to synthesize and secrete NTs, which in turn act on all these cell types within the CNS.
Among NTs, BDNF is considered to exert the most significant and extensive neurotrophic effects; therefore, BDNF is also the most extensively studied NT in healthy and diseased brains [86,87,88]. The expression and maturation of BDNF protein is a multistage process. The original BDNF protein is synthesized in the endoplasmic reticulum and folded into pre-pro-BDNF [89]. After pre-pro-BDNF is translocated into the Golgi apparatus, the precursor signal sequence is cleaved to produce the pro-BDNF isoform [90]. Then, pro-BDNF is further cleaved to form mature BDNF (mBDNF), which has full physiological functions [91]. There has been substantial evidence indicating that BDNF is fundamental in the effectuation of LTP, mediating both functional and structural plasticity in the CNS, and BDNF has been discovered to modulate neuronal survival and neurogenesis in human hippocampus [92, 93]. Accordingly, patients with neurodegenerative diseases tend to have lower BDNF concentrations detected in their blood and brain [94, 95]. Some researchers hypothesized that since neuroinflammation is known to affect several BDNF-related signaling pathways, abnormal BDNF levels in diseased brains might be due to the chronic inflammatory state [96, 97]. The current study of BDNF’s effects is largely focused on the effects of antidepressant treatment on BDNF-mediated neuronal plasticity [98].
Neurotrophin receptor signaling pathways
BDNF signaling in receptor cells is generally categorized into receptor tyrosine kinase B (TrkB) pathways and the relatively low-affinity p75 neurotrophin receptor (p75NTR) pathways [96]. BDNF-mediated neuronal survival signaling can be summarized as follows [92]: released mature dimeric BDNF binds to presynaptic and postsynaptic TrkB in paracrine or autocrine manners, which results in the autophosphorylation and activation of TrkB. Consequently, several signaling pathways including phospholipase C-γ (PLC-γ), phosphoinositide 3-kinase (PI3K)-AKT, and extracellular signal-regulated kinase (ERK) are activated. First, BDNF-dependent PLC-γ activation can increase the Ca2+ neuronal response by activating downstream inositol triphosphate (IP3) or protein kinase C (PKC) cascades; meanwhile, the TrkB–PLC-γ–PKC axis decreases the inflammation-related apoptosis cascade by inhibiting glycogen synthase kinase-3β (GSK-3β). Second, the PI3K–AKT axis can induce de novo BDNF expression by activating mTOR-dependent BDNF translation [99, 100]. Third, BDNF can activate the transcription factor cyclic AMP response-binding protein (CREB) by inducing the ERK pathway, which regulates the expression of genes mediating neuronal survival, growth, and LTP. On the other hand, even though both pro-BDNF and mBDNF can bind p75NTR which may trigger nuclear factor-kappa B (NF-κB) signaling, its overall neurotrophic effects remain controversial and currently have no clear evidence to reach a conclusion [101].
During neuroinflammation, BDNF might regulate NF-κB-dependent proinflammatory gene expression through yet unclear mechanisms. Upregulated proinflammatory cytokines directly induce the production of various neurotoxic factors, such as reactive oxygen species (ROS), and more proinflammatory cytokines like IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and chemokines [102, 103]. Mature BDNF is released into the synapse gaps from either presynaptic boutons or postsynaptic dendritic sites. Cis- or trans-activation of TrkB by BDNF plays different roles in synaptogenesis, Hebbian plasticity, and homeostatic plasticity. For example, TrkB activation at presynaptic sites contributes to presynaptic terminal formation whereas TrkB activation at postsynaptic sites promotes the formation of postsynaptic specializations [87].
Neurotrophin levels and neurogenesis during depression
In recent years, researchers have shown great interest in the measurement of peripheral BDNF levels associated with various mental diseases. Since significant reductions in serum BDNF levels were verified in both MDD animal models and patients [104, 105], it has been defined as a potential biomarker for depression assessment in some studies [106, 107]. For example, ameliorated depressive symptoms are in accordance with increased serum BDNF levels in MDD patients treated with effective antidepressants [108]. Treatment-resistant depression (TRD) patients tend to exhibit lower BDNF levels in corresponding brain structures such as the hippocampus [109]. Despite these facts, the time-course correlation between BDNF levels and antidepressant effects seems to be indirect: drugs and electroconvulsive therapies gradually increase serum BDNF levels in MDD patients, while their antidepressant effects appear quickly [110]. Moreover, it is noteworthy that the MDD-modulated BDNF levels were reported by Karege et al. to be initiated from reduced BDNF concentrations in the CNS rather than released from platelets [111]. Accordingly, synchronism between the release of BDNF in the CNS and serum has been recorded [112], while the relationship between serum BDNF levels and platelet-derived BDNF remains to be further investigated [113]. Also, the decreased serum BDNF levels measured in MDD patients were observed to synchronize with upregulation of the proinflammatory cytokines such as IL-6 and IL-8 [114, 115]. Altogether, neuroinflammation under chronically stressful conditions usually leads to reduced neurotrophin levels and thus inhibited neurogenesis during the course of depression, which can be partially mitigated with antidepressant treatments.
On the other hand, however, cell-to-cell communications within the CNS microenvironment could be more complicated. For instance, based on spatially patterned antibody microchips, the mediators of neuron–immune interactions were categorized by Deng et al. into 12 types of secretomes, including cytokines, NTs, and neuron-derived exosomes [116]. They also found that pairwise neuron–macrophage interactions and neuron–microglia interactions in AD models exerted distinct effects on modulating immune responses and neuron secretion [116], which highlighted the heterogeneity of secretion from single cells within the CNS under pathological conditions.
Neuroinflammation attenuates neurogenesis
Microglial differentiation during brain innate immunity
Microglia are CNS-specific immune cells that affect the development and maintenance of the neural environment as well as the response and repair to injury [117]. Transmembrane protein 119 has been identified as the unique biomarker to distinguish microglia from peripheral monocyte-derived macrophages [118]. Similar to peripheral macrophages, microglia exhibit diverse phenotypes under different conditions to maintain homeostasis of the CNS through phenotypic transformation. For example, microglia can transdifferentiate into the M1 (proinflammatory) phenotype that upregulates the inflammatory response by expressing proinflammatory cytokines, or into the M2 (immunoregulatory) phenotype that inhibits inflammation and regulates tissue repair [119]. In vitro, cultured microglia have been shown to be induced by LPS or interferon-γ (IFN-γ) to differentiate to the M1 (proinflammatory) phenotype and to be induced by IL-4 or IL-13 to differentiate to the M2 (immunoregulatory) phenotype in previous studies [120]. In addition, microglia with the proinflammatory phenotype can enhance neuroinflammation responses within lesion areas through various mechanisms (see below), including recruiting peripheral monocytes and activating astrocytes [121, 122]. Various studies have shown that inhibiting the reactivity of microglia by reversing the differentiation direction leads to improved neurogenesis and cognitive performance in animal models by inhibiting neuroinflammation [123, 124]. However, it is noteworthy that summarizing the morphological and functional diversities of microglia into a binary opposition of M1 and M2 phenotypes by past studies is oversimplified. More recent studies revealed the indispensable roles of microglia in maintaining brain immune homeostasis and regulating neuronal activity under physiological conditions [125]. In addition, the M1 or M2 phenotype designation is not sufficient to summarize microglial functions under chronic stress [17]; for example, immunologically reactive microglia were found to mediate tissue damage and repair processes simultaneously [126, 127], which reflects the complexity of neuroimmune systems.
The proinflammatory roles of microglial cells in depression
Microglial differentiation into the proinflammatory phenotype has long been recognized to be secondary to neuronal injuries [17]. To date, several biomarkers for the proinflammatory phenotype have been identified [118], including Iba-1, CD11b, CD68, HLA-DR, etc. According to Farooq et al., CD11b expression is upregulated in MDD-related encephalic regions including the hippocampus and amygdala of CUMS mice [128]. Later, they also discovered that CD11b upregulation could be partially reversed by the administration of the antidepressant fluoxetine, which is consistent with the alleviation of CUMS-induced behavioral disorders [129]. Similarly, Preez et al. modified the CUMS model by adding a subsequent 6 weeks of social isolation (referred to as UCMSI). This procedure led to increased microglial proinflammatory differentiation and astrocytosis in the DG of the hippocampus, along with inhibited DG neurogenesis and several depressive-like behavioral changes in UCMSI animals [130]. These results uniformly suggest the critical role of microglial cells in mediating neuroinflammation during MDD progression (Fig. 1C; the corresponding experimental methods were described in Additional file 1). Moreover, abnormal levels of various bioactive elements within the CNS have also been shown to be involved in the regulation of microglia-mediated innate neuroinflammation. Mao et al. discovered the downregulation of miR-195 in the hippocampus during the chronic brain hypoperfusion process. miR-195 alters the communication between neurons and microglia by regulating CX3CL1–CX3CR1 signaling. miR-195 overexpression inhibited the chronic brain hypoperfusion-induced microglial differentiation to the proinflammatory phenotype, manifested by increased proportions of CD206+/Iba-1+ signals in hippocampal microglia [131]. Microglial proinflammatory differentiation is also the main cause of perioperative neurocognitive disorders. Luo et al. discovered through metabolomics that the glycolytic inhibitor 2-deoxy-d-glucose negatively regulates the proinflammatory differentiation of microglia and thereby inhibiting neuroinflammation in the hippocampus, alleviating the cognitive impairment caused by perioperative neurocognitive disorders, which is indicated by the decreased CD86+CD206− ratio of microglia in the hippocampus [132].
