
Abstract
Cancer stem cells (CSCs), a small subset of cells in tumors that are characterized by self-renewal and continuous proliferation, lead to tumorigenesis, metastasis, and maintain tumor heterogeneity. Cancer continues to be a significant global disease burden. In the past, surgery, radiotherapy, and chemotherapy were the main cancer treatments. The technology of cancer treatments continues to develop and advance, and the emergence of targeted therapy, and immunotherapy provides more options for patients to a certain extent. However, the limitations of efficacy and treatment resistance are still inevitable. Our review begins with a brief introduction of the historical discoveries, original hypotheses, and pathways that regulate CSCs, such as WNT/β-Catenin, hedgehog, Notch, NF-κB, JAK/STAT, TGF-β, PI3K/AKT, PPAR pathway, and their crosstalk. We focus on the role of CSCs in various therapeutic outcomes and resistance, including how the treatments affect the content of CSCs and the alteration of related molecules, CSCs-mediated therapeutic resistance, and the clinical value of targeting CSCs in patients with refractory, progressed or advanced tumors. In summary, CSCs affect therapeutic efficacy, and the treatment method of targeting CSCs is still difficult to determine. Clarifying regulatory mechanisms and targeting biomarkers of CSCs is currently the mainstream idea.
Similar content being viewed by others

Introduction
Due to advances in cancer early detection and cancer treatments, cancer yearly mortality has been decreasing since 1995.1 However, cancers still caused more deaths than COVID-19 and ranked as the second cause of death in the United States in 2020 and 2021.1 The presence of cancer stem cells (CSCs) can be an essential factor that leads to failure of cancer treatments.
CSCs, first identified in 1990,2 are a small group of cancer cells that possess properties of normal stem cells, such as self-renewal and pluripotency.3 The CSC model, also known as the hierarchical model, provides a paradigm for people to understand intratumoral heterogeneity, as they can differentiate into various phenotypes of cancer cells and maintain their population.4 CSCs are also characterized by enhanced ability to initiate tumor growth, proliferate, invade, migrate, and resist therapeutic effects.3 This implies a crucial role of CSCs in cancer development and makes CSCs an evaluable target for anti-cancer treatments. Therapeutic agents, such as monoclonal antibodies, tyrosine kinase inhibitors, chimeric antigen receptors (CAR) T cells, and tumor vaccines, targeting CSCs have been developed and tested in clinical trials.5
In recent years, studies have added knowledge in the origin, features, and especially therapeutic aspects of CSCs. Here, we summarize the research history, origins, properties, molecular regulations, mechanisms for therapeutic resistance, and treatment strategies of CSCs.
The development of the CSCs theory
The discovery and controversy of the CSCs
As early as 1855, the work of pathologist Rudolph Virchow illuminated that tumors stem from existing normal cells, sparking a scientific discourse about the origin of tumors (Fig. 1). Julius Cohnheim disagreed with that and contributed to his “embryonal cell rests” hypothesis in 1867.6 This posited that dormant embryonic cells within tissues could awaken into tumors.7 Spanning the 19th and 20th centuries, burgeoning research into the genetic underpinnings of cancer has fostered the prevailing notion that cancer arises from the accumulation of mutations in susceptible cells. However, given the terminal differentiation and quiescence of most body cells, their lifespan seldom permits the accrual of the requisite mutations to become cancerous.8,9,10 Hence, cells endowed with the capacity for sustained proliferation are the likely precursors of tumors. This hypothesis gained traction and was bolstered with the discovery of Jacob Furth and Morton Kahn in 1937, which leukemia could be recapitulated in mice from single malignant cells.11

The Development of the CSC Theory. As early as 1855, in the discourse on the origins of tumors, Cohnheim posited that tumors stemmed from embryonic cells. Subsequent decades of genetic research concluded that tumor formation necessitates the accumulation of susceptibility genes, implying that the cells causing tumors must possess self-renewal capabilities. It wasn’t until 1937 that Furth demonstrated the potential of single malignant cells to induce tumors. This revelation spurred researchers to delve into the characteristics of such cells, encompassing self-renewal, aberrant differentiation, interaction with the microenvironment, and heightened plasticity. In 1997, John Dick identified leukemia stem cells. Since then, the theory of CSCs has basically taken shape. And people have begun to continuously isolate and prove CSCs from different tumor types
The understanding of this field deepened when James Till and Ernest McCulloch, in 1961, observed clone formation in the spleen during hematopoietic regeneration.12,13 Moreover, these clones could form additional clones in other mice, laying the groundwork for understanding the self-renewing and differentiative capabilities of stem cells.14,15 Schofield introduced the definition of “stem cell niche” in 1978, and further nuanced this, highlighting the role of microenvironment in nourishing and directing stem cells.16,17 From the 1960s to the early 1990s, debates about the origin of tumors had oscillated between non-genetic “induction” or “niche destruction” versus proliferative single-cell mutations.18,19,20
The landscape of stem cell research was revolutionized in 1997 when John and Bonnet first identified cells with extensive proliferative potential in acute myeloid leukemia (AML) and isolated CSCs characterized by the CD34+CD38− phenotype.2 This seminal breakthrough acknowledged the existence of leukemia stem cells and paved the way for the theory of “CSCs” in 2001.21 Tumors house a kind of rare cells with self-renewing potential named CSCs that drive tumorigenesis, akin to normal stem cells but with a role in cancer progression.22 The concept of CSCs has since expanded to various solid tumors. In 2003, Al-Hajj first isolated CD44+CD24−/low CSCs from breast tumors, capable of significant tumorigenicity in mice. Their study had shown that 200 such cells could form transplanted tumors in recipient mice within 12 weeks. In the same culture time, 10,000 non-special breast cancer cells cannot form tumors.23 In the same year, Sheila K Singh purificated a CD133+ CSC population from diverse brain tumors.23,24 The discovery of these potent CSCs across both hematologic and solid malignancies has substantiated the CSC theory, with subsequent findings in prostate, colorectal, pancreatic, nasopharyngeal cancers, and so on.25,26,27,28,29,30
Despite widespread support and experimental evidence for the theory of CSCs, xenotransplantation success rates and actual CSC percentages have fallen short of expectations.31 For instance, only a small fraction (<5%) of leukemia transplants in mice, as seen in Jacob Furth’s studies, successfully engrafted.11 Hewitt’s research further corroborates the scarcity of successful transplantation, with colony formation in murine spleens documented at a mere 1% to 4%.32 Moreover, Park et al. in vitro clonal cultures from myeloma cells extracted from murine ascites exhibited clonal colony formation in only 0.01% to 1%.33 Analogous low successful frequencies of tumorigenic cells are reported in vitro cultures of lung, ovarian, and neuroblastoma cancers.34 Concurrently, more and more research reveals the diversity and instability of CSCs, with variations in cell origin, proportion, genetic makeup, and even phenotypic and functional traits.35,36,37 Initially, CSCs exhibiting the CD34+CD38− phenotype were linked to the etiology of AML. Subsequent findings, however, indicated that CD34+CD38+ cells also possess tumor-initiating potential in NOD/SCID mice lacking the Interleukin 2 Receptor Subunit Gamma (IL2RG) chain, suggesting that such activity might be independent of CD38 expression.2,38 This phenomenon is mirrored in solid tumors, where certain CSC populations within the same tumor display distinct and non-overlapping marker profiles. Ginestier et al.‘s research in breast cancer revealed that cells exhibiting high Aldehyde Dehydrogenase (ALDH) activity not only demonstrated traits of tumorigenicity but also the capacity to self-renew and replicate the heterogeneity of the original tumor.39 These cells also displayed minimal overlap with the previously characterized CD44+CD24−/low phenotype breast CSCs, constituting less than 1% of the cancer cell population.39 Research suggests that CD133 is a marker capable of identifying CSC populations across various solid tumors, including different forms of brain cancer.40,41,42,43 However, subsequent studies have raised questions regarding the reliability of using CD133 to distinguish and isolate CSCs, indicating a degree of controversy in its application.44,45,46 Firstly, CD133 may serve as a marker for glandular epithelium in certain tissues, complicating the distinction between CSCs and non-stem-like cancer cells. Secondly, research has demonstrated that CD133+ cell populations fail to replicate the morphology of the original upon xenotransplantation, suggesting the possibility of expression of CD133 on normal differentiated cells. Lastly, studies have shown that CD133−/low populations have been shown to recapitulate the original tumor architecture, indicating that CD133 may not be the sole marker for identifying CSCs.47,48,49 Moreover, plasma cells expressing the CD138 phenotype were found to only induce multiple myeloma (MM) in SCID-hu mice, failing to generate comparable tumors in NOD/SCID mice.25,50,51,52 Similarly, analyses of samples from AML patients revealed distinct genetic and phenotypic characteristics of CSCs among individuals, highlighting the variability within the CSC population across different models and patient samples.53,54 The proportion of CSCs within primary tumors is also highly variable, ranging from 0.2% to 82.5%.35 For instance, CSCs with the CD34+ phenotype constitute less than 1% of AML cases, yet represent 82.5% in B-cell precursor acute lymphoblastic leukemia (ALL).2,38,55 Conversely, the proportion of CSCs expressing the CD133+ phenotype in lung cancer ranges from a mere 0.4% to 1.5%, while in brain tumors and colorectal cancer, it can escalate to as high as 20%.24,56,57 Furthermore, the frequency of CSCs may increase during tumor progression. Pece’s study found a higher proportion of CSCs in stage III breast cancer compared to stage I about 3 to 4 times on average.58 The origins of CSCs remain elusive, with evidence from myeloid leukemia and brain tumors suggesting they may arise from normal stem cells, while findings from MM and ALL suggest alternative origins.2,24,42,59,60 The definition of CSCs becomes increasingly nebulous, raising doubts about the model itself. However, the methods used at that time could not account for cellular heterogeneity and proliferative potential within different tumor cell populations.61,62 Moreover, there was heterogeneity in the analytical methods used.63,64 Thus, debates over the CSCs model will persist until direct empirical evidence is presented. Nonetheless, the validity of the model should not be discounted due to the diversity and complexity that continue to emerge in experimental evidence.
Advances in key technologies: from sorting to sequencing
The hypothesis of CSCs offers a pivotal theoretical framework for understanding tumor initiation and progression. Initially considered rare and dormant, forming a unidirectional hierarchy within tumors, CSCs were thought to generate all cell types within a tumor, occupying the apex of the tumor cell hierarchy.65 However, further research has revealed the model of CSCs to be more complex and dynamic. CSCs exhibit phenotypic plasticity, transforming in response to the microenvironment, leading to genetically heterogeneous tumors.66,67,68 Competitive interactions among various related but distinct subclones within tumors favor subpopulations with enhanced self-renewal capabilities and therapeutic resistance.35 Initial research propelled by traditional cell sorting techniques, advancements, especially in sequencing technologies, have continually enriched and evolved our understanding of the CSC model. The evolution of cell sorting technologies has progressed from utilizing physical properties of cells, such as size, density, adhesiveness, and refractivity, to targeting cell surface antigen phenotypes and functional characteristics like dye efflux, calcium ion concentration, and pH.69 Techniques include density gradient centrifugation, fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting (MACS), and side population (SP) cell sorting (Table 1).
While density gradient centrifugation was initially designed for isolating mononuclear peripheral blood cells, its application quickly extended to stem cell separation.70,71 Compared with a single-layer density gradient, the method of using a multi-layer discontinuous density gradient can separate CSCs more effectively. Percoll, a colloidal silica coated with polyvinylpyrrolidone (PVP), is preferred, though the toxicity of PVP in percoll limits its clinical safety.72
In isolating CSCs, methods based on cell surface markers are prevalent, notably FACS and MACS.73 MACS employs antibodies attached to magnetic beads to target cell membrane antigens, using magnetism to retain cells bound to beads within a column while unbound cells are washed away.74 Despite its minimal impact on cell viability and suitability for large-scale sorting, MACS is limited by its reliance on single antigens, complex operation, and high costs.75 FACS, on the other hand, utilizes fluorescently labeled antibodies to distinguish between CSCs and non-CSCs, offering higher specificity by screening multiple markers simultaneously. FACS can assess intracellular pathways and protein interactions and overcome the specificity challenges of CSC membrane antigens.76 However, FACS requires stringent experimental conditions and precise cell pretreatment to maintain cell viability, posing challenges in terms of equipment cost and operational requirements.75
The SP cell sorting method identifies CSCs using the Hoechst 33342 dye, capable of penetrating cell membranes.77 Since the discovery of Goodell et al. in bone marrow studies in 1996, this technique has proven effective across various tumor cell lines.78,79,80 SP cells, capable of asymmetric division and self-renewal, align with the characteristics of CSCs, suggesting SP sorting as a viable CSC enrichment strategy. Despite its simplicity, requiring only microscopy or flow cytometry to detect unstained cells, challenges include low separation efficiency and dye cytotoxicity. Nonetheless, its utility in sorting drug-resistant CSCs offers valuable insights for novel drug research. Researches indicated that when the activity of ABC transporters, such as ATP-Binding Cassette Transporter G2 (ABCG2), was inhibited, the SP phenotype cells decreased.81,82 Conversely, an increase in expression lead to an augmentation of the SP phenotype cells.83 Consequently, some researchers suggest that SP cells are not CSCs but rather a subset of cells capable of evading the cytotoxic effects of Hoechst dye.84
Beyond the aforementioned methods, alternative approaches for isolating and identifying CSCs exist.85 Drug selection separation gradually evolves cells towards drug resistance, isolating those capable of stable growth and passage, believed by some researchers to be CSCs.86,87 Western blot analysis serves as a traditional identification method, prized for its simplicity, universality, and cost-effectiveness, though it risks false positives or negatives if improperly executed, typically serving as a technique for validation. In 2021, Han et al. developed a novel label-free, microfluidic technology for CSC sorting based on physical characteristics like size, elasticity, and adhesiveness, enabling stable, rapid, and efficient CSC selection and enrichment, offering a new platform for targeted drug screening and functional identification.88
The integration of CSC sorting techniques with sequencing studies offers new insights into tumor complexity and heterogeneity. High-throughput technologies like RNA sequencing facilitate the monitoring of tumor microenvironment interactions and key gene expression dynamics. For instance, Chen discovered that under specific Trimethylation Of Lysine 4 On Histone H3 Protein Subunit (H3K4Me3) epigenetic modifications, the transcription factor MYC upregulates histidine decarboxylase, endowing glioblastoma stem cells (GSCs) with the ability to synthesize and secrete histamine. Histamine secreted by GSCs acts on the Histamine Type I Receptor (H1R) of vascular endothelial cells, activating the H1R/Ca2+/Nuclear Factor Kappa-B (NF-κB) signaling pathway to promote angiogenesis and advancing glioblastoma progression.89 The combination of advanced imaging, short hairpin RNA (shRNA) technology, and subgroup analysis tools has also highlighted the critical role of tumor-associated antigens in GSC differentiation.90,91 The development of multi-channel optical imaging systems has made it feasible to simultaneously monitor cell chemotaxis, proliferation, and NF-κB activity.92 In breast cancer, CSCs hyperactivate the Nuclear Respiratory Factors 2 (NRF2) pathway via the epigenetic reader Zinc Finger MYND-Type Containing 8 (ZMYND8), enhancing antioxidative capacity and evasion from oxidative damage and ferroptosis.93 Erythropoietin-Producing Hepatocellular Carcinoma Receptor B2 (EPHB2) and Lysine-Specific Histone Demethylase 1 (LSD1) are noted for their roles in promoting CSC generation and drug resistance in hepatocellular carcinoma and thyroid cancer, respectively. The sequencing technology found that they interact with the T Cell Factor 1(TCF1)/EPHB2/β-Catenin signal pathway and Wingless-Type MMTV Integration Site Family (WNT)/β-Catenin, respectively.94,95 Whole-genome sequencing of circular RNAs like circSLC4A7 has unveiled their interaction with Heat Shock Proteins 90 (HSP90), activating the Notch1 pathway and influencing gastric CSC progression.96 In addition to solid tumors, leukemia stem cells have also been found to undergo specific ribosomal RNA methylation (2’-O-methylation) modifications. This methylation pattern can reshape ribosome function and protein translation, allowing leukemia stem cells to preferentially translate amino acid transporters, which facilitates the cells’ uptake of amino acids in the environment, thus improving the self-renewal and function of leukemia stem cells.97 Sequencing studies were typically used to purify CSCs, focusing on molecular markers, which are involved in asymmetrical division, migration, and signaling pathways. Among them, MYC, Octamer-Binding Transcription Factor 4 (OCT4), Sex Determining Region Y 2 (SOX2), and ALDH are several key genes related to CSCs that are often focused on in research.98,99,100
The complexity of tumors transcends single malignant cells, encompassing a diverse array of cell types such as immune and stromal cells, thereby exhibiting significant intra- and inter-tumoral heterogeneity.101 While traditional transcriptomic analyses have provided valuable insights into tumor growth and evolution, they may overlook signals from crucial cell groups or states.102 These pivotal cellular states, including CSCs and immune cells relevant to treatment responses, are essential for understanding and treating tumors. To surmount this limitation, scientists are adopting advanced technologies like single-cell RNA analysis (scRNA-seq) and spatial transcriptomics. These methods offer a refined understanding at the cellular and molecular levels, unveiling new dimensions of complex interactions and heterogeneity within tumors, thereby opening new avenues for cancer research and therapeutic strategy development.103
Research into malignant gliomas has been at the forefront of single-cell analyses of brain tumor.104 Utilizing scRNA-seq, researchers have uncovered a spectrum of stemness and differentiation potential in primary glioblastoma cells, revealing the importance of expression programs like POU domain, class 3, transcription factor 2 (POU3F2), Nuclear Factor I A (NFIA), and NFIB in regulating stem-like phenotypes.105 Similar analyses of IDH mutant oligodendrogliomas and astrocytomas have disclosed comparable developmental hierarchies and gliogenic differentiation lineages, supporting the CSCs model. The model posits that the majority of cancer cells are well-differentiated, maintaining oligodendrocyte-like or astrocyte-like lineages, with a subset of undifferentiated cells exhibiting stem/progenitor traits.106,107 Interestingly, higher tumor grades are associated with an enrichment of proliferative stem-like glioma cells, suggesting a significant role for a minority of cancer cells in the growth and progression of IDH mutant gliomas.108,109 However, in primary H3K27M gliomas, lower differentiation correlates with a higher proportion of stem-like cells, indicating greater tumorigenic potential.110 Copy number variation (CNV) subclones and expression profiles inferred from scRNA-seq can also be used to study the relationship between genetic subclones and cellular state diversity within tumors.111,112 In IDH1 or IDH2 mutant human oligodendrogliomas, different CNV subclones exhibit similar cellular hierarchies, suggesting that cellular status is primarily determined by developmental programs.106 In contrast, IDH wild-type glioblastomas are characterized by four plastic and highly malignant cellular states, including neural progenitor cells (NPC-like), oligodendrocyte precursor-like cells (OPC-like), astrocytes like (AC-like) and mesenchymal-like (MES-like), and these states are not strictly determined by the CNV pattern.113
Beyond gliomas, CSCs-like subpopulations have been identified in other solid tumor types. In advanced prostate cancer, the growth of CSCs correlates with diminished androgen response and enhanced expression of cell cycle-related genes, promoting androgen-independent plasticity.114 In breast cancer, mesenchymal/stem-like tumor cells are present in patients who respond to Epidermal Growth Factor Receptor (EGFR) inhibitors, and an EGFR-high-expressing subpopulation displays enhanced stem-like characteristics, reflecting an EGFR-dependent hierarchy.115 Chung et al. found characterized expression features promoting metastatic progression in rare subgroups of primary triple-negative breast cancer via scRNA-seq, uncovering pronounced epithelial-mesenchymal transition (EMT) and stemness traits driving tumor advancement and metastasis.116 Similarly, metastatic breast cancer cells display overarching EMT and stemness characteristics, though with distinct marker gene expressions.117 The scRNA-seq analysis of hepatocellular carcinoma also reveals heterogeneity in phenotype, function, and transcriptome of CSCs.118 Velten et al. combined scRNA-seq with lineage tracing using nuclear and mitochondrial somatic mutations to identify leukemia stem cell gene expression programs in AML, marked by transcriptional dysregulation and co-expression of stem and myeloid priming genes.119 Differentiated AML cells express various immunoregulatory genes, inhibiting T-cell activity in vitro.120 In chronic myelogenous leukemia (CML), researchers identified unique molecular features of CSCs, revealing heterogeneity. A CSC subgroup in CML, characterized by distinct molecular traits, persists selectively during prolonged tyrosine kinase inhibitors (TKI) treatment, featuring quiescence-related gene expression and dysregulated genes and pathways.121 These insights deepen the understanding of cellular and molecular mechanisms underlying CML treatment resistance. Unlike other tumor cells, CSCs in head and neck tumors show extreme genomic instability, including chromosomal gains and losses.122 Ren et al. proposed a differentiation trajectory from CSC-like ductal cells to invasive ductal cells in pancreatic cancer, identifying five genes significantly associated with CSC prognosis.123 Wu et al. found common mutations in signaling pathway genes in different colorectal cancer cell clones, providing evidence for monoclonal CRC origin and subsequent subclonal evolution.124,125 Leung et al. demonstrated, through single-cell sequencing, exome sequencing, and targeted deep sequencing, that colorectal cancer metastasis follows a late dissemination model, with tumor cells evolving and acquiring mutations that enable clonal spread at the primary site.126
Research into CSCs is an evolving and deepening field. Despite challenges such as the lack of a clear definition for CSCs and the need for integrating various experimental methodologies, continuous research and technological advancements hold promise for a deeper understanding of cancer’s essence. This progress is anticipated to unveil novel strategies for cancer treatment, navigating through the complexities of tumor biology to illuminate new pathways for intervention.
Overview of cancer stem cells
Origin hypothesis of cancer stem cells
Differentiated cells
Dedifferentiation is a reversed process by which differentiated cells return to a less differentiated stage within the same lineage.127 Dedifferentiation represents a common biological phenomenon in several physiological processes, such as cardiac regeneration and wound healing.128 By dedifferentiation, cells can gain stem-like properties, such as self-renewal and pluripotency, so this process also implies CSC formation and tumorigenesis (Fig. 2).128 Taking advantage of scRNA-seq and lineage tracing techniques, a study reveals a trajectory of dedifferentiation that PROM-1+ hepatocellular CSC follows, which strongly supports the role of dedifferentiation in CSC formation.129

Origin, formation and/or maintenance of CSCs. CSCs originate from differentiated normal/cancer cells, stem/progenitor cells, or cell-cell fusion of cancer cells with stem cells or cancer cells with differentiated cells. The microenvironment of the CSC niche plays an essential role in the formation and maintenance of CSCs. MSCs, TAMs, MDSCs, and CAFs can secrete cytokines and chemokines that induce and/or maintain stem-like properties of cancer cells. Besides, CAFs can also modulate stemness by secreting EVs, and MSCs can regulate stemness through direct contact with CSCs. Finally, hypoxia and high nitric oxide (NO) concentration also support the CSC niche
Genetic or post-transcriptional alteration can lead to dedifferentiation of normal cells into CSCs. The combined loss of p16INK4a and p19ARF along with EGFR activation triggers the dedifferentiation of astrocytes and the genesis of glioblastoma.130 Besides astrocytes, terminally differentiated neurons can also undergo dedifferentiation to neural stem cell following shNF1-shp53 virus injection and induce gliomas.131 In intestinal crypts, ablation of Leucine-Rich Repeat-Containing G-Protein Coupled Receptor 5 (LGR5+) stem cells leads to dedifferentiation of daughter crypt cells and replenishment of stem cells, which is dependent on the transcription factor Achaete-Scute Homolog 2 (ASCL2).132 Mature pigment-producing melanocytes can also dedifferentiate into tumor progenitors of cutaneous melanoma induced by mutant BRAF.133 PGC7 induces promoter demethylation of transcription factors, such as GLI1 and MYCN, and facilitates dedifferentiation of hepatocellular cancer cells.134 Downregulation or loss of Bcl3 leads to dedifferentiation of pancreatic cancer cells and expansion of the CSC population.135 A study shows that RNA slicing also plays an important part in tumor cell dedifferentiation, as the splicing factor SRSF1 maintains stemness in colorectal cancer.136 Also, downregulated miR-613 expression is associated with liver cell dedifferentiation and CSC formation, which is mediated by increased SOX9 expression.137
Several signaling pathways are involved in the regulation of dedifferentiation in terms of CSC formation. For instance, activation of the WNT pathway and loss of Sterile Alpha Motif Domain (Smad4) drive differentiated intestinal epithelium to stem cell-like status and initiate colon cancer growth.138 The WNT pathway is also associated with the dedifferentiation of breast cancer bone metastases into CSCs.139 Activation of NF-κΒ leads to enhancement of the WNT signaling, which further supports dedifferentiation of intestinal villus cells and acquisition of stem cell markers and tumor-initiating capabilities of these cells.140 The activation of the Transforming Growth Factor β (TGF-β) signaling pathway can also convert colorectal cancer cells into CSCs, which is dependent on the transcription factor Twist-Related Protein 1 (TWIST1).141 The activation of Hypoxia-Inducible Factor 1α (HIF-1α)/Notch pathway leads to dedifferentiation of pancreatic cancer cells and formation of stem-like cells.142 Extracellular Signal-Regulated Kinase (ERK) inhibition promotes cancer cell dedifferentiation and expands the CSC population in non-small cell lung cancer (NSCLC).143 Fibroblast-released IL-6, activin-A, and Granulocyte Colony-Stimulating Factor (G-CSF) induce Signal Transducer And Activator Of Transcription (STAT3) and Smad activation, which consequently activate the WNT, Notch, and hedgehog pathways and induce dedifferentiation of lung carcinoma cells.144
Environmental factors, including hypoxia, cytokines, and NO, also relate to the dedifferentiation in CSC formation. Under hypoxia, glioma, lung cancer, and hepatoma cells express high levels of stemness-associated transcription factors and CSC markers.145 In lung adenocarcinoma, CSCs can be formed through dedifferentiation induced by Insulin-Like Growth Factor-II (IGF-II) secreted from cancer-associated fibroblasts, where the transcription factor Forkhead Box M1 (FOXM1) is involved.146,147 In nasopharyngeal carcinoma cells, Epstein-Barr virus (EBV) latent protein Latent Membrane Protein 1 (LMP1) induces dedifferentiation to form stem-like cells through transcriptional inhibition of CCAAT Enhancer Binding Protein Alpha (CEBPA).148 Exposure to progranulin leads to dedifferentiation of breast cancer cells and expansion of the CSC population.149 Exosomes from GSCs can cause dedifferentiation of surrounding non-CSCs by activating the Notch1 pathway.150 Finally, by stabilizing OCT4, a crucial transcription factor in CSCs, NO can induce the formation of lung or endometrial CSCs from differentiated cells.151,152
Non-malignant stem/progenitor cells
CSCs are generally functionally and structurally like normal stem cells, such as the ability of self-renewal and multipotent differentiation and similar transcriptional profiles.128 For instance, prostate CSCs share a conserved transcriptional program with normal prostate basal stem cells.153 Also, based on results from immunohistochemistry and double-fluorescence immunostaining, hepatocellular cholangiocarcinoma shares a similar set of markers with hepatic progenitor cells.154,155 These observations indirectly support that these CSCs can derive from tissue-resident stem/progenitor cells (Fig. 2).
Several studies have succeeded in the transformation from induced pluripotent stem cells (iPSCs) to CSCs. When cultured in a conditioned medium of mouse Lewis lung cancer,156,157 pancreatic carcinomas,158,159 hepatocellular carcinomas,160 or prostate cancer cell lines,161 iPSCs obtained CSC features and higher tumorigenicity in vivo. Similar results can be obtained by culturing iPSCs with Lewis cell-derived extracellular vesicles162 or recombinant human Fibroblast Growth Factor 2 (FGF2).163 Moreover, mouse embryonic stem cells can also get converted into CSCs in conditioned medium from mouse Lewis lung cancer or melanoma cells.164 These studies provide evidence that CSCs can be induced from normal stem cells, although iPSCs are not equivalent to normal somatic stem cells. Ewing Sarcoma Breakpoint Region 1 (EWS)-Friend Leukemia Integration 1 (FLI-1) fusion gene and miR-145 in human pediatric mesenchymal stem cells drive their reprogramming into CSCs by increasing the expression of SOX2.165 Although iPSCs do not fully represent adult stem cells, these studies show a possibility that CSCs can be induced from normal stem cells.
Some studies give more direct evidence that CSCs may originate from non-malignant adult stem cells. For instance, following Adenomatous Polyposis Coli (Apc) depletion, LGR5+ intestinal stem cells transform into CSCs, fueling the unimpeded growth of adenomas.166 Hepatocellular CSCs are found to derive from hepatic progenitor cells when the TGF-β or the WNT pathway is constantly activated in mice.167,168 Mouse primary hepatic stem/progenitor cells, when transduced with oncogenic genes, acquire CSC markers, self-renewal ability, and pluripotency.169,170 It is also notable that lineage-committed hepatoblasts and differentiated adult hepatocytes also gain stemness after the process.169 Finally, following deletion of Brca1, mouse mammary epithelial luminal progenitors get the ability to generate basal-like breast tumors.171
Cell-cell fusion
Cell-cell fusion is commonly involved in several physiological processes, including fertilization, muscle maturation, development of bones and placenta, and immune responses.172,173 For instance, a sperm and an egg fuse into a fertilized egg, a set of mononucleated myoblasts form a string of muscle fibers, trophoblasts fuse to form syncytiotrophoblasts, and several macrophages combine to make giant cells.172,173 Bone marrow cells can adopt the phenotype of other cells, such as embryonic stem cells, through cell-cell fusion.174 Similarly, cancer cells can also fuse with other cancer cells or non-malignant cells, forming tumor hybrid cells.175,176 Particularly, fusion of cancer cells with non-malignant cells often gives rise to their malignancy and potentiates tumor heterogeneity.172 For instance, melanoma cells can gain phenotypes of fibroblasts and monocytes by cell-cell fusion,177 and co-grafting of bone marrow-derived mesenchymal/stromal stem cells (BM-MSCs) and murine prostate cancer cells in vivo leads to enhanced tumor growth by cell-cell fusion.178 Clinically, a study suggests that the number of tumor hybrids (fusion of cancer cells and leukocytes) in peripheral blood correlates with cancer stage and patients’ survival.179
Given these properties of tumor hybrid cells, several studies support that cell-cell fusion can be one of the origins of CSCs (Fig. 2). MSCs have been recognized as an essential component in the tumor microenvironment and actively participate in tumor progression.180 Fusion between BM-MSCs or embryonic stem cells and cell lines of breast cancer, NSCLC, liver cancer, ovarian cancer, or gastric cancer upregulated their stem cell markers and enhanced tumorigenicity abilities in vitro.181,182,183,184,185,186 Similarly, hybrids from human/mice liver cancer, breast cancer, or lung cancer cells and BM-MSCs exhibited mesenchymal features and demonstrated enhanced stemness and metastatic capabilities in vivo.187,188,189 Besides solid tumors, cells of hematological malignancies, such as multiple myeloma, can also fuse with BM-MSC to gain stemness and stronger resistance to treatments.190 However, the fusion of BM-MSCs with cancer cells does not always produce CSCs, such as esophageal CSCs,191 indicating that cell fusion is not the only mechanism of CSC formation at least in certain tumor types. Plus, human umbilical cord MSCs can also fuse with gastric cancer cells to enhance cancer proliferation, migration, and stemness.192
In addition to MSCs, cancer cells can also fuse with other types of non-malignant cells and gain stem-like properties in the process. CD34+ liver CSCs can be formed by fusion of hepatobiliary stem/progenitor cells with CD34+ hematopoietic precursor-derived cells,193 suggesting that tissue-resident stem cells are also able to fuse with other cells and form CSCs. The fusion of prostate cancer cells with muscle cells increased the number of CD133+ stem-like cells.194 Hybrids from fusions of non-malignant human breast epithelial cells and breast cancer cells exhibit CSC properties,195 which is dependent on the transcription factor Zinc Finger E-Box Binding Homeobox 1 (ZEB1).196 Hybrids from tumor-associated macrophages and breast cancer cells also exhibit CSC phenotype and promote cancer metastases in a mouse model.197 Furthermore, CSCs can also fuse with other cells and obtain higher malignancy. For instance, hybrids of CSCs and monocytes gain highly invasive capacities,198 and those of BM-MSCs and SU3-RFP human glioma stem cells (GSCs) exhibited enhanced angiogenic effects compared to the parental cells.199
Additionally, a study shows that the fusion of two human lung fibroblast cell lines, E6E7 and RST, results in hybrids with elevated ALDH activity, which is a CSC marker.200 This study suggests that hybrids from two non-malignant cells may also lead to CSC formation.
Environmental factors in cancer stem cell formation and/or maintenance
It is commonly believed that CSCs reside in niches of tumors, and their microenvironment, which is generally characterized by hypoxia, aberrant angiogenesis, and chronic inflammation, has great impacts on the formation and maintenance of CSCs (Fig. 2).200
Hypoxia/angiogenesis
Hypoxia and aberrant angiogenesis have been identified as two crucial features of the tumor microenvironment.201,202 Tumor angiogenesis can be a consequence of hypoxia since hypoxia serves as a potent stimulus of Vascular Endothelial Growth Factor (VEGF) production, and disorganized vessels in tumors can aggravate hypoxia and vice versa.203 Under hypoxia, the HIF system is activated.204 And upregulated HIFs can promote the dedifferentiation of pancreatic cancer cells by activating the Notch pathway142 or that of melanoma cells by upregulating OCT4 expression.205 Also, hypoxia induces upregulation of SOX2, OCT4, KLF-4, Nanog, and Lin-28A, which are transcription factors contributing to dedifferentiation, and formation of stem-like cells in glioma, lung cancer, and hepatoma cells.145 Likewise, VEGF, an essential pro-angiogenic molecule, can interact with the VEGFR family or the neuropilin (NRP) family and promote stemness of skin/breast cancer cells and extend the CSC pool in the niche.206,207 Hypoxia can also indirectly regulate the stemness of cancer cells by altering functions of surrounding stromal cells, such as cancer-associated fibroblasts (CAFs)208,209 and myeloid-derived suppressor cells (MDSCs).210
Cancer-associated fibroblasts
CAFs are a group of interstitial cells of a mesenchymal lineage that are not epithelial, endothelial, or immune cells found in or adjacent to tumors.210 Compared to normal fibroblasts, CAFs are hyperproliferative and have a unique secretion pattern that contributes to tumor angiogenesis and metastases.211 Some believe that CAFs can also promote dedifferentiation and CSC formation by activating the WNT or the Notch pathway.4 WNT5a from surrounding CAFs induce dedifferentiation of ovarian cancer and gastric cancer cells and maintain the undifferentiated state of ovarian CSCs by activating a noncanonical WNT pathway.212,213
Moreover, several studies show that secretomes from CAFs promote the stemness of cancer cells. Head and neck squamous cell carcinoma and scirrhous gastric cancer cells express higher CSC markers when cultured in a CAF-derived conditioned medium compared to that from normal fibroblasts.214,215 CAFs secret IL-6, activin-A, G-CSF, and IGF-II that mediate the dedifferentiation of lung cancer cells into CSCs.144,146 Periostin from podoplanin-positive CAFs facilitates stem-like properties of gastric cancer cells by activating the Focal Adhesion Kinase (FAK)/Yes-Associated Protein (YAP) signaling.216 Leukemia Inhibitory Factor (LIF) and Gremlin 1 from CAFs can promote Nanog and OCT4 expression along with stem cell markers CD24−/CD44+ in breast cancer cells.217,218 CAF-derived HGF and IL-6 enhance the stemness of CD24+ liver cancer cells by activating the STAT3 pathway,219 and IL-6 from CAFs also induce Chromobox 4 (CBX4) expression, which is a CSC phenotype regulator, in skin squamous cell carcinoma.220 Additionally, Matrix Metallopeptidases (MMPs) from activated CAFs induce EMT and enhance the stemness of prostate cancer cells.221 However, when liver cancer or pancreatic ductal adenocarcinoma cell lines are cultured in a conditioned medium from CAFs, they have distinct expressions of CSC markers and aggressive phenotypes in a cell-line dependent manner,222,223 suggesting that effects of CAFs on stemness of cancer cells may vary depending on types and subtypes of cancers.
Moreover, CAF-derived exosomes also participate in CSC formation and/or maintenance. MiR-146a-5p in CAF-derived exosomes can promote the stemness of bladder cancer cells by activating the Mammalian Target Of Rapamycin (mTOR) pathway.224 CircHIF1A in exosomes from hypoxic CAFs can sponge miR-580-5p in breast cancer cells and increase their expression of CD44, which is a CSC marker for breast cancers.208 And small extracellular vesicles with low level of miR-7641 are associated with activation of the HIF-1α pathway, which promotes stemness of breast cancer cells.225 Likewise, loss of miR-34c-5p in exosomes from CAFs maintains the stemness of laryngeal cancer cells.226 In summary, CAFs regulate the stemness of cancer cells through paracrine mechanisms that involve cytokines or extracellular vesicles.
Mesenchymal stem cells
During tumor initiation and development, MSCs are believed to be constantly recruited to the tumor, making them an unneglectable group of cells in the tumor microenvironment (TME).227 Coculturing BM-MSCs with hypopharyngeal or prostate cancer cells induces expression of stemness markers in these cells,228,229 indicating MSCs can be not only the origin of CSCs but also an ally in CSC formation and maintenance. MSCs can induce fatty acid oxidation by upregulating mitofusin 2, a mitochondrial fusion-inducible factor, Carnitine Palmitoyl Transferase 1 (CPT1), and lncRNA MACC1-AS1 in gastric cancer, which finally leads to enhancement of stemness.230,231,232 Plus, the direct contact between MSCs and breast cancer cells upregulates the miR-199a in cancer cells, which subsequently represses the transcriptional regulator forkhead box P2 (FOXP2) and finally leads to higher stemness.233
Culturing colon cancer or melanoma cells in an MSC-derived conditioned medium increased the expressions of stemness markers of the cancer cells,234,235 indicating the secretomes of MSCs can induce stemness of cancer cells. Platelet-Derived Growth Factor (PDGF) from MSCs gives rise to ALDH+ CSCs in a model of ovarian malignant ascites.236 MSC-derived IL-8 can induce stem-like properties of gastric cells and blocking Programmed Cell Death-Ligand 1 (PD-L1) undermines this effect by reducing the expression of the transcription factor CTCF.237 Conditioned medium or just IL-6 from MSCs increases expression of stemness markers, such as OCT4, Nanog, and SOX2, via NF-κB activation in osteosarcoma cells.238,239 Prostaglandin E2 from MSCs also increases the level of ALDH-high CSCs in human colorectal carcinoma cells by activating the β-Catenin pathway.240 Exosomes from p53 deficient mouse BM-MSCs can internalize UBR2 into gastric cancer cells and increase their expression of CSC markers via the WNT/β-Catenin pathway.241 Adipose- and placenta-derived MSCs increase the proportion of CD133+/CD44+ colon CSCs via the IL-8/Mitogen-Activated Protein Kinase (MAPK) pathway.242
However, MSCs do not always facilitate the stemness of cancer cells. For instance, endometrium‑derived MSCs suppress the stemness of endometrial cancer by inhibiting the WNT/β‑Catenin signaling pathway.243 MSC-derived exosomes reduce the proliferation, migration, invasion, angiogenesis-stimulating, and self-renewal abilities of hepatocellular CSCs by inducing ERK phosphorylation244 or pancreatic CSCs by inhibiting the β-Catenin signaling.245 Altogether, MSCs are an important component in the TME, some of which can promote the stemness of their surrounding cancer cells through their secretomes or exosomes, while some MSCs may reduce the stemness of the surrounding cells.