Based on the effects of microglia described above, it is reasonable that some anti-inflammatory medicines, such as minocycline, have been reported to exert profound neuroprotective effects by diminishing microglial proinflammatory differentiation in the hippocampus, which is correlated with accelerated DG neurogenesis and better spatial memory under neuroinflammatory conditions [63, 133,134,135]. For example, in a rat model of cognitive impairment caused by sleep deprivation-induced neuroinflammation, the introduction of minocycline treatment was observed to improve the attenuated neurogenesis during sleep deprivation by inhibiting microglial proinflammatory differentiation and reducing the levels of microglia-released proinflammatory cytokines, which profoundly reduced the impairment of spatial memory [124]. As for the clinical application of minocycline, various trials have been conducted with depressed patients, in which minocycline showed promising potential to attenuate depressive symptoms in patients and improve their responses to antidepressants, even for TRD patients, while its long-term effects with large number of subjects remained ambiguous [136]. Taken together, microglial proinflammatory differentiation and the corresponding neuroinflammatory responses in the hippocampus during depression could be reversed by anti-inflammatory medicines, thereby facilitating hippocampal neurogenesis.
Microglial cells and P2X7-NLRP3 inflammasome signaling
Among different inflammasome cascades, the NLRP3 inflammasome signaling has received increasing attention in the study of brain innate immunity because of its relatively high expression in microglia [4]. Inside microglia, NLRP3 inflammasome activation was reported to be triggered by the purinergic P2X7 receptor bound to adenosine triphosphate (ATP), which induces potassium efflux to activate the downstream intracellular signaling [137]. Defined as “sterile inflammation” of the brain, this P2X7–NLRP3 inflammasome cascade in microglia is independent of the recognition of pathogen-associated molecular patterns [137, 138] and plays an important role in the pathophysiology of depression. In clinical observations, single nonsynonymous nucleotide polymorphisms in the human P2X7 receptor gene have been reported to be correlated with the risk of MDD aggravation in patients with affective mood disorders [139]. Furthermore, P2X7 receptor knockout in mice prevented the development of depressive-like symptoms under LPS treatment [140].
The NLRP3 inflammasome activates caspase-1, which subsequently cleaves pro-IL-1β and pro-IL-18 into their active, proinflammatory and releasable forms [141]. Under the steady state, IL-1β release induced by the microglia-related NLRP3 inflammasome was considered contributive to neuroprotection in excitotoxin-damaged mouse retinas [142]. However, IL-1β at high levels could be excitotoxic and hinder synaptic transmission [143]. For instance, hippocampal NLRP3 and IL-1β upregulation was detected in LPS-stimulated mice with depressive-like behaviors, and the subsequent ketamine treatment was found to exert the antidepressant effects via suppressing NLRP3 inflammasome signaling in the hippocampus [144]. Outside the CNS, the IL-1β upregulation induced by the NLRP3 inflammasome has been demonstrated to direct the differentiation of naïve CD4+ T cells into T helper 17 cells, which is known to promote the BBB permeability of peripheral immunocytes by weakening tight junctions, thus exacerbating neuroinflammation [145].
The neuroprotective functions implemented by microglial cells
Apart from mediating innate immunity in the CNS, increasingly extensive studies have revealed novel neuroprotective mechanisms implemented by highly specialized microglial subtypes under physiological conditions. Although microglia were classically viewed as being responsible for synaptic refinement and brain wiring in healthy brains [146, 147], not until recently have GABA-receptive microglial subtypes been discovered to selectively recognize and sculpt developing inhibitory circuits [148]. The dysfunction of this microglial subtype often leads to impaired synaptogenesis of GABAergic neurons and hyperexcitability [149, 150]. The activation of the complement (C3, C1q) pathway has long been considered an “eat me” signal to promote microglia-mediated identification of diseased synapses that need to be sculpted [151]. Meanwhile, certain “don’t eat me” counterparts, like CD47-related signaling, are required to balance the complement pathway [152] to avoid the excessive microglia-mediated phagocytosis of synapses, which plays an important role in synaptic loss and cortical functional connectivity disorders in depression [153]. In addition, the cytokine IL-33 derived from astrocytes and neurons was found to play a vital role in regulating microglial metabolic adaptation and synapse engulfment, which contributes to synapse remodeling, neural circuit development, and memory consolidation [147, 154, 155].
Moreover, Zhang et al. identified the Arg1+ microglial subtype located in the hippocampus, the proliferation, and maturation of which were driven by IL-4 signaling [156]. This Arg1+ microglia subtype is responsible for secreting additional BDNF which maintains microglial self-renewal, adult neurogenesis in the DG, and working memory under stress conditions [156, 157]. However, BDNF released from microglia was also reported to promote the transdifferentiation of astrocytes and microglia to release proinflammatory cytokines, causing aggravated neuroinflammation [158]. In conclusion, the neuroprotective effects exerted by microglia under both physiological and pathological conditions should not be neglected.
Astrocytes participate in neuroinflammation
Astrocytes provide nutrients to neurons and contribute to temporal integration of neural activity by ensuring the functioning of synapses and driving action potentials [159]. They have also been illustrated to modulate neurological diseases in various manners. Similar to microglial cells, astrocytes could be induced into a reactive neurotoxic A1 phenotype in response to proinflammatory mediators and CNS injuries [160]. This process is defined as astrocytosis and is fundamental to cope with immune attack, chronic neurodegenerative disease, or acute trauma [161, 162]. McAlpine et al. reported that amyloid-β (Aβ) deposition during AD progression leads to the upregulation of IL-3 in astrocytes and IL-3 receptor α in microglia. Microglia sensitized by astrocyte-derived IL-3 clear Aβ aggregates, thus rendering IL-3 among crucial mediators of astrocyte–microglia crosstalk to resist AD progression and cognitive decline [163]. Yshii et al. discovered that astrocyte-specific IL-2 delivery in mice resulted in enhanced proliferation of reactive astrocytes and brain-resident Treg cells, which alleviated pathological neuroinflammation without affecting the peripheral immunity [164]. Moreover, various astrocyte-derived cytokines and metabolites have been found to exert neuroprotective effects and facilitate neuronal survival, including glutamate and TGF-β [165, 166]. In addition, proinflammatory microglia have been discovered to mediate the conversion of astrocytes into the A1 phenotype in various neurological diseases by secreting IL-1α, TNF-α, and C1q [167], while the blockade of this microglia-mediated conversion exhibited neuroprotective effects in Parkinson's disease (PD) models [168].
Researchers have also focused on the roles of astrocytes in facilitating the crosstalk between peripheral immunity and the CNS parenchyma due to the close contact between astrocytes and BBB components such as endothelial cells and pericytes [169]. Gimsa et al. reported that the reduced glutamate internalization of reactive astrocytes mitigated the production of the gap junction constituent connexin 43, thus increasing the permeability of the BBB to peripheral lymphocytes [170]. Moreover, bidirectional communications between reactive astrocytes and peripheral immunocytes have been revealed to modulate the recruitment of peripheral immunocytes to bypass the BBB and infiltrate into the CNS parenchyma. Astrocyte-derived C–C motif chemokine ligand 2 (CCL2) and C–X–C motif chemokine ligand 10 (CXCL10) were reported to induce extravasation and infiltration of peripheral macrophages, monocytes, T lymphocytes and promote their subsequent transdifferentiation into proinflammatory phenotypes [171,172,173]. Similarly, astrocyte-derived CXCL12 was shown to be responsible for the recruitment of pathogenic B lymphocytes [174]. On the other hand, astrocytes were also found to respond to inflammatory cues from infiltrated peripheral immunocytes. For example, IFN-γ produced by peripheral Th1 lymphocytes was reported to upregulate IFN-γ receptor 1 and MHC-II expression in astrocytes in the experimental autoimmune encephalitomyelitis (EAE) mouse models, which enables astrocytes to perform antigen-presenting functions [175]. IL-17 secreted from pathogenic Th17 lymphocytes was revealed to trigger proinflammatory transcriptional activities in astrocytes in EAE and multiple sclerosis (MS) models, according to single-cell RNA sequencing [176, 177].