Macrophages
Tumor-associated macrophages (TAMs) represent one of the most abundant groups of immune cells in tumors.246 Based on their immune functions, TAMs can be simply classified into the M1 proinflammatory phenotype and the M2 anti-inflammatory phenotype, although this classification neglects the great diversity of TAMs.246 ScRNA-seq shows that the maintenance of stemness of hepatocellular cells is mainly based on M2 polarization rather than the recruitment of TAMs.247 In the spleen of a murine chronic myeloid leukemia model, red pulp macrophages provide a niche for leukemia stem cells and support their stemness.248
Coculturing of TAMs and pancreatic cancer cells or culturing oral squamous cell carcinoma in an M2 macrophage-derived conditioned medium promotes their expression of stemness-related genes.249,250 Conditioned medium from TAMs promotes stemness of lung cancer cells by upregulating Ubiquitin-Specific Peptidase 17 (USP17), which subsequently disrupts the TNFR-Associated Factor (TRAF) 2/TRAF3 complex.251 These results suggest that the secretomes of TAM are essential in the process. TAM-derived TGF-β1 promotes stem-like properties of esophageal squamous cancer cells,252 glioblastoma cells,253,254 pancreatic cancer cells,255 prostate cancer cells,256 hepatocellular cancer cells,257 and breast cancer cells.258 Additionally, M2-TAMs secretory pleiotrophin enlarges the CSC group in human diffuse large B lymphoma by upregulating the β-Catenin expression.259 TAM-derived interleukin-1β, TNF-α, and IL-6 promote stemness of Doublecortin Like Kinase 1 (Dclk+) colon tuft cells and initiate tumor growth.260 TAM-derived IL-6 also enriches breast CSCs by activating the STAT3 signaling,261 and it also activates WNT and promotes stemness of ovarian cancer cells in 3D engineered microenvironments.262 M2-TAM-derived IL-8 induces stemness of ovarian cancer cells in vitro by activating the STAT3 pathway.263 Macrophages can also secrete IL-10 to promote stemness of NSCLC cells by activating the JAK1/STAT1/NF-κB/Notch1 signaling.264 IL-33 can also recruit macrophages into the TME and stimulate the secretion of prostaglandin E2, which subsequently supports stemness of colon cancer cells.265 Inhibitor Of Differentiation 1 (ID1) from TAMs can inhibit transcription of two stemness inhibitory factors, SerpinB2 and CCL4, and lead to stemness enhancement.266 Besides, LSEC in TAMs can enhance breast cancer stemness by binding to Butyrophilin Subfamily 3 Member A3 (BTN3A3) on breast cancer cells,267 suggesting that direct cellular contact of TAMs and cancer cells can also enhance stemness. TAM secretory S100 calcium-binding protein can induce stemness of hepatocellular cancer cells by activating the NF-κB pathway in a calcium-dependent manner.268 M2-TAM secretory VEGF or EGF promotes stemness of breast cancer cells by activating the VEGF/NRP-1/GTPase Activating Protein And VPS9 Domains 1 (GAPVD1) axis or the EGFR/STAT3/SOX2 signaling, respectively.207,269 M2-TAM-derived IGF-1 and IGF-2 promotes thyroid cancer stemness by activating the PI3K/AKT/mTOR pathway.270 Macrophage-derived glycoprotein nonmetastatic B induces the production of IL-33, an IL-1-like cytokine, via CD44 in a mouse lung cancer model, which in turn induces the CSC properties of these cells.271 Likewise, in head and neck squamous cell carcinoma, macrophage-derived hyaluronic acid (HA) induces activation of the PI3K/Eukaryotic Translation Initiation Factor 4E-Binding Protein 1 (EIF4EBP1)/SOX2 signaling via CD44 and increases the density of CSCs in vitro.272 M2-TAM-derived exosomal miR-27a-3p and the miR-17-92 cluster promote stemness of hepatocellular cancer cells by upregulating Thioredoxin-Interacting Protein (TXNIP) or disturbing the balance of the TGF-β1/Bone Morphogenetic Protein 7 (BMP-7) pathways.273
Several chemokines and chemokine ligands are involved in TAM-induced CSC formation or maintenance as well. TAM-derived Chemokine C-C Motif Ligand 2 (CCL2) activates AKT and increases the expression of β-Catenin in triple-negative breast cancer cell lines, which eventually induces their CSC properties.274 CCL8 promotes stemness of glioblastoma cells by activating ERK1/2.275 CXCL12 and TGF-β from M2 TAMs elevate DNA Topoisomerase II Alpha (TOP2A) expression and enhance stemness of hepatocellular cancer cells via the TOP2A/β-Catenin/YAP1 axis.276 Also, CXCL12 from M2 macrophages activate the WNT/β-Catenin pathway to facilitate stemness of colorectal cancer cells.277 Macrophage-derived CXCL7 fosters glioma stemness.278 Macrophage secretory IL-1β and CCL18 facilitate stemness of head and neck squamous carcinoma.279,280
Notably, M1 macrophages can also induce a subgroup of CD44high/CD24−/low or ALDH1+ breast CSCs in vitro, although prolonged coculture finally endows the macrophages with M2 properties.281 A study shows that breast CSCs respond more robustly to monocytes/macrophages than do differentiated non-stem cells through a juxtracrine mechanism, indicating monocytes/macrophages play an essential role in maintaining CSC niches.282
Myeloid-derived suppressor cells
MDSCs are a heterogeneous group of immune cells from the myeloid lineage that exert immunosuppressive effects.283 Coculture of ovarian cancer cells and MDSCs increases the expression of colony-stimulating factor 2 that activates the STAT3 and leads to upregulation of stemness markers.284 MDSCs can upregulate the expression of miR101 in ovarian cancer cells and subsequently repress the core-pressor gene C-terminal Binding Protein-2 (CtBP2), which restrains cancer stemness.285 Granulocytic MDSCs trigger piRNA-823 expression that promotes DNA methylation and maintains the stemness of multiple myeloma CSCs.286 Hypoxia can induce increased secretion of exosomes containing S100 Calcium-Binding Protein A9 (S100A9) from granulocytic MDSCs, which leads to enhanced stemness of colorectal cancer cells.210 MDSCs cultured in CAF-derived conditioned medium express a higher level of 5-Lipoxygenase (5-LO) that induces synthesis of leukotriene B4, which finally results in enhanced stemness of intrahepatic cholangiocarcinoma.287 MDSCs also endow stemness to breast cancer cells by secretory IL-6 and NO that activate the STAT3 and Notch pathways, respectively.288 The Notch pathway can also be activated by granulocytic MDSCs and contributes to stem maintenance in esophageal squamous cell carcinoma,289 and the STAT3 pathway is also activated in pancreatic cancer cells in the presence of MDSCs.290 MDSC-derived PGE2 also increases the stem cell-like properties in epithelial ovarian cancer.291
Besides, NO is frequently upregulated in cancers.292 NO disturbs the ubiquitin-mediated prosomal degradation of OCT4 and induces dedifferentiation of human lung cancer cells.151 NO also promotes stem-like properties of mouse glioma cells by activating the Notch pathway.293 Ionizing radiation is one of the inducers of CSCs’ formation across several cancer types,294 which will be discussed in more detail in the CSCs and sensitivity/resistance to radiotherapy section.
Features of cancer stem cells
Self-renewal and pluripotency
Since the first identification of CSCs in 1997,2 self-renewal and pluripotency have been considered two essential features of CSCs. This discovery comes from the observation that ALL cells are organized hierarchically, with a subset of cells that can replicate themselves and give rise to other malignant lineages, mimicking normal hematopoietic stem cells.2 In solid tumors, CSCs were first identified in breast cancer, in which the CD44+ CD24−/low lineage-cells underwent self-renewal and differentiation processes.23 These two properties of CSCs are also the basis to explain the formation of intratumoral heterogeneity in the CSC hypothesis or the hierarchical model of tumorigenesis4.
Cancer stem cells in cancer development
Based on the hierarchical model of carcinogenesis or the classical CSC hypothesis, CSCs, originating from normal stem cells or progenitors, are the cellular origins of cancers that can self-renew and give rise to the cellular hierarchies that explain the intratumoral heterogeneities.295 Nevertheless, the observation that some cancer cells can interchange between differentiated states and stem-like states does not favor this hypothesis.296 Plus, some cancers do not follow the CSC model.297 Therefore, although the terms tumor-initiating cell (TIC) and CSC have been used interchangeably, CSCs are not necessarily the cell origin of cancers based on the cellular plasticity model.4 That is, some malignant differentiated cells with oncogenic mutations can undergo dedifferentiation and form stem-like cells that cause intratumoral heterogeneities.4 However, this does not eliminate the role of CSCs in cancer initiation supported by many studies. AML cells originate from a subgroup of stem-like cells, as mentioned above.2 LGR5+ intestinal crypt stem cells, upon oncogenic mutations, serve as cells of origin of intestinal cancer.166 Injection of CD133+ human brain CSCs into non-obese diabetic, severe combined immunodeficient mice causes tumor formation, while that of CD133− does not.24 Conversely, deletion of SOX2, which is essential in maintaining the stemness of CSCs, decreases the formation of skin squamous-cell carcinoma.298 Also, decreasing MYC activity that sustains stemness of hepatocellular CSCs attenuates hepatocellular carcinoma initiation.299
CSCs are generally characterized by vigorous proliferation.3 Cancer proliferation is heavily dependent on the activation of the AKT, mTOR, and MAPK/ERK, which result in upregulated expression of proteins responsible for the cell cycle.300 The signaling pathways that involve these molecules are also major signaling pathways,5 which we will introduce in detail in the following section. Indeed, the acquisition of stemness is usually accompanied by enhancement of proliferation.301,302,303,304 Conversely, interventions that inhibit stemness also impair the proliferative potential of the cells.305,306
Cancer metastasis involves several biological processes that can be summarized into 5 essential steps, including cell escape, intravasation, survival maintenance, extravasation, and outgrowth.307 In epithelial malignancies, the EMT is a crucial event in metastasis.308 And the EMT can generate stem-like cells in human mammary epithelial cells,66 indicating a strong correlation between the EMT and CSCs. Molecularly, the WNT/β-Catenin, Notch, PI3K/AKT, hedgehog, and NF-κB signaling pathways are involved in the acquisition of mesenchymal properties of cancer cells. The pathways are also crucial in inducing and maintaining the stemness of CSCs.309
CSCs are often indicated as a reason for multi-drug resistance. This is partially attributed to their capability to maintain quiescence to avoid the therapeutic effects of anti-cancer treatments.310 For instance, CD13+ hepatocellular CSCs predominate in the G0 phase of the cell cycle and exhibit resistance to 5-fluorouracil treatment, as the mechanism of 5-fluorouracil primarily involves inhibition of DNA replication.311 In addition, CSCs can reduce intracellular accumulation of therapeutic agents by overexpressing ALDH and ATP-Binding Cassette (ABC) transporters.312 They also have better DNA repair capabilities and ROS clearance to avoid apoptosis induced by chemotherapy or radiation therapy stress.313 Finally, CSC supports an immunosuppressive niche that can exclude therapeutic agents and impair the efficacy of immunotherapy.4 Detailed mechanisms for CSC-induced chemoresistance, radioresistance, and resistance to targeted therapy and immunotherapy will be introduced in the following sections.
Biomarkers of cancer stem cells
One of the most efficient ways to identify CSCs in tumors is to use biomarkers for CSCs. Based on their cellular distribution, CSC markers can be classified into intracellular markers and cell-surface markers. Intracellular markers include transcription factors that function in the nucleus and markers found in the cytoplasm. Tables 2 and 3 summarize frequently used CSC markers in solid tumors and hematopoietic malignancies, respectively. Among them, generally accepted markers are introduced below.
OCT4, SOX2, and Nanog are the core transcription factors that regulate the embryonic stem cell state.314 OCT4, SOX2, and Nanog are encoded by the Sex-Determining Region Y (SRY) gene,315 the POU Domain, Class 5, Transcription Factor 1 (POU5F1),316 and the Nanog gene,317 respectively. They collaborate to positively regulate their promoters, activate the expression of genes necessary to maintain the embryonic stem cell state, and repress the expression of lineage-specific transcription factors.205,314 Similar stemness-maintaining functions of OCT4, SOX2, and Nanog have also been determined in adult stem cells.318,319,320,321 Expression of these transcription factors in cancer also endows stem-like properties to the cancer cells, unsurprisingly making them classical markers for CSCs.322,323,324 In addition, SALL4, encoded by a member of the Spalt-Like (SALL) gene family, SALL4,325 is also a transcription factor that regulates embryonic stem cell state by cooperating with Nanog.326 SALL4 expression is identified in several solid and hematopoietic malignancy types and correlates with CSC properties.325
Several cytoplasmic proteins are also identified as CSC markers. ALDHs refer to a group of enzymes that catalyze the oxidation of aldehydes to carboxylic acids, which can be further classified into 3 classes in mammals.327 Physiologically, ALDHs are present in most tissues of humans and have the highest concentration in livers, orchestrating drug metabolism.328 This also indicates an important role of ALDH in cancer drug resistance.329 ALDH activity has been considered a marker for not only normal stem cells but also CSCs of solid and hematopoietic malignancies.330
RNA-binding protein Musashi Homolog 1 and 2 (Musashi-1/2) are encoded by the MSI1 gene and the MSI2 gene, respectively.331 Both are RNA-binding proteins involved in post-transcriptional regulations of gene expressions and expressed in stem cells and progenitors to maintain their self-renewal.332 Musashi-2 also support hematopoiesis, which makes them a CSC marker in hematopoietic malignancies.333,334,335
Leucine Zipper-EF-Hand Containing Transmembrane Protein 1 (Letm1) is encoded by the Letm1 gene, which is a transmembrane protein located in the inner membrane of mitochondria and functions as a Ca2+/H+ antiporter.336 In gastric, colorectal, and lung cancer, studies reveal a positive correlation between Letm1 and stemness-related signatures.337,338,339 Furthermore, suppressing or elevating the Letm1 expression leads to inhibited or enhanced stemness of colorectal cancer or osteosarcoma cells, respectively.340,341
Alpha-Fetoprotein (AFP, α-fetoprotein) is encoded by the AFP gene in humans, which is produced by the fetal liver and the yolk sac. The serum level of AFP peaks during embryogenesis and rapidly decreases after birth but re-increases in the presence of hepatocellular cancer or germ cell tumors, making it an evaluable biomarker for these two types of malignancies.342 Cells with high AFP levels exhibit stem-like properties in pancreatic cancer, cholangiocarcinoma, and hepatocellular cancer, making it a potential CSC marker for these types of cancer.343,344,345
Polycomb complex protein BMI-1, also known as polycomb group RING Finger Protein 4 (PCGF4) or RING Finger Protein 51 (RNF51), is encoded by the BMI-1 gene. BMI-1 takes part in the repair of DNA double-strand breaks by homologous recombination346 and is essential for self-renewal in stem cells,347,348 which makes it also a marker for several types of solid tumor and hematopoietic CSCs.
Doublecortin-Like Kinase 1 (Dcamkl-1) is encoded by the DCLK1 gene,349 which is a microtubule-associated protein that was recently revealed to have a role in regulating inflammation.350 Also, a study reports that Dcamkl-1 marks intestinal CSCs but not normal CSCs, making it an ideal marker for colorectal CSCs.351
A large variety of cell-surface proteins can be applied as CSC markers for solid tumors. C-X-C Chemokine Receptor Type 4 (CXCR4), also known as CD184, is a CXC chemokine receptor encoded by the CXCR4 gene.352 The ligand for this receptor is CXCL12.353 CXCR4 is famous for its role as one of the receptors inducing the human immunodeficiency viruses (HIV) infection of T cells.354 CXCR4 is also involved in cancer progression for its role in activating the PI3K/AKT, PLC, hedgehog, ERK1/2, and JAK/STAT pathways.355
LGR5, also known as G-Protein Coupled Receptor 49 (GPR49) or G-Protein Coupled Receptor 67 (GPR67), is encoded by the LGR5 gene.356 LGR5 has been identified as a part of the WNT signaling complex to potentiate the WNT/β-Catenin signaling.357 Given the crucial role of the WNT signaling in cancer stemness, LGR5 has also been identified as a cell-surface marker for several solid tumor types (Table 2).
Epithelial Cell Adhesion Molecule (EpCAM), also known as CD326, is known for its role in cell-cell adhesion in the epithelia,358 but its roles exceed this in cancer. Upon cleavage, the intracellular domain of EpCAM forms a complex with FHL2 and β-Catenin, which, with interaction with Lef1, leads to transcription of oncogenes, such as c-Myc.359 Besides, EpCAM also facilitates EMT by inhibiting E-cadherin.359
CD24, also known as Heat Stable Antigen (HSA), is encoded by the CD24 gene in humans, which also functions as a cell-cell adhesion molecule.360 CD24 also mediates several signaling pathways that could lead to stemness enhancement of tumor cells.361 Likewise, CD44, also known as Homing Cell Adhesion Molecule (HCAM) and Phagocytic Glycoprotein-1 (Pgp-1)also induces cell-cell adhesion and interactions.362 It also takes part in activations of PI3K/AKT and Src/MAPK pathways and serves as a c-Met co-receptor.362 Both molecules can individually or combinedly mark CSCs in several solid tumor types. Moreover, the combination of CD44+/CD24− also marks CSCs in breast cancer, prostate cancer, head and neck squamous cell carcinoma, and ovarian cancer (Table 2).
CD133, also known as Prominin-1 (PROM1) and encoded by the PROM1 gene, belongs to the pentaspan transmembrane glycoproteins family.363 CD133 can activate the PI3K/AKT, Src, and β-Catenin signaling intracellularly to participate in cancer progression.363 CD133 is expressed in a wide range of human tissues and can serve as a CSC marker for various types of solid tumors and hematopoietic malignancies (Tables 2 and 3). The combined use of CD44 and CD133 as CSC markers has been reported in gallbladder cancer.364 Plus, CD44+/CD133− and CD44−/CD133+ cells both can represent CSCs in colorectal cancer.365,366
The differences between CSC markers for solid tumors and those for hematopoietic malignancies mainly lie in the variation of cell surface markers (Table 3) (Fig. 3). Interleukin-1 Receptor Accessory Protein (IL1RAP), encoded by the IL1RAP gene, is a receptor for interleukin-1.367,368 It has been identified as a CSC marker for myeloid leukemia.369 Similarly, CD25, a receptor for interleukin-2, and CD123, a receptor for interleukin-3, are also identified as CSC markers for AML or CML.370,371,372,373 CD70, expressed on the surface of various cells, and CD27, expressed on the T cell surface, are a pair of costimulatory molecules. The CD70/CD27 signaling is found activated in acute or chronic myeloid leukemia stem cells and contributes to the stemness formation of these cells by activating the WNT pathway.374,375,376 CD34+/CD38− is also identified as a marker for myeloid leukemia stem cells and has been widely used.2 Compared to myeloid leukemia, CSC markers for lymphoblastic leukemia are hardly reported. A study suggests that CD90 and CD110 correlate with stemness of ALL cells and might be a CSC marker.377

Biomarkers for CSCs in solid tumors and hematopoietic malignancies. Biomarkers for CSCs in solid tumors (left), hematopoietic malignancies (right), or both (center). The biomarkers can be classified into cell-surface markers and intracellular markers. Intracellular markers can be further classified into transcription factors that function in the nucleus and molecules that are found in the cytoplasm. Cell-surface markers make up the main differences between markers of solid tumors and those of hematopoietic malignancies
Notably, a single CSC marker or a pair of CSC markers might not be sufficient to identify CSCs. For instance, while CD133+, CD166+CD44+, and CD24+CD44+ phenotypes of human colorectal cells do not correlate with stem cell properties, these 3 sets of markers are reported as CSC-specific in colorectal cancer.378 Plus, ALDH1 alone does not correlate with stem cell-like features in hepatocellular cancer cells,379 but CD133+ALDH+ cells are significantly more tumorigenic than their CD133−ALDH+ or CD133−ALDH− counterparts,380 suggesting that combined use of CD133 and ALDH can better distinguish hepatocellular CSCs. Conversely, the absence of a CSC marker does not always indicate the absence of stemness. For instance, CD44− head and neck squamous carcinoma cells also have stem-like features, although CD44 is a CSC marker for this type of cancer.381 This phenomenon indicates that these CSCs may have distinct origins. Indeed, in glioblastoma, CD133+ and CD133− CSC respectively resemble fetal neural stem cells and adult neural stem cells, both of which exhibit stem-like properties.382 It is also noteworthy that certain CSC markers do not apply to every type of malignancy, even though it expressed in a wide range of tissues. For example, although ALDHs are present in most human tissues and represent a CSC marker for several cancer types, their activities play no functional role in stem cell-like properties in anaplastic thyroid cancer cells.383 Also, the CSC markers, CD133 and CD44, are generally overexpressed in gastrointestinal stromal tumors (GISTs) and cannot be used to distinguish CSCs from non-CSCs.384
Molecular regulations in CSCs
WNT/β-Catenin pathway
The WNT/β-Catenin signaling pathway, known for its involvement in various physiological processes and diseases, is evolutionarily conserved.385 Recent evidence highlights its crucial role in maintaining the stemness of CSCs. Chen et al. demonstrated its significance in converting mouse-iPSCs into CSCs.386 This pathway regulates stemness in CSCs across diverse cancer types, including lung, liver, thyroid, colorectal, cervical, and glioblastoma. For instance, in cervical cancer, cells with elevated Leucine-Rich Repeat-Containing G-Protein-Coupled Receptor 6 (LGR6) exhibit enhanced stemness, as LGR6 activates the WNT/β-Catenin pathway, forming a positive feedback loop with Transcription Factor 7-Like 2 (TCF7L2).387 Similarly, LSD1 maintains stemness in thyroid cancer by targeting Adenomatous Polyposis Coli 2 (APC2) or indirectly regulating Dickkopf WNT Signaling Pathway Inhibitor 1 (DKK1) via the HIF-1α/miR-146a axis to antagonize the WNT pathway.94 In liver cancer, EPHB2 sustains tumor stemness by activating the SRC/β-Catenin cascade. The WNT/β-Catenin pathway, in turn, upregulates EPHB2 expression in a TCF1-dependent manner, forming a positive feedback loop linked to liver CSCs.95 Furthermore, non-coding RNAs play a pivotal role in stemness maintenance. For example, Protein Kinase Membrane-Associated Tyrosine/Threonine 1 (PKMYT1) associated lncRNA sponges miR-485-5p to upregulate PKMYT1, inhibiting β-transducin repeat containing protein 1 (β-TrCP1)-mediated β-Catenin degradation and activating WNT signaling in NSCLC stem cells.388 Similarly, in liver CSCs, lncRNA Small Nucleolar RNA Host Gene 5 (lncSNHG5) activates the WNT/β-Catenin pathway by inhibiting Upstream Frameshift 1 (UPF1), sustaining stemness.302 Additionally, overexpression of LINC00839 in GSCs via Methyltransferase-Like 3 (METTL3)-mediated m6A modification enhances c-Src-driven phosphorylation of β-Catenin, activating WNT signaling and promoting stemness.389 Likewise, in colorectal cancer, Sec62, induced by METTL3-mediated m6A modification, enhances β-Catenin nuclear translocation, reducing its ubiquitination degradation and promoting cancer stemness.390
The involvement of the WNT/β-Catenin signaling pathway in CSCs contributes to malignant behaviors such as tumorigenesis and differentiation. Kim et al. showed that colorectal cancer cells expressing CD44 and CD133, markers of CSCs, exhibit strong tumor-initiating effects, accompanied by significant activation of the WNT/β-Catenin pathway.391 Furthermore, a CD44+Cellular Prion Protein (PrPc+) LGR4+ CSC subpopulation in colorectal cancer demonstrates high metastatic potential, with LGR4 and PrPC activating the WNT/β-Catenin pathway.392 Far Upstream Element-Binding Protein 1 (FUBP1) upregulation in colorectal cancer activates the WNT/β-Catenin cascade, enhancing stemness and potentially driving tumorigenesis.393 In breast cancer, Calreticulin (CALR) promotes a stem cell phenotype, with upregulation by HIF-1 activating the WNT/β-Catenin pathway to facilitate tumor initiation.394 Piwi-Like RNA-Mediated Gene Silencing 2 (Piwil2)-overexpressing cervical cancer cells exhibit strong stemness, partly attributed to the WNT/β-Catenin pathway, inhibition of which induces cell differentiation and suppresses tumorigenicity.395
The WNT/β-Catenin signaling pathway in CSCs is involved in the metastasis process. Husain et al. demonstrated that Farnesyl Dimethyl Chromanol (FDMC), an inhibitor of the WNT/β-Catenin pathway, suppresses the stemness and metastatic potential of colorectal CSCs, inducing their apoptosis.396 Colorectal cancer exhibits overexpression of Disheveled3 (DVL3), activating the WNT/β-Catenin/c-Myc/SOX2 signaling cascade, thereby enhancing stemness and metastatic potential.397 In gastric cancer, ST2+ serves as a functional marker of CSCs and activates the WNT signaling pathway, promoting metastasis through interaction with BCL-XL.398 Similarly, in pancreatic cancer, upregulated Frizzled-7 (FZD7) promotes CSC phenotype and liver metastasis via the canonical WNT/β-Catenin pathway.399 Additionally, polychlorinated biphenyls 2,3,5-trichloro-6-phenyl-[1,4]-benzoquinone (PCB29-pQ) activates the WNT/β-Catenin pathway, enhancing breast cancer stemness and metastasis.400
Most studies have consistently shown a positive correlation between the activation of the WNT/β-Catenin pathway and the malignant behavior of CSCs. However, in radioresistant glioblastoma, the expression of N-cadherin correlates positively with the inhibition of the WNT/β-Catenin signaling pathway. N-cadherin binds to β-Catenin in the cytoplasm, inhibiting neuronal differentiation mediated by the WNT signaling pathway and maintaining a stem-like phenotype.401 Conversely, in ameloblastoma, β-Catenin expression is negatively correlated with the CSCs’ marker SOX2. Exogenous activation of the WNT/β-Catenin signaling pathway leads to the inhibition of tumor stemness and invasiveness.402 These findings suggest that the role of the WNT/β-Catenin signaling pathway in CSCs is complex and may vary across different cancer types and states.