Oligodendrocytes participate in neuroinflammation
Oligodendrocytes were traditionally considered inactive in the process of neuroinflammation due to their highly differentiated and specialized nature for myelination [178]. However, various mechanisms of oligodendrocytes in CNS immunomodulation have been recently identified, which greatly improved the general understanding of the roles of oligodendrocytes in neuroinflammation [179,180,181]. It is noteworthy that many of these mechanisms depend on the intercommunication between oligodendrocytes and reactive astrocytes under pathological conditions [182]. For example, proinflammatory cytokines like TNF-α derived from A1 astrocytes have been found to initiate programmed death of adjacent oligodendrocytes, leading to mitigated remyelination and subsequent neuronal injuries [183, 184]. What’s more, the oligodendrocyte death and myelin loss might further induce adaptive autoimmune responses during the pathogenesis of MS [185]. On the other hand, certain inflammatory cues secreted from reactive astrocytes, such as CXCL1 and CCL2, also contribute to the mobilization and recruitment of oligodendrocyte progenitor cells (OPCs) to the inflammation sites [182, 186], which increases remyelination after the maturation of OPCs and helps the restoration of immunocompromised neural functions.
The diverse effects of oligodendrocyte–astrocyte communications might be attributed to the heterogeneity of reactive astrocytes. Oligodendrocytes have also been reported to be involved in CNS immunomodulation in astrocyte-independent manners. Papaneophytou et al. discovered that the downregulation of gap junction constituent connexin 47 in oligodendrocytes aggravates the disruption of blood–spinal cord barrier and the infiltration of peripheral immunocytes in MS models [187]. Similarly, aberrantly accumulated oligodendrocytes could interfere with astrocytes’ vascular interactions, disrupting the integrity of BBB in MS models [188]. In addition, diseased oligodendrocytes were found to secrete proinflammatory cytokines, including IL-1β, IL-6, and IL-17, which in turn modulate the functions of other neuroglial cells at inflammation sites [182, 189].
Neuroinflammation augmented by the aberrance of peripheral immunocytes attenuates neurogenesis
Interactions between peripheral immunity and MDD
The existence of peripheral immune cells within the CNS used to be considered negligible under normal physiological conditions. Anatomically, the brain and spinal cord are surrounded by the meninges, which are richly vascularized and innervated multilayer structures consisting of the outer dura mater, the middle arachnoid, and the inner pia mater [190]. CSF produced by choroid plexus epithelial cells flows both within the meninges [191] and into cervical lymph nodes [192], thus facilitating the communication between the meninges and peripheral lymphatic system. Therefore, when neuroinflammation occurs, peripheral immunocytes can respond to antigens presented in the CNS, selectively penetrate the BBB in different ways into the meninges or other cerebral immune-privileged sites [193, 194], and then contribute to sustained immune responses for both autoimmunity and infectious diseases of the CNS [195].
Blood leukocytes in MDD
The studies of immune cell trafficking between the meninges and peripheral lymphatic system have mainly focused on peripheral T lymphocytes [196, 197]. CD8+ T cells recognize linear epitopes presented by the MHC class I molecules expressed by almost all cells and differentiate into cytotoxic T lymphocytes to kill infected cells or tumor cells [198]. CD4+ T lymphocytes are composed of various subtypes according to their phenotypes, including helper T1 (Th1), Th2, Th17, and regulatory T (Treg) cells [199]. Treg cells can be further divided into thymus-derived Treg cells (tTreg cells) directly developed from CD4-single positive (CD4-SP) thymocytes or peripheral-derived Treg cells (pTreg cells) differentiated from naïve CD4+ T cells upon TCR ligation with TGF-β and IL-2 [200]. Other Th cells also differentiate from naïve CD4+ T cells upon TCR ligation, although with different cytokine requirements [201].
In MDD patients, alterations in peripheral immune cells at the systemic level have been frequently reported. Blood T cells in MDD patients exhibit reduced mitochondrial respiration and glycolytic capacity [202]. Furthermore, there is a link between the suicidality of MDD patients and premature Th cell aging and increased proportions of Th17 cells in the blood [200]. Also, a trend of lower blood CD8+ T cell numbers and lower CD4+/CD8+ T cell ratios is associated with MDD severity [203]. In addition, reduced Treg frequency or impaired Treg function has been observed in the blood of MDD patients [204,205,206]. These findings suggest that T cells are involved in MDD progression.
In MDD state, profound changes occur to blood leukocytes other than T cells. MDD patients exhibit higher neutrophil/lymphocyte and monocyte/lymphocyte ratios [207, 208]. Accordingly, increased numbers of proinflammatory monocytes have been observed in the blood of MDD patients [203, 209]. Gene expression analysis suggests mitochondrial dysfunction, premature aging, and high expression of proinflammatory cytokines [210]. Although the changes in total B cell numbers are not consistent in different studies [211], blood CD1d+CD5+ and CD24+CD38hi transitional B cells, which show immune-regulatory functions, are reduced in MDD patients [212]. Furthermore, blood B cells in MDD patients show shorter telomeres as well as T cells, indicating the accelerated loss of B-cell functions [213]. There are also reports of reduced numbers of CD56+CD16– regulatory NK cells in MDD patients [214].
The discovery of brain-resident leukocytes
The rediscovery of fluid exchange between the CSF and interstitial fluid of the brain could serve as a sufficient delivery mechanism for soluble factors released from meningeal leukocytes to modulate neurological diseases [215]. The origin of meningeal leukocytes has been intensely investigated [216,217,218,219,220]. It has been established that most meningeal myeloid cells and B cells are not blood-derived. As skull-to-dura channels exist, it is speculated that meningeal leukocytes transit from the skull through these specialized channels [216, 217, 220]. However, Schafflick et al. questioned this possibility: they labeled skull leukocytes and the fluorescence signal lasted up to 17 days but dura leukocytes remained unlabeled throughput the process [218]. Niu et al. proposed another possibility by identification of hematopoietic stem cells residing in the meninges of adult mice at steady state [219]. There are also various resident peripheral leukocytes in the CSF and parenchyma [20, 21, 216]. With a tissue-specific expression profile, meningeal hematopoietic stem cells can provide the CSF and parenchyma with a constant supply of leukocytes more adapted to the local microenvironment [219]. Since dura myeloid subsets showed similar changes to their femur bone marrow counterparts in a mouse CUMS model [221], it is possible that the aberrance in blood leukocytes of MDD patients also occurs in brain-resident leukocytes. Future studies are required to address this issue.
CD4+ T lymphocytes and hippocampal neurogenesis
It has now been established that T lymphocytes contribute to brain development and immune homeostasis under physiological conditions. In 2009, Wolf et al. [222] found that systemic clearance of CD4+ T cells in the brains of mice led to reduced neurogenesis in the hippocampus, impaired spatial memory indicated by the MWM test, and diminished BDNF expression. The clearance of CD8+ or B cells in the brain, however, did not inflict similar effects on neurogenesis [222]. Such a role has been confirmed by several studies. For example, Negrini et al. reported that BALB/c nude mice show social disorders, accompanied by decreased expression of IFN-γ in the prefrontal cortex and the absence of T cells in the meninges [223]. The absence of a residential CD4+CD69+ T lymphocyte subset in healthy murine brains resulted in suspended microglial activation, together with developmental defects of synapses and behavioral abnormalities [224]. Furthermore, Treg cells were identified to protect female mice from colony stimulating factor 1 (CSF1) intrathecal administration-induced neuropathic pain by suppressing microglial differentiation into proinflammtory phenotypes [225]. Together, these studies indicate protective roles of CD4+ T cells in maintaining immune homeostasis.
Cytokines for brain-resident T lymphocytes and hippocampal neurogenesis
Various cytokines in the CNS have been shown to affect the differentiation and functions of brain-resident T lymphocytes [226, 227]. For example, astrocyte-targeted IL-2 expression expanded brain-resident Treg cells under pathological conditions without impacting the peripheral immunity [164]. Moreover, exercise was found to induce the upregulation of CD4+CD25+ Treg cells and anti-inflammatory responses [228]. Derecki et al. reported that the accumulation of CD4+ T lymphocytes in the meninges of a mouse model after MWM training was in accordance with enhanced visuo-spatial memory. According to their results, the numbers of meningeal CD4+ T cells expressing CD69 increased remarkably after training, while meningeal CD8+ T cells did not exhibit the induction of the early activation marker CD69. Further detection showed that the proportion of CD4+CD25+Foxp3+ Treg subsets in meningeal cells of MWM-trained mice was elevated, accompanied by increased IL-4 secretion. Meanwhile, the subpopulations of myeloid cells in the meninges were also confirmed to respond. All these changes were correlated with improved visuo-spatial memory, suggesting they contribute to hippocampal neurogenesis [229].
For brain-resident CD4+ T lymphocytes, the CD4 coreceptor ligand IL-16 controls their trafficking and biological characteristics. IL-16 was originally identified as a T-cell chemoattractant in the 1980s. Subsequent researches have mainly focused on its roles in inflammation regulation [230]. In particular, Treg-associated IL-16 functions include accelerating the proliferation of CD4+CD25+ T lymphocytes, de novo induction of Foxp3 and migration of Foxp3+ T lymphocytes in the case of long-term culture with IL-2, the key factor of Treg differentiation and proliferation [231]. IL-16 also modulates neuronal excitability and synaptic activity in the mouse hippocampus ex vivo, serving as a neuroprotective factor against excitotoxicity [232, 233]. Notably, in the steady state, hippocampal and cerebellar granule neurons produce and secret considerable amounts of secretory neuronal interleukin-16 (NIL-16), a splice variant of IL-16, into the systemic circulation [234, 235]. NIL-16 is a cytoplasmic protein detected only in neurons in the cerebellum and hippocampus [236]. The N-terminus of NIL-16 selectively interacts with various neuronal ion channels, which is similar to the function of many other PDZ domain proteins as intracellular scaffold proteins [237]. The C-terminus of IL-16/NIL-16 can be recognized and cleaved by caspase-3, resulting in the release of secretory IL-16/NIL-16 [238]. The expression of the IL-16 receptor, namely the CD4 protein, also exists in granule neurons. These neurons treated with NIL-16 induced the expression of c-fos through the tyrosine phosphorylation signaling pathway, which indicates that NIL-16 could function in an autocrine manner. Conclusively, NIL-16 is a protein with dual functions in the nervous system, acting as both a secreted signaling molecule and a scaffold protein [236]. NIL-16-induced inhibition of neuroinflammation contributes to hippocampal neurogenesis, while promoted neurogenesis might in turn further elevate NIL-16 secretion.
Meanwhile, it is noteworthy that the effects of immune-regulatory cytokines during neuroinflammation could be source-specific and context-dependent [239]. By diverting IL-2 production, Whyte et al. identified the different secretory manners of IL-2 released from Treg cells, CD8+ T lymphocytes, and NK cells (autocrine or paracrine), as well as different downstream functional mechanisms [240]. Therefore, targeting cytokine production in specific cell types could be conducive to overcoming the adverse effects of anti-inflammatory treatment.
The effects of neuroinflammation on memory impairment via modulating the hypothalamic–pituitary–adrenal (HPA) axis
Abnormalities in the HPA axis during depression
The HPA axis has been known as one of the canonical stress hormone systems functioning in response to external pressures [241]. In both mice and humans, the corticotropin-releasing hormone (CRH) secreted from paraventricular neurons in the hypothalamus binds and activates the CRH receptor in cells of the pituitary. The above process causes adrenocorticotropic hormone (ACTH) secretion into peripheral circulation. Then, ACTH transported to adrenal glands induces the release of glucocorticoids (primarily corticosterone in mice and cortisol in humans) [242]. Glucocorticoids have been reported to act on nearly all parts of the body and function in various physiological processes by activating glucocorticoid receptors (GRs), exerting anti-inflammatory and immunosuppressive effects and regulating neuronal functions of the limbic system (including the hippocampus) [243, 244]. However, upregulated glucocorticoids also bind to GRs located in multiple brain regions including the hypothalamus and the pituitary, maintaining the homeostasis of the HPA axis through negative feedback loops [245].
The dysregulation and imbalance of the HPA axis have been observed in a prominent proportion of MDD clinical cases and are believed to be initiated from the malfunction of GR-dependent negative feedback of the HPA axis, which is defined as GR resistance [246, 247]. Meanwhile, the alleviation of GR resistance was discovered in MDD patients after receiving effective antidepressive therapies [248]. Genetic variations in the GR-encoding gene NR3C1 and mineralocorticoid receptor (MR)-encoding gene NR3C2 have been manifested in both bioinformatics and clinical studies to correlate with increased risks of cognitive impairments and HPA axis dysregulation [246, 249]. In specific, aberrant glucocorticoid levels have been found to participate in regulating hippocampal neuroinflammation [250, 251], and to interfere with the development, differentiation, and function of T lymphocytes through TCR signaling [252, 253]. On the other hand, certain inflammatory cytokines have been reported to stimulate HPA axis activity and glucocorticoid secretion, including IL-1, IL-6, IL-10, and TNF-α [254]. In addition, various genetic mouse models of MDD have been constructed by selective breeding. In these models, aberrant corticosterone levels were found to be correlated with the severity of depressive-like behaviors [255]. In conclusion, HPA axis dysregulation and GR resistance under chronic stress partially account for the exaggerated neuroinflammation observed during depression, in which glucocorticoids play the central role in exerting anti-inflammatory effects [256].
Hypercortisolism and neuroinflammation during depression
Nevertheless, GR resistance initiated from an imbalanced HPA axis also synchronizes with glucocorticoid hypersecretion observed in MDD patients, which was defined as hypercortisolism [246]. This prompted us to explore the potential explanation based on our understanding of the role of neuroinflammation in accelerating MDD progression and cognitive impairments [257]. To date, several possible mechanisms have been proposed, which have mainly focused on glucocorticoid-induced upregulation of proinflammatory genes. Toll-like receptor families (TLRs) are claimed to be upregulated and overactivated under hypercortisolism through multiple mechanisms, which subsequently augment the secretion of TNF-α [258]. Bullsilo et al. showed that glucocorticoids induce NLRP3 upregulation at both the mRNA and protein levels, and the NLRP3 inflammasome sensitizes the release of IL-1β, TNF-α, and IL-6 from mature macrophages [259]. Treatment with glucocorticoids and TNF-α synergistically increased the expression of the proinflammatory gene serpinA3 in vitro and in vivo [260].
Furthermore, persistent activation of the HPA axis and hypercortisolism during depression have been clarified to impair the immune surveillance mediated by peripheral lymphocytes [261]. Age-dependent dysregulation of the HPA axis leads to a decline in the peripheral pool of T lymphocytes in the thymus, since the signaling of hormone, neurotransmitter, and neuropeptide receptors expressed in thymocytes affects their maturation and function [262]. In addition, the effects of other brain-derived hormones and neurotransmitters on peripheral leukocytes have also been discovered; for instance, norepinephrine could bind to the β2 adrenergic receptors expressed on peripheral CD4+ T cells and B cells to regulate the corresponding immune responses [263]. Cytokines derived from peripheral leukocytes were also verified to bypass the BBB and modulate neuronal functions. Thus, the CNS and the peripheral immune system form bidirectional communications to promote the pathogenesis and progression of depression [264].
Serotonin dysfunctions and neuroinflammation during depression
Serotonin/5-hydroxytryptamine (5-HT) is an important neurotransmitter synthesized from tryptophan, and serotonin metabolism has been observed to be reduced during depression [265]. Decreased cerebral serotonin levels in MDD patients could be reversed with the treatment of antidepressants [266]. The current hypothesis is that the catabolism of serotonin into kynurenine is highly related to neuroinflammation. The metabolites of kynurenine could be either “excitotoxic” quinolinic acid or “neuroprotective” kynurenic acid, which are mainly synthetized by microglia and astrocytes within the CNS, respectively [267, 268]. The serotonin/kynurenine ratio in the plasma and CSF, which indicates the activation of the kynurenine pathway, has been found to correlate with MDD occurrence and the intensity of depressive symptoms, according to both animal models and clinical data [269,270,271]. Also, since microglial kynurenine mono-oxygenase is responsible for catabolizing the synthesis of quinolinic acid from kynurenine, a mono-oxygenase-knockout mouse model was constructed, which verified the irreplaceable role of mono-oxygenase in stirring the kynurenine pathway toward excitotoxic effects under the treatment of inflammatory inducers such as LPS [272].
Moreover, cross-interactions between the serotonin system and HPA axis have been elucidated. Serotonin binds 5-HT1A receptors in hippocampal NPCs and regulates their reactivities to glucocorticoids [273], while the sensitivity of serotonin to bind to the serotonin transporter could be markedly influenced by endocrine regulation of the HPA axis [274].
Conclusion
As reviewed above, the interplay between neuroinflammation and depression has been extensively explored in both preclinical and clinical studies. The impairments of the hippocampal structures and functions implemented by neuroinflammation during MDD progression correspond to attenuated memory performance. The current understanding could be summarized as follows: neurogenesis in the hippocampal dentate gyrus, the HPA axis, and the brain serotonin system are all closely related to the performance of multiple cognitive functions. In addition, the neuronal morphology, neural plasticity, and phenotypes of neuroglial cells within the same encephalic region also affect cognitive capabilities. Therefore, when neuroinflammation occurs in the course of depression, these factors are negatively regulated by excessive chronic neuroinflammatory responses, causing clinically observed cognitive impairments. Reactive microglia and astrocytes and peripheral proinflammatory cells that specifically penetrate the BBB into the CNS after receiving signaling molecules are involved in the mediation of neuroinflammation. By contrast, regulatory immune cells inhibit the neuroimmune responses after receiving specific stimulation, thereby protecting cognitive functions from damage (Fig. 2). The synthesis, secretion, and signal transduction of various types of secretory factors are indispensable in the effectuation of the cross-talks between abovementioned cell types (Table 1). All these mechanisms are supported by evidence from either clinical studies or animal models related to major depression, but more details still need to be further clarified, so as to achieve potentially better clinical therapeutic effects for the development of the corresponding treatments.