Hedgehog pathway
The classic hedgehog signaling pathway encompasses several cascades. Initially, Patched (PTCH) binds to the hedgehog ligand, relieving the inhibition of Smoothened (SMO). This event further facilitates the dissociation of the Suppressor of Fused (SuFu) from GLI, allowing GLI activators to regulate target genes.403 Yan et al. demonstrated that the interaction between glioma cells and endothelial cells activates the hedgehog pathway, promoting the transformation of glioma cells into a GSC phenotype.404 The regulation of the hedgehog pathway in CSCs is intricately linked to their emergence and various malignant biological behaviors.405
The hedgehog signaling pathway plays a pivotal role in maintaining the stemness of CSCs. Kelch Domain-Containing 8 A (KLHDC8A) has been identified in GSCs as an upstream factor involved in maintaining stemness by activating the hedgehog signaling pathway through ciliogenesis.406 Similarly, Liu et al. revealed the existence of the ISL1/sonic hedgehog (SHH)/GLI1 axis, which promotes GSCs’ stemness.407 The elimination of the liver CSCs’ stemness maintainer Ubiquitin-Like With PHD And Ring Finger Domains 1 (UHRF1) results in extensive DNA hypomethylation, ultimately upregulating CEBPA to inhibit the hedgehog pathway.408 Additionally, miR-324-5p weakens the function of multiple myeloma stem cells by inhibiting the hedgehog signaling pathway.409 Guen et al. demonstrated the connection between the EMT program and stemness, showing that tumor-initiating cells activate the hedgehog pathway through the EMT program to enhance stemness.410
Furthermore, the hedgehog signaling pathway is implicated in the tumor-initiating function of CSCs. In liver CSCs, the circIPO11/Topoisomerase 1 (TOP1)/GLI1 axis associated with liver cancer initiation has been identified. TOP1 is recruited to the GLI1 promoter by circIPO11 to activate the hedgehog pathway, promoting stemness and tumor initiation.411 Mok et al. revealed that cholesterol-related pathways are significantly upregulated in liver CSCs compared to normal stem cells. The hedgehog signaling pathway is activated in hepatic CSCs as a downstream factor for cholesterol synthesis mediated by the caspase-3/Sterol-Regulatory Element-Binding Protein 2 (SREBP2) axis, ultimately maintaining stemness and tumorigenicity.412 Similarly, TRNA Methyltransferase 6 (TRMT6)/TRMT61A-mediated N1-methyladenosine methylation in liver CSCs promotes cholesterol metabolism and activates the hedgehog pathway to maintain stemness and enhance tumorigenicity.413 In breast CSCs, the activated hedgehog signaling pathway is positively associated with stemness maintenance and tumorigenicity. Overexpression of Tetraspanin-8 (TSPAN8) relieves the inhibition of SMO by PTCH1 and phosphorylates SMO by promoting the binding of PTCH1 to SHH1, recruiting Ataxin-3 (ATXN3) to reduce the ubiquitination degradation of the SHH/PTCH1 complex, ultimately promoting GLI1 transcription.414 Additionally, Polypeptide N-Acetylgalactosaminyltransferase 1 (GALNT1)-mediated glycosylation of SHH in bladder cancer activates the hedgehog pathway, increasing the stemness and tumorigenicity of CSCs.415 Immunity may also play a significant role in influencing the effects of the hedgehog pathway in CSCs. IL-25, an intrinsic hedgehog pathway agonist, promotes CSCs’ function, increasing colitis-related tumorigenesis through the accumulation of GLI1.416
It is widely recognized that CSCs participate in the process of metastasis by activating the hedgehog signaling pathway.417 Upregulated Ubiquitin-Specific Peptidase 37 (USP37) in breast CSCs binds and stabilizes GLI1 to activate the hedgehog pathway, which further regulates the stemness and metastatic potential of CSCs.418 GLI1 was identified as a key regulatory gene for colorectal cancer stemness, and activation of the Hh/GLI1 signaling cascade was positively correlated with the invasiveness of colorectal CSCs.419 Disc Large Homolog 5 (DLG5), an activator of the hedgehog signaling pathway in glioblastoma. DLG5 prevents ubiquitination and degradation of GLI1 to promote the migration and stemness maintenance of GSCs.420
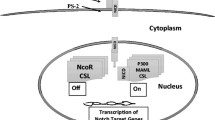
Notch pathway
The Notch pathway comprises several main components: the Notch receptor, Notch ligand, CBF-1, suppressor of hairless, Lag (CSL), DNA binding protein, and downstream target genes. Initially discovered by Drosophila,421 the Notch pathway has been shown to play a crucial role in promoting the formation of medulloblastoma stem cells.422 It is implicated in maintaining the stemness of CSCs, as evidenced by its upregulation in supratentorial ependymoma and mucoepidermoid carcinoma, where it correlates positively with the expression of CSCs’ markers.423,424 Additionally, syndecan-1 in inflammatory breast CSCs acts as a molecular marker maintaining their stem phenotype by activating the Notch pathway.425 While most studies support the positive relationship between Notch pathway activation and stemness maintenance, Högström et al. reported that upregulation of the Notch pathway attenuated the stemness of Prospero Homeobox 1 (PROX1+) colorectal cancer cells.426
Moreover, activation of the Notch pathway in CSCs has been associated with metastasis in various tumors such as breast cancer, glioma, renal cancer, and ovarian cancer. In breast cancer, Bone Morphogenetic Protein 4 (BMP-4) promotes stemness and EMT programs by activating the Notch pathway in a Smad4-dependent manner.427 Similarly, Signal Peptide CUB Domain And EGF-Like Domain Containing 2 (SCUBE2) overexpression in breast cancer cells enhances tumorigenicity and metastatic potential by activating the Notch pathway.428 Family With Sequence Similarity 129 Member A (FAM129A) prevents ubiquitination and degradation of Notch1, upregulating the Notch pathway to maintain the stemness and metastatic potential of GSCs.429 Notably, the upregulated Notch pathway in renal CSCs contributes to multiple malignant biological behaviors, including metastasis, stemness maintenance, and tumorigenesis.430 Additionally, glycosyltransferase GnT-III-mediated bisecting glycosylation of Notch1 effectively activates the Notch pathway, supporting stemness maintenance and metastasis.431
Activation of the Notch pathway in CSCs is associated with tumorigenesis, differentiation, and immune regulation. Liposarcoma cells with continuous activation of the Notch pathway exhibit overexpression of CSCs’ marker genes, leading to enhanced tumorigenesis compared to cells with normal Notch activity.432 Speckle-Type POZ Protein-Like (SPOPL), a stemness maintainer highly expressed in GSCs, activates the Notch pathway, thereby increasing tumorigenicity.433 Inhibition of the Notch pathway in GSCs induces significant neuronal differentiation and reduces stemness.434 Similarly, lncRNA FOXD2 Adjacent Opposite Strand RNA 1 (FOXD2-AS1) recruits TATA-Box Binding Protein Associated Factor 1 (TAF-1) to the promoter of Notch1, initiating the Notch signaling pathway in GSCs. Inhibition of FOXD2-AS1 induces the apoptosis and differentiation of GSCs while attenuating their stemness.435 Additionally, the Notch pathway plays a crucial role in immune system regulation. Expression of histone methyltransferase G9a in GSCs positively correlates with stemness characteristics. G9a binds to the Notch suppressor F-Box And WD Repeat Domain Containing 7 (FBXW7), upregulating the Notch pathway and enhancing the expression of PD-L1 in GSCs. This, in turn, weakens the function of T lymphocytes, creating an immunosuppressive microenvironment.436
NF-κB pathway
The NF-κB pathway, consisting of several cascades, is activated when cells encounter various stimuli, leading to the degradation of I-kappa B (IκB) protein by IκB kinase activation. This degradation releases NF-κB dimers, which are further activated through various post-translational modifications and translocated to the nucleus. There, they bind to target genes, promoting the transcription of these genes.437 NF-κB activation plays a critical role in the formation of breast CSCs.438 Pathway analysis of CSCs isolated from prostate cancer and NSCLC patient revealed the specific activation of the NF-κB pathway, suggesting its potential as an effective therapeutic target.439,440 Evaluation of the NF-κB signature in patient-derived GSCs can accurately predict the prognosis of low-grade glioma.441
Moreover, the NF-κB pathway is implicated in maintaining stemness. Calcium Calmodulin-Dependent Protein Kinase II γ (CaMKIIγ), identified as a marker of AML stem cells, maintains stemness by activating the 5-LO/NF-κB pathway.442 In ovarian CSCs, NF-κB pathway-related proteins are highly expressed, and inhibiting the NF-κB pathway reduces the CSC population.443 The lncRNA ASB16 Antisense RNA 1 (ASB16-AS1) cooperates with ATM kinase to phosphorylate Tripartite Motif Containing 37 (TRIM37), activating the NF-κB pathway and promoting gastric cancer cell stemness.444 Additionally, the Let-7a/Ras/NF-κB axis acts as a stemness antagonistic pathway in breast CSCs, with Let-7a inactivating the NF-κB pathway in a Ras-dependent manner.445 Overexpression of S100 Calcium-Binding Protein A4 (S100A4) activates the Inhibitor Of Kappa B Kinase (IKK)/NF-κB signaling pathway, contributing to the stemness maintenance of bladder CSCs.446
The activated NF-κB pathway in CSCs is intimately linked to tumorigenesis and metastasis. The transition from a proneural to mesenchymal phenotype (PMT) characterizes the conversion of less aggressive proneural GSCs into highly aggressive mesenchymal GSCs.447,448 Fos-Like Antigen 1 (FOSL1) has been identified as a key regulator of PMT, upregulating Ubiquitin-Conjugating Enzyme (UBC9) to enhance the SUMOylation of Cylindromatosis (CYLD). This process activates the NF-κB pathway, supporting the PMT program of GSCs.449 Similarly, Mixed Lineage Kinase 4 (MLK4) binds to phosphorylated IKKa, activating the NF-κB pathway and facilitating the transformation of GSCs into the mesenchymal phenotype.450 Upregulated BMI-1 in CD133+ liver CSCs enhances NF-κB activation and nuclear translocation, promoting CSC stemness and metastatic potential while inhibiting apoptosis.451 The estrogen metabolite 2-methoxy estradiol (2-ME2) disrupts the NF-κB/HIF-1 axis, reversing the EMT program and abolishing the metastatic potential of nasopharyngeal carcinoma stem cells.452 Stromal Cell-Derived Factor-1 (SDF-1) overexpression in breast cancer induces stemness and EMT phenotypes by activating the NF-κB pathway.453 Additionally, miR-221/222 inhibits Phosphatase And Tensin Homolog (PTEN), leading to NF-κB activation and enhanced stem cell characteristics, tumorigenesis, and metastasis in breast cancer cells.454 A positive feedback loop involving DiGeorge Syndrome Critical Region 8 (DGCR8)/circKPNB1/SPI1/DGCR8 promotes stemness in GSCs, with SPI1 upregulating the NF-κB pathway in a TNF-α-dependent manner, thereby promoting tumorigenesis.455
JAK/STAT pathway
The JAK/STAT pathway consists of three main components: tyrosine kinase-related receptors that receive signals, tyrosine kinase JAK that transmits signals, and transcription factors STAT.456 Upon binding of various stimulatory factors to the receptor, JAK is phosphorylated and activated, subsequently recruiting and phosphorylating the transcription factor STAT. This phosphorylated STAT then forms dimers and is translocated to the nucleus, where it binds to target genes, regulating downstream gene expression.457
Regulation of the JAK/STAT pathway is closely linked to the maintenance of stemness. Misra et al. demonstrated that selective inhibition of STAT3 significantly reduced the expression of stemness-related genes in breast CSCs.458 Alpha-casein acts as a STAT pathway antagonist, inhibiting the STAT3/HIF-1α axis and impairing the function of breast CSCs.459 Moreover, activation of the lipid metabolism-related STAT3/CPT1B/fatty acid β-oxidation (FAO) axis in breast CSCs correlates positively with stemness maintenance.460 Similarly, STAT pathway activation contributes to stemness maintenance in osteosarcoma, liposarcoma, and thyroid cancer.461,462,463 Immunity may play a significant role in JAK/STAT pathway regulation in CSCs. IL-17E/IL-25 secreted by non-CSCs binds to IL-17 Receptor B (IL-17RB) on CSCs, activating the JAK/STAT3 pathways to regulate liver CSCs’ stemness.464 Additionally, IL-6 is secreted by regulatory T cells, which upregulates the STAT3 pathway in glioma cells, maintaining the stemness-associated phenotype.465
Regulation of the JAK/STAT pathway in CSCs is intricately linked to tumorigenesis, metastasis, and metabolic reprogramming. Kanno et al. demonstrated that Von Hippel-Lindau (VHL) inhibits the JAK2/STAT3 signaling pathway, thereby reducing the tumorigenic ability of GSCs.466 In prostate CSCs, IL-6-mediated activation of the JAK/STAT pathway is a crucial event in tumorigenesis, and its inhibition eliminates tumor initiation.467 LIM Domain Only 2 (LMO2) acts as an endogenous agonist of the JAK/STAT pathway by forming a complex with LIM Domain-Binding 1 (LDB1) that phosphorylates STAT3, promoting the expression of ID1 and thereby upregulating the stemness and metastatic potential of GSCs.468 Leptin, an adipocyte-derived hormone, activates the JAK/STAT pathway in gastric cancer cells, maintaining their stemness and metastatic potential.469 Interferon-Induced Transmembrane Protein 3 (IFITM3), derived from GSCs, activates the JAK/STAT3 pathway to upregulate Basic Fibroblast Growth Factor (bFGF) expression, promoting angiogenesis in glioblastoma, a critical step in metastasis.470 Contrary to the STAT3/CPT1B/FAO axis, which is activated to maintain the stemness of breast CSCs, viperin overexpression in CSCs partially inhibits FAO through the JAK/STAT pathway, thereby reprogramming metabolism to promote tumor progression.460,471
TGF-β pathway
The TGF-β family ligands form a complex with receptors on the membrane, and the activated receptor kinase recruits and activates downstream Smad proteins, thereby inducing nuclear transfer of Smad proteins and exerting transcriptional regulation.472 The TGF-β pathway plays a pivotal role in embryonic development, immune surveillance, and maintenance of homeostasis.473 Dysregulation of the TGF-β pathway in CSCs is closely associated with the occurrence and progression of tumors.474 Nakano et al. demonstrated that stimulation of the TGF-β pathway triggers the conversion of CD44− non-colorectal CSCs into CD44+ colorectal CSCs. Moreover, sustained activation of the TGF-β pathway is crucial for colorectal CSCs to maintain an undifferentiated state.141 Similarly, activation of the TGF-β signaling pathway has been observed during breast CSC generation.475
Activation of the TGF-β pathway in CSCs is closely linked to tumorigenesis and stemness maintenance. The U2 Auxiliary Factor 65 (U2AF65)/circNCAPG/Ras-Responsive Element-Binding Protein 1 (RREB1) positive feedback loop was identified in GSCs, where U2AF65 binds to and stabilizes circNCAPG, thereby stabilizing RREB1 and promoting its nuclear translocation. Accumulated RREB1 activates the TGF-β1 pathway to maintain the stemness of GSCs and promote tumorigenesis.476 Similarly, Heat Shock Protein 47 (HSP47) induces the stemness and tumorigenesis of GSCs by activating the TGF-β pathway.477 Lymphoid Enhancer-Binding Factor 1 (LEF1) directly binds to and upregulates the expression of ID1, triggering the TGF-β pathway, which in turn promotes the stemness-associated phenotype and tumorigenicity of esophageal squamous cell carcinoma.478 Wang et al. discovered that CD51, a marker of colorectal CSCs, activates the TGF-β/Smad signaling pathway to support tumorigenesis.479 The Hematological And Neurological Expressed 1-Like (HN1L) overexpression triggers the TGF-β pathway by upregulating FOXP2, ultimately maintaining stemness and promoting tumorigenesis of prostate cancer.480
Activation of the TGF-β pathway in CSCs plays a pivotal role in tumor metastasis. Wen et al. demonstrated that targeted inhibition of the TGF-β/Smad pathway effectively eliminated the EMT program and metastatic potential of ovarian CSCs.481 FZD7 activates the TGF-β1/Smad3 pathway to confer stemness to pancreatic cancer cells. Further evidence suggests that upregulation of FZD7/TGF-β1/Smad3 promotes the EMT program to support pancreatic cancer liver metastasis.399 Similarly, Epithelial Membrane Protein 3 (EMP3) in lung CSCs interacts with TGF-β Receptor Type 2 (TGFBR2) to activate the TGF-β/Smad pathway, subsequently upregulating stemness and promoting the EMT program.482 Activation of the SIX Homeobox 1 (Six1)/Eyes Absent (EYA)/TGF-β pathway mediates CSC characteristics and EMT programs in breast cancer.483 Additionally, the interaction between miRNAs and the TGF-β pathway is a critical factor affecting tumor metastasis. MiR-495, identified as a stemness suppressor in oral squamous cell carcinoma, inhibits Homeobox C6 (HOXC6), thereby inhibiting the TGF-β pathway to prevent stemness characteristics and the EMT program of CSCs and induce their apoptosis.484 MiR-106b attenuates the expression of the inhibitory Smad protein Smad7 to trigger the TGF-β pathway and promote the EMT program.485 Angiogenesis is a crucial aspect of the metastatic cascade.486 Chen et al. identified Paired-Related Homeobox 1 (Prrx1) as a non-GSC stemness-promoting factor and a GSC stemness-maintaining factor in glioma. Prrx1 directly binds to the TGF-β1 promoter region to activate the TGF-β/Smad pathway, which in turn upregulates stemness and promotes vascularization in the tumor microenvironment.487
PI3K/AKT pathway
As a pivotal factor in the PI3K/AKT pathway, AKT undergoes structural changes and activation by PI3K, which subsequently modulates a cascade of downstream substrates to regulate various cellular behaviors.488 The mTOR is a classic downstream target of the PI3K/AKT pathway, while PTEN acts as a negative regulator by dephosphorylating AKT to suppress downstream signaling.489 The involvement of the PI3K/AKT pathway in driving the differentiation of normal stem cells into CSCs has been confirmed.490,491 Moreover, this pathway is closely associated with the maintenance of stemness in CSCs. Madsen et al. demonstrated a positive correlation between PI3K/AKT/mTOR pathway activation and breast cancer stemness score.492 Additionally, activation of the insulin/insulin-like growth factor signaling (IIS) pathway in breast CSCs further potentiates the PI3K/AKT pathway to sustain MYC expression, thereby enhancing the stemness traits of breast CSCs.493 Furthermore, PD-L1 contributes to the establishment of a suppressive immune microenvironment.494 Almozyan et al. revealed that the continuously activated PI3K/AKT pathway by PD-L1 is pivotal in maintaining the stemness of breast CSCs.495
Activation of the PI3K/AKT pathway in CSCs is intricately linked to tumorigenicity. Activation of the PI3K/AKT pathway and the MAPK/ERK pathway respectively promote and inhibit the stemness signatures and tumorigenic potential of lung cancer.496 The liver cancer tumor suppressor Connexin 32 (Cx32) attenuates the activity of the PI3K/AKT pathway, thereby suppressing stemness and tumorigenicity.497 The tumor suppressor miR-30a binds to and inhibits 5’-Nucleotidase Ecto (NT5E), thus downregulating the activity of the NT5E-mediated PI3K/AKT pathway, thereby impeding the stemness and tumorigenicity of GSCs.498 Non-coding RNAs also play a regulatory role in the PI3K/AKT pathway. Tumor suppressors miR-873 and miR-30a bind and inhibit patterns of Pleckstrin-2 (PLEK2) and NT5E respectively, leading to downregulation of the PLEK2 or NT5E-mediated PI3K/AKT pathway, thus hindering stemness and tumorigenicity of pancreatic CSCs and GSCs.498,499 Similarly, miR-3187-3p, which can be sponged by circ_0000745, inhibits Erb-B2 Receptor Tyrosine Kinase 4 (ERBB4), thereby attenuating the activity of the PI3K/AKT pathway, exerting a suppressive effect on the tumorigenicity and stemness of ovarian cancer.500
The activation of the PI3K/AKT pathway in CSCs is intricately linked to metastasis. AKT-mediated phosphorylation of Testis-Specific Y-Like Protein 5 (TSPYL5), a factor involved in stemness maintenance, impedes its ubiquitination and degradation. Phosphorylated TSPYL5 further inhibits negative regulators of the PI3K/AKT pathway, forming an AKT/TSPYL5/PTEN positive feedback loop that sustains the expression of stemness-related genes and promotes EMT programs.501 In head and neck squamous cell carcinoma, activation of the PI3K/AKT/mTOR pathway upregulates SOX2, promoting the maintenance of the stemness phenotype and the E-cadherin-mediated EMT program.502 Stress-Induced Phosphoprotein 1 (STIP1) in osteosarcoma enhances MMP-2 and MMP-9 by activating the PI3K/AKT and ERK1/2 pathways, ultimately promoting osteosarcoma CSC metastasis.503 In breast CSCs, Transmembrane And Coiled-Coil Domain Family 3 (TMCC3) binds AKT to activate the PI3K/AKT pathway, thereby supporting tumorigenesis and metastasis.504 CAFs are pivotal in supporting metastasis. CAFs upregulate TNF Receptor Superfamily Member 19 (TNFRSF19/TROY), a marker of liver CSCs, which activates the PI3K/AKT/T-Box Transcription Factor 3 (TBX3) pathway by promoting polyubiquitination of the PI3K inhibitory subunit p85α. Accumulated TBX3 maintains stemness and promotes metastasis.505 Similarly, CAFs-secreted periostin induces the phosphorylation of FAK to activate AKT, enriching CSCs in the gastric cancer cell population.216 Moreover, Liang et al. demonstrated that inhibition of the PI3K/AKT pathway attenuates the stemness characteristics and angiogenesis of endometrial cancer, which are closely associated with distant metastasis.506,507
PPAR pathway
PPARs, belonging to the nuclear hormone receptor family, are ligand-activated receptors that regulate various metabolic processes like fat and glucose metabolism. There are three main subtypes: PPARα, PPARδ/β (PPARD), and PPARγ (PPARG).508
The activation of the PPAR pathway plays a pivotal role in maintaining the stemness of CSCs. In liver CSCs, activation of the PPARα pathway and the enrichment of its downstream factor, Stearoyl-CoA Desaturase 1 (SCD1), contribute to the stemness characteristics.509 Similarly, increased PPARγ activity has been observed in melanoma stem cells.510 Co-culturing MSCs with gastric cancer cells leads to the enrichment of lncRNA Histocompatibility Leukocyte Antigen Complex P5 (HCP5) in MSC-stimulated gastric cancer cells, which sponges miR-3619-5p to promote the expression of PPARG Coactivator 1 Alpha (PPARGC1A). PPARGC1A accumulation triggers the PPAR Coactivator-1α (PGC1α)/CCAAT Enhancer Binding Protein Beta (CEBPB)/CPT1 axis, inducing FAO and stemness characteristics.230
Conversely, inhibition of the PPAR pathway has also been associated with maintaining CSC stemness. Activation of PPARγ induced by inhibiting TRAF2- and NCK-Interacting Protein Kinase (TNIK) correlates with the reduction of osteosarcoma cell stemness and their differentiation into adipocytes.511 Downregulation of PPARD in the acidic microenvironment of colorectal cancer inhibits Vitamin D Receptor (VDR) expression, promoting the emergence of a CSC phenotype.512 Similarly, PPARγ activation effectively inhibits the stem cell phenotype of bladder cancer.513 Moreover, besides fat metabolism, the PPAR pathway also regulates CSC characteristics through glucose metabolism. Low expression of PPARα in AML CSCs inversely correlates with their stemness characteristics. PPARα binds to HIF1α, inhibiting the expression of its downstream Phosphoglycerate Kinase 1 (PGK1) gene, ultimately weakening glucose metabolism activity and inhibiting stemness.514
Activation of the PPAR pathway in CSCs is closely associated with tumorigenicity, differentiation, and metastasis. GSCs exhibit overexpression of PPARα compared to normal neural stem cells. Knockdown of PPARα significantly reduces the expression of stemness-related genes and fat metabolism-related genes in GSCs, leading to a decrease in tumorigenicity.515 In hepatic CSCs, fatty acid 4-phenylbutyric acid (4-PBA) upregulates the expression of PPARα, preventing its degradation, thereby promoting the initiation and tumorigenicity of hepatic CSCs.516 N1-methyladenosine methylation-driven expression of PPARδ in hepatic CSCs activates the PPAR pathway, regulating cholesterol metabolism to maintain stemness and enhance tumorigenicity.413 Activation of PPARδ has been implicated in colorectal cancer liver metastasis induced by a high-fat diet, where it increases Nanog transcription.517 Moreover, PPARα activation is positively correlated with the invasive and stemness phenotypes of GSCs518. Conversely, stimulation of PPARγ may inhibit the migration ability of GSCs.519 Activation of PPARγ has also been shown to downregulate the stemness of brain CSCs and induce the expression of differentiation-related genes such as Collagen Type II Alpha 1 (COL2A1) and Motor Neuron And Pancreas Homeobox 1 (HLXB9).520 Similarly, PPARγ activation reduces the activity of SOX2 and YAP1 genes, inhibiting the stemness of osteosarcoma stem cells and promoting their differentiation.521
Molecular crosstalks
The formation and maintenance of CSCs involve complex interactions between multiple signaling pathways. For instance, SCD1 has been identified as a target for colorectal CSCs, inhibiting both WNT and Notch signaling pathways simultaneously to maintain the stemness-associated phenotype.522 Similarly, NK6 Homeobox 1 (NKX6-1) in leiomyosarcoma upregulates stemness by activating Notch and SHH pathways.523 Tumors with high expression of Notch and hedgehog signaling pathways exhibit stronger stemness, often associated with a hypoxic microenvironment and activation of regulatory T cells.524 Protein kinase CK2 activates AKT, NF-κB, and STAT3 pathways to maintain the stemness of AML cells.525 Breast CSCs overexpressing Cyclooxygenase-2 (COX-2) activate PI3K/AKT, Notch, and WNT pathways via E-type Prostaglandin Receptor 4 (EP4), contributing to breast cancer metastasis.526 Moreover, Frizzled10 (FZD10) activation in liver CSCs through N6-methyladenosine methylation mediated by METTL3 stimulates the WNT and Hippo pathways, critical for hepatic CSC self-renewal.527
The formation and the stemness of CSCs are supported by crosstalk between multiple pathways. IL-6 and NO secreted by MDSCs activate STAT3 and Notch signaling in breast cancer cells, collectively inducing CSC formation.288 Notch signaling can drive NF-κB pathway-related gene expression in skin CSCs (Fig. 4a).528 Notably, NF-κB pathway upregulation also activates the Notch pathway to support breast CSC expansion (Fig. 4b).529 Additionally, activated PPARγ inhibits the STAT5 pathway, downregulating HIF2α and Cbp/P300 Interacting transactivator with Glu/Asp-Rich Carboxy-Terminal Domain 2 (CITED2) expression, which are protectors of CML CSCs (Fig. 4c).530 Lastly, Breast Cancer Susceptibility Gene 1-Associated Protein (BRAP) inhibits the TGF-β/PI3K/AKT/mTOR pathway, weakening the stem cell properties of GSCs (Fig. 4d).531 LncROPM exerts a direct binding effect on Phospholipase A And Acyltransferase 3 (PLA2G16), thereby augmenting its expression and facilitating phospholipid metabolism. This process subsequently activates the PI3K/AKT, WNT/β-Catenin, and Hippo/YAP pathways to maintain the characteristics of breast CSCs (Fig. 4e).532

Crosstalk of signaling pathways in CSCs. a, b The Notch pathway can be activated by the NF-κB pathway while activating the NF-κB pathway. c PPARγ inhibits the STAT5 pathway to downregulate the expression of HIF2α and CITED2, ultimately attenuating the stemness characteristics of CSCs. d BRAP inhibits the TGF-β/PI3K/AKT/mTOR axis to reduce the stemness of CSCs. e LncROPM upregulates PLA2G16 expression to facilitate phospholipid metabolism, which subsequently activates the PI3K/AKT, WNT/β-Catenin, and Hippo/YAP pathways to maintain the stemness of CSCs. f Amplified miR-139 through the miR-139/PDE2A/Notch1 loop, inhibits the WNT pathway to attenuate the tumorigenicity of CSCs. g LINC00115, upregulated by the TGF-β pathway, sponges miR-200s to activate the ZNF596/EZH2/STAT3 axis to promote the stemness and tumorigenesis of CSCs. h Activation of the TLR4/NANOG axis subsequently upregulates the YAP1/SMAD3 and IGF2BP3/AKT/mTOR/SMAD3 pathways to inhibit the nuclear transfer and phosphorylation of SMAD3, ultimately attenuating the tumorigenicity of CSCs. i WNT/β-Catenin pathway downstream effector PROX1 inhibits each other with Notch1, thereby elevating the stemness of CSCs and hindering their differentiation. j PROX1, which can be activated by the WNT/β-catenin pathway, inhibits each other with Notch1, thereby enhancing the stemness of tumor cells and hindering their differentiation. k SOX9/PROM1 positive feedback loop in inhibits differentiation by activating the CSC program, which positively correlates with WNT pathway and negatively correlates with TGF-β pathway. l The PI3K/AKT pathway, activated by CBX7, further stimulating the NF-κB/miR-21 axis, and ultimately promoting the stemness characteristics and metastasis of tumor cells
The tumorigenicity, differentiation, and metastasis capabilities of CSCs are regulated by the intricate crosstalk among multiple signaling pathways. In GSCs, the miR-139/Phosphodiesterase 2 (PDE2A)/Notch1 loop inhibits stemness and tumorigenicity by suppressing WNT signaling (Fig. 4f).533 Chronic hypoxia-induced HIF-2α overexpression activates WNT and Notch pathways, leading to enhanced stemness-associated phenotype and tumorigenesis in breast CSCs.534 Additionally, LINC00115, activated by the TGF-β pathway, upregulates ZEB1 and Zinc Finger Protein 596 (ZNF596) expression to activate ZNF596/Enhancer Of Zeste Homolog 2 (EZH2)/STAT3, promoting the stemness and tumorigenesis of GSCs (Fig. 4g).535 Activation of the AKT and YAP pathways in CSCs may inversely correlate with tumorigenesis. In liver cancer-initiating stem cells, the Toll-like receptor 4 (TLR4)/Nanog/YAP1/insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) axis inhibits TGF-β pathway activity and tumor-initiating ability, which can be counteracted by TGF-β pathway activation (Fig. 4h).536 The interaction between the WNT pathway and hedgehog, Notch, and TGF-β pathways influences the differentiation of colorectal CSCs. Hedgehog signaling negatively regulates WNT signaling, while PTCH1-dependent non-canonical hedgehog signaling positively regulates WNT signaling, contributing to CSCs’ differentiation (Fig. 4i).537 PROX1, a downstream effector gene of the WNT/β-Catenin signaling pathway, can inhibit each other with Notch1, thereby enhancing the stemness of colorectal cancer cells and hindering their differentiation (Fig. 4j).426 Moreover, the SOX9/PROM1 positive feedback loop in colorectal cancer inhibits differentiation by activating the stem cell program, positively correlating with WNT pathway activation and negatively correlating with TGF-β pathway activation (Fig. 4k).538 In lung CSCs, the HIF-1ɑ/miR-1275 axis co-activates WNT/β-Catenin and Notch pathways, enhancing stemness and metastatic potential.539 In gastric cancer, Chromobox Protein Homolog 7 (CBX7) upregulates the PI3K/AKT pathway, activating the NF-κB pathway to promote miR-21 expression, enhancing the CSC-associated phenotype and metastasis (Fig. 4l).540
Clinical predictive values of CSCs
Using CSC markers, investigators have revealed a negative correlation between the presence of CSCs and patients’ survival in various types of cancers.541 Moreover, given the close correlation between CSCs and multi-drug resistance, it is also reasonable to use CSC as a parameter to predict patients’ prognosis after a specific type of anti-cancer treatment.
CSC markers can be used to predict response and survival after chemotherapy. In 47 patients with esophageal squamous cell carcinoma who receive neoadjuvant chemotherapy followed by radical esophagectomy, those with a high pre-chemotherapy expression of CD133 have significantly shorter survival compared to those with low CD133 expression, while the difference in survival is not significant between patients with high CD44 expression and low CD44 expression.542 This study also reveals that CD44high/CD133high expression is associated with significantly poorer survival compared to those with CD44low or CD44high/CD133low, suggesting that the combined use of CSC markers can provide better predictive values.542 In 112 patients with advanced NSCLC treated with platinum-based chemotherapy, high Nanog levels were independently associated with shorter PFS (hazard ratio (HR) = 3.09, 95% confidence interval (CI) 2.01−4.76) and OS (HR = 3.00, 95% CI 1.98−4.54).543 Likewise, overexpression of CXCR4 correlates with poorer PFS and OS in 124 patients with epithelial ovarian cancer receiving cisplatin-based chemotherapy.544 Some studies also investigated the predictive value of single-nucleotide polymorphisms (SNPs) of CSC markers, which can influence the transcription, translation, and splicing of these proteins.545 For instance, LGR5 rs17109924 is associated with prolonged time to recurrence (TTR) (HR 0.38, 95%CI 0.19−0.79; P = 0.006) based on data from 391patients with colon cancer treated with adjuvant 5-fluorouracil-based chemotherapy.546 However, this correlation does not occur in patients treated with surgery alone, indicating that the correlation is mainly attributed to the impact of LGR5 rs17109924 on adjuvant chemotherapy.
CSC markers or stemness-related gene signatures also correlate with response to radiation and can be used to predict patients’ prognosis after radiotherapy. A study suggests that the CSC marker CD44 expression can be used to predict local recurrence of larynx cancer based on data from 19 patients.547,548 Patients with rectal cancer and high expressions of CSC markers, CD133, OCT4, and SOX2, are prone to develop distant recurrence compared to those with low expressions of the genes.549 A systemic review identifies a series of CSC markers, including CD133, CD44, ALDH1, LGR5, and G9a, as indicators for the prognosis of patients with rectal cancer receiving radiotherapy.550 Using a machine learning method and data from the TCGA database, a model based on five tumor stemness and immune-related signatures, including Carbamoyl Phosphate Synthetase I (CPS1), CCR2, NT5E, Anillin (ANLN), and ATP-Binding Cassette Sub-Family C Member 2 (ABCC2), demonstrates favorable predictive values predicting radiotherapy responses.551 A study suggests that P16INK4A expression is negatively associated with CSC markers and predicts poor survival of patients with cervical cancer after radiotherapy.552 GSC markers, CD133 and O6-methylguanine-DNA methyltransferase, are also associated with patients’ responses to radiotherapy.553
Some studies attempt to predict patients’ response to or survival after targeted therapy and immunotherapy using CSC markers or stemness-related gene signatures. For instance, a study shows that head and neck squamous cell carcinoma patients with low CD44, a CSC marker, have a significantly better HR for OS than those with high CD44 expression when comparing nimotuzumab plus cisplatin-radiation (NCRT) with cisplatin-radiation (CRT), suggesting that CD44 might be a favorable reference for whether to use nimotuzumab.554 Using a five tumor stemness and immune‐specific‐gene (CPS1, CCR2, NT5E, ANLN, and ABCC2) signature, a study constructs a machine-learning model that can predict therapeutic responses in melanoma patients receiving adoptive T cell therapy (area under ROC curve (AUC) = 0.717) and immune checkpoint blockades (AUC = 0.703).551 The AUCs of this signature were higher than those of PD-1 (0.687 for adoptive T cell therapy and 0.505 for immune checkpoint blockades).551
Cancer stem cells and tumor chemotherapy resistance
The clinical significance of chemotherapy resistance
Chemotherapy remains the cornerstone of current clinical oncology, offering significant reductions in tumor burden and enhanced patient survival, thereby securing its widespread clinical application.555 However, patients initially responsive to chemotherapy inevitably evolve into drug resistance. This phenomenon, termed acquired resistance, poses a significant challenge to both clinicians and researchers. Research has progressively revealed that acquired resistance is intricately linked to intratumoral heterogeneity.300 Chemotherapy selectively eliminates sensitive subpopulations, allowing resistant cells to prevail and drive disease progression. The contribution of CSCs to intratumoral heterogeneity and acquired resistance has been a focal point of research for decades. This chapter explores the mechanisms through which CSCs mediate resistance and their ramifications for chemotherapy strategies.
Mechanism of resistance of CSCs to chemotherapy
In cancer biology, CSCs exhibit dynamic states of proliferation and quiescence. During dormancy, CSCs reduce their metabolic activity, enabling prolonged survival in a quiescent state. Upon extracellular stimulation, however, CSCs may re-enter the cell cycle, regaining proliferative capacity.556,557 This duality poses significant challenges for chemotherapy, as quiescent CSCs exhibit resistance to such treatments, and often develop more resistant phenotypes.558,559,560 This resistance is largely attributed to the mechanism of conventional chemotherapy, which targets rapidly dividing cells and acts in a cell cycle-specific manner (Fig. 5).561 However, CSCs, with their slow division rates, often residing in the G1 or S phase, exhibit resistance to a variety of chemotherapeutic agents including cisplatin, taxol, and doxorubicin.562,563 For instance, the overexpression of Zinc Finger E-Box-Binding Homeobox 2 (ZEB2) increases the proportion of colorectal CSCs in G0/G1 phase, leading to platinum resistance.564 Distinguishing quiescent CSCs from proliferative CSCs remains a challenge due to the lack of specific surface markers and common genotypic and phenotypic characteristics.565 CD13 has been proposed as a marker for quiescent hepatic CSCs, which have been proven capable of neutralizing chemotherapy-induced ROS and DNA damage.311 Moreover, epigenetic modifications also play crucial roles in regulating the quiescent state of CSCs. For example, SET Domain-Containing Protein 4 (SETD4) promotes breast CSC dormancy through the trimethylation of histone H4 lysine 20, facilitating heterochromatin formation.566 Elevated levels of miR-135a reduce the methylation at the CG5 site of the Nanog promoter by directly targeting DNA Methyltransferases 1 (DNMT1). Then, the combination of SET And MYND Domain Containing 4 (SMYD4) and unmethylated Nanog promoter will activate the expression of Nanog in those Nanog-negative tumor cells, thus promoting the switch of CSCs.567 Endothelial cells, by expressing miR-126, can induce dormancy in CML stem cells. Concretely, by targeting the PI3K/AKT/mTOR signaling pathway, miR-126 blocks the cell cycle progression of CSCs.568,569 Soluble growth factor/receptor pathways, such as CXCL1/CXCL12, Bone Morphogenetic Protein-4 (BMP4), and LIF, have also been shown to regulate the quiescent state of activated CSCs. For instance, CXCL1 induces liver CSC quiescence via mTORC1 kinase activation, while knocking out CXCL12 downregulates quiescence-associated genes, such as TGF-β and STAT3, facilitating the exit of leukemia stem cells from dormancy.570,571 BMP4 directly regulates the quiescent state of CML leukemia stem cells through a JAK/STAT3 pathway dependent on BMPR1B kinase activity, and the LIF Receptor (LIFR) correlates with the expression of quiescence-associated genes in CSCs, such as TGFβ2 and Notch1. Knockout of LIFR promotes the proliferation of breast CSCs and enhances their capacity for bone destruction.572,573 More and more evidence suggest the involvement of extracellular vesicles (EVs) in CSC quiescence regulation. EVs from CAFs of hormone therapy-resistant breast cancer patients promote estrogen receptor-independent oxidative phosphorylation and hormone therapy resistance.574 Furthermore, CAFs create a resistant niche through close interactions with CSCs, secreting factors like IL-6 and IL-8 that support CSC survival.575 In colorectal cancer, CAF-derived EVs trigger resistance to 5-fluorouracil in CSCs, which is the standard of care.576 Endothelial cells can promote resistance in GSCs through the secretion of NO, enhancing Notch signaling, or by releasing CD44 ligands.

Mechanism of resistance of CSCs to chemotherapy. CSCs possess the ability to maintain a quiescent state and reduce metabolic activity, thereby exhibiting resistance to chemotherapy. Furthermore, CSCs are capable of metabolic reprogramming, utilization of ABC transport proteins, and activation of DNA repair pathways, which allows them to evade chemotherapy. Additionally, the microenvironment plays a crucial role in supporting CSC survival. The balance between ROS and anti-apoptotic versus pro-apoptotic signals, along with exosomes secreted by tumor-associated fibroblasts, dynamically regulates CSCs
The HIF pathway emerges as one of the most pivotal regulators of the quiescent state in CSCs.577 With the identification of quiescent CSCs in MM, the expression levels of TRIM44 were elevated. This E3 ubiquitin ligase facilitates the deubiquitination and stabilization of HIF-1α under both normoxic and hypoxic conditions, underscoring the intricacy of oxygen sensing in tumorigenesis.578,579 Furthermore, the significance of HIF2α in the stability and transformation of CSCs in glioblastoma also highlights the critical role of oxygen levels in CSC biology.580 The markers of CSCs such as OCT4, Nanog, SOX2, Krüppel-Like Factor 4 (KLF4), c-Myc, and miR-302 are induced in hypoxic environments further supporting the adaptive responses of CSCs to oxygen deprivation.581 Notably, hypoxia not only modulates cell plasticity but also stimulates the proliferation and expansion of pre-existing CSC pools, suggesting a dynamic interplay between CSC quiescence and activation.582,583,584 In conclusion, CSCs exhibit long-term stability and a quiescent phenotype under hypoxia, characterized by low metabolism and reduced oxidative phosphorylation. Conversely, the presence of oxygen triggers the activation of tricarboxylic acid (TCA) cycle enzymes and oxidative phosphorylation, transitioning CSCs into a proliferative state.585,586
Metabolic reprogramming stands as one of the hallmarks of the bioenergetics of CSCs.587 ALDH enzymes serve as potential inducers of metabolic reprogramming, thereby promoting chemotherapy resistance. For example, ALDH enzymes mitigate aldehyde accumulation by converting them into less toxic carboxylic acids, which play a crucial role in detoxification within CSCs.588 Moreover, ALDH enzymes maintain a low level of ROS by consuming these reactive aldehydes induced by ROS.589 The increased activity of ALDH1A1 and ALDH3A1 subtypes enables CSCs to metabolize cyclophosphamide and its analogs, such as 4-hydroperoxycyclophosphamide, ifosfamide, and etoposide, and detoxify their intermediate products aldophosphamide into carboxyphosphoramide.590,591 ALDH also contributes to the synthesis of retinoic acid and neurotransmitter γ-aminobutyric acid (GABA), essential for the homeostasis and differentiation of CSCs.330,592 Inhibition of ALDH activity using all-trans retinoic acid (ATRA) significantly improves prognosis in leukemia, highlighting the enzyme’s involvement in cell differentiation and survival pathways including Notch, mTOR, and PI3K/AKT.593,594,595 The mediating role of ALDH activity in therapy resistance has been established across various cancers, including breast, pancreatic, lung, Ewing’s sarcoma, stomach, glioblastoma, head and neck, ovarian, and colorectal cancers in recent years. Types of chemotherapy agents covered include doxorubicin, paclitaxel, gemcitabine, gefitinib, temozolomide, doxorubicin, and platinum, implicating it as a key marker of CSC drug resistance.596,597,598,599,600,601,602,603,604 Among the 19 ALDH family members, ALDH1 is considered most closely associated with CSCs.605,606 However, the complex role of ALDH in CSC biology and therapy resistance warrants further investigation.
The ABC transporter superfamily, encoded within the human genome, represents the largest group of transmembrane proteins. These transporters are categorized into seven subfamilies, ABC-A to ABC-G, based on the similarity or disparity of their domain structures.607 Recent studies have revealed a significant upregulation of ABC transporters in CSCs, highlighting their pivotal role in mediating chemotherapeutic resistance by extruding harmful toxins and xenobiotic compounds from cells, thereby reducing intracellular drug concentrations.83,608 ABCB1, also known as Multidrug Resistance Protein 1 (MDR1) or P-glycoprotein, was the first member of the ABC transporter family identified in humans.609 Wright et al. demonstrated that ABCB1 expression serves as a crucial marker for doxorubicin resistance in breast CSCs.610 Various members of the ABC transporter family, especially ABCB1, ABCC1, and ABCG2, are recognized for their heightened expression in CSCs and their involvement in MDR mechanisms.611,612,613 Certain cancers, such as melanoma, might exhibit specific ABC transporter profiles, with ABCB5 playing a significant role.614,615 From the structure and properties of ABC transporter, ABCB1, characterized by two ATP-binding sites, exhibits enhanced drug transport capabilities.616 Distinct ABC transporters have been implicated in various chemotherapeutic resistances. For example, ABCC1 is primarily associated with resistance to anthracycline drugs, whereas ABCG2 exhibits the broadest spectrum of drug resistance.617,618,619 New roles for ABC transporters in CSCs have been uncovered. ABCB5 possesses the ability to regulate IL-8-dependent CSC maintenance in melanoma and promote the invasion of tumor cells in colorectal cancer. ABCG2 also plays a role in the enhancement of CSC tumorigenic potential.83,620 It should be noted that the ABC transporter family is intimately linked with signaling pathways. The ABCB1 gene promoter contains multiple targets for the β-Catenin complex, suggesting a reciprocal relationship where the WNT/β-Catenin signaling pathway targets ABCB1 activity.621 Activation of the WNT/β-Catenin pathway can induce ABCB1 expression, facilitating chemotherapy resistance in CSCs.622,623 ABCG2 is also involved in the WNT/β-Catenin signaling cascade.613,624 And its expression can be regulated by the Notch pathway as well.625 Furthermore, the Hippo pathway effector YAP1 promotes the drug resistance of CSCs through ABCG2.626,627 The PI3K/AKT pathway regulates ABCG2 in GSCs at the plasma membrane, instead of the mTOR pathway.628 Inhibition of the PI3K/AKT pathway results in the downregulation of ABCG2 in CML cells.629 Despite these insights, clinical successes with specific ABC transporter inhibitors remain scarce, underscoring the ongoing need for mechanistic exploration.
Several studies have elucidated that the mechanisms of DNA-damaging chemotherapeutic agents underlying tumor cells, which include DNA crosslinkers (cisplatin, carboplatin, oxaliplatin), DNA synthesis inhibitors (methotrexate), and topoisomerase inhibitors (doxorubicin, daunorubicin).630 These agents predominantly target the S phase of tumor cells, where DNA replication occurs, exploiting the diminished DNA repair capacity in tumor cells, which culminates in genomic instability and subsequent apoptosis. Notably, in CSCs, DNA damage checkpoints are activated, facilitating repair mechanisms that enhance cell survival. Sequencing data reveal an upregulation in the majority of DNA damage response and repair genes within CSCs, indicating a superior DNA repair efficiency.23,631 The p53 signaling pathway and apoptosis are pivotal to DNA damage repair. Upon detrimental DNA damage, the ATM and Ataxia Telangiectasia And Rad3-Related (ATR) kinase complex with Poly ADP-Ribose Polymerase 1 (PARP-1) and BRCA1, phosphorylate CHK1 and CHK2, thereby activating p53, which leads to cell cycle arrest, DNA repair, or the execution of apoptosis.632 Remarkably, genes like p53, which induce cell death, often harbor mutations or are dysregulated in CSCs, and inhibiting p53 aggregation can restore sensitivity to platinum-based treatments.633 DNA repair proteins directly or indirectly linked to CSCs’ drug resistance include CHK1, CHK2, ATR, MSI1, RAD50, and RAD51, with RAD51 playing a significant role in resistance to PARP inhibitors.634,635,636,637
Furthermore, CSCs can prevent DNA damage through effective ROS clearance.638 A highly compatible ROS scavenging system has evolved in the CSCs of some tumors to maintain low ROS concentrations. Antioxidant enzymes like superoxide dismutase, glutathione peroxidase, and catalase are markedly active in CSCs.639,640,641 NRF2, a transcription factor, mediates CSC drug resistance by not only regulating the expression of genes involved in the cellular antioxidant response, but also by stimulating drug efflux through raising ATP Binding Cassette Subfamily F Member 2 (ABCF2) expression among other functions.642,643 ROS overload or elevated ROS levels induced by chemotherapy have been implicated in the activation of HIFs, which can trigger the activation of pro-survival and developmental pathways such as Notch, WNT, and hedgehog. These pathways in turn contribute to the sustenance of CSCs’ survival.644 Additionally, studies have identified a negative feedback loop between ROS and COX-2 within CSCs, where elevated ROS levels induce COX-2 expression, which in turn mitigates ROS accumulation, fostering CSC enrichment and metastasis.645,646 Autophagy, a critical biological process for cellular homeostasis, has been recognized as a pivotal resistance mechanism in metastatic prostate CSCs, significantly contributing to ROS scavenging.647 To sum up, CSCs exhibit heightened sensitivity to any alteration in the oxidant/antioxidant balance, acquiring resistance under both low and elevated ROS levels.
One of the primary approaches of chemotherapeutic agents is the induction of apoptosis.648 The balance between pro-apoptotic (BCL2-Associated X Protein (BAX), BCL2 Antagonist/Killer (BAK), BCL2 Asociated Death Promoter (BAD)) and anti-apoptotic (BCL2, BCL-XL, MCL1) proteins constitutes a focal point of cellular response to apoptosis.649 In CSCs with chemotherapy resistance, the balance tips towards anti-apoptotic proteins. It has been shown that compared to tumor cells, CSCs exhibit higher levels of anti-apoptotic gene expression (such as BCL2, BCL-XL).650,651 Knockdown of the biomarkers of CSCs, such as CD44, increases apoptosis, evidenced by elevated expression of pro-apoptotic proteins BAX and caspases-3, -8, and -9, while the levels of anti-apoptotic proteins BCL2 and BCL-XL decrease.652 Further analysis by Konopleva et al. demonstrated that the overexpression of anti-apoptotic genes BCL-XL and BCL2 could also induce a quiescent state in CSCs.653 Moreover, the upregulation of specific cell surface receptors (such as EGFR, Fibroblast Growth Factor Receptor (FGFR), HER2R) in CSCs can inhibit apoptosis by downregulating the pro-apoptotic protein BAD.654 Additionally, CSCs can evade apoptosis by prolonging the G2/M phase in the cell cycle through upregulation of G2/M checkpoint proteins CHK1 and CHK2.650 Some CSCs overcome apoptosis by upregulating the expression of Inhibitors Of Apoptosis Proteins (IAPs).655 The expression of Cellular FLICE-Like Inhibitory Protein (C-FLIP) also affects the apoptosis receptor initiation pathway, inhibiting caspase activation and thereby hindering the apoptotic process. Studies have shown that different splice variants of c-FLIP are associated with resistance to chemotherapeutic drugs.656,657 CSCs employ various indirect or direct mechanisms to evade apoptosis, such as endoplasmic reticulum stress. It has been discovered that several components directly involved in endoplasmic reticulum protein processing are dysregulated in CSCs.658 In vitro CSC models observed the inactivation of IRE1 (XBP-1 splicing) and the activation of the PERK (elF2α phosphorylation) pathway, both key conduits of the endoplasmic reticulum stress response.659 Mitochondrial integrity is crucial for the survival and maintenance of CSCs, with its dysregulation having profound effects on autophagy and apoptosis.660 Studies indicate mitochondrial alterations in CSCs of CML compared to normal stem cells. Resistant CSC subpopulations can be identified by higher mitochondrial mass and increased endopeptidase activity.661
In conclusion, the chemoresistance mechanisms of CSCs constitute an interactive network (Fig. 5). Targeting individual components may not eliminate the resistance posed by CSCs. To devise accurate CSC-targeted treatments that enhance sensitivity, further exploration in the domain of resistance is warranted.
Clinical trials targeting CSCs combined with chemotherapy
With a profound understanding of the pivotal role CSCs play in chemotherapy resistance, researchers have initiated a series of targeted clinical trials aimed at exploring potential therapeutic strategies for CSCs (Table 4). These trials broadly fall into three categories. The first category, guided by the ChemoID assay, identifies subsequent treatment regimens using patient biopsy samples before treatment to enrich stem cells and test their response to chemotherapy drugs. Results from a phase III clinical trial, exemplified by NCT03632135, demonstrated a significant reduction in patient mortality risk in the ChemoID assay-guided group, suggesting that the ChemoID assay could become a routine diagnostic and treatment method akin to genetic testing in the future. The second category involves the development of specific inhibitors targeting mechanisms by which CSCs contribute to chemotherapy resistance. This category encompasses most clinical trials, such as those using vismodegib or PF-04449913 to inhibit the hedgehog pathway, LGK974 targeting the WNT pathway, and OMP-52M51 against DLL4 in the Notch pathway. Although γ-secretase inhibitors are the largest group of drugs targeting the Notch pathway, trial outcomes have not been disclosed yet. Additionally, the development of the drugs RO4929097 and PF-03084014 has been halted for various reasons. NCT04137627 evaluated melatonin as an antioxidant in combination with neoadjuvant chemotherapy for changes in tumor stemness expression in oral squamous cell carcinoma, but results showed no statistical difference despite a reduction in miR-210 and CD44 expression, implying the tumor microenvironment might play a role in CSC resistance mechanisms but may not be the dominant factor. In theory, PARP inhibitors involved in DNA repair could also be effective against CSCs, but current clinical trials have not measured changes in CSCs or biomarkers before and after treatment, necessitating further exploration of their effect on CSCs. The third category targets markers specific to CSCs for treatment, such as CD44v6. Bivatuzumab mertansine, an antibody-drug conjugate (ADC) targeting CD44v6, enhances the specificity of chemotherapy and has shown promising results in various cancer treatments. However, its effectiveness against CSCs, as indicated by NCT02254005, remains inconclusive. These results indicate that we cannot yet conclusively determine whether targeting CSCs can reverse chemotherapy resistance. We look forward to the anticipated outcomes of these clinical trials in the coming years.
Cancer stem cells and tumor immunotherapy resistance
CSCs and immune evasion
Immune cells within the tumor microenvironment play a pivotal role throughout the oncogenesis and progression of tumors. Unlike their counterparts in normal tissues, these immune cells often exhibit attenuated inflammatory responses or enhanced suppressive functions, thereby facilitating tumor immune evasion. This section aims to provide an overview of the principal roles played by TAMs, MDSCs, NK cells, T cells, and B cells in mediating immune escape of CSCs. Their complex interplay and the mechanisms through which they contribute to the immunological cloak that shields CSCs from the host’s immune defense are critical to understanding and developing novel therapeutic strategies.
Research across various tumor types has revealed that TAMs often constitute up to 50% of the immune cell population, positioning them at one of the forefront research fields of immune cells within the microenvironment.662 The heterogeneity of TAMs emerges as a critical mechanism behind immunotherapy resistance. Engagement of damage-associated molecular patterns (DAMPs) with specific pattern recognition receptors on macrophages, such as TLR4, triggers pro-inflammatory signaling and polarization towards the M1 phenotype.663 M1-TAMs, characterized as classically activated macrophages, exhibit enhanced pathogen phagocytosis capabilities, thereby exerting anti-tumoral properties. However, the tumor microenvironment promotes polarization towards the M2-TAMs, which possess pro-tumoral potential, supporting and sustaining CSCs and therapy resistance through the secretion of chemokines and activation of stemness pathways, such as sonic hedgehog ligands. For instance, GSCs can recruit M2-TAMs by secreting periostin.664 CSCs may also elevate M2–TAM levels through chemokines like CCL2 and macrophage colony-stimulating factor 1 (CSF1).665 Drug-resistant lung CSCs activate the Interferon Regulatory Factor 5 (IRF5)/M-CSF pathway to promote the production of M2-TAMs from CD14+ monocytes.666 In turn, M2-TAMs secrete factors like milk fat globule-EGF factor 8 (MFG-E8), activating STAT3 and sonic hedgehog signaling in CSCs, thereby enhancing treatment resistance.667 Moreover, TAMs secrete substantial amounts of TGF-β1, maintaining CSC characteristics and promoting EMT.257,668 Lu et al. demonstrated that CSCs undergoing EMT upregulate CD90/Thy1 and EphA4, crucial proteins mediating physical interactions between CSCs and TAMs. EphA4 receptor activation secretes Src and NF-κB, inducing CSCs to secrete various cytokines maintaining stem cell status.282 ScRNA-seq has clarified the bidirectional feedback mechanisms between CSCs and TAMs. CSCs secrete S100A11 protein to promote TAM polarization towards the M2 phenotype, which in turn enhances CSC self-renewal and metastatic capabilities.247 The crosstalk between CSCs and macrophages is intricate, wherein CSCs not only polarize macrophages towards a tumorigenic state but also employ protective mechanisms to avoid macrophage phagocytosis. Elevated expression of CD47, observed in CSCs from both hematological malignancies like AML and solid tumors like pancreatic, liver, and lung cancers, interacts with Signal Regulatory Protein α (SIRPα) on TAMs, broadcasting a “don’t eat me” signal to protect them from macrophage engulfment.669,670,671,672,673,674 Recent studies have also identified TAMs as “iron donors” within the tumor microenvironment, fulfilling the high iron demand of CSCs and playing a crucial role in influencing iron homeostasis.675 Although the mediators involved in this crosstalk may vary with tumor pathology, such interactions may one day become potential therapeutic targets against CSCs.
Tumor-infiltrating myeloid cells represent a heterogeneous lineage that includes TAMs, MDSCs, and so on, the latter being a focal point of research due to their impact on limiting the efficacy of immunotherapy.676 MDSCs, immature myeloid cells derived from the bone marrow, are categorized into polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs), expressing CD15 and CD14, respectively.677,678 These cells exert immunosuppressive effects through distinct mechanisms, with the ratio of PMN-MDSCs to M-MDSCs in peripheral blood being crucial.679 In murine models of melanoma, prostate, and cervical cancers, CSCs promote PMN-MDSC infiltration by overexpressing G-CSF, CXCL5, and TGFβ.680,681,682 In turn, PMN-MDSCs increase STAT3 phosphorylation, CD133 and CD44 expression, and sphere formation of colorectal CSCs in vitro by secreting S100A9 protein.210 PMN-MDSCs also enhance the ratio of CSCs, spheroid-forming ability, and expression of stemness-related genes in myeloma cells by inducing piRNA-823 expression.286 Moreover, studies have identified M-MDSCs as primary drivers of the CSC phenotype in pancreatic and breast cancers.288,290 In breast tumor models, M-MDSCs comprise the majority of tumor-infiltrating MDSCs. Mechanistic analysis has shown that NO produced by M-MDSCs promotes the CSC phenotype through activation of Notch signaling and sustained STAT3 phosphorylation in cancer.288,677 The relationship between CSCs and MDSCs is bidirectional, as CSCs also recruit MDSCs to limit T cell activity, creating a favorable environment for tumor growth. MDSCs in peripheral lymphoid organs are predominantly PMN-MDSCs, which exhibit relatively mild immunosuppressive activity compared to M-MDSCs. PMN-MDSCs primarily produce high levels of ROS to exhibit immunosuppressive activity, which are unstable and transiently, requiring antigen-specific interactions with T cells to ultimately induce tumor-specific T cell tolerance.683 In contrast, M-MDSCs produce substantial amounts of NO, arginase 1, and immunosuppressive cytokines with longer half-lives, effectively inhibiting nonspecific T cell responses without the need for direct contact between MDSCs and T cells.679 It is noteworthy, however, that despite functional annotation and transcriptomic profiles widely recognizing PMN-MDSCs as distinct from inflammatory neutrophils, tumor-associated neutrophils, and PMN-MDSCs share overlaps in markers and suppressive functions, suggesting a close phenotypic and functional relationship.684
T cells are the most pivotal immune effector cells in the anti-tumor response, executing cytotoxic effects on tumor cells through classical pathways such as perforin/granzyme release, death receptor engagement, and induction of apoptosis.685 Studies have revealed that CSCs evade T cell-mediated immune rejection by downregulating key components of the antigen processing and presentation machinery and suppressing T cell anti-tumor functionality.686 Tumor antigens are broadly classified into two categories: (1) tumor-specific antigens (TSAs), encoded by mutated or rearranged genes, and (2) tumor-associated antigens (TAAs), encoded by genes specific to the normal cellular lineage.687 CSCs may selectively avoid expressing differentiation-related TAAs, thus resisting T cell-mediated rejection.688,689 Another mechanism of immune evasion involves the downregulation or loss of MHC-I by CSCs.690 As MHC-I play a crucial role in immune recognition, their absence or reduced expression can limit T cell-mediated lysis of CSCs.689,691 The induction of T cell tolerance by CSCs is another key strategy in escaping immune surveillance. A primary mechanism of tolerance induction involves the clonal deletion of antigen-reactive T cells through apoptosis or death, with the Factor-Related Apoptosis (Fas)/Fas-L pathway serving as a significant mediator.692,693 CSCs may actively destroy T cells through the expression of Fas-L, moreover, the autocrine secretion of soluble Fas-L protects CSCs from cytotoxic T cell-mediated Fas killing.693,694,695 Furthermore, CSCs can evade immune attack by downregulating Fas.693 The tumor-expressed ligand Receptor-Binding Cancer Antigen Expressed On SiSo Cells (RCAS1) has also been found to induce apoptosis in T, B, and NK cells expressing its receptor.696 Immunogenic tolerance can be achieved through non-deletional processes, such as the inactivation of antigen-reactive cells.697 The secretion of TGF-β underpins the inhibition of T cell proliferation mediated by MSCs.698 CSCs produce TGF-β and IL-10, directly suppressing T cells to avoid immune-mediated destruction.699,700,701 The TGF-β signaling pathway is also specifically activated in CSCs, with secreted morphogens of the TGF-β superfamily and their receptors preferentially expressed by CSCs.702,703,704 T cell activation in the immune response requires two signals.705 The first signal comes from the T Cell Receptor (TCR) recognizing the MHC/antigen peptide complex, conveying an antigen-specific recognition signal.706 The second signal is provided by co-stimulatory molecules of antigen-presenting cells (APCs), offering a non-specific synergistic co-stimulation signal.707 CSCs may reduce T cell responsiveness to tumor antigens by actively modulating the activation state of APCs and may express negative co-stimulatory molecules to disrupt anti-tumor immune responses.708,709 PD-1/PD-L1-mediated negative co-stimulatory signal transduction is the most common way of inhibiting lymphocyte activation.710,711 Other mechanisms include the induction or active recruitment of regulatory T (Treg) cells, which can effectively suppress the activation, proliferation, and cytokine production of other T cells, crucial for maintaining immune self-tolerance and homeostasis.712,713,714,715 In summary, CSCs employ a multitude of processes to drive tumor escape from immune-mediated rejection responses.
In the era of immune checkpoint inhibitors (ICIs) and adoptive T cell therapies, the pivotal role of T cells in anti-tumor immunity has become indisputable. However, these advancements have also exposed numerous limitations of T cells, underscoring the urgent need to unravel immunological mechanisms. With the advancement of scRNA-seq technology, the subpopulations and states of B cells within the tumor microenvironment are increasingly scrutinized. For instance, in melanoma, genes associated with early B cell stages are extensively expressed.716 In breast cancer, B cells predominantly exist as naive B cells, memory B cells, with fewer plasma cells and germinal center cell clusters observed. Notably, compared to peripheral blood B cells, tumor-associated B cells exhibit higher levels of somatic mutations and greater clonal expansion.717,718 Another distinctive function of B cells was observed in ovarian cancer, where B cells preferentially express IgA, while in breast cancer, B cells mainly express IgM and IgG. This IgA can target antigens and be internalized by tumor cells in an antigen-independent manner through Polymeric Immunoglobulin Receptor (PIGR), sensitizing tumors to T cell.719 Current research indicates that the states of tumor tissue-associated B cells vary across different types of tumors, but largely remain in a pre-antibody class-switched state. Some studies suggest that in the presence of ongoing tumors, exhausted or dysfunctional CD8+ and CD4+ T cells seek the aid of B cells in the microenvironment, through the expression of CXCL13, to form tertiary lymphoid structures (TLS).720,721,722 Research across multiple cancers demonstrates that the presence of TLS and B cells in tumor tissues correlates with better prognoses, and the anti-tumor efficacy of T cells is enhanced in the presence of B cells.723,724,725,726 Interestingly, TLS can also be exploited by tumor cells under certain conditions to promote lymphatic infiltration of tumor cells, leading to lymphatic metastasis.727 However, few studies have revealed a direct significant correlation between the CSC phenotype and both TLS and B cells.728 Tumors with low TLS infiltration may present higher CSC characteristics, with increased proliferation and metastatic potential.729 In summary, the presence of TLS is considered a crucial component of anti-tumor immunity. With the development of scRNA-seq and spatial transcriptomics, research into the functions of TLS within tumors and their relationship with CSCs is expected to mature and refine further.
NK cells represent a crucial component of the innate immune system, constituting the third major lymphocyte type, following T cells and B cells. They play a complementary role to T cells by eliminating reduced or absent MHC class I expression tumor cells which evade CD8+ T cell detection, and can also recruit dendritic cells to indirectly enhance T cell-mediated responses.730 Emerging evidence suggests that CSCs may be particularly susceptible to NK cell-mediated targeting. In colorectal cancer models, CSCs exhibit increased vulnerability to NK cell cytotoxicity, associated with the upregulation of natural cytotoxicity receptors, especially NKp30 and NKp44.731 Intriguingly, GSCs demonstrate resistance to unstimulated NK cells but exhibit heightened sensitivity in co-culture models following pre-treatment with IL-2 and IL-15.732 This preferential susceptibility might be mediated by increased expression of Natural-Killer Group 2 Member D (NKG2D) ligands UL16 Binding Protein 1 (ULBP1), ULBP2, and MHC Class I Chain-Related Protein A (MICA) on CSCs.733 Beyond their capacity to directly eliminate CSCs, NK cells can also induce their differentiation. In the presence of CSCs and IL-2, the cytotoxicity of NK cells is suppressed, and cytokine production is enhanced, a state referred to as “split energy”.734 These split anergic NK cells secrete high levels of Interferon-γ (IFN-γ), which induces the expression of MHC-I, differentiation receptors, and PD-L1 while reducing CD44 levels on CSCs. This induction of CSC differentiation subsequently leads to slowed tumor growth and decreased metastatic spread.735 Therefore, NK cells appear to counter tumor progression through a dual-step mechanism: initially eliminating a portion of CSCs and then, following a phase of split energy inducing cellular differentiation within the remaining CSC population.736 However, the local tumor microenvironment can directly inhibit NK cell effector mechanisms. Tregs suppress NK cell functions in a TGF-β-dependent manner, while CAFs inhibit NK cell functions through cell-cell communication and the release of PGE2.737,738,739 CSCs can also impede NK immune responses through various inhibitory mechanisms. In metastatic melanoma, the expression of Indoleamine-2,3-Dioxygenase (IDO) and/or production of PGE2 can modulate the expression of NKp30, NKp44, and NKG2D.740 In neuroblastoma, TGF-β suppresses NK cell functions by regulating the expression of activation receptors and chemokine receptor repertoires, chiefly interfering with their migration and accumulation within tumor nests.741,742 In ovarian tumors, the expression of Macrophage Migration Inhibitory Factor (MIF) and the glycoprotein MUC-16 can downregulate NKG2D and disrupt the formation of synapses between tumor cells and NK cells.743,744 Additionally, CSCs evade immune surveillance and reduce NK cell-mediated killing by actively shedding MICA and MICB and recruiting Tregs.745,746 Kryczek et al. observed that IL-22 promotes the CSC phenotype in preclinical and patient-derived models, with IL-22 being produced by NK and T cells.747
In summary, immune cells’ fight against tumors mainly goes through three stages: immune elimination, immune equilibrium, and immune evasion. In the initial phase, T cells and NK cells identify and eradicate proliferating CSCs before they develop into full-blown cancer. Consequently, CSCs with high immunogenicity are gradually eliminated by the immune system, such as high MHC-I or NKG2D, leaving behind those with low immunogenicity or those in a quiescent state to survive into the second phase. Ultimately, these selected CSCs expand uncontrollably with the help of immunosuppressive effectors, and the immune system becomes incapable of suppressing them.
Immunotherapy targets cancer stem cells
Tumor immunotherapy represents a therapeutic approach that harnesses the immune system to generate tumor-specific immune responses, aimed at suppressing and eliminating tumor cells. Based on the different mechanisms of the immune response against tumors, tumor immunotherapy can be categorized into “active immunotherapy” and “passive immunotherapy”.748
The core of passive immunotherapy hinges on administering immune effectors with antitumor activity, such as tumor-specific T cells and antibodies. This method offers rapid action but fails to elicit a lasting immune response.748 Tumor-specific monoclonal antibodies (mAbs) represent the most well-known form of immunotherapy, widely utilized in clinical practice.749 The mechanisms for mAbs primarily encompass (1) specific recognition of molecules expressed on tumor cell surfaces, leading to tumor cell death via phagocytosis, complement system activation, and antibody-dependent cell-mediated cytotoxicity (ADCC); (2) disruption of signaling pathways essential for tumor cell progression and survival, or inducing death signals by binding to surface receptors; and (3) conjugation with cytotoxic drugs or radioactive isotopes for specific delivery to tumors.750 Adoptive cell immunotherapy (ACI or AIT) involves infusing immune cells with anticancer activity back into the patient. This includes chimeric antigen receptor T-cell (CAR-T) therapy, tumor-infiltrating lymphocytes (TILs) therapy, NK cell therapy, and cytokine-induced killer (CIK) cell therapy.751,752 The principle behind these therapies is the isolation of immune cells with cytotoxic potential from the patient. CAR-T therapy involves genetically engineering isolated T cells to bind tumor cell antigens;753,754 CIK cells, expressing both CD3 and CD56 membrane proteins, are a novel type of immune cell known as NK-like T lymphocytes with potent anticancer activity.755,756 In contrast to passive immunotherapy, active immunotherapy only exerts anticancer effects after activating the host’s immune system. Initial attempts to enhance antitumor immunity relied on non-specific immune stimulation, such as the local administration of inflammatory molecules (such as pathogen-associated molecular patterns (PAMPs) and DAMPs) and immunostimulatory cytokines (such as G-CSF, GM-CSF, TNF-α, IFN-α, IL-2).757 Unlike non-specific immunostimulants, antitumor vaccine inoculation offers high tumor specificity.758 Notably, immunomodulatory mAbs, such as ICIs, fall under active immunotherapy. These drugs activate new or restore pre-existing host immune responses by blocking the interactions between tumor cells expressing immune checkpoints and immune cells.
Over the past two decades, mAbs have emerged as effective therapeutic agents for cancers, significantly enhancing the survival rates and quality of life.759 CSCs can be identified by combinations of positive and negative expression of surface markers, with novel mAbs becoming increasingly potent and specific drugs targeting CSCs (Fig. 6a). Among these, CD44 has been recognized as one of the well-known CSC markers, playing a crucial role in EMT as well as in the initiation, progression, and metastasis of tumors.760,761 RG7356, an anti-CD44 mAb, has shown promise in preclinical models, demonstrating activation of macrophages and good tolerance in both solid and hematological malignancies.762,763,764 However, the ADC targeting CD44v6, bivatuzumab mertansine, was prematurely discontinued due to life-threatening off-target skin toxicity.765 Other CSC markers that have entered clinical trials include CD24, CD47, CD123, EpCAM, CD9 and so on.759,766,767,768,769 However, only CD105-targeting crituximaband EpCAM-targeting edrecolomab have entered phase III clinical trials.770,771 Beyond ADCs, bispecific antibodies, which target two different CSC antigens simultaneously, have shown superior efficacy in preclinical models compared to agents targeting a single antigen.772 However, the clinical trial success of these bispecific antibodies remains to be further observed.

Immunotherapy targets CSCs. a Targeted therapy using antigens of CSCs, such as CAR-T and monoclonal antibodies, etc. b Leverage the innate immune cells’ natural cytotoxic activity to circumvent antigen presentation and nonspecifically target CSCs, such as NK cells or CIK cells. c Active immunization strategies involve the use of DC vaccines loaded with CSC lysates, or the reinvigoration of T cells through targeting immune checkpoints. d γδ T cells exhibit the dual capacity to directly attack CSCs and indirectly stimulate NK cells or DCs to target CSCs
Another promising therapeutic strategy targeting CSCs is CAR-T cell therapy (Fig. 6a).773 This approach holds a distinct advantage over TILs therapy or ex vivo activation of autologous unmodified T cells, as CSCs often exhibit reduced antigen presentation capabilities due to the downregulation of MHC and/or antigen-processing machinery (APM) molecules. While, tumor cell recognition by CAR-T cells without relying on the MHC complex.774,775 Various CAR-T cell therapies have been developed for GSCs. For instance, research by Zhu et al. demonstrated that CAR-T cells targeting CD133 effectively kill CD133+ CSCs in glioma patients, both in vitro and in vivo.776 However, such therapies have not succeeded in completely eradicating tumors, possibly due to tumor cell-induced terminal differentiation or senescence of CAR-T cells. When CD57+ glioma cells interact with CAR-T cells, an increase in the expression of the T cell senescence marker CD57 on CAR-T cells is observed.776 Similarly, CAR-T cells targeting the epidermal growth factor receptor variant III (EGFRvIII) have been effective in killing target cells. EGFRvIII has been identified as a tumor-specific antigen for GSCs.777 Yet, in a phase I clinical trial for glioblastoma patients, EGFRvIII-targeted CAR-T cells induced downregulation of tumor antigens and significant upregulation of inhibitory molecules.778 These findings underscore the need for further efforts to enhance the efficacy of CAR-T cell therapy. In recent years, numerous CAR-T therapies targeting antigens associated with CSCs, including CD22, CD123, and ALDH, etc., have been developed. CAR-T therapy remains a leading trend in future research endeavors.
As previously mentioned, most normal cells expressing MHC-I molecules are not targeted by NK cells. However, tumor cells and CSCs that downregulate MHC-I molecules while upregulating activating ligands become primary targets for NK cell-mediated cytotoxicity.779,780 The imbalance in the expression of MHC-I and NK activating ligands on CSCs leads to increased sensitivity to NK cell killing.781 This pattern of NK ligand expression and sensitivity to its cytotoxic effects has been reported across multiple tumor types, including gliomas, colorectal cancer, melanoma, pancreatic cancer, oral squamous cell carcinoma, breast cancer, and Ewing’s sarcoma.732,733,734,782,783,784 Nonetheless, multiple studies have also shown that the targeting capability of NK cells can be influenced by the tumor microenvironment, underscoring the need for further research to identify the appropriate subtypes of NK cells as carriers and to effectively target and kill tumors (Fig. 6b).740,785,786
In recent years, γδ T cells, a subset of non-conventional T cells characterized by their expression of heterodimeric T-cell receptors (comprising γ and δ chains) and their non-restrictive antigen recognition, have garnered significant interest within immunotherapy.787 Present in the immune infiltration of human cancers, γδ T lymphocytes have been shown to play a role in antitumor immune responses.788 Specifically, Vγ9Vδ2 T cells have demonstrated the capability to kill various tumor cells in vitro and in vivo, independent of the tumor cells’ MHC molecule expression levels.789 The antitumor activity of Vγ9Vδ2 T cells is exerted through two main mechanisms: direct induction of cytotoxic mechanisms akin to those of CD8+ T cells and indirect stimulation of other immune cells such as NK cells and cytotoxic T lymphocytes (CTLs).790,791 Notably, the activation of Vγ9Vδ2 T cells can be induced by bisphosphonates, drugs associated with bone metastasis.792 Treatment of CSCs with zoledronic acid stimulates Vγ9Vδ2 T cells to secrete cytokines like IFN-γ, express pro-apoptotic molecules such as TNF-Related Apoptosis-Inducing Ligand (TRAIL), and release cytotoxic granules, ultimately inducing CSC death through a TCR-dependent mechanism (Fig. 6d).793,794,795 Moreover, chemotherapy drugs like doxorubicin and 5-fluorouracil can induce the expression of TRAIL and NKG2D activating ligands on CSCs, rendering them sensitive to Vγ9Vδ2 T cell-mediated killing.796 Vγ9Vδ2 T cells can also enhance the chemosensitivity of ovarian CSCs by reducing the expression of multidrug resistance components ABCG2, topoisomerase 2a, and 2b.797 Clinical trials involving Vγ9Vδ2 T cells have been conducted in various tumors, including breast cancer, prostate cancer, lung cancer, and head and neck cancer.788 Although these therapies can reduce tumor burden, only modest improvements in long-term survival rates have been observed, highlighting the importance of further research into mechanisms regulating CSC sensitivity to γδ T cells and considering these mechanisms in the design of new clinical trials.
Additionally, CIK cells have demonstrated the ability to kill CSCs in preclinical models of melanoma, sarcoma, and liver cancer (Fig. 6b).798,799 In liver CSCs, CIK cells induce caspase-3-dependent apoptosis and G2/M arrest.800 In melanoma and sarcoma, CIK cells exert direct cytotoxic effects.798,799 CIK cells emerge as promising candidates for targeting CSCs in immunotherapy for two main reasons: their cost-effectiveness compared to other immune cell populations and their sensitivity to CSCs resistant to chemotherapy and targeted therapies, with easy derivation from patients who have undergone these treatments.801 Therefore, combining CIK therapy with chemotherapy or molecular-targeted therapies may represent a future direction for immunotherapy.
Dendritic cells (DCs)-based antitumor vaccines, a widely applied immunotherapeutic strategy targeting CSCs, primarily operate by loading DCs with proteins or mRNA from tumor lysates, thereby activating specific tumor immune responses (Fig. 6c).802 The nature of the antigen (such as peptides, whole proteins, or mRNA) impacts the resultant immune response, with whole proteins capable of activating both CD8+ and CD4+ T cells, whereas mRNA encoding antigens induces only CD8+ T cell responses.803 Pellegatta et al. pioneered the construction of DC vaccines using lysates from GSCs, demonstrating that CSC-based DC vaccines exhibit higher efficacy compared to non-CSC-based DC vaccines (utilizing glioma cells).804 Moreover, therapeutic tumor vaccines, as adjunct therapy post-radiotherapy or surgical resection, show more potential benefits than prophylactic vaccination. Qiao et al.‘s series of studies confirmed that adjuvant therapy with DC vaccines based on ALDH+ cells significantly reduces local tumor recurrence, inhibits spontaneous lung metastasis, and prolongs host survival in lung cancer or melanoma patients, outcomes not achieved with DCs loaded with non-CSCs or an unselected cancer cell population.805,806 This advantage likely stems from the CSC-specific humoral and cellular immune responses generated by DCs loaded with CSCs.805,806 These encouraging preclinical results have propelled CSC-loaded DC vaccines into the clinical application phase. The first clinical trial of a CSC-loaded DC vaccine in glioblastoma patients, although limited to seven patients, reported extended PFS compared to historical controls.807 Additional clinical trials in lung and pancreatic cancer patients have not shown significant adverse side effects, confirming the safety of CSC-targeted DC vaccines.808,809 However, it is noteworthy that these clinical studies did not compare outcomes with DC vaccines loaded with non-CSCs or unsorted cells, leaving the replicability of preclinical success in humans in question. Furthermore, while CSC-targeted vaccines offer a cost-effective advantage over other immunotherapies, they may increase economic burdens.
Immune checkpoints such as PD-L1 play a crucial role in the AKT signaling pathway, impacting the expression of embryonic stem cell transcription factors OCT4A, Nanog, and the stem cell factor BMI1.495 Concurrently, the downregulation of PD-L1 impairs the self-renewal capabilities of breast CSCs. Interaction between PD-L1 and PD-1 enhances the proliferative capacity of gastric cancer stem-like cells.810 Similarly, CTLA-4 exhibits analogous functions. ALDH+ melanoma stem cells express CTLA-4, indicating its ability to support cellular proliferation and inhibit apoptosis in vitro. Blocking CTLA-4 can suppress both in vitro and in vivo self-renewal and tumorigenic capabilities by depleting ALDH+ cells.811 Consequently, in CSC-targeted therapy, the application of ICIs holds particular appeal. Preclinical studies have demonstrated that anti-CSC vaccines combined with anti-PD-L1 therapy, as adjuvant treatment following surgical resection of squamous cell carcinoma, significantly inhibit tumor recurrence and prolong survival compared to monotherapy.806 Moreover, a triple regimen combining anti-PD-L1 with anti-CTLA-4 and an anti-CSC vaccine is more effective in promoting tumor regression in melanoma-bearing mice than the anti-CSC vaccine alone.812 These antitumor effects are attributed to the significant depletion of ALDH+ CSCs following combination therapy, associated with T cell expansion, suppression of TGF-β secretion, increased IFN-γ secretion, and notably enhanced host-specific CD8+ T cell responses against CSCs.813 Researchers conclude that combining anti-CSC vaccines with PD-1 blockade can enhance the functionality of tumor-specific CTLs and protect mice from secondary challenges by CSCs.813
Clinical trials of targeting CSCs combined with immunotherapy
In recent years, vaccination against CSCs has garnered increasing attention in the clinical research domain (Table 5). These trials encompass various cancer types, including pancreatic cancer (NCT02074046), nasopharyngeal carcinoma (NCT02115958), breast cancer (NCT02063893), hepatocellular carcinoma (NCT02089919), lung cancer (NCT02084823), colorectal cancer (NCT02176746), and ovarian cancer (NCT02178670). Although these trials are listed as completed on ClinicalTrials.gov, the research outcomes have yet to be reported. On another front, with the advancement of CAR-T cell technology, an increasing number of clinical trials are exploring CAR-T cell therapies using CSC biomarkers. These cells can bypass the antigen presentation process and directly target CSCs, exerting anti-tumor effects. NCT02541370, a single-arm phase II trial, demonstrated promising anti-tumor activity and manageable safety for CD133-targeted CAR-T cells in advanced hepatocellular carcinoma. Additionally, Catumaxomab, a bispecific antibody targeting EpCAM and CD3, has been proven effective in eliminating malignant ascites in several clinical trials. However, due to its high cost and potential adverse reactions from targeting CD3, the drug was withdrawn from the market in 2017. While therapies like ICIs and NK cells have shown some efficacy in combating tumors, they struggle to effectively distinguish between tumor cells and CSCs. Hence, despite the promising strategy of employing immunotherapy to target CSCs, further research is crucial for its broader clinical application.