Schematic summary of the cell types that participate in the CNS immunoregulation and impact on memory functions by modulating hippocampal neurogenesis in the DG during neuroinflammation. As presented in this figure, under pathological conditions, the differentiation of microglia, astrocytes, and other neuroglia into reactive phenotypes modulates the CNS neuroinflammation and affects the DG neurogenesis. Brain-resident leukocytes in the meninges have been reported to influence the DG neurogenesis and memory functions. In addition, more blood immune cells bypass the BBB and infiltrate into the CNS. The intercommunication between these cell types, as well as neurons, greatly relies on various cytokines and neurotrophins
Availability of data and materials
Not applicable.
References
Abdoli N, et al. The global prevalence of major depressive disorder (MDD) among the elderly: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2022;132:1067–73.
Tonhajzerova I, et al. Major depressive disorder at adolescent age is associated with impaired cardiovascular autonomic regulation and vasculature functioning. Int J Psychophysiol. 2022;181:14–22.
Jeong HG, et al. Role of severity and gender in the association between late-life depression and all-cause mortality. Int Psychogeriatr. 2013;25(4):677–84.
Troubat R, et al. Neuroinflammation and depression: a review. Eur J Neurosci. 2021;53(1):151–71.
Eliwa H, et al. Adult hippocampal neurogenesis: is it the alpha and omega of antidepressant action? Biochem Pharmacol. 2017;141:86–99.
Samuels BA, Hen R. Neurogenesis and affective disorders. Eur J Neurosci. 2011;33(6):1152–9.
Harro J. Animal models of depression: pros and cons. Cell Tissue Res. 2019;377(1):5–20.
Planchez B, et al. Animal models of major depression: drawbacks and challenges. J Neural Transm (Vienna). 2019;126(11):1383–408.
Petković A, Chaudhury D. Encore: behavioural animal models of stress, depression and mood disorders. Front Behav Neurosci. 2022;16: 931964.
Song J, Kim YK. Animal models for the study of depressive disorder. CNS Neurosci Ther. 2021;27(6):633–42.
Hu Y, et al. Memory and processing speed impairments in first-episode drug-naïve patients with major depressive disorder. J Affect Disord. 2022;322:99–107.
Beblo T, et al. Memory deficits in patients with major depression: yes, they are trying hard enough! Expert Rev Neurother. 2020;20(5):517–22.
Sakaguchi Y, Sakurai Y. Left-right functional difference of the rat dorsal hippocampus for short-term memory and long-term memory. Behav Brain Res. 2020;382: 112478.
Kandel ER, et al. Principles of neural science. 6th ed. New York: McGraw Hill; 2021. p. 1291–4.
Jeneson A, Squire LR. Working memory, long-term memory, and medial temporal lobe function. Learn Mem. 2011;19(1):15–25.
Lech RK, Suchan B. The medial temporal lobe: memory and beyond. Behav Brain Res. 2013;254:45–9.
Woodburn SC, et al. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J Neuroinflammation. 2021;18(1):258.
Forrester JV, et al. CNS infection and immune privilege. Nat Rev Neurosci. 2018;19(11):655–71.
Engelhardt B, et al. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–31.
Korin B, et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 2017;20(9):1300–9.
Korin B, et al. Mass cytometry analysis of immune cells in the brain. Nat Protoc. 2018;13(2):377–91.
Chausse B, et al. Microglia and lipids: how metabolism controls brain innate immunity. Semin Cell Dev Biol. 2021;112:137–44.
Castellani G, et al. Transforming the understanding of brain immunity. Science. 2023;380(6640):eabo7649.
Korn T, Kallie A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17(3):179–94.
Gate D, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404.
Congdon KL, et al. Effective effectors: how T cells access and infiltrate the central nervous system. Pharmacol Ther. 2019;197:52–60.
Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Zelic M, et al. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021;35(6): 109112.
Calcia MA, et al. BloomfieldStress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology. 2016;233(9):1637–50.
Yao H, et al. Gut microbiota regulates chronic ethanol exposure-induced depressive-like behavior through hippocampal NLRP3-mediated neuroinflammation. Mol Psychiatry. 2022;28(2):919–30.
Zhang J, et al. LPS activates neuroinflammatory pathways to induce depression in Parkinson’s disease-like condition. Front Pharmacol. 2022;13: 961817.
Won E, et al. Associations between melatonin, neuroinflammation, and brain alterations in depression. Int J Mol Sci. 2021;23(1):305.
Rush AJ, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–17.
Pan Z, et al. Cognitive impairment in major depressive disorder. CNS Spectr. 2019;24(1):22–9.
Lewis LR, et al. Affective biases and their interaction with other reward-related deficits in rodent models of psychiatric disorders. Behav Brain Res. 2019;372: 112051.
Pfau ML, Russo SJ. Neuroinflammation regulates cognitive impairment in socially defeated mice. Trends Neurosci. 2016;39(6):353–5.
Liu Q, et al. Akebia saponin D protects hippocampal neurogenesis from microglia-mediated inflammation and ameliorates depressive-like behaviors and cognitive impairment in mice through the PI3K-Akt pathway. Front Pharmacol. 2022;13: 927419.
Liu H, et al. Muscone with attenuation of neuroinflammation and oxidative stress exerts antidepressant-like effect in mouse model of chronic restraint stress. Oxid Med Cell Longev. 2022;2022:3322535.
Schultz C, Engelhardt M. Anatomy of the hippocampal formation. Front Neurol Neurosci. 2014;34:6–17.
Fares J, et al. Neurogenesis in the adult hippocampus: history, regulation, and prospective roles. Int J Neurosci. 2019;129(6):598–611.
Toda T, et al. The role of adult hippocampal neurogenesis in brain health and disease. Mol Psychiatry. 2019;24(1):67–87.
Apple DM, et al. The role of adult neurogenesis in psychiatric and cognitive disorders. Brain Res. 2017;1655:270–6.
Christian KM, et al. Adult neurogenesis and the dentate gyrus: predicting function from form. Behav Brain Res. 2020;379: 112346.
Falk S, Götz M. Glial control of neurogenesis. Curr Opin Neurobiol. 2017;47:188–95.
Song J, et al. Neuronal circuitry mechanisms regulating adult mammalian neurogenesis. Cold Spring Harb Perspect Biol. 2016;8(8): a018937.
Domínguez-Rivas E, et al. Adult hippocampal neurogenesis in the context of lipopolysaccharide-induced neuroinflammation: a molecular, cellular and behavioral review. Brain Behav Immun. 2021;97:286–302.
Yao B, et al. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci. 2016;17(9):537–49.
Vicidomini C, et al. Communication, cross talk, and signal integration in the adult hippocampal neurogenic niche. Neuron. 2020;105(2):220–35.
Vieira MS, et al. Neural stem cell differentiation into mature neurons: mechanisms of regulation and biotechnological applications. Biotechnol Adv. 2018;36(7):1946–70. https://doi.org/10.1016/j.biotechadv.2018.08.002.
Bond AM, et al. Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell. 2015;17(4):385–95.
Obernier K, Alvarez-Buylla A. Neural stem cells: origin, heterogeneity and regulation in the adult mammalian brain. Development. 2019;146(4): dev156059.
Gebara E, et al. Heterogeneity of radial glia-like cells in the adult hippocampus. Stem Cells. 2016;34(4):997–1010.
Lazutkin A, et al. Modes of division and differentiation of neural stem cells. Behav Brain Res. 2019;374: 112118.
Abbott LC, Nigussie F. Adult neurogenesis in the mammalian dentate gyrus. Anat Histol Embryol. 2020;49(1):3–16.
Luo C. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia. 2016;64(9):1508–17.
Kempermann G, et al. Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell. 2018;23(1):25–30.
Kohman RA, Rhodes JS. Neurogenesis, inflammation and behavior. Brain Behav Immun. 2013;27(1):22–32.
Horgusluoglu E, et al. Adult neurogenesis and neurodegenerative diseases: a systems biology perspective. Am J Med Genet B Neuropsychiatr Genet. 2017;174(1):93–112.
Ip CW, et al. Stereological estimation of dopaminergic neuron number in the mouse substantia nigra using the optical fractionator and standard microscopy equipment. J Vis Exp. 2017;127:56103.
Tamura Y, Kataoka Y. PET imaging of neurogenic activity in the adult brain: Toward in vivo imaging of human neurogenesis. Neurogenesis (Austin). 2017;4(1): e1281861.
Haapakoski R, et al. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–15.
Su K, et al. Interferon-alpha-induced depression: Comparisons between early- and late-onset subgroups and with patients with major depressive disorder. Brain Behav Immun. 2019;80:512–8.
Köhler-Forsberg O, et al. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand. 2019;139(5):404–19.
Stankiewicz AM, et al. The effect of acute and chronic social stress on the hippocampal transcriptome in mice. PLoS ONE. 2015;10(11): e0142195.