Cancer stem cells and sensitivity/resistance to radiotherapy
CSCs and radiotherapy sensitivity/resistance
Radiotherapy, or radiation therapy, is one of the most common and important therapeutic strategies in terms of solid tumor treatment. Radiotherapy exerts its cell-killing effects mainly by inducing DNA damage that is beyond repair, which consequently leads to cell cycle arrest, apoptosis, autophagy, or senescence of target cells.294 In this process, ROS is considered a critical mediator (Fig. 7).814

Radioresistance induced by CSCs. a As for radiosensitive cancer cells, radiation can induce the production of ROS, which subsequently leads to the accumulation of cytochrome C and apoptosis, and DNA damage that causes various types of cell death. b CSCs can be radioresistant due to their high expression of DNA damage repair-associated molecules and powerful radical scavenging system
Research indicates that radiation can induce the formation of CSCs and that CSCs are less radiosensitive than other cancer cells.294,815,816,817,818,819,820,821,822 For instance, radiation-induced radioresistant NSCLC cell line has increased expressions of CSC markers, including SOX2, CD133, and ALDH compared to radiosensitive cells, and upregulating SOX2, a DNA repair regulator, results in more robust radioresistance of these cells.823 FOXM1 can also induce SOX2 expression in glioblastoma and induce radioresistance.824 In response to radiation, CD133+ GSCs exhibit radioresistance by preferentially activating DNA damage checkpoints and repairing DNA damage more effectively.825 CD24−/low/CD44+ breast CSC-enriched mammospheres are also more radioresistant than monolayers breast cancer cells,826 and the CD24−/low/CD44+ breast CSCs contain less ROS levels compared to non-tumorigenic cells.641 Likewise, CD133+ hepatocellular CSCs are more resistant to radiation than CD133− cells, and suppression of CD133 sensitizes these cells to radiation by breaking cell-cycle arrest and inducing apoptosis.827 On the contrary, knocking down CSC markers can sensitize the cells to radiation.828,829,830 This suggests that the acquisition of stem-like properties and radioresistance can be two sides of the same coin. Indeed, studies show that radioresistance is companied by enlarged CSC population in glioma/glioblastoma,824,831,832,833,834,835,836 breast cancer,828,837,838,839,840,841 colorectal cancer,842,843,844,845 lung cancer,846 salivary adenoid cystic carcinoma,847 oral squamous carcinoma,848 head and neck squamous cell carcinoma,849 neuroblastoma,850 cervical cancer,841,851 esophageal squamous cell carcinoma,852 ovarian cancer,853 and gastric cancer.854
The induction mechanism of CSC properties by radiation has not been fully revealed, and some researchers believe CSC properties are acquired through radiation-inducible EMT.294 Also, some studies provide insights into the relationship between radiation and CSC formation. Following DNA damage, senescence-associated secretory phenotype (SASP) is released and promotes the emergence of CSCs in MM.855 Similarly, High Mobility Group Box 1(HMGB1), a DAMP, is released after radiation and subsequently activates the HIF-1α signaling in pancreatic cancer cells which leads to the acquisition of CSC properties.856 Another study regarding glioblastoma shows that the radiation-inducible activation of the K-RAS/ERK/CD44 axis facilitates the stemness of the cells.857 And the miR-603 in extracellular vesicles of glioblastoma after radiation targets IGF1 and IGF1R that promote CSC state.835 Additionally, following radiation, non-CSCs of breast cancer are converted into CSCs, which can be prevented by Notch inhibition, suggesting a crucial role of the Notch pathway in this transition.858
The radioresistance of CSCs depends on their enhanced abilities to repair DNA damage and maintain ROS levels (Fig. 7). MYCN-amplified neuroblastoma cells exhibit increased c-Myc expression, dysregulated DNA repair pathway, stable ROS level after radiation, and CSC properties.850 c-Myc plays an important role in radioresistance of nasopharyngeal carcinoma CSCs by upregulating DNA damage checkpoint Checkpoint Kinase 1 (CHK1) and CHK2.859 OCT4, a CSC marker, endows radioresistance to head and neck squamous cell carcinoma cells by regulating the homologous recombination factors PSMC3IP and RAD54L, and either upregulation or downregulation of OCT4 diminishes radioresistance of the cells.860 THOC2 and THOC5 play an important role in the radioresistance of triple-negative breast cancer cells by upregulating SOX2.837 SOX2 can lead to radioresistance by inducing cell cycle arrest to avoid DNA damage checkpoints.861 Ubiquitination-Specific Protease 1 (USP1), which is upregulated in GSCs, stabilizes DNA damage response regulators and induces radioresistance of these cells.862 Musashi1, a CSC marker, regulates the expression of a DNA-protein kinase catalytic subunit to induce enhanced DNA repair response, which finally endows radioresistance to GSCs.863
Several signaling pathways are involved in the acquisition of both stemness and radioresistance. The activation of the JAK2/STAT3 pathway promotes colorectal cancer stemness characterized by increased expression of cyclin D2, which also maintains low levels of DNA damage accumulation.842 The TGF-β pathway activation or the WNT/β-Catenin pathway also enhances not only stemness but also radioresistance of breast cancer, salivary adenoid cystic carcinoma, colon cancer, cervical cancer, or gastric cancer.839,844,847,851,854,864 MiR-19b can downregulate FBXW7 expression and consequently activate the WNT/β-Catenin pathway, which eventually leads to stemness enhancement and radioresistance.843 SFRP2 is downregulated in glioma patients treated with radiotherapy, and a study shows that SFRP2 diminishes stemness and radioresistance of glioma cells by inhibiting the WNT/β-Catenin signaling.833 Besides, the Forkhead Box Q1 (FOXQ1)/Sirtuin 1 (SIRT1)/β-Catenin axis and the Ecotropic Virus Integration Site 1 (EVI1)/β-Catenin can also mediate stemness and radioresistance of colorectal cancer.845,865 The activation of the PI3K/AKT/mTOR pathway decreases apoptosis thus inducing radioresistance of prostate CSCs.866 Also, Tribble 2 activates the mTOR pathway and induces stemness and radioresistance in esophageal squamous cell carcinoma.852 In glioblastoma, the cyclin-like protein Spy1 endows the cancer cells with self-renewal abilities and downregulates CAP-Gly Domain-Containing Linker Protein 3 (CLIP3) whose expression leads to the glycolytic flux that induces radioresistance.832,867 The Proliferating Cell Nuclear Antigen (PCNA)-Associated Factor (PAF) supports GSC maintenance and promotes radioresistance by inducing translesion DNA synthesis.868 Activation of NRP1 not only improves stemness but also potentiates radioresistance of breast cancer cells by reducing radiation-mediated apoptosis.840 Integrin β1 increases stemness of oral squamous carcinoma cells and induces radioresistance by suppressing radiation-induced apoptosis.869
Preclinical studies on improving radiosensitivity by targeting CSCs
Efforts have been made to restore radiosensitivity by inhibiting CSCs in preclinical studies. DNA-Dependent Protein Kinase (DNA-PK) stabilizes SOX2 and maintains the stemness of GSCs, and NU7441, a DNA-PK inhibitor, can effectively reduce the stem cell sphere formation and sensitize the tumor to radiotherapy in vivo.831 Combining radiotherapy with glimepiride, an agent to treat type 2 diabetes, can disturb GSC maintenance and sensitize the tumor to radiation by reducing glycolysis.832 MiR-7-5p can reduce stemness of colorectal CSCs and sensitize these cells to radiation by downregulating the stemness-associated transcription factor, KLF4.870 Delivery of miR-145 that targets multiple stemness-related transcriptional factors reduces stemness and reverse radioresistance of colorectal CSCs.871 The lncRNA Transmembrane Phosphatase With Tensin Homology Pseudogene 1 (TPTEP1) interacts with miR-106a-5p and thus activates the P38/MAPK pathway that suppresses stemness and radioresistance of glioma cells.834 An Oncostatin M Receptor (OSMR) promotes mitochondrial respiration in GSCs, and suppression of this receptor sensitizes the cells to ionizing radiation.872 Apigenin can attenuate stemness of glioblastoma by downregulating HIF-1α and NF-κB and sensitizing the cells to radiotherapy due to reduced glycolysis.873 MiR-146b-5p can target the Hu antigen R and increase lncR-p21 which leads to inhibition of β-Catenin.874 This process attenuates stemness and increases apoptosis and radiosensitivity of the cells.874 Silencing Human Telomerase Reverse Transcriptase (hTERT) abolishes telomerase activity, reduces stemness, and reverses the radioresistance of a radioresistant nasopharyngeal carcinoma cell line.875 Given that miR-210 induces hypoxia adaption and maintains stemness of GSCs, knockdown of miR-210 abolishes CSC markers and endows radiosensitivity to these cells.876 Inhibition of integrin a6 leads to reduced DNA damage response and normalizes cell cycle pathways, which eventually helps overcome radioresistance and diminish stemness of the GSCs.836 Methyltransferase-like 14 and miR-99a-5p can downregulate Tribble 2, and the Tribble 2-induced activation of the mTOR pathway can be inhibited by an H-Istone Deacetylase 2 (HDAC2) inhibitor and restore radiosensitivity of the esophageal squamous CSCs.852 Restoration of E3 ubiquitin ligase C Terminus Of HSC70-Interacting Protein (CHIP) not only reduces expression of stemness of NSCLC cells but also sensitizes the cells to radiotherapy by improving apoptosis via inhibition of the PBK/ERK axis.877 BEZ235, a dual PI3K/mTOR inhibitor, can effectively sensitize prostate CSCs to radiotherapy by reducing the stemness of the cells.866
Clinical trials targeting CSCs combined with radiotherapy
Despite the efforts made to increase radiosensitivity by targeting CSCs in preclinical trials, few clinical trials that combine radiotherapy and CSC-targeting therapies are carried out (Table 6). A study tried to set the periventricular stem cell niche as additional target volumes in newly diagnosed high-grade glioma to eliminate the potential CSC pool. However, all 4 enrolled patients had adverse events and did not complete the study (NCT02039778). Another phase I study (NCT01068327) evaluated the safety and efficacy of nelfinavir, an Akt inhibitor, plus stereotactic body radiotherapy in treating locally advanced borderline or unresectable pancreatic adenocarcinoma. Among the 46 patients enrolled, sixteen patients experienced grade ≥2 adverse events, and grade 3−4 adverse events only occurred in 1 patient. The median overall survival of all the patients was 14.4 months. This trial concludes that concurrent stereotactic body radiation therapy (SBRT) (40 Gy) plus nelfinavir (1250 mg BID) was tolerable and safe for patients with locally advanced pancreatic cancer, but the efficacy of this combination still required investigations.878
Cancer stem cells and targeted therapy
Targeted therapy for tumors, a pivotal component of precision medicine, entails identifying specific carcinogenic sites at the molecular level and employing drugs to selectively target these areas, thereby achieving therapeutic objectives.879 Due to its notable advantages in prolonging patient survival, targeted therapy has garnered increasing attention, with a considerable number of treatments earning Food and Drug Administration (FDA) approval for tumor management.880 However, resistance to targeted therapy remains a significant consideration during treatment, emphasizing the critical role of CSCs.881
Thoracic tumors
Resistance of thoracic tumors to targeted drugs such as gefitinib, osimertinib, erlotinib, afatinib, palbociclib, and lapatinib can be partially attributed to the presence of a rare subset of CSCs. NSCLC stem cells with elevated expression levels of ALDH1A1 and CD44 demonstrate heightened resistance to gefitinib. Notably, ALDH1A1 activity can be neutralized by ATRA, restoring sensitivity (Fig. 8a).882 Osimertinib-resistant lung cancer cells exhibit increased stemness traits. Ginsenoside Rg3 has been identified as a sensitizing factor for osimertinib by activating the Hippo pathway (Fig. 8b).883 Furthermore, NSCLC cells resistant to erlotinib and afatinib demonstrate enhanced CSCs-related characteristics.884,885 The CSCs’ marker ALDH1A1 has been identified as a critical gene for erlotinib resistance in lung cancer cells. In ALDH1A1-positive cells, the anti-ROS system is activated, leading to significant upregulation of its associated enzymes Superoxide Dismutase 2 (SOD2) and Glutathione Peroxidase 4 (GPX4) during ALDH1A1-induced erlotinib resistance (Fig. 8c).886 Additionally, besides their intrinsic resistance to targeted therapy, CSCs confer drug resistance to non-CSCs by secreting vesicles. Vesicles originating from lung CSCs augment Apurinic Endonuclease 1 (APE1) expression in NSCLC, subsequently activating the IL-6/STAT3 axis, thus contributing to erlotinib resistance (Fig. 8d).887 Further, Fibroblast Growth Factor Receptor 1 (FGFR1), which promotes breast cancer stemness through the WNT/β-Catenin pathway, was identified as a key factor in palbociclib resistance, a CDK4/6-related targeted drug (Fig. 8e).888 While the majority of studies suggest that heightened stemness in thoracic tumors fosters resistance to targeted drugs, Huang et al. reported a contrasting finding. Specifically, they demonstrated that overexpression of Thymocyte Expressed Molecule Involved In Selection 2 (THEMIS2) in breast cancer promotes the binding of Protein-Tyrosine Phosphatases 1B (PTP1B) to MET, leading to MET activation, and ultimately sustaining stemness characteristics. Interestingly, THEMIS2 expression was positively associated with lapatinib sensitivity and inversely correlated with chemotherapy sensitivity (Fig. 8f).889

Targeted drug resistance of CSCs (except liver cancer). (a, k) gefitinib resistance (b) osimertinib resistance (c, d, j) erlotinib resistance (e) palbociclib resistance (f) capmatinib resistance (g, h) vemurafenib resistance (i) sunitinib resistance
Liver cancer
CSCs play a pivotal role in the resistance of liver cancer to various targeted drugs, including sorafenib, trametinib, lenvatinib, and regorafenib. Chang et al. uncovered a negative correlation between the expression of YAP1, a promoter of stemness-related genes SOX2 and OCT4, and the sensitivity of liver cancer cells to sorafenib.890 Viral infection-associated hepatocellular carcinoma cells (vHCC) exhibit resistance to sorafenib.891 The activated Interferon-Gamma Receptor (IFNGR)/JAK2/STAT1/Poly(ADP-Ribose) Polymerase 1 (PARP1) pathway in vHCC maintains stemness, leading to resistance to sorafenib. Conversely, the JAK2 inhibitor momelotinib reverses vHCC drug resistance (Fig. 9a).892 Integration of the hepatitis B virus gene HBx-ΔC contributes to liver cancer stemness and resistance to sorafenib and 5-fluorouracil.893

Targeted drug resistance of liver CSCs. a IFNGR stimulation of the JAK2/STAT1/PARP1 pathway is responsible for stemness maintenance and sorafenib resistance, and can be reversed by the JAK2 inhibitor momelotinib. b MSI2 binds LFNG to stimulate the Notch1 pathway to upregulate tumor cell’ stemness and sorafenib resistance. c Wortmannin inactivates the TROY/PI3K/AKT axis triggered by CAFs to inhibit the stemness of tumor cells and restore their sensitivity to sorafenib. d FZD10 contributes to stemness maintenance and lenvatinib resistance by activating the β-Catenin/c-Jun/MEK/ERK axis. e CD73 upregulates the c-Myc/SOX9 axis and inhibits GSK3β to hinder the ubiquitination and degradation of SOX9, ultimately maintaining the stemness characteristics and lenvatinib resistance of tumor cells. f CSCs release exosomes to upregulate Nanog expression in a RAB27A-dependent manner, promoting stemness characteristics and regorafenib resistance of tumor cells
Activation of Notch and PI3K/AKT pathways in CSCs is pivotal in developing resistance to targeted drugs. CD44v6 serves as a marker of liver CSCs positively associated with sorafenib resistance. Musashi2 (MSI2) overexpression in CD44v6-positive liver CSCs contributes to sorafenib resistance by binding Lunatic Fringe (LFNG) to activate the Notch1 pathway (Fig. 9b).894 Highly expressed TROY in liver cancer correlates with stemness characteristics and sorafenib resistance, while wortmannin inactivates the TROY-induced PI3K/AKT pathway, restoring sensitivity to sorafenib (Fig. 9c).505 Plasma-activated medium (PAM) enhances the efficacy of trametinib and sorafenib in CSC-rich liver cancer cell populations by inducing various forms of cell death.895 FZD10 expression significantly increases in lenvatinib-resistant liver cancer cells, maintaining liver CSC characteristics by activating the WNT/β-Catenin pathway and β-Catenin/c-Jun/MEK/ERK axis, thereby contributing to lenvatinib resistance (Fig. 9d).527 CD73, a marker of liver CSCs, upregulates the c-Myc/SOX9 axis, inhibiting GSK3β and the ubiquitination and degradation of SOX9, thereby conferring stemness characteristics to liver cancer (Fig. 9e).896
Cytokines and exosomes are critical factors in conferring resistance to targeted drugs in liver cancer cells. Kahraman et al. demonstrated that the application of targeted drugs, such as sorafenib and regorafenib, enriches liver CSCs, indirectly suggesting resistance to targeted therapy. Further mechanistic studies showed that IL-8 derived from the liver cancer niche maintains the stemness phenotype and inhibits sensitivity to sorafenib.897 CSCs can also confer resistance to targeted drugs to differentiated malignant cells. Exosomes released by hepatic CSCs in Ras-Related Protein Rab-27A (RAB27A)-dependent manner confer regorafenib resistance to differentiated hepatoma cells by inducing the upregulation of Nanog expression (Fig. 9f).898
Other tumors
The relationship between resistance to targeted therapy and CSCs is confirmed in various tumors, including melanoma, colorectal cancer, renal cell carcinoma, osteosarcoma, and oral squamous cell carcinoma. Vemurafenib-resistant melanoma cells exhibit higher expression of CSCs-related markers such as CD271 and fibronectin.899 The Mechanistic Target Of Rapamycin Complex 2 (mTORC2) confers stemness characteristics to melanoma-initiating cells in Rapamycin-Insensitive Companion Of MTOR (RICTOR)-dependent manner, promoting melanoma cell resistance to vemurafenib (Fig. 8G).900 Overexpression of the stemness-related gene SOX2 in melanoma correlates closely with vemurafenib resistance. SOX2 binds to the promoter of CD24 to upregulate its expression, activating Src and STAT3 and conferring adaptive resistance rather than acquired resistance to melanoma cells against targeted therapy (Fig. 8h).901 Additionally, the NRG-1β/ErbB-3 axis and the AKT pathway are critical for colon CSC resistance to vemurafenib.902
The antipsychotic drug penfluridol inhibits Dopamine Receptor D2 (DRD2) to eliminate the CSCs associated phenotype of renal cell carcinoma mediated by the hedgehog pathway, inducing apoptosis and autophagy, and enhancing the efficacy of the targeted drug sunitinib (Fig. 8i).903 However, the use of sunitinib also enriches CSC subsets in renal cell carcinoma. Sunitinib enhances Estrogen Receptor β (ERβ) expression by upregulating lncRNA-ECVSR, activating HIF-2α, and promoting the emergence of a CSC phenotype.904 MiR-499a suppresses resistance to the EGFR inhibitor erlotinib in CD166+ osteosarcoma stem cells. TGFβ-induced enhancement of Snail1 and Zeb1 expression suppresses the miR-499a/SHKBP1 axis, enhancing stemness characteristics and erlotinib resistance (Fig. 8j).905 Conversely, highly expressed Lysyl Oxidase-Like 2 (LOXL2) in oral squamous cell carcinoma correlates positively with the activation of the EMT program and the maintenance of stemness. LOXL2 promotes the expression of stemness-related genes and EGFR in an IFIT1- and IFIT3-dependent manner, ultimately rendering oral squamous cell carcinoma more sensitive to the EGFR inhibitor gefitinib (Fig. 8k).906
There is a scarcity of ongoing or completed clinical trials specifically targeting CSCs and their resistance to targeted therapies. Clinical trial NCT01215487 aims to investigate whether the content of CML stem cells can serve as a predictor of efficacy in CML patients undergoing imatinib therapy. Another trial, NCT03481868, is centered on epigenetics and resistance to tyrosine kinase inhibitors in CML stem cells. However, no results from these trials have been reported.
Therapeutic strategies targeting CSCs
Targeting classic markers of CSCs
Markers of CSCs, whether they are cell surface markers like CD13, CD44, and CD133, or intracellular markers such as Nanog, ALDH1, and SOX2, are effective molecules for identifying the rare population of CSCs and represent important targets for eliminating their various malignant biological behaviors. For instance, CD13 expression in liver CSCs positively correlates with the activation of the TGF-β-mediated EMT program, which enhances stemness characteristics while inhibiting ROS accumulation. Inhibiting CD13 induces apoptosis of liver CSCs.907 Liposomes modified with CD44 monoclonal antibodies exhibit enhanced anti-tumor efficacy by effectively targeting CSCs.908 Similarly, the plant extract emodin serves as a specific inhibitor of the liver CSCs marker CD44, exerting anti-tumor effects.909 Targeting CD133+ CSCs in gastric cancer with anti-CD133 CAR-T cells significantly inhibits CSCs-mediated tumor progression and treatment resistance.910 Targeting intracellular stemness-related marker Nanog effectively reduces the stemness of breast CSCs.911 Inhibiting the expression of intracellular stemness-related marker ALDH1 using the cell cycle regulatory kinase wee1 inhibitor MK1775 eliminates the stemness characteristics of MM.912 Moreover, FDA-approved drugs like ATRA and Suberoylanilide Hydroxamic acid (SAHA) can specifically target CSCs based on the expression of cell surface marker CD133 and intracellular marker Nanog. Their combination relieves the inhibition of Tet Methylcytosine Dioxygenase 2 (TET2) and PTEN by inactivating the lncRNA MIR22HG/miR‐22 axis, ultimately attenuating the stemness characteristics of liver cancer and inducing apoptosis of liver CSCs.913 Similarly, ATRA effectively inhibits the expression of cell surface marker CD44 and stemness-related genes ALDH, SOX2, and KLF4 to target gastric CSCs and hinder gastric cancer progression.914
Cell surface markers, such as CD123, and intracellular markers, such as Nanog, represent commonly utilized targets in these clinical trials, predominantly through CAR-T cells, specific antibodies, and targeted drugs (NCT04272125, NCT02232646). However, the majority of these trials are in phase I or phase II, with few reporting outcomes. Limited survival data from completed trials make it challenging to draw definitive conclusions regarding the clinical efficacy of targeting CSC markers in relapsed or refractory tumors.
Targeting the classic pathway of CSCs
The malignant biological behavior of CSCs is underpinned by multiple interacting signaling pathways, hinting at the potential significance of targeting classic pathways in CSCs (Fig. 10). Preclinical investigations have validated the feasibility of targeting signaling pathways within CSCs. For instance, ICG-001, a WNT pathway inhibitor, effectively eliminates the stemness and metastasis phenotypes of colorectal cancer cells by suppressing the downstream gene of the WNT pathway, Myeloid Ecotropic Viral Insertion Site 1 (MEIS1).915 Similarly, a complex comprising [PdCl(terpy)](sac)2H2O and niclosamide, designed by Karakas et al., enhances the therapeutic efficacy against breast cancer by inhibiting the WNT pathway and inducing apoptosis of CSCs.916 The sonic hedgehog pathway, when activated in pancreatic CSCs, can be attenuated by sulforaphane (SFN), derived from cruciferous vegetables, which reduces GLI activity, suppresses stemness, and induces apoptosis.917 Additionally, silencing the Notch2 pathway significantly inhibits the stemness and metastatic phenotypes of bladder cancer cells, revealing a promising target to impede bladder cancer progression.918 The inhibitory effects of nonsteroidal anti-inflammatory drugs on colorectal CSCs could be attributed to the inactivation of the Notch pathway and the activation of the PPARγ pathway.919 The JAK2-specific inhibitor CYT387 markedly suppresses the paclitaxel-induced enhancement of stemness characteristics in ovarian cancer by attenuating the activity of the JAK2/STAT3 pathway.920 Natural products such as curcumin from turmeric and epigallocatechin-3-gallate (EGCG) from green tea have demonstrated inhibition of breast CSCs activity by deactivating the JAK/STAT and NF-κB pathways.921 Moreover, celastrus orbiculatus extract had demonstrated the capability to deactivate the TGF-β/Smad pathway by inhibiting Smad3/4, which ultimately results in the suppression of gastric CSCs.922 Hongwiangchan et al. synthesized hydroquinone 5-O-cinnamoyl ester of renieramycin M (CIN-RM), which exhibits inhibitory effects on lung CSCs by deactivating the AKT/PI3K pathway and downstream c-Myc.923 Furthermore, GSK-458 effectively disrupts the stemness characteristics of CSCs and induces caspase-3-mediated cell death by inactivating the PI3K/mTOR pathway.924

Targeting CSCs through classical signaling pathways. a WNT/β-Catenin pathway. Commonly developed targets include WNT/Frizzled complex, β-Catenin/TCF, CK1α, tankyrase, and COX. b Hedgehog pathway. Commonly developed targets include SHH-PTCH interaction, SMO, and GLI. c Notch pathway. Commonly developed targets include Notch, Dll3/4, γ-secretase, ADAM. d NF-κB pathway. Commonly developed targets include NF-κB complex, IκB, IKKα/β/γ, NF-κB inducing kinase (NIK). e JAK/STAT pathway. Commonly developed targets include JAK1/2/3, STAT1/2/3/4/5. f TGF-β pathway. Commonly developed targets include TGF-β1/β2/β3, TβRI/II, Smad3/4/5. g PI3K/AKT pathway. Commonly developed targets include PI3K complex, AKT1/2/3, mTORC1/2. h PPAR pathway. Common targets that have been developed include PPARα/γ/δ
While preclinical data indicate the feasibility of targeting signaling pathways within CSCs to eliminate them, there are currently limited corresponding clinical trials. Most ongoing clinical trials solely utilize signaling pathway inhibitors in patients. However, discerning whether the anti-tumor effect necessitates targeting CSCs remains challenging (NCT00106145, NCT01608867, NCT00844064). More sophisticated clinical trial designs are imperative to ascertain the effectiveness of targeting signaling pathways within CSCs.
Targeting the niche of CSCs
The supportive niche surrounding CSCs represents another crucial protective factor in maintaining their stemness characteristics.4 In diffuse large B-cell lymphoma, a significant positive correlation was observed between the stemness score and the scores of immune cells and stromal cells, highlighting the importance of targeting the niche as a key strategy for eliminating CSCs.925 Components of the CSCs niche, including the hypoxic microenvironment, acidic microenvironment, TAMs, CAFs, and cytokines, have been shown to closely influence the stemness maintenance and survival of CSCs.
Hypoxia-induced HIF-2α serves as a key factor in maintaining stemness in breast cancer. Mechanistically, inhibition of HIF-2α effectively attenuates the stemness phenotypes through inactivating the PI3K/AKT/CD44 pathway.926 Acidosis, a hallmark of the tumor microenvironment, acts as a promoting factor for the stemness phenotype in melanoma, prostate cancer, colorectal cancer, and gastric cancer.927 The extracellular acidic microenvironment may become another promising target for the treatment of CSCs. Doherty et al. proposed significant inactivation of the IFN pathway in breast CSCs, suggesting that IFN-β could serve as a new targeted therapy for breast CSCs. Exogenous IFN-β induces breast CSCs to transition toward a non-stemness phenotype and promotes lymphocyte infiltration.928 Further mechanistic studies have indicated that overexpression of non-phosphorylated IFN-Stimulated Gene Factor 3 (ISGF3) in breast CSCs is responsible for their stemness phenotype and invasive behavior. Exogenous IFN-β therapy significantly phosphorylates ISGF3 to suppress the stemness characteristics of breast CSCs.929 Chemokines are implicated in tumor progression as components of the tumor microenvironment. C-X-C Motif Chemokine Receptor 2 (CXCR2), upregulated by Galectins-3 (Gal-3), has been identified as a renal cell cancer stemness maintenance factor.930 IL-8 transactivates the EGFR/HER2 pathway through CXCR1 and CXCR2 activation in SRC-dependent manner, ultimately enhancing the stemness characteristics of breast CSCs.931 This further suggests that chemokines can serve as potential targets for controlling CSCs.
Highly expressed CD51 in TAMs maintains the M2 polarized phenotype and promotes TGF-β1 secretion. Niche-derived TGF-β1 further activates the TGF-β/Smad pathway to sustain the stemness characteristics of pancreatic cancer. CD51-based TAM-targeted therapy may become another option to control pancreatic CSCs.255 TAMs-derived CCL2 effectively activates the AKT pathway in breast cancer cells, facilitating the nuclear transfer of β-Catenin and ultimately sustaining the stemness and EMT-related phenotypes of breast cancer.274 Similarly, TAMs-derived CCL22 was identified as a factor promoting stemness and invasiveness in esophageal squamous cell carcinoma. CCL22 in the tumor microenvironment activates the FAK/AKT axis to bind and phosphorylate GLI1, thereby activating the hedgehog pathway.932 Additionally, M1-TAMs secrete IL-6 to upregulate the STAT3/Thrombospondin-1 (THBS1) axis, maintaining the stemness of oral squamous cell carcinoma.933 The interaction of TAMs and CAFs with non-CSCs populations can promote the transformation of CD44+CD24+ non-CSCs into CD44+CD24− breast CSCs. Mechanistically, Rab13 supports the stimulation of IL-8 derived from the breast CSC niche to promote membrane translocation of CXCR1/2, ultimately upregulating the stemness of breast cancer. Upon inhibition of Rab13 with bardoxolone-methyl, a notable suppression of breast CSCs was observed.934 IL-6 and IL-33 secreted by CAFs significantly enhance the activity of 5-LO in MDSCs, stimulating downstream Leukotriene B4 (LTB4)/Leukotriene B4 Receptor Type 2 (BLT2) axis to promote stemness and chemoresistance of intrahepatic cholangiocarcinoma.287 Furthermore, IL-6 and IL-8 derived from myofibroblasts in the tumor microenvironment activate the Notch/Hairy And Enhancer Of Split 1 (HES1) and STAT3 pathways, enhancing the CSCs population in early colorectal cancer.935 This further suggests that CAFs in the niche are another promising target for controlling CSCs. Unlike most evidence supporting CAFs as protectors of CSCs, McAndrews et al. reported that the presence of αSMA+ CAFs was associated with suppressed activity of LGR5+ colorectal CSCs, increased regulatory T cells and decreased CD8+ T cells.936
While preclinical studies indicate the feasibility of targeting the niche to affect CSCs, there remains a notable absence of relevant clinical trials. Current ongoing clinical trials involve interventions such as the use of autologous activated T cells and CSC vaccines comprising dendritic cells, T cells, B cells, and CSC-derived antigens to reprogram the CSC niche (NCT05341947, NCT02074046, NCT00846456). However, the majority of these trials are in phases I and II, with no conclusive experimental outcomes reported yet.
Targeting CSC through other approaches
In addition to targeting the markers, signaling pathways, and niches of CSCs, other potential approaches to eliminate CSCs include modulating stemness-related genes, abnormal metabolism, non-coding RNA, etc. The Protein Arginine Methyltransferase Family (PRMTs) has emerged as a key player in tumor progression.937,938 Feng et al. highlighted PRMTs as crucial enzymes regulating ovarian cancer stemness, suggesting that PRMT inhibitors could serve as potential targeted therapeutics for ovarian CSCs.939 Dysregulation of iron metabolism, lipid metabolism, and mitochondrial function contributes to stemness maintenance. Katsura’s team demonstrated a close association between imbalanced iron metabolism and tumor stemness. Deferasirox application effectively downregulates stemness in esophageal cancer and oral cancers.940 High-fat diets activate lipid metabolism via PPARα and PPARδ, enhancing intestinal stem cell function and tumorigenesis.941 Activation of the FOXM1/PRDX3 axis in mitochondria is essential for endometrial CSCs’ survival, suggesting mitochondria as a feasible CSC target.942 Similarly, the mitochondria function-associated FOXM1/PRDX3 pathway is indispensable for colorectal CSCs survival, with its induced upregulation of CD133 expression significantly contributing to colorectal stemness.943
Non-coding RNAs play a crucial role in the intricate regulatory network governing tumor progression and the stemness maintenance of CSCs.944,945 Utilizing a delivery vector termed human telomerase reverse transcriptase promoter-driven VISA (TV), circular RNA RANBP2-Like And GRIP Domain-Containing Protein 6 (circRGPD6) is transported to breast CSCs to impede their tumor initiation and metastasis potential. Mechanistically, TV-circRGPD6 acts as a sponge for miR-26b, alleviating its suppression of yes-associated factor 2.946 The DGCR8/circKPNB1/SPI1 positive feedback loop, persistently activated in glioblastoma, sustains the upregulation of circKPNB1, which subsequently activates the SPI1/TNF-α/NF-κB axis, maintaining stemness of glioblastoma.455 Elevated circ_0007385 in NSCLC functions as a stemness-promoting factor by sponging miR-493-3p to alleviate its inhibition of ras-related protein Rab-22A.947 Similarly, lung CSCs-secreted lncRNA Mir100hg is delivered via exosomes to non-CSCs, targeting miR-15a-5p and miR-31-5p, thereby promoting lung cancer progression.948 MiR-148a, inversely correlated with the expression of stemness-related genes SOX2, OCT4, and Nanog, attenuates the stemness of esophageal squamous cell carcinoma by inhibiting Activin A Receptor, Type I (ACVR1).949 Non-coding RNA emerges as a promising therapeutic avenue for targeting CSCs.
While preclinical studies have shown promise in targeting CSCs through alternative pathways, their clinical efficacy remains to be established. We provide a summary of pertinent clinical trials in Table 7. Interventions in these trials encompass CSC vaccines, repurposing of existing drugs (such as metformin), and targeting of genes potentially linked to stemness (NCT02084823, NCT01440127, NCT03298763). However, most investigations are in early phases (phase I and phase II), with limited comparison between treatment strategies targeting CSCs and standard therapies. The available results from a few clinical trials do not conclusively demonstrate significant patient benefit from CSC-targeted treatments (NCT01579812, NCT02001974, NCT02001974).
Drug delivery system for targeting CSCs
Therapies targeting CSCs face several obstacles. Traditional CSC-targeting drugs have shown significant progress, yet they suffer from shortcomings such as poor solubility, stability, and dose-limiting toxicity.950 Additionally, CSCs present a unique challenge in cancer treatment, displaying heightened resistance compared to ordinary tumor cells due to their robust capability of drug concentration regulation and metabolism.951,952 Addressing these challenges, a notable trend in targeted CSCs therapy involves drug delivery systems to optimize therapeutic effects and overcome treatment resistance.953,954,955 Targeting CSCs treatment predominantly utilizes nanoparticles, liposomes, and polymer micelle, while pH-sensitive capsules, and aptamers are also prevalent (Fig. 11).956,957,958 Moreover, other treatments include echogenic PEGylated PEI-loaded microbubble, virus preparations, multinuclear complexes, etc.959,960,961,962 Nanobiotechnology not only aids in early detection and tumor diagnosis but also offers several advantages in CSC treatment, including precise targeting, high-dose administration, multiple drug delivery, and controlled drug release.963

Drug delivery systems in targeting CSCs therapy. The utilization of drug delivery systems, predominantly nanomaterials, plays a pivotal role in targeting CSCs therapy. Traditional passive targeting relies on the leakage of immature blood vessels. However, advancements in technology have enabled the attainment of active targeting of nanoparticles through surface modifications. Nanomaterials, serving as carriers, offer the capacity to encapsulate therapeutic agents such as small interfering RNA (siRNA) and drugs, thus safeguarding against drug degradation. Moreover, active targeting facilitated by nanomaterials enhances drug concentration and enables precise identification of CSCs. Furthermore, nanoparticles can be stimulated both internally and externally to trigger drug release, with these triggering factors potentially doubling as therapeutic strategies
Precise targeting is crucial in nanotechnology-based therapies. Previously, passive targeting of tumor cells relied on exploiting vascular leakage within the tumor microenvironment to facilitate the accumulation and release of nanomaterials at tumor sites. However, advancements in technology have enabled active targeting strategies, wherein nanoparticles are equipped with targeting ligands for specific recognition of tumor cells. This active targeting mechanism enhances nanoparticle accumulation in close proximity to the tumor, thereby augmenting cellular uptake of therapeutic agents. Such precision targeting serves as the foundation for achieving localized, high-dose drug delivery.964,965 For instance, HA-mediated Fe3O4 nanocubes exhibit selective recognition of liver CSCs via the HA-CD44 receptor ligand pathway, effectively inhibiting their migration and proliferation.966 Wang et al. have developed a peptide-based drug delivery system characterized by deep tissue penetration and enhanced cellular uptake. This system, when combined with platinum, enhances radiation-induced DNA damage, thereby overcoming CSC-mediated radiation resistance.967 Moreover, nanocomposites such as H-MnO2@(ICG + ISL)@HA were a monodispersed hollow structure of MnO2 with a continuously modified mesoporous shell structure of HA. These nanocomposites can effectively deliver isoliquiritigenin at high concentration to CD44+ CSCs. H-MnO2@(ICG + ISL)@HA nanocomposites integration with chemotherapy or phototherapy synergistically enhances tumor eradication with minimal side effects.968 Conjugation with antibodies, peptides or aptamers improves CSCs recognition. For instance, Toshiyama et al. developed a poly (ethylene glycol)-poly (lysine) block copolymer-ubenimex conjugate, which enhances the production of ROS to selectively eradicate CSCs by inhibiting aminopeptidase N.969 Micellar nanomedicine of cisplatin, coupled with cyclic Arg-Gly-Asp peptide, exhibits enhanced inhibition of CSCs.970 To address challenges like off-target effects and rapid degradation, Xu et al. engineered peptide-modified nanoparticles for targeted delivery to laryngeal CSCs.971 Similarly, activated carbon nanoparticles loaded with metformin effectively elevate drug concentrations within liver CSCs, enhancing therapeutic efficacy.972 Furthermore, nucleic acid aptamers, often referred to as “chemical antibodies”, possess specific tertiary structures that bind molecular targets with high affinity. Due to their lower immunogenicity and small volume, aptamers have emerged as promising tools for CSCs targeting, especially when combined with siRNA and miRNA.973 Beyond precise targeting, nanoparticles can also induce the expression of tumor molecules, offering additional avenues for therapeutic intervention.974
High-dose administration. Nanomaterials provide a relatively stable environment for drugs, siRNA, etc., which enable prolonged drug circulation within the body. For example, CD44v6-targeted polymeric micelles loaded with niclosamide exhibit tumor-specific accumulation, allowing for increased intravenous dosages without a corresponding increase in adverse events.975 Furthermore, studies by Yuan et al. have demonstrated that although the plasma concentration of albumin nanoparticles carrying paclitaxel is 3–5 times lower than that of free paclitaxel, the tumor/plasma concentration ratio can reach up to 10 times higher. This underscores the specific tumor targeting capability of albumin nanoparticles and provides robust evidence supporting their suitability for high-dose administration.976 In vivo delivery of therapeutic molecules such as siRNA and miRNA face numerous challenges, including enzymatic degradation, interactions with blood components, and non-specific cellular uptake. In addition to enhancing therapeutic efficacy through nano-loaded drugs, loading therapeutic miRNAs like miR34a and miR200c can further augment treatment outcomes.977,978
Controlled drug release mechanisms rely on both endogenous and exogenous stimuli. Endogenous stimuli responses encompass pH variations, redox reactions, enzyme activity, etc. pH-sensitive nanomaterials maintain stability under physiological conditions but rapidly degrade in the acidic tumor microenvironment, facilitating targeted drug release and enhancing therapeutic efficacy.979 For instance, nanoparticles encapsulating SchB exhibit pH-sensitive release properties and reverse multidrug resistance in breast CSCs by inhibiting P-Glycoprotein.957 pH-sensitive core-shell nanoparticles can simultaneously target GSCs and differentiated cells, significantly reducing the proportion of CSCs.980 Exogenous stimuli, including temperature, light, and ionizing radiation etc., serve as triggers for nanomaterials and can exert therapeutic effects on tumors. Combination therapy, integrating conventional anticancer treatments with anti-CSC drugs, represents a prudent approach to enhance treatment effect.981,982 Photothermal or photodynamic therapy offers higher selectivity, lower toxicity, and improved reproducibility.951 For instance, Zhu et al. engineered nanoparticles with sheddable PEG shells and acid-activatable pro-penetration peptides to deliver a diradical-featured croconium-based photothermal agent and a natural cytotoxic HSP inhibitor to CSCs, achieving synergistic thermo-chemotherapy.983 Fernandes et al. designed magnetic nanoparticles released via hyperthermia to exert potent inhibitory effects on colorectal CSCs when combined with chemotherapy.984
Enhancing drug delivery systems to accommodate multiple drugs is crucial for inhibiting CSCs. CSCs possess unique metabolic pathways and often exhibit overexpression of drug efflux pumps, leading to multidrug resistance. Multiple dosing strategies can increase drug concentrations and target CSCs through diverse mechanisms. For example, coating cisplatin and disulfiram with hydroxypropyl-β-cyclodextrin enhances solubility, inhibiting tumor stemness and improving chemotherapy resistance.985 Liposomes coated with bufalin and doxorubicin effectively suppress the self-renewal of breast CSCs.986 Dual-targeting nanoparticles, characterized by excellent biocompatibility and precise CSC recognition, can simultaneously deliver doxorubicin and siRNA cocktails, exerting potent anti-CSC effects.987 Zhang et al. devised mesoporous silica nanoparticles co-loaded with multiple siRNAs, which effectively treat leukemia when combined with chemotherapy drugs.988 Furthermore, the combined delivery of salinomycin and docetaxel via dual-targeting gelatinase nanoparticles demonstrates significant inhibition of cervical CSCs.989 Nanoparticles incorporating penetration peptide RW9, an Histone Deacetylase (HDAC) inhibitor warhead, and 5-fluorouracil, along with AS1411, enhance inhibitory efficiency against stem-like cells.990 These advancements in drug delivery systems hold promise for combating CSC-mediated resistance and improving cancer treatment outcomes.
Various drug delivery systems exhibit both advantages and limitations.991 In preclinical research, nanotechnology is frequently employed to target CSCs markers such as CD44 and CD133, as well as signaling pathways like WNT/β-Catenin, Notch, and hedgehog.991,992 However, in current clinical trials, the utilization of inhibitors of CSCs markers or related molecular pathways is more prevalent, with limited investigations focusing on enhancing CSCs-targeted therapy through drug delivery systems. Given the heterogeneity of CSCs and the complexity of the tumor microenvironment, achieving precise targeting of CSCs remains a critical challenge. Accurate targeting not only enhances efficacy but also mitigates side effects. Furthermore, leveraging computer technology to assist in setting specific triggers for controlling drug release and identifying precise and efficient targets, employing multi-target, multi-function, and multi-drug combination strategies, will enhance the efficiency of CSC targeting.993,994,995,996,997,998 Designing diverse nanomaterials based on the five fundamental characteristics of nanoparticle therapy—long circulation, tumor accumulation, deep penetration, cellular internalization, and controlled drug release—remains the prevailing research paradigm.999 Although most studies are currently confined to preclinical investigations, optimized therapeutic strategies targeting CSCs via drug delivery systems hold significant promise.952,1000
In tackling brain tumors, particularly gliomas and brain metastases, optimizing drug delivery systems is essential due to the inherent limitations of chemotherapy, including lack of specificity, harmful side effects, low efficacy, and limited transport.1001 The blood-brain barrier (BBB) high selectivity for permeating substances, the unique brain microenvironment, and the deep-seated location of GSCs, possess robust chemotherapy resistance for GSCs.1002 Overcoming these obstacles and effectively targeting CSCs is pivotal. Knauer et al. demonstrated in vitro inhibition of GSCs and modulation of tumor cell surface markers such as PD-L1, TIM-3, and CD47 using a polycationic phosphorus dendrimer-based approach for siRNAs.1003 Aptamer technology has also shown promise in GSCs. Behrooz et al. reported that B19 aptamer-conjugated PAMAM G4C12 dendrimer nanoparticles simultaneously deliver paclitaxel and temozolomide into U87 CSCs, effectively eliminating U87 CSCs without toxic side effects.1004 Multi-drug therapy by employing nanotechnology is gaining traction. Smiley et al. utilized functionalized nanoparticles to co-deliver TMZ and the MDM2 inhibitor idasanutlin to target GSCs.1005 Gold nanoparticles releasing retinoic acid and TMZ upon low-intensity ultrasound stimulation sensitize GSCs to chemotherapy.1006 Nanostructured lipid carriers co-deliver paclitaxel and doxorubicin to inhibit GSCs proliferation via PI3K/AKT/mTOR signal pathway.1007 Peptides also exhibit anti-GSCs properties. Multifunctional tandem peptide R8-c (RGD) destroys vasculogenic mimicry to suppress GSCs proliferation.1008 Additionally, functional curcumin liposomes, layered double hydroxide nanoparticles, and other formulations demonstrate therapeutic efficacy against GSCs.1009
Moreover, nanoparticles or liposomes capable of penetrating the BBB may play a pivotal role. Engineered high-density lipoprotein-mimetic nanoparticles effectively deliver SHH inhibitors to stem-like cells in medulloblastoma.1010 Lu et al. synthesized folic acid-modified albumin nanoparticles to enhance BBB permeability and cellular uptake. These nanoparticles loaded with paclitaxel and autophagy inhibitor chloroquine effectively inhibit GSCs.1011 Curcumin-loaded chitosan-poly (lactic-co-glycolic acid) nanoparticles, processing with sialic acid to enhance BBB permeability and target the brain CSCs via anti-ALDH, demonstrate therapeutic potential.1012 Furthermore, liposomes capable of crossing the BBB induce necrosis, apoptosis, and autophagy in glioma and GSCs.1013 These advancements offer promising avenues for combating brain tumors and targeting CSCs effectively.
Séhédic et al. reported that radiopharmaceutical nanoparticles penetrated the BBB and demonstrated therapeutic efficacy against glioblastoma in mice.1014 Despite numerous drug delivery systems proving effective in inhibiting tumors by traversing the BBB, studies targeting CSCs remain relatively scarce.1015,1016 Although few treatments targeting GSCs have reached clinical trials, continued research into potential pathways and treatment strategies is imperative.1001 Mechanical or chemical disruption of the BBB via MRI-guided focused ultrasound, convection-enhanced diffusion, microdialysis catheters, hypertonic agents, hydrophilic surfactants, and other methods have been explored to modulate BBB permeability. However, the BBB serves as a highly selective diffusion barrier, shielding the brain from toxins and other blood compounds.1017 Balancing the beneficial opening of the BBB for drug delivery with the preservation of its protective barrier function poses a critical question. Brain tumors such as GBM, brain parenchymal metastasis, and leptomeningeal metastasis exhibit high malignancy, with patients experiencing extremely short survival times. While technologies like intrathecal injection and Omaya reservoir enable localized treatment of the nervous system and increase drug concentration, their potential combination with CSCs-targeting approaches requires further investigation.1018,1019,1020 Phase I/II clinical trials (NCT03566199) have demonstrated the safety of the panobinostat nanoparticle formulation MTX110 for newly-diagnosed diffuse intrinsic pontine glioma. However, nanotechnology specifically targeting brain CSCs remains an area lacking in research.
Challenges in cancer stem cell research
Given the pivotal role of CSCs in tumor relapse and resistance mechanisms, extensive research efforts are being dedicated to the task of identifying and targeting CSCs. However, the identification of CSC-specific antigens or biomarkers remains a formidable challenge. Potential CSC biomarkers, identified through aberrant signaling and metabolic pathways, can be broadly classified into two categories: cell surface markers and intracellular markers. Cell surface markers, particularly transport proteins, and signaling receptors have garnered attention for their potential to facilitate the diagnostic and precise delivery of therapeutic agents to CSCs.1021,1022 Yet, the non-specificity and low abundance of these markers pose significant obstacles to their practical application. The surface markers identified to date lack specificity for any single CSC type, as they are also expressed on non-CSCs or healthy cells, albeit at lower levels.1023 Large libraries of intracellular molecules may reveal concentration differences between CSCs and other cell populations, overexpressed intracellular enzymes in CSCs remain key molecular targets for CSC-specific strategies. These enzymes, exemplified by ALDH, can be targeted with prodrugs activated in the presence of specific enzymes, thereby preferentially killing CSCs.1024 Additionally, transcription factors regulating CSC proliferation and differentiation, such as BMI-1 and c-Myc, offer avenues for the design of inhibitors to induce CSC apoptosis.1025,1026 Other crucial CSC-related transcription factors, essential for maintaining CSC tumorigenicity and stemness, like OCT3/4 and SOX2, have also garnered widespread interest.1027,1028 However, a critical issue is that these intracellular transcription factors are not unique to CSCs, as most signaling and metabolic pathways are shared among CSCs, non-stem cells, and healthy cells.
Secondly, although existing CSCs-target therapy shows promise in cancer treatment, numerous limitations persist. CD133, as a potential molecular target, poses challenges in terms of reliable detection and specific antibody recognition.47 Its expression is influenced by various factors, including oxygen levels, cell density, and cell cycle, all of which can affect its protein expression within the microenvironment. Currently, detection of CD133 primarily relies on immunohistochemistry and flow cytometry, both of which require specific antibodies. However, CD133 is sensitive to glycosylation modifications, potentially impacting antibody binding. Commonly used CD133 antibody clones, including CD133/1 (AC133 or W6B3C1) and CD133/2 (AC141 or 293C3), recognize different glycosylated epitopes in the CD133 EC3 region. Yet, glycosylation differences may lead to selective splicing and masking of epitope binding sites, thereby reducing detection accuracy.40 Additionally, it is noteworthy that both CD133+/− cancer cells can initiate tumors, raising questions about the validity of current CSC biomarkers as true tumor-initiating cells.1029 Recent studies have also discovered that CSCs exhibit high plasticity, capable of phenotypic transitions under specific conditions. For instance, in xenograft mouse cancer organoids, gene knockout of LGR5+ CSCs can limit tumor growth but not eliminate it. Tumors can be sustained by proliferative LGR5− cells and, upon cessation of the knockout, LGR5+ CSCs reemerge, leading to rapid tumor regeneration.1030 This suggests that tumor cells with a higher degree of differentiation, following CSC depletion, possess plasticity to revert to the CSC state to compensate for CSC loss. Furthermore, CSC niches created by different cells within the microenvironment can facilitate the evolution of distinct CSC dominant clones.1031 However, current research on the microenvironment and CSCs relies heavily on tumor implantation analyses in mouse models, which cannot fully replicate the microenvironment of primary tumors and the interactions between human CSCs and their microenvironment, thus introducing certain limitations.
Given the challenges associated with CSCs-target therapy, combination therapy emerges as a promising strategy to eradicate CSCs and thereby improve patient outcomes.1032 Combination therapy is recognized for its potency, as it targets multiple pathways to effectively address tumor heterogeneity and enhance efficacy. Moreover, the concurrent use of multiple drugs can tackle drug resistance, aiding in the elimination of CSCs.1033 Although traditional chemotherapy may not directly target CSCs, its foundational and critical role in treating various cancers, especially in early-stage patients, cannot be overlooked. Perhaps their combination with other CSCs-target therapy could overcome the issue of relapse.1034 When applying combination therapy in clinical practice, several issues need to be considered.1035 Firstly, drug interactions may affect efficacy, with one drug potentially interfering with the metabolic activity of another, thereby reducing overall effectiveness. Additionally, the pharmacokinetics of concurrent administration become exceedingly complex due to differences in drug metabolism and uptake. Secondly, the combined use of multiple drugs might provoke cumulative side effects, complicating the assessment of treatment dosages. If two drugs have similar side effects, this could negatively impact patient survival. Identifying the specific drug responsible for side effects also poses a challenge, sometimes necessitating the cessation of all drugs. Lastly, CSCs might acquire an MDR phenotype through mechanisms such as overexpression of drug efflux pumps, alterations in DNA repair mechanisms, and modulation of cell death pathways, rendering combination therapy ineffective.611,612,613 However, a judicious sequence of combination therapy could delay the onset of resistance, thus eliminating CSCs before they become drug-resistant. Therefore, developing a rational sequence of combination therapy is crucial. Moreover, devising novel and effective methods for targeting CSCs remains a focus of research.1036 Nanotherapy, as a potential strategy, offers possibilities for sustained treatment by enhancing drug specificity for CSCs, reducing off-target effects, increasing drug load, optimizing penetration of biological barriers, and controlling drug release.1037 Additionally, nanomaterials can carry multiple therapeutic agents, achieving synergistic effects and potentially reducing resistance. Improved pharmacokinetic properties and protection against enzymatic degradation further consolidate the status of nanomaterials as an effective and versatile platform for targeting CSCs.
Currently, the most prominent and likely direction for the next decade revolves around identifying novel CSC-specific biomarkers and leveraging them for cancer treatment. There’s an active exploration into introducing artificial biomarkers into CSCs. Metabolic glycoengineering of unnatural sugars offers a straightforward tool for incorporating artificial chemical receptors into the cell membrane for subsequent targeting purposes. One such example, the azidosugar AAMCO, can label cells in an ALDH1A1-activated manner, thus preferentially tagging CSCs overexpressing ALDH1A1 with azide groups. This method transforms intracellular ALDH1A1 into a clickable tag on the cell surface, paving a new pathway for developing CSC-targeting technologies.1038 In principle, other unnatural sugars that can be reactivated by other overexpressed CSC enzymes could also be used for CSC labeling and targeting. However, a challenge is the relatively low labeling efficiency of AAMCO, as the ALDH1A1 response is partly at the expense of overall metabolic labeling efficiency. A delicate balance between labeling efficiency and selectivity is necessary. This issue could be mitigated by deploying strategies to improve the delivery of enzyme-activatable unnatural sugars to tumors, such as using nanoparticles. Metabolic lipid labeling could also serve as an alternative method for chemical labeling of CSCs.1039 Given the aberrant lipid metabolism in CSCs, rational design of unnatural lipids based on structural lipids (like dioleoylphosphatidylcholine, DOPC) or signaling lipids (like ceramides, phosphatidylinositol lipids) could achieve preferential labeling of CSCs over non-stem cancer cells or healthy cells. Chemical tags, such as azide groups, could then target therapeutic drugs to CSCs.1040 Compared to metabolic glycoengineering pathways involving multiple reactions, lipid metabolism pathways involve fewer steps and may lead to higher labeling efficiency.1041 Similar to metabolic glycan labeling, strategies to improve the delivery of unnatural lipids to tumors and enhance CSC uptake of unnatural lipids could further increase labeling efficiency and subsequent CSC targeting efficacy.
Despite the numerous challenges faced in CSC research, ongoing studies and technological advancements offer hope for a deeper understanding of the nature of cancer. Such progress promises to unveil novel cancer treatment strategies by navigating the complexities of tumor biology to reveal new avenues of intervention.
Conclusion
Although CSCs originate from differentiated cells, normal stem/progenitor cells, or hybrids from cell-cell fusion, environmental factors in CSC niches are essential in the formation and maintenance of CSCs. Characterized by self-renewal and pluripotency, CSCs play pivotal roles in cancer initiation, proliferation, metastasis, and therapeutic resistance. Identification of CSCs relies on specific biomarkers, including intracellular and cell-surface markers, which serve as tools to predict patients’ prognosis regarding specific treatments. Multiple signaling pathways are excessively activated in CSCs, with intricate crosstalk among them. Extensive evidence indicates that the activation of pathways such as WNT/β-Catenin, hedgehog, Notch, NF-κB, JAK/STAT, TGF-β, PI3K/AKT, and PPAR contributes to various malignant behaviors of CSCs, offering potential therapeutic targets.
Despite facing numerous challenges, researchers remain dedicated to exploring innovative approaches to eradicate CSCs, thereby enhancing the responsiveness to chemotherapy. In-depth research into the characteristics of CSCs, including novel markers, signaling pathways, and the microenvironment, is expected to be a hot theme in the coming decades. Although there is currently a lack of sufficient high-quality clinical trials to confirm the efficacy of these strategies, the deepening of research gives us reason to anticipate the discovery of more effective methods for eliminating CSCs to overcome chemotherapy resistance in the future. The rapid advancement of immunotherapy has ushered in a new era in anti-tumor treatment. In-depth investigations into the immunological characteristics of CSCs have laid a theoretical foundation for immunotherapy targeting CSCs and validated its technical feasibility. Extensive preclinical research has thoroughly demonstrated the potential benefits of CSC-targeted immunotherapy. However, to translate this approach into clinical practice, we still face a host of challenges. These include identifying specific antigens of CSCs, unraveling the mechanisms by which CSCs evade immune surveillance, and understanding the impact of the immunosuppressive tumor microenvironment on therapy. Addressing these issues may pave the way toward successful anti-tumor treatment.
Radiation can induce CSC formation, and CSCs are generally radioresistant. Although it is mechanically reasonable to target CSCs to improve radiosensitivity, few clinical studies succeeded. While preclinical studies consistently demonstrate that the presence of CSCs correlates positively with resistance to targeted therapies, their eradication has the potential to reverse this resistance. However, clinical trials currently lack supportive evidence for this proposition. Therapeutic approaches aimed at eliminating CSCs primarily involve targeting CSC markers, signaling pathways, and their microenvironmental niche. Although numerous clinical trials have been undertaken, substantial, high-quality clinical evidence supporting the efficacy of these strategies remains elusive.
CSCs, a rare subset within tumors, exert significant influence over various malignant processes, including tumor initiation, proliferation, and metastasis. Notably, CSCs exhibit resistance to diverse therapeutic interventions, encompassing chemotherapy, immunotherapy, radiotherapy, and targeted therapy. Comprehensive understanding of CSCs’ involvement in treatment resistance, coupled with strategies to specifically target CSCs, is crucial for advancing patient outcomes. Preliminary findings from preclinical studies indicate the potential efficacy of CSC-targeted interventions in overcoming treatment resistance. Moreover, the forthcoming results from ongoing clinical trials hold promise for further advancing the understanding and therapeutic management in this field.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Bonnet, D. & Dick, J. E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737 (1997).
Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017).
Plaks, V., Kong, N. & Werb, Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238 (2015).
Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct Target Ther. 5, 8 (2020).
Houghton, J., Morozov, A., Smirnova, I. & Wang, T. C. Stem cells and cancer. Semin. Cancer Biol. 17, 191–203 (2007).
Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 51, 1–28 (2004).
Shimkin, M. B. The written word and cancer–some personal involvements, 1940-1977: autobiographical essay. Cancer Res. 38, 241–252 (1978).
Cairns, J. Mutation selection and the natural history of cancer. Nature 255, 197–200 (1975).
Fisher, J. C. & Hollomon, J. H. A hypothesis for the origin of cancer foci. Cancer 4, 916–918 (1951).
Furth, J., Kahn, M. C. & Breedis, C. The transmission of Leukemia of Mice with a Single Cell1. Am. J. Cancer 31, 276–282 (1937).
Till, J. E. & Mc, C. E. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res. 14, 213–222 (1961).
Siminovitch, L., McCulloch, E. A. & Till, J. E. The distribution of colony-forming cells among spleen colonies. J. Cell Comp. Physiol. 62, 327–336 (1963).
Potten, C. S. & Loeffler, M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110, 1001–1020 (1990).
Orkin, S. H. & Zon, L. I. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644 (2008).
Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4, 7–25 (1978).
Zhang, J. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841 (2003).
Jassim, A., Rahrmann, E. P., Simons, B. D. & Gilbertson, R. J. Cancers make their own luck: theories of cancer origins. Nat. Rev. Cancer 23, 710–724 (2023).
Riva, L. et al. The mutational signature profile of known and suspected human carcinogens in mice. Nat. Genet. 52, 1189–1197 (2020).
Soto, A. M. & Sonnenschein, C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. Bioessays 33, 332–340 (2011).
Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001).
Clarke, M. F. Clinical and therapeutic implications of cancer stem cells. N Engl. J. Med. 380, 2237–2245 (2019).
Al-Hajj, M. et al. Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. USA 100, 3983–3988 (2003).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
Matsui, W. et al. Characterization of clonogenic multiple myeloma cells. Blood 103, 2332–2336 (2004).
Collins, A. T. et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65, 10946–10951 (2005).
Fang, D. et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 65, 9328–9337 (2005).
O’Brien, C. A., Pollett, A., Gallinger, S. & Dick, J. E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110 (2007).
Ricci-Vitiani, L. et al. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115 (2007).
Li, C. et al. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037 (2007).
Rahman, M. et al. The cancer stem cell hypothesis: failures and pitfalls. Neurosurgery 68, 531–545 (2011). discussion 545.
Hewitt, H. B. Studies of the dissemination and quantitative transplantation of a lymphocytic leukaemia of CBA mice. Br. J. Cancer 12, 378–401 (1958).
Park, C. H., Bergsagel, D. E. & McCulloch, E. A. Mouse myeloma tumor stem cells: a primary cell culture assay. J. Natl Cancer Inst. 46, 411–422 (1971).
Hamburger, A. W. & Salmon, S. E. Primary bioassay of human tumor stem cells. Science 197, 461–463 (1977).
Baccelli, I. & Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 198, 281–293 (2012).
Valent, P. et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat. Rev. Cancer 12, 767–775 (2012).
Quintana, E. et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523 (2010).
Taussig, D. C. et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 112, 568–575 (2008).
Ginestier, C. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007).
Grosse-Gehling, P. et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J. Pathol. 229, 355–378 (2013).
Jaksch, M. et al. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 68, 7882–7886 (2008).
Singh, S. K. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 (2003).
Maitland, N. J. & Collins, A. T. Prostate cancer stem cells: a new target for therapy. J. Clin. Oncol. 26, 2862–2870 (2008).
LaBarge, M. A. & Bissell, M. J. Is CD133 a marker of metastatic colon cancer stem cells? J. Clin. Investig. 118, 2021–2024 (2008).
Cheng, J. X., Liu, B. L. & Zhang, X. How powerful is CD133 as a cancer stem cell marker in brain tumors? Cancer Treat Rev. 35, 403–408 (2009).
Wu, Y. & Wu, P. Y. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev. 18, 1127–1134 (2009).
Glumac, P. M. & LeBeau, A. M. The role of CD133 in cancer: a concise review. Clin. Transl. Med. 7, 18 (2018).
Meng, X. et al. Both CD133+ and CD133- subpopulations of A549 and H446 cells contain cancer-initiating cells. Cancer Sci. 100, 1040–1046 (2009).
Alamgeer, M. et al. Cancer stem cells in lung cancer: evidence and controversies. Respirology 18, 757–764 (2013).
Matsui, W. et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 68, 190–197 (2008).
Yaccoby, S. & Epstein, J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood 94, 3576–3582 (1999).
Yaccoby, S., Barlogie, B. & Epstein, J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood 92, 2908–2913 (1998).
Eppert, K. et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 17, 1086–1093 (2011).
Gibbs, K. D. Jr et al. Decoupling of tumor-initiating activity from stable immunophenotype in HoxA9-Meis1-driven AML. Cell Stem Cell 10, 210–217 (2012).
Kong, Y. et al. CD34+CD38+CD19+ as well as CD34+CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia 22, 1207–1213 (2008).
Odoux, C. et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 68, 6932–6941 (2008).
Eramo, A. et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15, 504–514 (2008).
Pece, S. et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 140, 62–73 (2010).
Lapidot, T. et al. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science 255, 1137–1141 (1992).
Hemmati, H. D. et al. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl Acad. Sci. USA 100, 15178–15183 (2003).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl. J. Med. 366, 883–892 (2012).
Campbell, P. J. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113 (2010).
Ying, X. et al. AC133 expression associated with poor prognosis in stage II colorectal cancer. Med. Oncol. 30, 356 (2013).
Wang, D. et al. Detection of CD133 expression in U87 glioblastoma cells using a novel anti-CD133 monoclonal antibody. Oncol. Lett. 9, 2603–2608 (2015).
Fulawka, L., Donizy, P. & Halon, A. Cancer stem cells–the current status of an old concept: literature review and clinical approaches. Biol. Res. 47, 66 (2014).
Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Wellner, U. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 (2009).
Abbaszadegan, M. R. et al. Isolation, identification, and characterization of cancer stem cells: a review. J. Cell Physiol. 232, 2008–2018 (2017).
Insausti, C. L. et al. Isolation and characterization of mesenchymal stem cells from the fat layer on the density gradient separated bone marrow. Stem Cells Dev. 21, 260–272 (2012).
Fong, C. Y. et al. The use of discontinuous density gradients in stem cell research and application. Stem Cell Rev. Rep. 5, 428–434 (2009).
Chen, M. J. & Bongso, A. Comparative evaluation of two density gradient preparations for sperm separation for medically assisted conception. Hum. Reprod. 14, 759–764 (1999).
Tárnok, A. Cancer and cytometry. Cytometry A 95, 257–258 (2019).
Zhang, D. G. et al. Isolation, cultivation and identification of human lung adenocarcinoma stem cells. Oncol. Lett. 9, 47–54 (2015).
Liu, L. & Borlak, J. Advances in liver cancer stem cell isolation and their characterization. Stem Cell Rev. Rep. 17, 1215–1238 (2021).
Greve, B. et al. Flow cytometry in cancer stem cell analysis and separation. Cytometry A 81, 284–293 (2012).
Goodell, M. A. et al. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 183, 1797–1806 (1996).
Kondo, T., Setoguchi, T. & Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl Acad. Sci. USA 101, 781–786 (2004).
Hirschmann-Jax, C. et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl Acad. Sci. USA 101, 14228–14233 (2004).
Chiba, T. et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 44, 240–251 (2006).
Unno, K., Jain, M. & Liao, R. Cardiac side population cells: moving toward the center stage in cardiac regeneration. Circ. Res. 110, 1355–1363 (2012).
Zhou, S. et al. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc. Natl Acad. Sci. USA 99, 12339–12344 (2002).
Zhou, S. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 7, 1028–1034 (2001).
Wu, A. et al. Persistence of CD133+ cells in human and mouse glioma cell lines: detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 17, 173–184 (2008).
Jariyal, H. et al. Advancements in cancer stem cell isolation and characterization. Stem Cell Rev. Rep. 15, 755–773 (2019).
Brown, H. K., Tellez-Gabriel, M. & Heymann, D. Cancer stem cells in osteosarcoma. Cancer Lett. 386, 189–195 (2017).
Ghanei, Z., Jamshidizad, A., Joupari, M. D. & Shamsara, M. Isolation and characterization of breast cancer stem cell-like phenotype by Oct4 promoter-mediated activity. J. Cell Physiol. 235, 7840–7848 (2020).
Jia, Y. et al. Microfluidic tandem mechanical sorting system for enhanced cancer stem cell isolation and ingredient screening. Adv. Healthcare Mat.er 10, e2100985 (2021).
Chen, J. et al. Glioblastoma stem cell-specific histamine secretion drives pro-angiogenic tumor microenvironment remodeling. Cell Stem Cell 29, 1531–1546.e1537 (2022).
Parashar, D. et al. Targeted biologic inhibition of both tumor cell-intrinsic and intercellular CLPTM1L/CRR9-mediated chemotherapeutic drug resistance. NPJ Precis. Oncol. 5, 16 (2021).
Low, J. et al. Knockdown of cancer testis antigens modulates neural stem cell marker expression in glioblastoma tumor stem cells. J. Biomol. Screen. 15, 830–839 (2010).
Huang, D. et al. Multichannel-optical imaging for in vivo evaluating the safety and therapeutic efficacy of stem cells in tumor model in terms of cell tropism, proliferation and NF-κB activity. Biomaterials 307, 122510 (2024).
Luo, M. et al. ZMYND8 protects breast cancer stem cells against oxidative stress and ferroptosis through activation of NRF2. J. Clin. Investig. 134, e171166 (2024).
Zhang, W. et al. KDM1A promotes thyroid cancer progression and maintains stemness through the Wnt/β-catenin signaling pathway. Theranostics 12, 1500–1517 (2022).
Leung, H. W. et al. EPHB2 activates β-Catenin to enhance cancer stem cell properties and drive sorafenib resistance in hepatocellular carcinoma. Cancer Res. 81, 3229–3240 (2021).
Hui, Y. et al. circSLC4A7 accelerates stemness and progression of gastric cancer by interacting with HSP90 to activate NOTCH1 signaling pathway. Cell Death Dis. 14, 452 (2023).
Zhou, F. et al. A dynamic rRNA ribomethylome drives stemness in acute myeloid leukemia. Cancer Discov. 13, 332–347 (2023).
Luo, H. T. et al. Dissecting the multi-omics atlas of the exosomes released by human lung adenocarcinoma stem-like cells. NPJ Genom. Med. 6, 48 (2021).
Robinson, M. et al. Characterization of SOX2, OCT4, and NANOG in Ovarian Cancer Tumor-Initiating Cells. Cancers 13, 262 (2021).
Mitchell, K. et al. WDR5 represents a therapeutically exploitable target for cancer stem cells in glioblastoma. Genes Dev. 37, 86–102 (2023).
Cavalli, F. M. G. et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31, 737–754.e736 (2017).
Northcott, P. A. et al. The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317 (2017).
Gohil, S. H. et al. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 244–256 (2021).
Gisina, A. et al. Glioma stem cells: novel data obtained by single-cell sequencing. Int. J. Mol. Sci. 23, 14224 (2022).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Couturier, C. P. et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 11, 3406 (2020).
Wang, L. et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 9, 1708–1719 (2019).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
Dirkse, A. et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 (2019).
Bhaduri, A. et al. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 26, 48–63.e46 (2020).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e821 (2019).
Horning, A. M. et al. Single-Cell RNA-seq reveals a subpopulation of prostate cancer cells with enhanced cell-cycle-related transcription and attenuated androgen response. Cancer Res. 78, 853–864 (2018).
Savage, P. et al. A targetable EGFR-dependent tumor-initiating program in breast cancer. Cell Rep. 21, 1140–1149 (2017).
Chung, W. et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 8, 15081 (2017).
Lawson, D. A. et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526, 131–135 (2015).
Zheng, H. et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 68, 127–140 (2018).
Velten, L. et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat. Commun. 12, 1366 (2021).
van Galen, P. et al. Single-Cell RNA-Seq reveals aml hierarchies relevant to disease progression and immunity. Cell. 176, 1265–1281.e1224 (2019).
Giustacchini, A. et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 23, 692–702 (2017).
Xiao, M. et al. Complex interaction and heterogeneity among cancer stem cells in head and neck squamous cell carcinoma revealed by single-cell sequencing. Front. Immunol. 13, 1050951 (2022).
Ren, X. et al. Single-cell RNA-seq reveals invasive trajectory and determines cancer stem cell-related prognostic genes in pancreatic cancer. Bioengineered 12, 5056–5068 (2021).
Frank, M. H., Wilson, B. J., Gold, J. S. & Frank, N. Y. Clinical implications of colorectal cancer stem cells in the age of single-cell omics and targeted therapies. Gastroenterology 160, 1947–1960 (2021).
Wu, H. et al. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene 36, 2857–2867 (2017).
Leung, M. L. et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 27, 1287–1299 (2017).
Yao, Y. & Wang, C. Dedifferentiation: inspiration for devising engineering strategies for regenerative medicine. NPJ Regen. Med. 5, 14 (2020).
Pérez-González, A., Bévant, K. & Blanpain, C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 4, 1063–1082 (2023).
Zhou, L. et al. Lineage tracing and single-cell analysis reveal proliferative Prom1+ tumour-propagating cells and their dynamic cellular transition during liver cancer progression. Gut 71, 1656–1668 (2022).
Bachoo, R. M. et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269–277 (2002).
Friedmann-Morvinski, D. et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012).
Murata, K. et al. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 26, 377–390.e376 (2020).
Köhler, C. et al. Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 21, 679–693.e676 (2017).
Yan, Q. et al. PGC7 promotes tumor oncogenic dedifferentiation through remodeling DNA methylation pattern for key developmental transcription factors. Cell Death Differ. 28, 1955–1970 (2021).
Ai, J. et al. Bcl3 couples cancer stem cell enrichment with pancreatic cancer molecular subtypes. Gastroenterology 161, 318–332.e319 (2021).
Hall, A. E. et al. RNA splicing is a key mediator of tumour cell plasticity and a therapeutic vulnerability in colorectal cancer. Nat. Commun. 13, 2791 (2022).
Li, B. et al. miR-613 inhibits liver cancer stem cell expansion by regulating SOX9 pathway. Gene 707, 78–85 (2019).
Perekatt, A. O. et al. SMAD4 suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res. 78, 4878–4890 (2018).
Sandiford, O. A. et al. Mesenchymal stem cell-secreted extracellular vesicles instruct stepwise dedifferentiation of breast cancer cells into dormancy at the bone marrow perivascular region. Cancer Res. 81, 1567–1582 (2021).
Schwitalla, S. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38 (2013).
Nakano, M. et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 38, 780–793 (2019).
Mu, R. et al. Hypoxia promotes pancreatic cancer cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential by the HIF-1α/Notch signaling pathway. Pancreas 50, 756–765 (2021).
Cai, S. et al. ERK inactivation enhances stemness of NSCLC cells via promoting Slug-mediated epithelial-to-mesenchymal transition. Theranostics 12, 7051–7066 (2022).
Rodrigues, C. F. D. et al. Stroma-derived IL-6, G-CSF, and Activin-A mediated dedifferentiation of lung carcinoma cells into cancer stem cells. Sci. Rep. 8, 11573 (2018).
Wang, P. et al. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 3, 16105 (2017).
Chen, W. J. et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun 5, 3472 (2014).
Pan, Z. et al. Single-cell transcriptomics unveils the dedifferentiation mechanism of lung adenocarcinoma stem cells. Int. J. Mol. Sci. 24, 482 (2022).
Xie, J. et al. Targeting cancer cell plasticity by HDAC inhibition to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma. Signal Transduct Target Ther. 6, 333 (2021).
Rhost, S. et al. Sortilin inhibition limits secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion. Breast Cancer Res. 20, 137 (2018).
Sun, Z. et al. Glioblastoma stem cell-derived exosomes enhance stemness and tumorigenicity of glioma cells by transferring Notch1 protein. Cell Mol. Neurobiol. 40, 767–784 (2020).
Maiuthed, A. et al. Nitric oxide promotes cancer cell dedifferentiation by disrupting an Oct4:caveolin-1 complex: A new regulatory mechanism for cancer stem cell formation. J. Biol. Chem. 293, 13534–13552 (2018).
Lin, T. C. et al. Oct-4 induces cisplatin resistance and tumor stem cell-like properties in endometrial carcinoma cells. Taiwan J. Obstet Gynecol. 62, 16–21 (2023).
Smith, B. A. et al. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl Acad. Sci. USA 112, E6544–6552 (2015).
Zhang, F. et al. Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: immunohistochemical and double-fluorescence immunostaining evidence. Histopathology 52, 224–232 (2008).
Kim, H. et al. Primary liver carcinoma of intermediate (hepatocyte-cholangiocyte) phenotype. J. Hepatol. 40, 298–304 (2004).
Chen, L. et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One 7, e33544 (2012).
Du, J. et al. Signaling INHIBITORS ACCELERATE THE CONVERSION of mouse iPS cells into cancer stem cells in the tumor microenvironment. Sci. Rep. 10, 9955 (2020).
Calle, A. S. et al. A new PDAC mouse model originated from iPSCs-converted pancreatic cancer stem cells (CSCcm). Am. J. Cancer Res. 6, 2799–2815 (2016).
Hassan, G., Zahra, M. H., Seno, A. & Seno, M. The significance of ErbB2/3 in the conversion of induced pluripotent stem cells into cancer stem cells. Sci. Rep. 12, 2711 (2022).
Afify, S. M. et al. A novel model of liver cancer stem cells developed from induced pluripotent stem cells. Br. J. Cancer 122, 1378–1390 (2020).
Xu, N. et al. Induction of cells with prostate cancer stem-like properties from mouse induced pluripotent stem cells via conditioned medium. Am. J. Cancer Res. 8, 1624–1632 (2018).
Yan, T. et al. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J. Cancer 5, 572–584 (2014).
Sheta, M. et al. Chronic exposure to FGF2 converts iPSCs into cancer stem cells with an enhanced integrin/focal adhesion/PI3K/AKT axis. Cancer Lett. 521, 142–154 (2021).
Seno, A. et al. Cancer stem cell induction from mouse embryonic stem cells. Oncol. Lett. 18, 2756–2762 (2019).
Riggi, N. et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 24, 916–932 (2010).
Barker, N. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009).
Wu, K. et al. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 56, 2255–2267 (2012).
Mokkapati, S. et al. β-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res. 74, 4515–4525 (2014).
Holczbauer, Á. et al. Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterology 145, 221–231 (2013).
Chiba, T. et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 133, 937–950 (2007).
Molyneux, G. et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7, 403–417 (2010).
Bjerkvig, R. et al. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat. Rev. Cancer 5, 899–904 (2005).
Ogle, B. M., Cascalho, M. & Platt, J. L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 6, 567–575 (2005).
Terada, N. et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416, 542–545 (2002).
Dittmar, T. Generation of cancer stem/initiating cells by cell-cell fusion. Int. J. Mol. Sci. 23, 4514 (2022).
Clawson, G. A. et al. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS One 10, e0134320 (2015).
Niu, N., Mercado-Uribe, I. & Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 36, 4887–4900 (2017).
Luo, F. et al. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget 7, 30924–30934 (2016).
Gast, C. E. et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 4, eaat7828 (2018).
Li, X. et al. Mesenchymal/stromal stem cells: necessary factors in tumour progression. Cell Death Discov. 8, 333 (2022).
Dörnen, J., Myklebost, O. & Dittmar, T. Cell fusion of mesenchymal stem/stromal cells and breast cancer cells leads to the formation of hybrid cells exhibiting diverse and individual (stem cell) characteristics. Int. J. Mol. Sci. 21, 9636 (2020).
Xu, M. H. et al. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS One 9, e87893 (2014).
Wang, R. et al. Fusion with stem cell makes the hepatocellular carcinoma cells similar to liver tumor-initiating cells. BMC Cancer 16, 56 (2016).
Lee, J. et al. Engineering liver microtissues to study the fusion of HepG2 with mesenchymal stem cells and invasive potential of fused cells. Biofabrication. 14, 014104 (2021).
Ramakrishnan, M., Mathur, S. R. & Mukhopadhyay, A. Fusion-derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 73, 5360–5370 (2013).
He, X. et al. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer 15, 24 (2015).
Li, H. et al. Fusion of HepG2 cells with mesenchymal stem cells increases cancer‑associated and malignant properties: an in vivo metastasis model. Oncol. Rep. 32, 539–547 (2014).
Zhang, L. N. et al. Fusion with mesenchymal stem cells differentially affects tumorigenic and metastatic abilities of lung cancer cells. J. Cell Physiol. 234, 3570–3582 (2019).
Melzer, C., von der Ohe, J. & Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun Signal. 16, 2 (2018).
Wang, Z. et al. Impact of cell fusion in myeloma marrow microenvironment on tumor progression. Oncotarget 9, 30997–31006 (2018).
Fan, H. & Lu, S. Fusion of human bone hemopoietic stem cell with esophageal carcinoma cells didn’t generate esophageal cancer stem cell. Neoplasma 61, 540–545 (2014).
Xue, J. et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 15, 793 (2015).
Zeng, C. et al. CD34(+) liver cancer stem cells were formed by fusion of hepatobiliary stem/progenitor cells with hematopoietic precursor-derived myeloid intermediates. Stem Cells Dev. 24, 2467–2478 (2015).
Uygur, B. et al. Interactions with muscle cells boost fusion, stemness, and drug resistance of prostate cancer cells. Mol. Cancer Res. 17, 806–820 (2019).
Gauck, D. et al. Hybrid clone cells derived from human breast epithelial cells and human breast cancer cells exhibit properties of cancer stem/initiating cells. BMC Cancer 17, 515 (2017).
Merckens, A., Sieler, M., Keil, S. & Dittmar, T. Altered phenotypes of breast epithelial × breast cancer hybrids after ZEB1 Knock-Out. Int. J. Mol. Sci. 24, 17310 (2023).
Ding, J. et al. Tumor associated macrophage × cancer cell hybrids may acquire cancer stem cell properties in breast cancer. PLoS One 7, e41942 (2012).
Aguirre, L. A. et al. Tumor stem cells fuse with monocytes to form highly invasive tumor-hybrid cells. Oncoimmunology 9, 1773204 (2020).
Cooper, K. E. et al. Changes in body temperature and vasopressin content of brain neurons, in pregnant and non-pregnant guinea pigs, during fevers produced by Poly I:Poly C. Pflugers Arch. 412, 292–296 (1988).
Merle, C., Lagarde, P., Lartigue, L. & Chibon, F. Acquisition of cancer stem cell capacities after spontaneous cell fusion. BMC Cancer 21, 241 (2021).
Chen, Z. et al. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal. Transduct Target Ther. 8, 70 (2023).
Liu, Z. L. et al. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal. Transduct Target Ther. 8, 198 (2023).
Tian, W., Cao, C., Shu, L. & Wu, F. Anti-Angiogenic Therapy In The Treatment Of Non-small Cell Lung Cancer. Onco Targets Ther. 13, 12113–12129 (2020).
Acker, T. & Plate, K. H. Hypoxia and hypoxia inducible factors (HIF) as important regulators of tumor physiology. Cancer Treat Res. 117, 219–248 (2004).
Kumar, S. M. et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 31, 4898–4911 (2012).
Beck, B. et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478, 399–403 (2011).
Wang, L. et al. VEGFA/NRP-1/GAPVD1 axis promotes progression and cancer stemness of triple-negative breast cancer by enhancing tumor cell-macrophage crosstalk. Int. J. Biol. Sci. 20, 446–463 (2024).
Zhan, Y. et al. Carcinoma-associated fibroblasts derived exosomes modulate breast cancer cell stemness through exonic circHIF1A by miR-580-5p in hypoxic stress. Cell Death Discov. 7, 141 (2021).
Kinugasa, Y., Matsui, T. & Takakura, N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells 32, 145–156 (2014).
Wang, Y. et al. Granulocytic Myeloid-derived Suppressor Cells Promote The Stemness Of Colorectal Cancer Cells Through Exosomal S100A9. Adv. Sci. 6, 1901278 (2019).
Wu, F. et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther. 6, 218 (2021).
Fang, Y. et al. Cancer associated fibroblasts serve as an ovarian cancer stem cell niche through noncanonical Wnt5a signaling. NPJ Precis. Oncol. 8, 7 (2024).
Kanzawa, M. et al. WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology 80, 235–244 (2013).
Álvarez-Teijeiro, S. et al. Factors Secreted By Cancer-associated Fibroblasts That Sustain Cancer Stem Properties In Head And Neck Squamous Carcinoma Cells As Potential Therapeutic Targets. Cancers 10, 334 (2018).
Hasegawa, T. et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-β signaling. Int. J. Cancer 134, 1785–1795 (2014).
Zhao, Z. et al. Periostin secreted from podoplanin-positive cancer-associated fibroblasts promotes metastasis of gastric cancer by regulating cancer stem cells via AKT and YAP signaling pathway. Mol. Carcinog. 62, 685–699 (2023).
Vaziri, N. et al. Cancer-Associated Fibroblasts Regulate The Plasticity Of Breast Cancer Stemness Through The Production Of Leukemia Inhibitory Factor. Life 11, 1298 (2021).
Ren, J. et al. Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 21, 109 (2019).