Papakostas GI, Culpepper L. Understanding and managing cognition in the depressed patient. J Clin Psychiatry. 2015;76(4):418–25.
Noworyta K, et al. Neuromolecular underpinnings of negative cognitive bias in depression. Cells. 2021;10(11):3157.
McKim DB, et al. Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci. 2016;36(9):2590–604.
Garrido-Mesa N, et al. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337–52.
Ryan SM, Nolan YM. Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition: can exercise compensate? Neurosci Biobehav Rev. 2016;61:121–31.
Leal G, et al. BDNF and hippocampal synaptic plasticity. Vitam Horm. 2017;104:153–95.
Alam MJ, et al. Adult neurogenesis conserves hippocampal memory capacity. J Neurosci. 2018;38(31):6854–63.
van Praag H, et al. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2(3):266–70.
Creer DJ, et al. Running enhances spatial pattern separation in mice. Proc Natl Acad Sci U S A. 2010;107(5):2367–72.
Hötting K, Röder B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci Biobehav Rev. 2013;37(9 Pt B):2243–57.
Brown J, et al. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci. 2003;17(10):2042–6.
Stranahan AM, et al. Running induces widespread structural alterations in the hippocampus and entorhinal cortex. Hippocampus. 2007;17(11):1017–22.
Tang K, et al. Exercise-induced VEGF transcriptional activation in brain, lung and skeletal muscle. Respir Physiol Neurobiol. 2010;170(1):16–22.
Wu C, et al. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J Neurochem. 2007;103(6):2471–81.
Wu M, et al. Adult murine hippocampal neurogenesis is inhibited by sustained IL-1β and not rescued by voluntary running. Brain Behav Immun. 2012;26(2):292–300.
Barrientos RM. Voluntary exercise as an anti-neuroinflammatory therapeutic. Brain Behav Immun. 2011;25(6):1061–2.
Ayari S, et al. A systematic review of exercise modalities that reduce pro-inflammatory cytokines in humans and animals’ models with mild cognitive impairment or dementia. Exp Gerontol. 2023;175: 112141.
Skaper SD. Nerve growth factor: a neuroimmune crosstalk mediator for all seasons. Immunology. 2017;151(1):1–15.
Huang E, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736.
Skaper SD. Neurotrophic factors: an overview. Methods Mol Biol. 2018;1727:1–17.
Shohayeb B, et al. Factors that influence adult neurogenesis as potential therapy. Transl Neurodegener. 2018;7:4.
Giacobbo LB, et al. Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol Neurobiol. 2019;56(5):3295–312.
Wang CA, et al. BDNF signaling in context: from synaptic regulation to psychiatric disorders. Cell. 2022;185(1):62–76.
Correia AS, et al. BDNF unveiled: exploring its role in major depression disorder serotonergic imbalance and associated stress conditions. Pharmaceutics. 2023;15(8):2081.
Foltran RB, Diaz SL. BDNF isoforms: a round trip ticket between neurogenesis and serotonin? J Neurochem. 2016;138(2):204–21.
Mizui T, et al. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol Res. 2016;105:93–8.
Kowiański P, et al. BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. 2018;38(3):579–93.
Leal G, et al. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2014;76(Pt C):639–56.
Numakawa T, et al. Actions of brain-derived neurotrophin factor in the neurogenesis and neuronal function, and its involvement in the pathophysiology of brain diseases. Int J Mol Sci. 2018;19(11):3650.
Mousten IV, et al. Cerebrospinal fluid biomarkers in patients with unipolar depression compared with healthy control individuals: a systematic review and meta-analysis. JAMA Psychiat. 2022;79(6):571–81.
Ventriglia M, et al. Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed Res Int. 2013;2013: 901082.
Park H, Poo M. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14(1):7–23.
Bazzari AH, Bazzari FH. BDNF therapeutic mechanisms in neuropsychiatric disorders. Int J Mol Sci. 2022;23(15):8417.
Colucci-D’Amato L, et al. Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci. 2020;21(20):7777.
Dieni S, et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. 2012;196(6):775–88.
Snow WM, et al. Roles for NF-κB and gene targets of NF-κB in synaptic plasticity, memory, and navigation. Mol Neurobiol. 2014;49(2):757–70.
Chao M. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309.
Sochal M, et al. The role of brain-derived neurotrophic factor in immune-related diseases: a narrative review. J Clin Med. 2022;11(20):6023.
Mehterov N, et al. Interactions among brain-derived neurotrophic factor and neuroimmune pathways are key components of the major psychiatric disorders. Mol Neurobiol. 2022;59(8):4926–52.
Teng Z, et al. Low BDNF levels in serum are associated with cognitive impairments in medication-naïve patients with current depressive episode in BD II and MDD. J Affect Disord. 2021;293:90–6.
Larsen MH, et al. Regulation of brain-derived neurotrophic factor (BDNF) in the chronic unpredictable stress rat model and the effects of chronic antidepressant treatment. J Psychiatr Res. 2010;44(13):808–16.
Molendijk ML, et al. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol Psychiatry. 2014;19(7):791–800.
Klein AB, et al. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol. 2011;14(3):347–53.
Mosiołek A, et al. Effects of antidepressant treatment on neurotrophic factors (BDNF and IGF-1) in patients with major depressive disorder (MDD). J Clin Med. 2021;10(15):3377.
Brunoni AR, et al. BDNF plasma levels after antidepressant treatment with sertraline and transcranial direct current stimulation: results from a factorial, randomized, sham-controlled trial. Eur Neuropsychopharmacol. 2014;24(7):1144–51.
Allen AP, et al. Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: a comparison of ketamine and ECT. J Affect Disord. 2015;186:306–11.
Karege F, et al. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry. 2005;57(9):1068–72.
Lorenzetti V, et al. Brain-derived neurotrophic factor association with amygdala response in major depressive disorder. J Affect Disord. 2020;267:103–6.
Serra-Millàs M. Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J Psychiatry. 2016;6(1):84–101.
Lang UE, Borgwardt S. Molecular mechanisms of depression: perspectives on new treatment strategies. Cell Physiol Biochem. 2013;31(6):761–77.
Lotrich FE, et al. Brain-derived neurotrophic factor serum levels and genotype: association with depression during interferon-α treatment. Neuropsychopharmacology. 2013;38(6):985–95.
Deng J, et al. Mapping secretome-mediated interaction between paired neuron-macrophage single cells. Proc Natl Acad Sci U S A. 2022;119(44): e2200944119.
Borst K, et al. Microglia: immune and non-immune functions. Immunity. 2021;54(10):2194–208.
Bennett ML, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738-1746. https://doi.org/10.1073/pnas.1525528113.
Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19(10):622–35. https://doi.org/10.1038/s41583-018-0057-5.
Orihuela R, et al. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649–65. https://doi.org/10.1111/bph.13139.
Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015;131:65–86.
Liddelow SA, et al. Microglia and astrocytes in disease: dynamic duo or partners in crime? Trends Immunol. 2020;41(9):820–35.
Takamura R, et al. All-trans retinoic acid improved impaired proliferation of neural stem cells and suppressed microglial activation in the hippocampus in an Alzheimer’s mouse model. J Neurosci Res. 2017;95(3):897–906.
Wadhwa M, et al. Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. J Neuroinflammation. 2017;14(1):222.
Cserép C. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367(6477):528–37.
Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–91.
Estes ML, et al. Alterations in immune cells and mediators in the brain: it’s not always neuroinflammation! Brain Pathol. 2014;24(6):623–30.
Farooq RK, et al. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav Brain Res. 2012;231(1):130–7.
Farooq RK, et al. A P2X7 receptor antagonist reverses behavioural alterations, microglial activation and neuroendocrine dysregulation in an unpredictable chronic mild stress (UCMS) model of depression in mice. Psychoneuroendocrinology. 2018;97:120–30.
Preez AD, et al. Chronic stress followed by social isolation promotes depressive-like behaviour, alters microglial and astrocyte biology and reduces hippocampal neurogenesis in male mice. Brain Behav Immun. 2021;91:24–47.
Mao M, et al. MicroRNA-195 prevents hippocampal microglial/macrophage polarization towards the M1 phenotype induced by chronic brain hypoperfusion through regulating CX3CL1/CX3CR1 signaling. J Neuroinflammation. 2020;17(1):244.
Luo G, et al. Metabolic reprogramming mediates hippocampal microglial M1 polarization in response to surgical trauma causing perioperative neurocognitive disorders. J Neuroinflammation. 2021;18(1):267.
Brites D, Fernandes A. Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci. 2015;9:476.
Belarbi K, Rosi S. Modulation of adult-born neurons in the inflamed hippocampus. Front Cell Neurosci. 2013;7:145.
Liu Z, et al. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke. 2007;38(1):146–52.
Qiu Y, et al. Efficacy and tolerability of minocycline in depressive patients with or without treatment-resistant: a meta-analysis of randomized controlled trials. Front Psychiatry. 2023;14:1139273.
Cassel SL, Utterwala FS. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol. 2010;40(3):607–11.