Li, Y. et al. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J. Mol. Med. 97, 243–255 (2019).
Fisher, M. L. et al. Cancer-associated fibroblasts promote cancer stemness by inducing expression of the chromatin-modifying protein CBX4 in squamous cell carcinoma. Carcinogenesis 44, 485–496 (2023).
Giannoni, E. et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 70, 6945–6956 (2010).
Ding, S. M. et al. MRC-5 Cancer-associated Fibroblasts Influence Production Of Cancer Stem Cell Markers And Inflammation-associated Cell Surface Molecules, In Liver Cancer Cell Lines. Int. J. Med. Sci. 16, 1157–1170 (2019).
Novák, Š. et al. Desmoplastic Crosstalk In Pancreatic Ductal Adenocarcinoma Is Reflected By Different Responses of Panc-1, MIAPaCa-2, PaTu-8902, and CAPAN-2 Cell Lines To Cancer-associated/normal Fibroblasts. Cancer Genomics Proteomics 18, 221–243 (2021).
Zhuang, J. et al. Cancer-Associated Fibroblast-Derived miR-146a-5p Generates a Niche That Promotes Bladder Cancer Stemness and Chemoresistance. Cancer Res. 83, 1611–1627 (2023).
Liu, Y. et al. Carcinoma associated fibroblasts small extracellular vesicles with low miR-7641 promotes breast cancer stemness and glycolysis by HIF-1α. Cell Death Discov. 7, 176 (2021).
Wang, M. et al. Loss of exosomal miR-34c-5p in cancer-associated fibroblast for the maintenance of stem-like phenotypes of laryngeal cancer cells. Head Neck 44, 2437–2451 (2022).
Sun, Z., Wang, S. & Zhao, R. C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 7, 14 (2014).
Li, D. et al. Bone Marrow Mesenchymal Stem Cells Promote The Stemness Of Hypopharyngeal Cancer Cells. Cell Reprogram. 22, 269–276 (2020).
Jiménez, G. et al. Mesenchymal stem cell’s secretome promotes selective enrichment of cancer stem-like cells with specific cytogenetic profile. Cancer Lett. 429, 78–88 (2018).
Wu, H. et al. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1α/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 11, 233 (2020).
Chen, Z. et al. MSC-NPRA loop drives fatty acid oxidation to promote stemness and chemoresistance of gastric cancer. Cancer Lett. 565, 216235 (2023).
He, W. et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 38, 4637–4654 (2019).
Cuiffo, B. G. et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 15, 762–774 (2014).
Luo, J. et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene 33, 2768–2778 (2014).
Du, Y. et al. Intracellular Notch1 Signaling In Cancer-associated Fibroblasts Dictates The Plasticity And Stemness Of Melanoma Stem/initiating Cells. Stem Cells 37, 865–875 (2019).
Raghavan, S. et al. Carcinoma-Associated Mesenchymal Stem Cells Promote Chemoresistance In Ovarian Cancer Stem Cells via PDGF Signaling. Cancers 12, 2063 (2020).
Sun, L. et al. Gastric cancer mesenchymal stem cells regulate PD-L1-CTCF enhancing cancer stem cell-like properties and tumorigenesis. Theranostics 10, 11950–11962 (2020).
Avnet, S. et al. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-κB activation. Int. J. Cancer 140, 1331–1345 (2017).
Cortini, M. et al. Tumor-activated mesenchymal stromal cells promote osteosarcoma stemness and migratory potential via IL-6 secretion. PLoS One 11, e0166500 (2016).
Li, H. J., Reinhardt, F., Herschman, H. R. & Weinberg, R. A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2, 840–855 (2012).
Mao, J. et al. UBR2 enriched in p53 deficient mouse bone marrow mesenchymal stem cell-exosome promoted gastric cancer progression via Wnt/β-catenin pathway. Stem Cells 35, 2267–2279 (2017).
Ma, X. et al. Mesenchymal stem cells maintain the stemness of colon cancer stem cells via interleukin-8/mitogen-activated protein kinase signaling pathway. Exp. Biol. Med. 245, 562–575 (2020).
Xu, Y. et al. Endometrium-derived mesenchymal stem cells suppress progression of endometrial cancer via the DKK1-Wnt/β-catenin signaling pathway. Stem Cell Res. Ther. 14, 159 (2023).
Gu, H. et al. Mesenchymal stem cell-derived exosomes block malignant behaviors of hepatocellular carcinoma stem cells through a lncRNA C5orf66-AS1/microRNA-127-3p/DUSP1/ERK axis. Hum. Cell 34, 1812–1829 (2021).
Yao, X. et al. Exosomal circ_0030167 derived from BM-MSCs inhibits the invasion, migration, proliferation and stemness of pancreatic cancer cells by sponging miR-338-5p and targeting the Wif1/Wnt8/β-catenin axis. Cancer Lett. 512, 38–50 (2021).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
Zhang, Q. et al. Reciprocal interactions between malignant cells and macrophages enhance cancer stemness and M2 polarization in HBV-associated hepatocellular carcinoma. Theranostics 14, 892–910 (2024).
Bührer, E. D. et al. Splenic red pulp macrophages provide a niche for CML stem cells and induce therapy resistance. Leukemia 36, 2634–2646 (2022).
Meng, F. et al. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life 66, 835–846 (2014).
Li, X. et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp. Cell Res. 378, 131–138 (2019).
Lu, C. H. et al. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 37, 6327–6340 (2018).
Yang, K. et al. M2 tumor-associated macrophage mediates the maintenance of stemness to promote cisplatin resistance by secreting TGF-β1 in esophageal squamous cell carcinoma. J. Transl. Med. 21, 26 (2023).
Nusblat, L. M., Carroll, M. J. & Roth, C. M. Crosstalk between M2 macrophages and glioma stem cells. Cell Oncol. 40, 471–482 (2017).
Liu, Z., Kuang, W., Zhou, Q. & Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 42, 3395–3403 (2018).
Zhang, B. et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-β1/smad2/3 axis in pancreatic cancer. Cancer Lett. 459, 204–215 (2019).
Cui, F., Xu, Z., Hu, J. & Lv, Y. Spindle pole body component 25 and platelet-derived growth factor mediate crosstalk between tumor-associated macrophages and prostate cancer cells. Front. Immunol. 13, 907636 (2022).
Fan, Q. M. et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 352, 160–168 (2014).
Kundu, P. & Shankar, B. S. Macrophage induced ERK-TGF-β1 signaling in MCF7 breast cancer cells result in reversible cancer stem cell plasticity and epithelial mesenchymal transition. Biochim. Biophys. Acta Gen. Subj. 1866, 130215 (2022).
Wei, X. et al. Tumor-associated macrophages increase the proportion of cancer stem cells in lymphoma by secreting pleiotrophin. Am. J. Transl. Res. 11, 6393–6402 (2019).
Shin, A. E. et al. F4/80(+)Ly6C(high) Macrophages Lead to Cell Plasticity and Cancer Initiation in Colitis. Gastroenterology 164, 593–609.e513 (2023).
Radharani, N. N. V. et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer Cell Int. 22, 122 (2022).
Raghavan, S. et al. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 7, 190 (2019).
Ning, Y. et al. Co-culture of ovarian cancer stem-like cells with macrophages induced SKOV3 cells stemness via IL-8/STAT3 signaling. Biomed. Pharmacother. 103, 262–271 (2018).
Yang, L. et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 145, 1099–1110 (2019).
Fang, M. et al. IL33 promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res. 77, 2735–2745 (2017).
Shang, S. et al. ID1 expressing macrophages support cancer cell stemness and limit CD8(+) T cell infiltration in colorectal cancer. Nat. Commun. 14, 7661 (2023).
Liu, D. et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res. 29, 365–378 (2019).
Wei, R. et al. S100 calcium-binding protein A9 from tumor-associated macrophage enhances cancer stem cell-like properties of hepatocellular carcinoma. Int. J. Cancer 148, 1233–1244 (2021).
Yang, J. et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 31, 248–258 (2013).
Lv, J. et al. M2‑like tumour‑associated macrophage‑secreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol. Med. Rep. 24, 604 (2021).
Liguori, M. et al. The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL-33. Cell Mol. Immunol. 18, 711–722 (2021).
Gomez, K. E. et al. Cancer Cell CD44 mediates macrophage/monocyte-driven regulation of head and neck cancer stem cells. Cancer Res. 80, 4185–4198 (2020).
Li, W. et al. Exosomes secreted by M2 macrophages promote cancer stemness of hepatocellular carcinoma via the miR-27a-3p/TXNIP pathways. Int. Immunopharmacol. 101, 107585 (2021).
Chen, X. et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/β-catenin signaling. Cell Commun. Signal. 20, 92 (2022).
Zhang, X. et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Invest. 100, 619–629 (2020).
Zhao, H. C. et al. CD168(+) macrophages promote hepatocellular carcinoma tumor stemness and progression through TOP2A/β-catenin/YAP1 axis. iScience 26, 106862 (2023).
Shi, X. et al. Exosome-derived miR-372-5p promotes stemness and metastatic ability of CRC cells by inducing macrophage polarization. Cell Signal. 111, 110884 (2023).
Yan, J. et al. FGL2-wired macrophages secrete CXCL7 to regulate the stem-like functionality of glioma cells. Cancer Lett. 506, 83–94 (2021).
Hsieh, C. Y. et al. Macrophage secretory IL-1β promotes docetaxel resistance in head and neck squamous carcinoma via SOD2/CAT-ICAM1 signaling. JCI Insight. 7, e157285 (2022).
She, L. et al. Tumor-associated macrophages derived CCL18 promotes metastasis in squamous cell carcinoma of the head and neck. Cancer Cell Int. 18, 120 (2018).
Guo, L. et al. Induction of breast cancer stem cells by M1 macrophages through Lin-28B-let-7-HMGA2 axis. Cancer Lett. 452, 213–225 (2019).
Lu, H. et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 16, 1105–1117 (2014).
Li, K. et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal. Transduct Target Ther. 6, 362 (2021).
Li, X. et al. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. Febs J. 287, 5218–5235 (2020).
Cui, T. X. et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39, 611–621 (2013).
Ai, L. et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 18, 88 (2019).
Lin, Y. et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology 75, 28–42 (2022).
Peng, D. et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 76, 3156–3165 (2016).
Yue, D. et al. NEDD9 promotes cancer stemness by recruiting myeloid-derived suppressor cells via CXCL8 in esophageal squamous cell carcinoma. Cancer Biol. Med. 18, 705–720 (2021).
Panni, R. Z. et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 63, 513–528 (2014).
Komura, N. et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol. Immunother. 69, 2477–2499 (2020).
Muntané, J. & Bonavida, B. Special collection: nitric oxide in cancer. Redox Biol. 6, 505–506 (2015).
Charles, N. et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6, 141–152 (2010).
Lee, S. Y. et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 16, 10 (2017).
Kreso, A. & Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291 (2014).
Kreso, A. et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 339, 543–548 (2013).
Magee, J. A., Piskounova, E. & Morrison, S. J. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21, 283–296 (2012).
Boumahdi, S. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250 (2014).
Wang, F. et al. SCARB2 drives hepatocellular carcinoma tumor initiating cells via enhanced MYC transcriptional activity. Nat. Commun. 14, 5917 (2023).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Ter Steege, E. J. et al. R-spondin-3 promotes proliferation and invasion of breast cancer cells independently of Wnt signaling. Cancer Lett. 568, 216301 (2023).
Li, Y. et al. LncRNA SNHG5 promotes the proliferation and cancer stem cell-like properties of HCC by regulating UPF1 and Wnt-signaling pathway. Cancer Gene Ther. 29, 1373–1383 (2022).
Planells-Palop, V. et al. Human germ/stem cell-specific gene TEX19 influences cancer cell proliferation and cancer prognosis. Mol. Cancer 16, 84 (2017).
García-Gómez, P. et al. NOX4 regulates TGFβ-induced proliferation and self-renewal in glioblastoma stem cells. Mol. Oncol. 16, 1891–1912 (2022).
Li, Q. S. & Zheng, P. S. ESRRB inhibits the TGFβ signaling pathway to drive cell proliferation in cervical cancer. Cancer Res. 83, 3095–3114 (2023).
Lu, H. et al. Targeting cancer stem cell signature gene SMOC-2 Overcomes chemoresistance and inhibits cell proliferation of endometrial carcinoma. EBioMedicine 40, 276–289 (2019).
Majidpoor, J. & Mortezaee, K. Steps in metastasis: an updated review. Med. Oncol. 38, 3 (2021).
Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 13, 395–412 (2018).
Liao, W. T. et al. Metastatic cancer stem cells: from the concept to therapeutics. Am. J. Stem Cells 3, 46–62 (2014).
Chen, W. et al. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 1740936 (2016).
Haraguchi, N. et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 120, 3326–3339 (2010).
Huang, T. et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 10, 8721–8743 (2020).
Skvortsov, S., Debbage, P., Lukas, P. & Skvortsova, I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin. Cancer Biol. 31, 36–42 (2015).
Young, R. A. Control of the embryonic stem cell state. Cell 144, 940–954 (2011).
Sinclair, A. H. et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346, 240–244 (1990).
Takeda, J., Seino, S. & Bell, G. I. Human Oct3 gene family: cDNA sequences, alternative splicing, gene organization, chromosomal location, and expression at low levels in adult tissues. Nucleic Acids Res. 20, 4613–4620 (1992).
Heurtier, V. et al. The molecular logic of Nanog-induced self-renewal in mouse embryonic stem cells. Nat. Commun. 10, 1109 (2019).
Masui, S. et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 9, 625–635 (2007).
Suh, H. et al. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 1, 515–528 (2007).
Tai, M. H. et al. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis 26, 495–502 (2005).
Pierantozzi, E. et al. Pluripotency regulators in human mesenchymal stem cells: expression of NANOG but not of OCT-4 and SOX-2. Stem Cells Dev. 20, 915–923 (2011).
Novak, D. et al. SOX2 in development and cancer biology. Semin Cancer Biol. 67, 74–82 (2020).
Villodre, E. S., Kipper, F. C., Pereira, M. B. & Lenz, G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat Rev. 51, 1–9 (2016).
Najafzadeh, B. et al. The oncogenic potential of NANOG: An important cancer induction mediator. J. Cell Physiol. 236, 2443–2458 (2021).
Tatetsu, H. et al. SALL4, the missing link between stem cells, development and cancer. Gene 584, 111–119 (2016).
Wu, Q. et al. Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J. Biol. Chem. 281, 24090–24094 (2006).
Crabb, D. W., Matsumoto, M., Chang, D. & You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 63, 49–63 (2004).
Goedde, H. W. & Agarwal, D. P. Pharmacogenetics of aldehyde dehydrogenase (ALDH). Pharmacol. Ther. 45, 345–371 (1990).
Januchowski, R., Wojtowicz, K. & Zabel, M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharmacother. 67, 669–680 (2013).
Ma, I. & Allan, A. L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. Rep. 7, 292–306 (2011).
Good, P. et al. The human Musashi homolog 1 (MSI1) gene encoding the homologue of Musashi/Nrp-1, a neural RNA-binding protein putatively expressed in CNS stem cells and neural progenitor cells. Genomics 52, 382–384 (1998).
Glazer, R. I., Vo, D. T. & Penalva, L. O. Musashi1: an RBP with versatile functions in normal and cancer stem cells. Front. Biosci. 17, 54–64 (2012).
Kharas, M. G. et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 16, 903–908 (2010).
Park, S. M. et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J. Clin. Investig. 125, 1286–1298 (2015).
Sureda-Gómez, M. et al. Tumorigenic role of Musashi-2 in aggressive mantle cell lymphoma. Leukemia 37, 408–421 (2023).
Waldeck-Weiermair, M. et al. Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286, 28444–28455 (2011).
Piao, L. et al. LETM1 is a potential biomarker of prognosis in lung non-small cell carcinoma. BMC Cancer 19, 898 (2019).
Piao, L. et al. LETM1 is a potential cancer stem-like cell marker and predicts poor prognosis in colorectal adenocarcinoma. Pathol. Res. Pract. 215, 152437 (2019).
Li, H., Piao, L., Xu, D. & Xuan, Y. LETM1 is a potential biomarker that predicts poor prognosis in gastric adenocarcinoma. Exp. Mol. Pathol. 112, 104333 (2020).
Che, N. et al. Suppression of LETM1 inhibits the proliferation and stemness of colorectal cancer cells through reactive oxygen species-induced autophagy. J. Cell Mol. Med. 25, 2110–2120 (2021).
Shi, Y. et al. Aberrant LETM1 elevation dysregulates mitochondrial functions and energy metabolism and promotes lung metastasis in osteosarcoma. Genes Dis. 11, 100988 (2024).
Jang, S. et al. Elevated alpha-fetoprotein in asymptomatic adults: Clinical features, outcome, and association with body composition. PLoS One 17, e0271407 (2022).
Sasaki, N. et al. Alpha-fetoprotein-producing pancreatic cancer cells possess cancer stem cell characteristics. Cancer Lett. 308, 152–161 (2011).
Ishii, T. et al. Alpha-fetoprotein producing cells act as cancer progenitor cells in human cholangiocarcinoma. Cancer Lett. 294, 25–34 (2010).
Zhu, M. et al. HBx drives alpha fetoprotein expression to promote initiation of liver cancer stem cells through activating PI3K/AKT signal pathway. Int. J. Cancer 140, 1346–1355 (2017).
Fitieh, A. et al. BMI-1 regulates DNA end resection and homologous recombination repair. Cell Rep. 38, 110536 (2022).
Lessard, J. & Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423, 255–260 (2003).
Molofsky, A. V. et al. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 19, 1432–1437 (2005).
Omori, Y. et al. Expression and chromosomal localization of KIAA0369, a putative kinase structurally related to Doublecortin. J. Hum. Genet. 43, 169–177 (1998).
Luo, W. et al. Doublecortin-like kinase 1 activates NF-κB to induce inflammatory responses by binding directly to IKKβ. Cell Death Differ. 30, 1184–1197 (2023).
Nakanishi, Y. et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 45, 98–103 (2013).
Caruz, A. et al. Genomic organization and promoter characterization of human CXCR4 gene. FEBS Lett. 426, 271–278 (1998).
Bleul, C. C. et al. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 184, 1101–1109 (1996).
Huang, L. S. M., Snyder, E. Y. & Schooley, R. T. Strategies and progress in CXCR4-Targeted Anti-Human Immunodeficiency Virus (HIV) therapeutic development. Clin. Infect. Dis. 73, 919–924 (2021).
Bianchi, M. E. & Mezzapelle, R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front. Immunol. 11, 2109 (2020).
McDonald, T. et al. Identification and cloning of an orphan G protein-coupled receptor of the glycoprotein hormone receptor subfamily. Biochem. Biophys. Res. Commun. 247, 266–270 (1998).
Carmon, K. S. et al. “LGR5 Interacts and Cointernalizes with Wnt Receptors To Modulate Wnt/β-Catenin Signaling”. Mol. Cell Biol. 37, 2054–2064 (2017).
Litvinov, S. V. et al. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J. Cell Biol. 125, 437–446 (1994).
Maetzel, D. et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 11, 162–171 (2009).
Hough, M. R. et al. Mapping of CD24 and homologous sequences to multiple chromosomal loci. Genomics 22, 154–161 (1994).
Yang, Y., Zhu, G., Yang, L. & Yang, Y. Targeting CD24 as a novel immunotherapy for solid cancers. Cell Commun. Signal. 21, 312 (2023).
Chen, C., Zhao, S., Karnad, A. & Freeman, J. W. The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol. 11, 64 (2018).
Moreno-Londoño, A. P. & Robles-Flores, M. Functional roles of CD133: more than stemness associated factor regulated by the microenvironment. Stem Cell Rev. Rep. 20, 25–51 (2024).
Shi, C. et al. CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol. Ther. 10, 1182–1190 (2010).
Wang, C. et al. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 28, 1301–1308 (2012).
Zhang, X. et al. Single-cell sequencing reveals CD133(+)CD44(-)-originating evolution and novel stemness related variants in human colorectal cancer. EBioMedicine 82, 104125 (2022).
Dale, M., Hammond, D. W., Cox, A. & Nicklin, M. J. The human gene encoding the interleukin-1 receptor accessory protein (IL1RAP) maps to chromosome 3q28 by fluorescence in situ hybridization and radiation hybrid mapping. Genomics 47, 325–326 (1998).
Ågerstam, H. et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc. Natl Acad. Sci. USA 112, 10786–10791 (2015).
Landberg, N. et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia 30, 253–257 (2016).
Aoki, T. et al. High IL2RA/CD25 expression is a prognostic stem cell biomarker for pediatric acute myeloid leukemia without a core-binding factor. Pediatr. Blood Cancer 71, e30803 (2024).
Nguyen, C. H. et al. IL2RA promotes aggressiveness and stem cell-related properties of acute myeloid leukemia. Cancer Res. 80, 4527–4539 (2020).
Sadovnik, I. et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin. Cancer Res. 22, 2051–2061 (2016).
Jordan, C. T. et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 14, 1777–1784 (2000).
Riether, C. et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J. Exp. Med. 214, 359–380 (2017).
Riether, C. et al. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci. Transl. Med. 7, 298ra119 (2015).
Schürch, C. et al. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J. Clin. Investig. 122, 624–638 (2012).
Yamazaki, H. et al. CD90 and CD110 correlate with cancer stem cell potentials in human T-acute lymphoblastic leukemia cells. Biochem. Biophys. Res. Commun. 383, 172–177 (2009).
Muraro, M. G. et al. CD133+, CD166+CD44+, and CD24+CD44+ phenotypes fail to reliably identify cell populations with cancer stem cell functional features in established human colorectal cancer cell lines. Stem Cells Transl. Med. 1, 592–603 (2012).
Suzuki, E. et al. Aldehyde dehydrogenase 1 is associated with recurrence-free survival but not stem cell-like properties in hepatocellular carcinoma. Hepatol. Res. 42, 1100–1111 (2012).
Ma, S. et al. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol. Cancer Res. 6, 1146–1153 (2008).
Oh, S. Y. et al. CD44-negative cells in head and neck squamous carcinoma also have stem-cell like traits. Eur. J. Cancer 49, 272–280 (2013).
Lottaz, C. et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 70, 2030–2040 (2010).
Shimamura, M., Kurashige, T., Mitsutake, N. & Nagayama, Y. Aldehyde dehydrogenase activity plays no functional role in stem cell-like properties in anaplastic thyroid cancer cell lines. Endocrine 55, 934–943 (2017).
Chen, J. et al. CD133 and CD44 are universally overexpressed in GIST and do not represent cancer stem cell markers. Genes Chromosomes Cancer 51, 186–195 (2012).
Rim, E. Y., Clevers, H. & Nusse, R. The Wnt pathway: from signaling mechanisms to synthetic modulators. Annu. Rev. Biochem. 91, 571–598 (2022).
Chen, L. et al. Up-regulation of Dsg2 confered stem cells with malignancy through wnt/β-catenin signaling pathway. Exp. Cell Res. 422, 113416 (2023).
Feng, Q. et al. LGR6 activates the Wnt/β-catenin signaling pathway and forms a β-catenin/TCF7L2/LGR6 feedback loop in LGR6(high) cervical cancer stem cells. Oncogene 40, 6103–6114 (2021).
He, Y. et al. LncRNA PKMYT1AR promotes cancer stem cell maintenance in non-small cell lung cancer via activating Wnt signaling pathway. Mol. Cancer 20, 156 (2021).
Yin, J. et al. METTL3-mediated m6A modification of LINC00839 maintains glioma stem cells and radiation resistance by activating Wnt/β-catenin signaling. Cell Death Dis. 14, 417 (2023).
Liu, X. et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 40, 132 (2021).
Kim, J. et al. Wnt/β-catenin Signaling Inhibitors suppress the Tumor-initiating properties of a CD44(+)CD133(+) subpopulation of Caco-2 cells. Int. J. Biol. Sci. 17, 1644–1659 (2021).
Cheng, Q. et al. LGR4 cooperates with PrPc to endow the stemness of colorectal cancer stem cells contributing to tumorigenesis and liver metastasis. Cancer Lett. 540, 215725 (2022).
Yin, H. et al. FUBP1 promotes colorectal cancer stemness and metastasis via DVL1-mediated activation of Wnt/β-catenin signaling. Mol. Oncol. 15, 3490–3512 (2021).
Liu, X. et al. HIF-1-regulated expression of calreticulin promotes breast tumorigenesis and progression through Wnt/β-catenin pathway activation. Proc. Natl Acad. Sci. USA 118, e2109144118 (2021).
Feng, D. et al. CBP-mediated Wnt3a/β-catenin signaling promotes cervical oncogenesis initiated by Piwil2. Neoplasia 23, 1–11 (2021).
Husain, K., Coppola, D., Yang, C. S. & Malafa, M. P. Farnesyl dimethyl chromanol targets colon cancer stem cells and prevents colorectal cancer metastasis. Sci. Rep. 11, 2185 (2021).
Li, Z. et al. Disheveled3 enhanced EMT and cancer stem-like cells properties via Wnt/β-catenin/c-Myc/SOX2 pathway in colorectal cancer. J. Transl. Med. 21, 302 (2023).
Kwon, J. W. et al. A synergistic partnership between IL-33/ST2 and Wnt pathway through Bcl-xL drives gastric cancer stemness and metastasis. Oncogene 42, 501–515 (2023).
Zhang, Z. & Xu, Y. FZD7 accelerates hepatic metastases in pancreatic cancer by strengthening EMT and stemness associated with TGF-β/SMAD3 signaling. Mol. Med. 28, 82 (2022).
Qin, Q. et al. Polychlorinated biphenyl quinone induced the acquisition of cancer stem cells properties and epithelial-mesenchymal transition through Wnt/β-catenin. Chemosphere 263, 128125 (2021).
Osuka, S. et al. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Investig. 131, e136098 (2021).
Kim, H. Y. et al. Activation of Wnt signalling reduces the population of cancer stem cells in ameloblastoma. Cell Prolif. 54, e13073 (2021).
Zhang, Y. & Beachy, P. A. Cellular and molecular mechanisms of Hedgehog signalling. Nat. Rev. Mol. Cell Biol. 24, 668–687 (2023).
Yan, G. N. et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 234, 11–22 (2014).
Merchant, A. A. & Matsui, W. Targeting Hedgehog–a cancer stem cell pathway. Clin. Cancer Res. 16, 3130–3140 (2010).
Lee, D. et al. Superenhancer activation of KLHDC8A drives glioma ciliation and hedgehog signaling. J. Clin. Investig. 133, e163592 (2023).
Liu, Y. et al. ISL1 promotes human glioblastoma-derived stem cells’ self-renewal by activation of sonic Hedgehog/GLI1 function. Stem Cells Dev. 31, 258–268 (2022).
Wang, Y. et al. UHRF1 inhibition epigenetically reprograms cancer stem cells to suppress the tumorigenic phenotype of hepatocellular carcinoma. Cell Death Dis. 14, 381 (2023).
Tang, B. et al. MicroRNA-324-5p regulates stemness, pathogenesis and sensitivity to bortezomib in multiple myeloma cells by targeting hedgehog signaling. Int. J. Cancer 142, 109–120 (2018).
Guen, V. J. et al. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl Acad. Sci. USA 114, E10532–e10539 (2017).
Gu, Y. et al. Circular RNA circIPO11 drives self-renewal of liver cancer initiating cells via Hedgehog signaling. Mol. Cancer 20, 132 (2021).
Mok, E. H. K. et al. Caspase-3-Induced Activation of SREBP2 drives drug resistance via promotion of cholesterol biosynthesis in hepatocellular carcinoma. Cancer Res. 82, 3102–3115 (2022).
Wang, Y. et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat. Commun. 12, 6314 (2021).
Zhu, R. et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 10, 2863 (2019).
Li, C. et al. GALNT1-mediated glycosylation and activation of sonic hedgehog signaling maintains the self-renewal and tumor-initiating capacity of bladder cancer stem cells. Cancer Res. 76, 1273–1283 (2016).
Liu, J. et al. IL25 enhanced colitis-associated tumorigenesis in mice by upregulating transcription factor GLI1. Front. Immunol. 13, 837262 (2022).
Ruiz i Altaba, A. Hedgehog signaling and the Gli code in stem cells, cancer, and metastases. Sci. Signal. 4, pt9 (2011).
Qin, T. et al. Abnormally elevated USP37 expression in breast cancer stem cells regulates stemness, epithelial-mesenchymal transition and cisplatin sensitivity. J. Exp. Clin. Cancer Res. 37, 287 (2018).
Zhou, Y. et al. Garcinone C suppresses tumorsphere formation and invasiveness by Hedgehog/Gli1 signaling in colorectal cancer stem-like cells. J. Agric. Food Chem. 70, 7941–7952 (2022).
Kundu, S. et al. The scaffolding protein DLG5 promotes glioblastoma growth by controlling Sonic Hedgehog signaling in tumor stem cells. Neuro. Oncol. 24, 1230–1242 (2022).
Bray, S. J. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689 (2006).
Wang, Y., Wang, Y., Chen, H. & Liang, Q. Endothelial cells promote formation of medulloblastoma stem-like cells via Notch Pathway activation. J. Mol. Neurosci. 63, 152–158 (2017).
de Almeida Magalhães, T. et al. Notch pathway in ependymoma RELA-fused subgroup: upregulation and association with cancer stem cells markers expression. Cancer Gene Ther. 27, 509–512 (2020).
Ni, W. et al. Targeting Notch and EGFR signaling in human mucoepidermoid carcinoma. Signal. Transduct. Target Ther. 6, 27 (2021).
Ibrahim, S. A. et al. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 16, 57 (2017).
Högström, J. et al. Transcription factor PROX1 suppresses notch pathway activation via the nucleosome remodeling and deacetylase complex in colorectal cancer stem-like cells. Cancer Res. 78, 5820–5832 (2018).
Choi, S. et al. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 9, 11724 (2019).
Chen, J. H. et al. Upregulated SCUBE2 expression in breast cancer stem cells enhances triple negative breast cancer aggression through modulation of notch signaling and epithelial-to-mesenchymal transition. Exp. Cell Res. 370, 444–453 (2018).
Liu, G. et al. FAM129A promotes self-renewal and maintains invasive status via stabilizing the Notch intracellular domain in glioma stem cells. Neuro. Oncol. 25, 1788–1801 (2023).
Xiao, W. et al. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J. Exp. Clin. Cancer Res. 36, 41 (2017).
Allam, H. et al. The glycosyltransferase GnT-III activates Notch signaling and drives stem cell expansion to promote the growth and invasion of ovarian cancer. J. Biol. Chem. 292, 16351–16359 (2017).
Tien, P. C., Quan, M. & Kuang, S. Sustained activation of notch signaling maintains tumor-initiating cells in a murine model of liposarcoma. Cancer Lett. 494, 27–39 (2020).
Hu, T. et al. SPOPL induces tumorigenicity and stemness in glioma stem cells by activating Notch signaling. J. Neurooncol. 164, 157–170 (2023).
Rajakulendran, N. et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 33, 498–510 (2019).
Wang, Y. et al. Overexpression of FOXD2-AS1 enhances proliferation and impairs differentiation of glioma stem cells by activating the NOTCH pathway via TAF-1. J. Cell Mol. Med. 26, 2620–2632 (2022).
Cao, Y. et al. G9a promotes immune suppression by targeting the Fbxw7/Notch pathway in glioma stem cells. CNS Neurosci. Ther. 29, 2508–2521 (2023).
Napetschnig, J. & Wu, H. Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468 (2013).
Choi, H. S., Kim, J. H., Kim, S. L. & Lee, D. S. Disruption of the NF-κB/IL-8 Signaling Axis by Sulconazole Inhibits Human Breast Cancer Stem Cell Formation. Cells 8, 1007 (2019).
Witte, K. E. et al. Analysis of Several Pathways For Efficient Killing Of Prostate Cancer Stem Cells: A Central Role of NF-κB RELA. Int. J. Mol. Sci. 22, 8901 (2021).
Windmöller, B. A. et al. Novel primary human cancer stem-like cell populations from non-small cell lung cancer: inhibition of cell survival by targeting NF-κB and MYC signaling. Cells 10, 1024 (2021).
Ius, T. et al. An NF-κB signature predicts low-grade glioma prognosis: a precision medicine approach based on patient-derived stem cells. Neuro Oncol. 20, 776–787 (2018).
Cheng, J. H. et al. CaMKIIγ regulates the viability and self-renewal of acute myeloid leukaemia stem-like cells by the Alox5/NF-κB pathway. Int. J. Lab. Hematol. 43, 699–706 (2021).
Gonzalez-Torres, C. et al. NF-κB participates in the stem cell phenotype of ovarian cancer cells. Arch. Med. Res. 48, 343–351 (2017).
Fu, T. et al. ASB16-AS1 up-regulated and phosphorylated TRIM37 to activate NF-κB pathway and promote proliferation, stemness, and cisplatin resistance of gastric cancer. Gastric Cancer 24, 45–59 (2021).
Xu, C. et al. Let-7a regulates mammosphere formation capacity through Ras/NF-κB and Ras/MAPK/ERK pathway in breast cancer stem cells. Cell Cycle 14, 1686–1697 (2015).
Zhu, Y. et al. S100A4 suppresses cancer stem cell proliferation via interaction with the IKK/NF-κB signaling pathway. BMC Cancer 18, 763 (2018).
Fedele, M. et al. Proneural-mesenchymal transition: phenotypic plasticity to acquire multitherapy resistance in glioblastoma. Int. J. Mol. Sci. 20, 2746 (2019).
Nakano, I. Stem cell signature in glioblastoma: therapeutic development for a moving target. J. Neurosurg. 122, 324–330 (2015).
Chen, Z. et al. FOSL1 promotes proneural-to-mesenchymal transition of glioblastoma stem cells via UBC9/CYLD/NF-κB axis. Mol. Ther. 30, 2568–2583 (2022).
Kim, S. H. et al. Serine/Threonine Kinase MLK4 Determines Mesenchymal Identity in Glioma Stem Cells in an NF-κB-dependent Manner. Cancer Cell 29, 201–213 (2016).
Ma, D. Q. et al. Effect of Bmi-1-mediated NF-κB signaling pathway on the stem-like properties of CD133+ human liver cancer cells. Cancer Biomark. 22, 575–585 (2018).
Wu, S. L. et al. 2-Methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-κB/HIF-1 signaling pathway inactivation and EMT reversal. Oncol. Rep. 37, 793–802 (2017).
Kong, L. et al. Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int. J. Onco.l 48, 1085–1094 (2016).
Li, B. et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-κB/COX-2 activation. Chem. Biol. Interact 277, 33–42 (2017).
Jiang, Y. et al. CircKPNB1 mediates a positive feedback loop and promotes the malignant phenotypes of GSCs via TNF-α/NF-κB signaling. Cell Death Dis. 13, 697 (2022).
Xue, C. et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signa.l Transduct Target Ther. 8, 204 (2023).
Hu, X. et al. The JAK/STAT signaling pathway: from bench to clinic. Signal. Transduct Target Ther. 6, 402 (2021).
Misra, S. K., De, A. & Pan, D. Targeted delivery of STAT-3 modulator to breast cancer stem-like cells downregulates a series of stemness genes. Mol. Cancer Ther. 17, 119–129 (2018).
Garner, K. E. L. et al. The milk protein alpha-casein suppresses triple negative breast cancer stem cell activity Via STAT and HIF-1alpha signalling pathways in breast cancer cells and fibroblasts. J. Mammary Gland Biol. Neoplasia 24, 245–256 (2019).
Wang, T. et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 27, 136–150.e135 (2018).
Subramaniam, D. et al. Suppressing STAT5 signaling affects osteosarcoma growth and stemness. Cell Death Dis. 11, 149 (2020).
Dolatabadi, S. et al. JAK-STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int. J. Cancer 145, 435–449 (2019).
Shiraiwa, K. et al. JAK/STAT3 and NF-κB Signaling Pathways Regulate Cancer Stem-Cell Properties in Anaplastic Thyroid Cancer Cells. Thyroid 29, 674–682 (2019).
Luo, Y. et al. Non-CSCs nourish CSCs through interleukin-17E-mediated activation of NF-κB and JAK/STAT3 signaling in human hepatocellular carcinoma. Cancer Lett. 375, 390–399 (2016).
Liu, S. et al. Regulatory T cells promote glioma cell stemness through TGF-β-NF-κB-IL6-STAT3 signaling. Cancer Immunol. Immunother. 70, 2601–2616 (2021).
Kanno, H. et al. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int. J. Oncol. 42, 881–886 (2013).
Kroon, P. et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 73, 5288–5298 (2013).
Park, C. G. et al. Cytoplasmic LMO2-LDB1 Complex Activates STAT3 Signaling through Interaction with gp130-JAK in Glioma Stem Cells. Cells 11, 2031 (2022).
Park, K. B. et al. Leptin stimulates migration and invasion and maintains cancer stem‑like properties in gastric cancer cells. Oncol. Rep. 48, 162 (2022).
Xiong, Z. et al. IFITM3 promotes glioblastoma stem cell-mediated angiogenesis via regulating JAK/STAT3/bFGF signaling pathway. Cell Death Dis. 15, 45 (2024).
Choi, K. M. et al. The interferon-inducible protein viperin controls cancer metabolic reprogramming to enhance cancer progression. J. Clin. Investig. 132, e157302 (2022).
Hata, A. & Chen, Y. G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 (2016).
Massagué, J. & Sheppard, D. TGF-β signaling in health and disease. Cell 186, 4007–4037 (2023).
Colak, S. & Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 3, 56–71 (2017).
Futakuchi, M. et al. The effects of TGF-β signaling on cancer cells and cancer stem cells in the bone microenvironment. Int. J. Mol. Sci. 20, 5117 (2019).
Li, H. et al. The U2AF65/circNCAPG/RREB1 feedback loop promotes malignant phenotypes of glioma stem cells through activating the TGF-β pathway. Cell Death Dis. 14, 23 (2023).
Jiang, X. et al. HSP47 Promotes Glioblastoma Stemlike Cell Survival by Modulating Tumor Microenvironment Extracellular Matrix through TGF-β Pathway. ACS Chem. Neurosci. 8, 128–134 (2017).
Zhao, Y. et al. The transcription factor LEF1 promotes tumorigenicity and activates the TGF-β signaling pathway in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 38, 304 (2019).
Wang, J. et al. CD51 correlates with the TGF-beta pathway and is a functional marker for colorectal cancer stem cells. Oncogene 36, 1351–1363 (2017).
Nong, S. et al. HN1L promotes stem cell-like properties by regulating TGF-β signaling pathway through targeting FOXP2 in prostate cancer. Cell Biol. Int. 46, 83–95 (2022).