Ratajczak MZ, et al. ATP-Nlrp3 inflammasome-complement cascade axis in sterile brain inflammation in psychiatric patients and its impact on stem cell trafficking. Stem Cell Rev Rep. 2019;15(4):497–505.
Roger S, et al. Single nucleotide polymorphisms that were identified in affective mood disorders affect ATP-activated P2X7 receptor functions. J Psychiatr Res. 2010;44(6):347–55.
Csölle C, et al. Neurochemical changes in the mouse hippocampus underlying the antidepressant effect of genetic deletion of P2X7 receptors. PLoS ONE. 2013;8(6): e66547.
Alishahi M, et al. NLRP3 inflammasome in ischemic stroke: as possible therapeutic target. Int J Stroke. 2019;14(6):574–91.
Todd L, et al. Reactive microglia and IL1β/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J Neuroinflammation. 2019;16(1):118.
Rossi S, et al. Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol Neurodegener. 2014;9:56.
Li JM, et al. Ketamine may exert antidepressant effects via suppressing NLRP3 inflammasome to upregulate AMPA receptors. Neuropharmacology. 2019;146:149–53.
Acosta-Rodriguez EV, et al. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–9.
Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18(11):658–70.
Nguyen PT, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell. 2020;182(2):388-403.e15.
Favuzzi E, et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. 2021;184(15):4048-4063.e32.
Gallo NB, et al. Microglia regulate chandelier cell axo-axonic synaptogenesis. Proc Natl Acad Sci U S A. 2022;119(11): e2114476119.
Lui H, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–35.
Hammond TR, et al. Microglia and the brain: complementary partners in development and disease. Annu Rev Cell Dev Biol. 2018;34:523–44.
Ding X, et al. Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nat Commun. 2021;12(1):2030.
Wang J, et al. Microglia-dependent excessive synaptic pruning leads to cortical underconnectivity and behavioral abnormality following chronic social defeat stress in mice. Brain Behav Immun. 2023;109:23–36.
Vainchtein ID, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–73.
He D, et al. Disruption of the IL-33-ST2-AKT signaling axis impairs neurodevelopment by inhibiting microglial metabolic adaptation and phagocytic function. Immunity. 2022;55(1):159-173.e9.
Zhang JQ, et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv. 2021;7(12):eabb9888.
Harley SBR, et al. Selective ablation of BDNF from microglia reveals novel roles in self-renewal and hippocampal neurogenesis. J Neurosci. 2021;41(19):4172–86.
Ding H, et al. BDNF promotes activation of astrocytes and microglia contributing to neuroinflammation and mechanical allodynia in cyclophosphamide-induced cystitis. J Neuroinflammation. 2020;17(1):19.
Deemyad T, et al. Astrocytes integrate and drive action potential firing in inhibitory subnetworks. Nat Commun. 2018;9(1):4336.
Guttenplan KA, Liddelow SA. Astrocytes and microglia: models and tools. J Exp Med. 2019;216(1):71–83.
Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67.
Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94(4):1077–98.
McAlpine CS, et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature. 2021;595(7869):701–6.
Yshii L, et al. Astrocyte-targeted gene delivery of interleukin 2 specifically increases brain-resident regulatory T cell numbers and protects against pathological neuroinflammation. Nat Immunol. 2022;23(6):878–91.
Miyazaki I, Asanuma M. Neuron-astrocyte interactions in Parkinson’s disease. Cells. 2020;9(12):2623.
Phatnani H, Maniatis T. Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol. 2015;7(6): a020628.
Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9(1):42.
Linnerbauer M, et al. Astrocyte crosstalk in CNS inflammation. Neuron. 2020;108(4):608–22.
Gimsa U, et al. Immune privilege as an intrinsic CNS property: astrocytes protect the CNS against T-cell-mediated neuroinflammation. Mediators Inflamm. 2013;2013: 320519.
Liddelow SA, Hoyer D. Astrocytes: adhesion molecules and immunomodulation. Curr Drug Targets. 2016;17(16):1871–81.
Ko E, et al. Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J Neuroinflammation. 2014;11:105.
Moreno M, et al. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J Neurosci. 2014;34(24):8175–85.
Sanmarco LM, et al. Functional immune cell-astrocyte interactions. J Exp Med. 2021;218(9): e20202715.
Yang J, et al. Characterization of the interaction between astrocytes and encephalitogenic lymphocytes during the development of experimental autoimmune encephalitomyelitis (EAE) in mice. Clin Exp Immunol. 2012;170(3):254–65.
Wheeler MA, et al. MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578(7796):593–9.
Wheeler MA, et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell. 2019;176(3):581-596.e18.
Kuhn S, et al. Oligodendrocytes in development, myelin generation and beyond. Cells. 2019;8(11):1424.
Falcão AM, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24(12):1837–44.
Jäkel S, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566(7745):543–7.
Gibson EM, et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell. 2019;176(1–2):43-55.e13.
Moyon S, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci. 2015;35(1):4–20.
Valentin-Torres A, et al. Blockade of sustained tumor necrosis factor in a transgenic model of progressive autoimmune encephalomyelitis limits oligodendrocyte apoptosis and promotes oligodendrocyte maturation. J Neuroinflammation. 2018;15(1):121.
Madsen PM, et al. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci. 2016;36(18):5128–43.
Traka M, et al. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci. 2016;19(1):65–74.
Domingues HS, et al. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71.
Papaneophytou CP, et al. Regulatory role of oligodendrocyte gap junctions in inflammatory demyelination. Glia. 2018;66(12):2589–603.
Niu J, et al. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat Neurosci. 2019;22(5):709–18.
Ramesh G, et al. A possible role for inflammation in mediating apoptosis of oligodendrocytes as induced by the Lyme disease spirochete Borrelia burgdorferi. J Neuroinflammation. 2012;9:72.
Coles JA, et al. Where are we? The anatomy of the murine cortical meninges revisited for intravital imaging, immunology, and clearance of waste from the brain. Prog Neurobiol. 2017;156:107–48.
Da Mesquita S, et al. The meningeal lymphatic system: a new player in neurophysiology. Neuron. 2018;100(2):375–88.
Li X, et al. Meningeal lymphatic vessels mediate neurotropic viral drainage from the central nervous system. Nat Neurosci. 2022;25(5):577–87.
Shechter R, et al. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. 2013;13(3):206–18.
Su Y, et al. Meningeal immunity and neurological diseases: new approaches, new insights. J Neuroinflammation. 2023;20(1):125.
Ma T, et al. Meningeal immunity: structure, function and a potential therapeutic target of neurodegenerative diseases. Brain Behav Immun. 2021;93:264–76.
Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41.
Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33(12):579–89.
Benallegue N, et al. Neuroinflammation: extinguishing a blaze of T cells. Immunol Rev. 2022;311(1):151–76.
Wang J, et al. (2022) The role of CD4+ T cells in the immunotherapy of brain disease by secreting different cytokines. j Neuroimmune Pharmacol. 2022;17(3–4):409–22.
Schiweck C, et al. Depression and suicidality: a link to premature T helper cell aging and increased Th17 cells. Brain Behav Immun. 2020;87:603–9.
Sekiya T, Yoshimura A. In vitro Th differentiation protocol. Methods Mol Biol. 2016;1344:183–91.
Gamradt S, et al. Reduced mitochondrial respiration in T cells of patients with major depressive disorder. iScience. 2021;24: 103312.
Zhou D, et al. A novel joint index based on peripheral blood CD4+/CD8+ T cell ratio, albumin level, and monocyte count to determine the severity of major depressive disorder. BMC Psychiatry. 2022;22:248.
Li Y, et al. Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J Affect Disord. 2010;124(1–2):68–75.
Grosse L, et al. Deficiencies of the T and natural killer cell system in major depressive disorder. T regulatory cell defects are associated with inflammatory monocyte activation. Brain Behav Immun. 2016;54:38–44.
Duggal NA, et al. Development of depressive symptoms post hip fracture is associated with altered immunosuppressive phenotype in regulatory T and B lymphocytes. Biogerontology. 2016;17(1):229–39.
Mazza MG, et al. Neutrophil/lymphocyte ratio and platelet/lymphocyte ratio in mood disorders: a meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2018;84(Pt A):229–36.
Wei Y, et al. Neutrophil/lymphocyte, platelet/lymphocyte and monocyte/ lymphocyte ratios in patients with affective disorders. J Affect Disord. 2022;309:221–8.
Nowak W, et al. Pro-inflammatory monocyte profile in patients with major depressive disorder and suicide behaviour and how ketamine induces anti-inflammatory M2 macrophages by NMDAR and mTOR. EBioMedicine. 2019;50:290–305.
Simon MS, et al. Monocyte mitochondrial dysfunction, inflammaging, and inflammatory pyroptosis in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111: 110391.
Su L, et al. Development and validation of a nomogram based on lymphocyte subsets to distinguish bipolar depression from major depressive disorder. Front Psychiatry. 2022;13:1017888.
Ahmetspahic D, et al. Altered B cell homeostasis in patients with major depressive disorder and normalization of CD5 surface expression on regulatory B cells in treatment responders. J Neuroimmune Pharmacol. 2018;13(1):90–9.
Karabatsiakis A, et al. Telomere shortening in leukocyte subpopulations in depression. BMC Psychiatry. 2014;14:192.
Suzuki H, et al. Altered populations of natural killer cells, cytotoxic T lymphocytes, and regulatory T cells in major depressive disorder: association with sleep disturbance. Brain Behav Immun. 2017;66:193–200.
Da Mesquita S, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185–91.
Cugurra A, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 2021;373(6553):eabf7844.
Brioschi S, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science. 2021;373(6553):eabf9277.
Schafflick D, et al. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat Neurosci. 2021;24(9):1225–34.
Niu C, et al. Identification of hematopoietic stem cells residing in the meninges of adult mice at steady state. Cell Rep. 2022;41: 111592.
Wang Y, et al. Early developing B cells undergo negative selection by central nervous system-specific antigens in the meninges. Immunity. 2021;54:2784-2794.e6.
Xu G, et al. Nonerythropoietic erythropoietin mimetic peptide ARA290 ameliorates chronic stress-induced depression-like behavior and inflammation in mice. Front Pharmacol. 2022;13: 896601.
Wolf SA, et al. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. 2009;182(7):3979–84.
Negrini GB, et al. The role of T-cells in neurobehavioural development: Insights from the immunodeficient nude mice. Behav Brain Res. 2022;418: 113629.
Pasciuto E, et al. Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell. 2020;182(3):625-640.e24.
Kuhn JA, et al. Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife. 2021;10: e69056.
Ren H, et al. IL-21 from high-affinity CD4 T cells drives differentiation of brain-resident CD8 T cells during persistent viral infection. Sci Immunol. 2020;5(51):eabb5590.
Xie L, et al. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol. 2015;45(1):180–91.
Dorneles GP, et al. Physical fitness modulates the expression of CD39 and CD73 on CD4+ CD25- and CD4+ CD25+ T cells following high intensity interval exercise. J Cell Biochem. 2019;120(6):10726–36.
Derecki NC, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207(5):1067–80.
Cruikshank W, Little F. lnterleukin-16: the ins and outs of regulating T-cell activation. Crit Rev Immunol. 2008;28(6):467–83.
Skundric DS, et al. Role of IL-16 in CD4+ T cell-mediated regulation of relapsing multiple sclerosis. J Neuroinflammation. 2015;12:78.
Shrestha R, et al. Lymphocyte-mediated neuroprotection in in vitro models of excitotoxicity involves astrocytic activation and the inhibition of MAP kinase signalling pathways. Neuropharmacology. 2014;76(Pt A):184–93.
Hridi SU, et al. Interleukin-16 inhibits sodium channel function and GluA1 phosphorylation via CD4- and CD9-independent mechanisms to reduce hippocampal neuronal excitability and synaptic activity. Mol Cell Neurosci. 2019;95:71–8.
Simpson S, et al. Thermal stability of cytokines: a review. Cytokine. 2020;125: 154829.
Croq F, et al. A homologous form of human interleukin 16 is implicated in microglia recruitment following nervous system injury in leech Hirudo medicinalis. Glia. 2010;58(14):1649–1462.
Kurschner C, Yuzaki M. Neuronal interleukin-16 (NIL-16): a dual function PDZ domain protein. J Neurosci. 1999;19(18):7770–80.
Fenster CP, et al. Modulation of Kv4.2 K+ currents by neuronal interleukin-16, a PDZ domain-containing protein expressed in the hippocampus and cerebellum. Brain Res. 2007;1162:19–31.
Bannert N, et al. PDZ Domain-mediated interaction of interleukin-16 precursor proteins with myosin phosphatase targeting subunits. J Biol Chem. 2003;278(43):42190–9.
Rankin L, Gray D. Location, location, location! Rewiring IL-2 circuits defines context-specific outcomes. Immunol Cell Biol. 2022;100(8):585–7.
Whyte CE, et al. Context-dependent effects of IL-2 rewire immunity into distinct cellular circuits. J Exp Med. 2022;219(7): e20212391.
Kim YK, et al. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:277–84.
Joseph DN, Whirledge S. Stress and the HPA axis: balancing homeostasis and fertility. Int J Mol Sci. 2017;18(10):2224.
Panettieri RA, et al. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci. 2019;40(1):38–49.
Schaaf MJ, et al. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813(1):112–20.
Menke A. Is the HPA axis as target for depression outdated, or is there a new hope? Front Psychiatry. 2019;10:101.
Keller J, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry. 2017;22(4):527–36.
Leistner C, Menke A. How to measure glucocorticoid receptor’s sensitivity in patients with stress-related psychiatric disorders. Psychoneuroendocrinology. 2018;91:235–60.
Ising M, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression—a potential biomarker? Biol Psychiatry. 2007;62(1):47–54.
Plieger T, et al. The role of genetic variation in the glucocorticoid receptor (NR3C1) and mineralocorticoid receptor (NR3C2) in the association between cortisol response and cognition under acute stress. Psychoneuroendocrinology. 2018;87:173–80.
Salehzadeh M, Soma KK. Glucocorticoid production in the thymus and brain: Immunosteroids and neurosteroids. Brain Behav Immun Health. 2021;18: 100352.
Schweingruber N, et al. Mechanisms of glucocorticoids in the control of neuroinflammation. J Neuroendocrinol. 2012;24(1):174–82.
Taves MD, Ashwell JD. Glucocorticoids in T cell development, differentiation and function. Nat Rev Immunol. 2021;21(4):233–43.
Carrillo-de Sauvage MÁ, et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ. 2013;20(11):1546–1457.
Dunn AJ. The HPA axis and the immune system: a perspective. NeuroImmune Biology. 2007;7:3–15.
Nandam LS, et al. Cortisol and major depressive disorder-translating findings from humans to animal models and back. Front Psychiatry. 2020;10:974.
Heffner KL. Neuroendocrine effects of stress on immunity in the elderly: implications for inflammatory disease. Immunol Allergy Clin North Am. 2011;31(1):95–108.
Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63.
Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol. 2007;275(1–2):30–42.
Busillo JM, et al. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. 2011;286(44):38703–13.
Lannan EA, et al. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology. 2012;153(8):3701–12.
Dai S, et al. Chronic stress promotes cancer development. Front Oncol. 2020;10:1492.
Hannestad J, et al. Age-dependent changes in the nervous and endocrine control of the thymus. Microsc Res Tech. 2004;63(2):94–101.
Simon T, et al. The cholinergic anti-inflammatory pathway inhibits inflammation without lymphocyte relay. Front Neurosci. 2023;17:1125492.
ThyagaRajan S, Priyanka HP. Bidirectional communication between the neuroendocrine system and the immune system: relevance to health and diseases. Ann Neurosci. 2012;19(1):40–6.
Masi G, Brovedani P. The hippocampus, neurotrophic factors and depression: possible implications for the pharmacotherapy of depression. CNS Drugs. 2011;25(11):913–31.
Shulman KI, et al. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27(10):789–97.
O’Farrell K, Harkin A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology. 2017;112(Pt B):307–23.
Schwarcz R, Stone TW. The kynurenine pathway and the brain: challenges, controversies and promises. Neuropharmacology. 2017;112(Pt B):237–47.
Laugeray A, et al. Peripheral and cerebral metabolic abnormalities of the tryptophan-kynurenine pathway in a murine model of major depression. Behav Brain Res. 2010;210(1):84–91. https://doi.org/10.1016/j.bbr.2010.02.014.
Ogawa S, et al. Plasma L-tryptophan concentration in major depressive disorder: new data and meta-analysis. J Clin Psychiatry. 2014;75(9):e906-915.
Raison CL, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15(4):393–403.
Parrott JM, et al. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl Psychiatry. 2016;6(10): e918.
Huang GJ, Herbert J. The role of 5-HT1A receptors in the proliferation and survival of progenitor cells in the dentate gyrus of the adult hippocampus and their regulation by corticoids. Neuroscience. 2005;135(3):803–13.
Pompili M, et al. The hypothalamic-pituitary-adrenal axis and serotonin abnormalities: a selective overview for the implications of suicide prevention. Eur Arch Psychiatry Clin Neurosci. 2010;260(8):583–600.
Funding
This study is supported by grants from the National Natural Science Foundation of China (81930027).
Author information
Authors and Affiliations
Contributions
Anbiao. Wu and Jiyan. Zhang wrote the main manuscript text and prepared figures 1-2. All authors reviewed the manuscript and participated in the revision.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The materials and methods corresponding to the results presented in Fig. 1 are described Additional file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, A., Zhang, J. Neuroinflammation, memory, and depression: new approaches to hippocampal neurogenesis. J Neuroinflammation 20, 283 (2023). https://doi.org/10.1186/s12974-023-02964-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02964-x