Wen, H. et al. Inhibiting of self-renewal, migration and invasion of ovarian cancer stem cells by blocking TGF-β pathway. PLoS One 15, e0230230 (2020).
Kahm, Y. J., Kim, R. K., Jung, U. & Kim, I. G. Epithelial membrane protein 3 regulates lung cancer stem cells via the TGF‑β signaling pathway. Int. J. Oncol. 59, 80 (2021).
Farabaugh, S. M. et al. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-β signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 31, 552–562 (2012).
You, X. et al. MicroRNA-495 confers inhibitory effects on cancer stem cells in oral squamous cell carcinoma through the HOXC6-mediated TGF-β signaling pathway. Stem Cell Res. Ther. 11, 117 (2020).
Yu, D., Shin, H. S., Lee, Y. S. & Lee, Y. C. miR-106b modulates cancer stem cell characteristics through TGF-β/Smad signaling in CD44-positive gastric cancer cells. Lab. Investig. 94, 1370–1381 (2014).
Gerstberger, S., Jiang, Q. & Ganesh, K. Metastasis. Cell 186, 1564–1579 (2023).
Chen, Z. et al. Prrx1 promotes stemness and angiogenesis via activating TGF-β/smad pathway and upregulating proangiogenic factors in glioma. Cell Death Dis. 12, 615 (2021).
Glaviano, A. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 22, 138 (2023).
Jafari, M., Ghadami, E., Dadkhah, T. & Akhavan-Niaki, H. PI3k/AKT signaling pathway: erythropoiesis and beyond. J. Cell Physiol. 234, 2373–2385 (2019).
Hassan, G. et al. Cancer stem cell generation by silenced MAPK enhancing PI3K/AKT signaling. Med. Hypotheses. 141, 109742 (2020).
Minematsu, H. et al. Cancer stem cells induced by chronic stimulation with prostaglandin E2 exhibited constitutively activated PI3K axis. Sci. Rep. 12, 15628 (2022).
Madsen, R. R. et al. Positive correlation between transcriptomic stemness and PI3K/AKT/mTOR signaling scores in breast cancer, and a counterintuitive relationship with PIK3CA genotype. PLoS Genet. 17, e1009876 (2021).
Lee, J. S. et al. The insulin and IGF signaling pathway sustains breast cancer stem cells by IRS2/PI3K-mediated regulation of MYC. Cell Re.p 41, 111759 (2022).
Yi, M. et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol. Cancer 21, 28 (2022).
Almozyan, S. et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J Cancer 141, 1402–1412 (2017).
Li, J. et al. Characteristics of the PI3K/AKT and MAPK/ERK pathways involved in the maintenance of self-renewal in lung cancer stem-like cells. Int. J. Biol. Sci. 17, 1191–1202 (2021).
Li, H. et al. Connexin32 regulates expansion of liver cancer stem cells via the PI3K/Akt signaling pathway. Oncol. Rep. 48, 166 (2022).
Peng, L. et al. MicroRNA-30a suppresses self-renewal and tumorigenicity of glioma stem cells by blocking the NT5E-dependent Akt signaling pathway. Faseb J. 34, 5128–5143 (2020).
Yang, X. L. et al. microRNA-873 inhibits self-renewal and proliferation of pancreatic cancer stem cells through pleckstrin-2-dependent PI3K/AKT pathway. Cell Signal. 84, 110025 (2021).
Wang, S. et al. RNA-binding protein IGF2BP2 enhances circ_0000745 abundancy and promotes aggressiveness and stemness of ovarian cancer cells via the microRNA-3187-3p/ERBB4/PI3K/AKT axis. J. Ovarian Res. 14, 154 (2021).
Kim, I. G. et al. Targeting therapy-resistant lung cancer stem cells via disruption of the AKT/TSPYL5/PTEN positive-feedback loop. Commun. Biol. 4, 778 (2021).
Keysar, S. B. et al. Regulation of head and neck squamous cancer stem cells by PI3K and SOX2. J. Natl Cancer Inst. 109, djw189 (2017).
Wang, J. H. et al. Knockdown of STIP1 inhibits the invasion of CD133‑positive cancer stem‑like cells of the osteosarcoma MG63 cell line via the PI3K/Akt and ERK1/2 pathways. Int. J. Mol. Med. 46, 2251–2259 (2020).
Wang, Y. H. et al. Transmembrane and coiled-coil domain family 3 (TMCC3) regulates breast cancer stem cell and AKT activation. Oncogene 40, 2858–2871 (2021).
Liu, B. et al. Targeting TROY-mediated P85a/AKT/TBX3 signaling attenuates tumor stemness and elevates treatment response in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 41, 182 (2022).
Liang, C., Jiang, Y. & Sun, L. Vitexin suppresses the proliferation, angiogenesis and stemness of endometrial cancer through the PI3K/AKT pathway. Pharm. Biol. 61, 581–589 (2023).
Chandler, K. B., Costello, C. E. & Rahimi, N. Glycosylation in the tumor microenvironment: implications for tumor angiogenesis and metastasis. Cells 8, 544 (2019).
Montaigne, D., Butruille, L. & Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 18, 809–823 (2021).
Ma, X. L. et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer 19, 760 (2019).
Giampietri, C. et al. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 18, 1271 (2017).
Hirozane, T. et al. Direct conversion of osteosarcoma to adipocytes by targeting TNIK. JCI Insight. 6, e137245 (2021).
Hu, P. S. et al. VDR-SOX2 signaling promotes colorectal cancer stemness and malignancy in an acidic microenvironment. Signal. Transduct. Target The.r 5, 183 (2020).
Wang, Y. et al. The combinatory effects of PPAR-γ agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int. J. Mol. Med. 34, 262–268 (2014).
Zhou, H. et al. Therapeutic inhibition of PPARα-HIF1α-PGK1 signaling targets leukemia stem and progenitor cells in acute myeloid leukemia. Cancer Lett. 554, 215997 (2023).
Haynes, H. R. et al. shRNA-mediated PPARα knockdown in human glioma stem cells reduces in vitro proliferation and inhibits orthotopic xenograft tumour growth. J. Pathol. 247, 422–434 (2019).
Chen, S. Z. et al. 4-phenylbutyric acid promotes hepatocellular carcinoma via initiating cancer stem cells through activation of PPAR-α. Clin. Transl. Med. 11, e379 (2021).
Wang, D. et al. PPARδ mediates the effect of dietary fat in promoting colorectal cancer metastasis. Cancer Res. 79, 4480–4490 (2019).
Binello, E. et al. Characterization of fenofibrate-mediated anti-proliferative pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. J. Neurooncol. 117, 225–234 (2014).
Clancy, H. et al. Glioblastoma cell migration is directed by electrical signals. Exp. Cell Res. 406, 112736 (2021).
Pestereva, E., Kanakasabai, S. & Bright, J. J. PPARγ agonists regulate the expression of stemness and differentiation genes in brain tumour stem cells. Br. J. Cancer 106, 1702–1712 (2012).
Basu-Roy, U. et al. PPARγ agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget 7, 60954–60970 (2016).
Yu, Y. et al. Targeting a lipid desaturation enzyme, SCD1, selectively eliminates colon cancer stem cells through the suppression of Wnt and NOTCH Signaling. Cells 10, 106 (2021).
Su, P. H. et al. NKX6-1 mediates cancer stem-like properties and regulates sonic hedgehog signaling in leiomyosarcoma. J. Biomed. Sci. 28, 32 (2021).
Chang, W. H. & Lai, A. G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 121, 666–678 (2019).
Quotti Tubi, L. et al. Protein kinase CK2 regulates AKT, NF-κB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia. 31, 292–300 (2017).
Majumder, M. et al. COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 34, 2290–2305 (2016).
Wang, J. et al. N6-Methyladenosine-Mediated Up-Regulation of FZD10 regulates liver cancer stem cells’ properties and Lenvatinib Resistance Through WNT/β-Catenin and hippo signaling pathways. Gastroenterology 164, 990–1005 (2023).
Quan, X. X. et al. Targeting Notch1 and IKKα Enhanced NF-κB Activation in CD133(+) Skin Cancer Stem Cells. Mol. Cancer Ther. 17, 2034–2048 (2018).
Zhang, W. & Grivennikov, S. I. Top Notch cancer stem cells by paracrine NF-κB signaling in breast cancer. Breast Cancer Res. 15, 316 (2013).
Prost, S. et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 525, 380–383 (2015).
Wang, B. et al. BRCA1-associated protein inhibits glioma cell proliferation and migration and glioma stem cell self-renewal via the TGF-β/PI3K/AKT/mTOR signalling pathway. Cell Oncol. 43, 223–235 (2020).
Liu, S. et al. A novel lncRNA ROPM-mediated lipid metabolism governs breast cancer stem cell properties. J. Hematol. Oncol. 14, 178 (2021).
Li, S. Z. et al. miR-139/PDE2A-Notch1 feedback circuit represses stemness of gliomas by inhibiting Wnt/β-catenin signaling. Int. J. Biol. Sci. 17, 3508–3521 (2021).
Yan, Y. et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 37, 256 (2018).
Tang, J. et al. TGF-β-activated lncRNA LINC00115 is a critical regulator of glioma stem-like cell tumorigenicity. EMBO Rep. 20, e48170 (2019).
Chen, C. L. et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J. Clin. Investig. 123, 2832–2849 (2013).
Regan, J. L. et al. Non-canonical hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 21, 2813–2828 (2017).
Liang, X. et al. An enhancer-driven stem cell-like program mediated by SOX9 blocks intestinal differentiation in colorectal cancer. Gastroenterology 162, 209–222 (2022).
Jiang, N. et al. HIF-1ɑ-regulated miR-1275 maintains stem cell-like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/β-catenin and Notch signaling. Theranostics 10, 2553–2570 (2020).
Ni, S. J. et al. CBX7 regulates stem cell-like properties of gastric cancer cells via p16 and AKT-NF-κB-miR-21 pathways. J. Hematol. Oncol. 11, 17 (2018).
Lathia, J., Liu, H. & Matei, D. The clinical impact of cancer stem cells. Oncologist 25, 123–131 (2020).
Okamoto, K. et al. Expression status of CD44 and CD133 as a prognostic marker in esophageal squamous cell carcinoma treated with neoadjuvant chemotherapy followed by radical esophagectomy. Oncol. Rep. 36, 3333–3342 (2016).
Chang, B. et al. NANOG as an adverse predictive marker in advanced non-small cell lung cancer treated with platinum-based chemotherapy. Oncol. Targets Ther. 10, 4625–4633 (2017).
Li, J. et al. Overexpression of CXCR4 is significantly associated with cisplatin-based chemotherapy resistance and can be a prognostic factor in epithelial ovarian cancer. BMB Rep. 47, 33–38 (2014).
Coate, L. et al. Germline genetic variation, cancer outcome, and pharmacogenetics. J. Clin. Oncol. 28, 4029–4037 (2010).
Szkandera, J. et al. LGR5 rs17109924 is a predictive genetic biomarker for time to recurrence in patients with colon cancer treated with 5-fluorouracil-based adjuvant chemotherapy. Pharmacogenomics J. 15, 391–396 (2015).
de Jong, M. C. et al. CD44 expression predicts local recurrence after radiotherapy in larynx cancer. Clin. Cancer Res. 16, 5329–5338 (2010).
Baumann, M. & Krause, M. CD44: a cancer stem cell-related biomarker with predictive potential for radiotherapy. Clin. Cancer Res. 16, 5091–5093 (2010).
Saigusa, S. et al. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann. Surg. Oncol. 16, 3488–3498 (2009).
Mare, M. et al. Cancer stem cell biomarkers predictive of radiotherapy response in rectal cancer: a systematic review. Genes 12, 1502 (2021).
Shi, H. et al. Tumor stemness and immune infiltration synergistically predict response of radiotherapy or immunotherapy and relapse in lung adenocarcinoma. Cancer Med. 10, 8944–8960 (2021).
Fu, H. C. et al. Low P16(INK4A) expression associated with high expression of cancer stem cell markers predicts poor prognosis in cervical cancer after radiotherapy. Int. J. Mol. Sci. 19, 2541 (2018).
He, J. et al. Expression of glioma stem cell marker CD133 and O6-methylguanine-DNA methyltransferase is associated with resistance to radiotherapy in gliomas. Oncol. Rep. 26, 1305–1313 (2011).
Patel, U. et al. Prognostic and predictive roles of cancer stem cell markers in head and neck squamous cell carcinoma patients receiving chemoradiotherapy with or without nimotuzumab. Br. J. Cancer 126, 1439–1449 (2022).
Chabner, B. A. & Roberts, T. G. Jr Timeline: chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72 (2005).
Vira, D. et al. Cancer stem cells, microRNAs, and therapeutic strategies including natural products. Cancer Metastasis Rev. 31, 733–751 (2012).
Schmidt-Kittler, O. et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc. Natl Acad. Sci. USA 100, 7737–7742 (2003).
Lytle, N. K. et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 177, 572–586.e522 (2019).
Was, H. et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 8, 9303–9322 (2017).
Luo, M. et al. Stem cell quiescence and its clinical relevance. World J. Stem Cells 12, 1307–1326 (2020).
Kwon, M. J. & Shin, Y. K. Regulation of ovarian cancer stem cells or tumor-initiating cells. Int. J. Mol. Sci. 14, 6624–6648 (2013).
Gao, M. Q. et al. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 29, 2672–2680 (2010).
Szotek, P. P. et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl Acad. Sci. USA 103, 11154–11159 (2006).
Francescangeli, F. et al. A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J. Exp. Clin. Cancer Res. 39, 2 (2020).
Arnold, C. R., Mangesius, J., Skvortsova, I. I. & Ganswindt, U. The role of cancer stem cells in radiation resistance. Front. Oncol. 10, 164 (2020).
Ye, S. et al. SET domain-containing protein 4 epigenetically controls breast cancer stem cell quiescence. Cancer Res. 79, 4729–4743 (2019).
Liu, S. et al. Methylation status of the nanog promoter determines the switch between cancer cells and cancer stem cells. Adv. Sci. 7, 1903035 (2020).
Zhang, B. et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 24, 450–462 (2018).
Lechman, E. R. et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell 29, 214–228 (2016).
Wolf, B. et al. Inducing differentiation of premalignant hepatic cells as a novel therapeutic strategy in hepatocarcinoma. Cancer Res. 76, 5550–5561 (2016).
Agarwal, P. et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 24, 769–784.e766 (2019).
Jeanpierre, S. et al. The quiescent fraction of chronic myeloid leukemic stem cells depends on BMPR1B, Stat3 and BMP4-niche signals to persist in patients in remission. Haematologica 106, 111–122 (2021).
Johnson, R. W. et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 18, 1078–1089 (2016).
Sansone, P. et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl Acad. Sci. USA 114, E9066–e9075 (2017).
Su, S. et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856.e816 (2018).
Hu, Y. et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One 10, e0125625 (2015).
Velasco-Hernandez, T. et al. Hif-1α deletion may lead to adverse treatment effect in a mouse model of MLL-AF9-Driven AML. Stem Cell Rep. 12, 112–121 (2019).
Chen, Z. et al. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 123, 2204–2208 (2014).
Chen, Z. et al. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1α stabilization. Leukemia 33, 469–486 (2019).
Heddleston, J. M. et al. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284 (2009).
Mathieu, J. et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 71, 4640–4652 (2011).
Blazek, E. R., Foutch, J. L. & Maki, G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133- cells, and the CD133+ sector is enlarged by hypoxia. Int. J. Radiat Oncol. Biol. Phys. 67, 1–5 (2007).
Das, B. et al. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 26, 1818–1830 (2008).
Soeda, A. et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28, 3949–3959 (2009).
Gao, C. et al. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS One 11, e0154576 (2016).
Garcia-Mayea, Y. et al. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 60, 166–180 (2020).
Yoshida, G. J. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 34, 111 (2015).
Tomita, H., Tanaka, K., Tanaka, T. & Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 7, 11018–11032 (2016).
Yaghjyan, L. et al. Associations of mammographic breast density with breast stem cell marker-defined breast cancer subtypes. Cancer Causes Control 30, 1103–1111 (2019).
Magni, M. et al. Induction of cyclophosphamide-resistance by aldehyde-dehydrogenase gene transfer. Blood 87, 1097–1103 (1996).
Parajuli, B., Fishel, M. L. & Hurley, T. D. Selective ALDH3A1 inhibition by benzimidazole analogues increase mafosfamide sensitivity in cancer cells. J. Med. Chem. 57, 449–461 (2014).
Young, S. Z. & Bordey, A. GABA’s control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology 24, 171–185 (2009).
Mu, X. et al. Notch signaling is associated with ALDH activity and an aggressive metastatic phenotype in murine osteosarcoma cells. Front. Oncol. 3, 143 (2013).
Moreb, J. S. et al. ALDH isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 7, 87 (2008).
Mu, X. et al. Rapamycin inhibits ALDH activity, resistance to oxidative stress, and metastatic potential in murine osteosarcoma Cells. Sarcoma 2013, 480713 (2013).
Sládek, N. E., Kollander, R., Sreerama, L. & Kiang, D. T. Cellular levels of aldehyde dehydrogenases (ALDH1A1 and ALDH3A1) as predictors of therapeutic responses to cyclophosphamide-based chemotherapy of breast cancer: a retrospective study. Rational individualization of oxazaphosphorine-based cancer chemotherapeutic regimens. Cancer Chemother. Pharmacol. 49, 309–321 (2002).
Kida, K. et al. Effect of ALDH1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Res. Treat 156, 261–269 (2016).
Landen, C. N. Jr et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther 9, 3186–3199 (2010).
Kozovska, Z. et al. ALDH1A inhibition sensitizes colon cancer cells to chemotherapy. BMC Cancer 18, 656 (2018).
Huang, C. P. et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 328, 144–151 (2013).
Nishikawa, S. et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 42, 1437–1442 (2013).
Awad, O. et al. High ALDH activity identifies chemotherapy-resistant Ewing’s sarcoma stem cells that retain sensitivity to EWS-FLI1 inhibition. PLoS One 5, e13943 (2010).
Schäfer, A. et al. Aldehyde dehydrogenase 1A1–a new mediator of resistance to temozolomide in glioblastoma. Neuro. Oncol. 14, 1452–1464 (2012).
Bertrand, G. et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev. Rep. 10, 114–126 (2014).
Mazor, G. et al. The lncRNA TP73-AS1 is linked to aggressiveness in glioblastoma and promotes temozolomide resistance in glioblastoma cancer stem cells. Cell Death Dis. 10, 246 (2019).
Chefetz, I. et al. A Pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep. 26, 3061–3075.e3066 (2019).
Liu, X. ABC family transporters. Adv. Exp. Med. Biol. 1141, 13–100 (2019).
Abdullah, L. N. & Chow, E. K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2, 3 (2013).
Ashley, N., Ouaret, D. & Bodmer, W. F. Cellular polarity modulates drug resistance in primary colorectal cancers via orientation of the multidrug resistance protein ABCB1. J. Pathol. 247, 293–304 (2019).
Wright, M. H. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 10, R10 (2008).
Britton, K. M. et al. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 323, 97–105 (2012).
Doyle, L. A. et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl Acad. Sci. USA 95, 15665–15670 (1998).
Chau, W. K. et al. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin-ATP-binding cassette G2 signaling. Oncogene 32, 2767–2781 (2013).
Welte, Y., Adjaye, J., Lehrach, H. R. & Regenbrecht, C. R. Cancer stem cells in solid tumors: elusive or illusive? Cell Commun. Signal. 8, 6 (2010).
Cui, H., Zhang, A. J., Chen, M. & Liu, J. J. ABC transporter inhibitors in reversing multidrug resistance to chemotherapy. Curr. Drug Targets 16, 1356–1371 (2015).
El-Awady, R. et al. The role of eukaryotic and prokaryotic abc transporter family in failure of chemotherapy. Fron.t Pharmacol. 7, 535 (2016).
Peterson, B. G., Tan, K. W., Osa-Andrews, B. & Iram, S. H. High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharmacol. Res. 119, 313–326 (2017).
Polgar, O., Robey, R. W. & Bates, S. E. ABCG2: structure, function and role in drug response. Expert. Opin. Drug Metab. Toxicol. 4, 1–15 (2008).
Mao, Q. & Unadkat, J. D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. Aaps J. 17, 65–82 (2015).
Guo, Q. et al. ATP-binding cassette member B5 (ABCB5) promotes tumor cell invasiveness in human colorectal cancer. J. Biol. Chem. 293, 11166–11178 (2018).
Gottesman, M. M., Fojo, T. & Bates, S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002).
Corrêa, S. et al. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 12, 303 (2012).
Zhang, Z. M. et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/β-catenin pathway. Oncogene 35, 4787–4797 (2016).
Wang, Z. et al. Caveolin-1 mediates chemoresistance in breast cancer stem cells via β-catenin/ABCG2 signaling pathway. Carcinogenesis 35, 2346–2356 (2014).
Bhattacharya, S., Das, A., Mallya, K. & Ahmad, I. Maintenance of retinal stem cells by Abcg2 is regulated by notch signaling. J. Cell Sci. 120, 2652–2662 (2007).
Basu-Roy, U. et al. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 6, 6411 (2015).
Escoll, M. et al. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 36, 3515–3527 (2017).
Bleau, A. M. et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4, 226–235 (2009).
Nakanishi, T., Shiozawa, K., Hassel, B. A. & Ross, D. D. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 108, 678–684 (2006).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Zhang, M. et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 68, 4674–4682 (2008).
Chen, J. et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012).
Jahanban-Esfahlan, R. et al. The herbal medicine Melissa officinalis extract effects on gene expression of p53, Bcl-2, Her2, VEGF-A and hTERT in human lung, breast and prostate cancer cell lines. Gene 613, 14–19 (2017).
Venere, M. et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 21, 258–269 (2014).
Manic, G. et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 67, 903–917 (2018).
Gallmeier, E. et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 29, 418–429 (2011).
Liu, Y. et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin. Cancer Res. 23, 514–522 (2017).
Peitzsch, C. et al. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 108, 378–387 (2013).
Mizuno, T. et al. Cancer stem-like cells of ovarian clear cell carcinoma are enriched in the ALDH-high population associated with an accelerated scavenging system in reactive oxygen species. Gynecol. Oncol. 137, 299–305 (2015).
Chandimali, N., Jeong, D. K. & Kwon, T. Peroxiredoxin II regulates cancer stem cells and stemness-associated properties of cancers. Cancers 10, 305 (2018).
Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 (2009).
Kim, D., Choi, B. H., Ryoo, I. G. & Kwak, M. K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 9, 896 (2018).
Ryoo, I. G., Lee, S. H. & Kwak, M. K. Redox Modulating NRF2: a potential mediator of cancer stem cell resistance. Oxid. Med. Cell Longev. 2016, 2428153 (2016).
Cojoc, M., Mäbert, K., Muders, M. H. & Dubrovska, A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin. Cancer Biol. 31, 16–27 (2015).
Wang, D. et al. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 149, 1884–1895.e1884 (2015).
Kim, E. H. et al. 15-Deoxy-Delta12,14-prostaglandin J2 induces COX-2 expression through Akt-driven AP-1 activation in human breast cancer cells: a potential role of ROS. Carcinogenesis 29, 688–695 (2008).
Balvan, J. et al. Oxidative stress resistance in metastatic prostate cancer: renewal by self-eating. PLoS One 10, e0145016 (2015).
Tsuruo, T. et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 94, 15–21 (2003).
Kim, R., Emi, M. & Tanabe, K. Role of mitochondria as the gardens of cell death. Cancer Chemother. Pharmacol. 57, 545–553 (2006).
Liu, G. et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67 (2006).
Todaro, M. et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 1, 389–402 (2007).
Park, Y. S., Huh, J. W., Lee, J. H. & Kim, H. R. shRNA against CD44 inhibits cell proliferation, invasion and migration, and promotes apoptosis of colon carcinoma cells. Oncol. Rep. 27, 339–346 (2012).
Konopleva, M. et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br. J. Haematol. 118, 521–534 (2002).
Economopoulou, P., Kaklamani, V. G. & Siziopikou, K. The role of cancer stem cells in breast cancer initiation and progression: potential cancer stem cell-directed therapies. Oncologist 17, 1394–1401 (2012).
Schwickart, M. et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107 (2010).
Day, T. W., Najafi, F., Wu, C. H. & Safa, A. R. Cellular FLICE-like inhibitory protein (c-FLIP): a novel target for Taxol-induced apoptosis. Biochem. Pharmacol. 71, 1551–1561 (2006).
Piggott, L. et al. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res. 13, R88 (2011).
Po, A. et al. Sonic Hedgehog Medulloblastoma Cancer Stem Cells Mirnome and Transcriptome Highlight Novel Functional Networks. Int. J. Mol. Sci. 19, 2326 (2018).
Fujimoto, A. et al. Inhibition of endoplasmic reticulum (ER) stress sensors sensitizes cancer stem-like cells to ER stress-mediated apoptosis. Oncotarget 7, 51854–51864 (2016).
De Luca, A. et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 6, 14777–14795 (2015).
Farnie, G., Sotgia, F. & Lisanti, M. P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 6, 30472–30486 (2015).
van Ravenswaay Claasen, H. H., Kluin, P. M. & Fleuren, G. J. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab. Investig. 67, 166–174 (1992).
Kalbasi, A., June, C. H., Haas, N. & Vapiwala, N. Radiation and immunotherapy: a synergistic combination. J. Clin. Investig. 123, 2756–2763 (2013).
Zhou, W. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182 (2015).
Wang, J. et al. Krüppel like factor 6 splice variant 1 (KLF6-SV1) overexpression recruits macrophages to participate in lung cancer metastasis by up-regulating TWIST1. Cancer Biol. Ther. 20, 680–691 (2019).
Yamashina, T. et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 74, 2698–2709 (2014).
Jinushi, M. et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl Acad. Sci. USA 108, 12425–12430 (2011).
Li, S. et al. Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. Oncoimmunology 7, e1380765 (2018).
Theocharides, A. P. et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 209, 1883–1899 (2012).
Cioffi, M. et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin. Cancer Res 21, 2325–2337 (2015).
Lee, T. K. et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 60, 179–191 (2014).
Liu, L. et al. Anti-CD47 antibody as a targeted therapeutic agent for human lung cancer and cancer stem cells. Front. Immunol. 8, 404 (2017).
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009).
Feng, Q. et al. Nebulized therapy of early orthotopic lung cancer by iron-based nanoparticles: macrophage-regulated ferroptosis of cancer stem cells. J. Am. Chem. So.c 145, 24153–24165 (2023).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e723 (2021).
Ouzounova, M. et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 8, 14979 (2017).
Bronte, V. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 7, 12150 (2016).
Kumar, V., Patel, S., Tcyganov, E. & Gabrilovich, D. I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 37, 208–220 (2016).
Wang, G. et al. Targeting YAP-Dependent MDSC infiltration impairs tumor progression. Cancer Discov. 6, 80–95 (2016).
Shidal, C., Singh, N. P., Nagarkatti, P. & Nagarkatti, M. MicroRNA-92 Expression in CD133(+) melanoma stem cells regulates immunosuppression in the tumor microenvironment via integrin-dependent activation of TGFβ. Cancer Res. 79, 3622–3635 (2019).
Kuroda, H. et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget 9, 36317–36330 (2018).
Haverkamp, J. M. et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 41, 947–959 (2014).
Antuamwine, B. B. et al. N1 versus N2 and PMN-MDSC: a critical appraisal of current concepts on tumor-associated neutrophils and new directions for human oncology. Immunol. Rev. 314, 250–279 (2023).
St Paul, M. & Ohashi, P. S. The Roles of CD8(+) T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 30, 695–704 (2020).
Clara, J. A., Monge, C., Yang, Y. & Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat. Rev. Clin. Oncol. 17, 204–232 (2020).
Rosenberg, S. A. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 10, 281–287 (1999).
Lee, P. P. et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685 (1999).
Khong, H. T., Wang, Q. J. & Rosenberg, S. A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J. Immunother. 27, 184–190 (2004).
Le Blanc, K. et al. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 31, 890–896 (2003).
Guerry, D. T. et al. HLA-DR histocompatibility leukocyte antigens permit cultured human melanoma cells from early but not advanced disease to stimulate autologous lymphocytes. J. Clin. Investig. 73, 267–271 (1984).
Ramsdell, F. & Fowlkes, B. J. Clonal deletion versus clonal anergy: the role of the thymus in inducing self tolerance. Science 248, 1342–1348 (1990).
Strand, S. et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells–a mechanism of immune evasion? Nat. Med. 2, 1361–1366 (1996).
Andreola, G. et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 195, 1303–1316 (2002).
Hallermalm, K. et al. Autocrine secretion of Fas ligand shields tumor cells from Fas-mediated killing by cytotoxic lymphocytes. Cancer Res. 64, 6775–6782 (2004).
Nakashima, M., Sonoda, K. & Watanabe, T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat. Med. 5, 938–942 (1999).
Ramsdell, F., Lantz, T. & Fowlkes, B. J. A nondeletional mechanism of thymic self tolerance. Science 246, 1038–1041 (1989).
Taylor, A. et al. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 117, 433–442 (2006).
Chen, Q., Daniel, V., Maher, D. W. & Hersey, P. Production of IL-10 by melanoma cells: examination of its role in immunosuppression mediated by melanoma. Int. J. Cancer 56, 755–760 (1994).
Gorelik, L. & Flavell, R. A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 7, 1118–1122 (2001).
Inge, T. H. et al. Inhibition of tumor-specific cytotoxic T-lymphocyte responses by transforming growth factor beta 1. Cancer Res. 52, 1386–1392 (1992).
Shipitsin, M. et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 11, 259–273 (2007).
Piccirillo, S. G. et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765 (2006).
Schatton, T. et al. Identification of cells initiating human melanomas. Nature 451, 345–349 (2008).
Rothstein, D. M. & Sayegh, M. H. T-cell costimulatory pathways in allograft rejection and tolerance. Immunol. Rev. 196, 85–108 (2003).
Frank, M. H. et al. Specific MDR1 P-glycoprotein blockade inhibits human alloimmune T cell activation in vitro. J. Immunol. 166, 2451–2459 (2001).
Pendse, S. S. et al. P-glycoprotein functions as a differentiation switch in antigen presenting cell maturation. Am. J. Transplant. 6, 2884–2893 (2006).
Sotomayor, E. M. et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood 98, 1070–1077 (2001).
Sotomayor, E. M. et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 5, 780–787 (1999).
Dong, H. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 (2002).
Fong, L. & Small, E. J. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J. Clin. Oncol. 26, 5275–5283 (2008).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Smith, T. R. & Kumar, V. Revival of CD8+ Treg-mediated suppression. Trends Immunol. 29, 337–342 (2008).
Wang, L. et al. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc. Natl Acad. Sci. USA 105, 9331–9336 (2008).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Hu, Q. et al. Atlas of breast cancer infiltrated B-lymphocytes revealed by paired single-cell RNA-sequencing and antigen receptor profiling. Nat. Commun. 12, 2186 (2021).
Hollern, D. P. et al. B Cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell 179, 1191–1206.e1121 (2019).
Biswas, S. et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 591, 464–470 (2021).
Thommen, D. S. et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004 (2018).
Workel, H. H. et al. A transcriptionally distinct CXCL13(+)CD103(+)CD8(+) T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol. Res. 7, 784–796 (2019).
Li, H. et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789.e718 (2019).
Kinker, G. S. et al. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front. Cell Dev. Biol. 9, 678127 (2021).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680 (2020).
Rodriguez, A. B. & Engelhard, V. H. Insights into tumor-associated tertiary lymphoid structures: novel targets for antitumor immunity and cancer immunotherapy. Cancer Immunol. Res. 8, 1338–1345 (2020).
Mustapha, R., Ng, K., Monypenny, J. & Ng, T. Insights into unveiling a potential role of tertiary lymphoid structures in metastasis. Front. Mol. Biosci. 8, 661516 (2021).
Wei, H. et al. Breast cancer stem cells phenotype and plasma cell-predominant breast cancer independently indicate poor survival. Pathol. Res. Pract. 212, 294–301 (2016).
Xu, W. et al. Prognostic value, DNA variation and immunologic features of a tertiary lymphoid structure-related chemokine signature in clear cell renal cell carcinoma. Cancer Immunol. Immunother. 71, 1923–1935 (2022).
Vivier, E. et al. Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49 (2011).
Tallerico, R. et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol. 190, 2381–2390 (2013).
Castriconi, R. et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 182, 3530–3539 (2009).
Yin, T. et al. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 300, 41–45 (2016).
Tseng, H. C. et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One 5, e11590 (2010).
Jewett, A. & Tseng, H. C. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J. Cancer 2, 443–457 (2011).
Kaur, K. et al. Natural killer cells target and differentiate cancer stem-like cells/undifferentiated tumors: strategies to optimize their growth and expansion for effective cancer immunotherapy. Curr. Opin. Immunol. 51, 170–180 (2018).
Ghiringhelli, F. et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 202, 1075–1085 (2005).
Li, T. et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 318, 154–161 (2012).
Balsamo, M. et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl Acad. Sci. USA 106, 20847–20852 (2009).
Pietra, G. et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 72, 1407–1415 (2012).
Castriconi, R. et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J. Immunol. 190, 5321–5328 (2013).
Regis, S. et al. TGF-β1 Downregulates the Expression of CX(3)CR1 by Inducing miR-27a-5p in Primary Human NK Cells. Front. Immunol. 8, 868 (2017).
Groh, V., Wu, J., Yee, C. & Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419, 734–738 (2002).
Kaiser, B. K. et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 447, 482–486 (2007).
Patel, S. A. et al. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-beta. J. Immunol. 184, 5885–5894 (2010).
Wang, B. et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 74, 5746–5757 (2014).
Kryczek, I. et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40, 772–784 (2014).
Parish, C. R. Cancer immunotherapy: the past, the present and the future. Immunol. Cell Biol. 81, 106–113 (2003).
Galluzzi, L. et al. Classification of current anticancer immunotherapies. Oncotarget 5, 12472–12508 (2014).
Weiner, G. J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 (2015).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Schmitt, T. M., Ragnarsson, G. B. & Greenberg, P. D. T cell receptor gene therapy for cancer. Hum. Gene Ther. 20, 1240–1248 (2009).
Singh, H. et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 68, 2961–2971 (2008).
Guo, Y., Feng, K., Wang, Y. & Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: a potential and curable approach for cancer treatment. Protein Cell 9, 516–526 (2018).
Schmidt-Wolf, I. G. et al. Propagation of large numbers of T cells with natural killer cell markers. Br. J. Haematol. 87, 453–458 (1994).
Guo, Y. & Han, W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin. J. Cancer 34, 99–107 (2015).
Lee, S. & Margolin, K. Cytokines in cancer immunotherapy. Cancers 3, 3856–3893 (2011).
Calogero, R. A. et al. Oncoantigens as anti-tumor vaccination targets: the chance of a lucky strike? Cancer Immunol. Immunother. 57, 1685–1694 (2008).
Naujokat, C. Monoclonal antibodies against human cancer stem cells. Immunotherapy 6, 290–308 (2014).
Hassn Mesrati, M., Syafruddin, S. E., Mohtar, M. A. & Syahir, A. CD44: a multifunctional mediator of cancer progression. Biomolecules 11, 1850 (2021).
Zhang, H. et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 33, 166–179 (2019).
Vugts, D. J. et al. Preclinical evaluation of 89Zr-labeled anti-CD44 monoclonal antibody RG7356 in mice and cynomolgus monkeys: Prelude to Phase 1 clinical studies. MAbs 6, 567–575 (2014).
Vey, N. et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 7, 32532–32542 (2016).
Menke-van der Houven van Oordt, C. W. et al. First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors. Oncotarget. 7, 80046–80058 (2016).
Tijink, B. M. et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 12, 6064–6072 (2006).
Chao, M. P., Weissman, I. L. & Majeti, R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immuno.l 24, 225–232 (2012).
Jäger, M. et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 72, 24–32 (2012).
Murayama, Y., Oritani, K. & Tsutsui, S. Novel CD9-targeted therapies in gastric cancer. World J. Gastroenterol. 21, 3206–3213 (2015).
Chivu-Economescu, M. et al. Gastrointestinal cancer stem cells as targets for innovative immunotherapy. World J. Gastroenterol. 26, 1580–1593 (2020).
Masoumi, J. et al. Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: challenges and prospects. Acta Pharm. Sin B 11, 1721–1739 (2021).
Eyvazi, S. et al. Antibody Based EpCAM targeted therapy of cancer, review and update. Curr. Cancer Drug Targets 18, 857–868 (2018).
Emlet, D. R. et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 74, 1238–1249 (2014).
Zheng, P. P., Kros, J. M. & Li, J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 23, 1175–1182 (2018).
June, C. H. et al. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Brudno, J. N. & Kochenderfer, J. N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 34, 45–55 (2019).
Zhu, X. et al. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 6, 171–184 (2015).
Morgan, R. A. et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 23, 1043–1053 (2012).
O’Rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Mandal, A. & Viswanathan, C. Natural killer cells: in health and disease. Hematol. Oncol. Stem Cell Ther. 8, 47–55 (2015).
Voutsadakis, I. A. Expression and function of immune ligand-receptor pairs in NK cells and cancer stem cells: therapeutic implications. Cell Oncol. 41, 107–121 (2018).
Tallerico, R., Garofalo, C. & Carbone, E. A new biological feature of natural killer cells: the recognition of solid tumor-derived cancer stem cells. Front. Immuno.l 7, 179 (2016).
Bae, J. H. et al. Susceptibility to natural killer cell-mediated lysis of colon cancer cells is enhanced by treatment with epidermal growth factor receptor inhibitors through UL16-binding protein-1 induction. Cancer Sci. 103, 7–16 (2012).
Pietra, G. et al. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 21, 793–801 (2009).
Ames, E. et al. NK Cells Preferentially Target Tumor Cells With A Cancer Stem Cell Phenotype. J. Immunol. 195, 4010–4019 (2015).
Dianat-Moghadam, H. et al. Natural killer cell-based immunotherapy: from transplantation toward targeting cancer stem cells. J. Cell Physiol. 234, 259–273 (2018).
Carlsten, M. et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 183, 4921–4930 (2009).
Gammaitoni, L. et al. Immunotherapy of cancer stem cells in solid tumors: initial findings and future prospective. Expert Opin. Biol. Ther. 14, 1259–1270 (2014).
Pauza, C. D. et al. Gamma Delta T cell therapy for cancer: it is good to be local. Front. Immunol. 9, 1305 (2018).
Harly, C., Peigné, C. M. & Scotet, E. Molecules and mechanisms implicated in the peculiar antigenic activation process of human Vγ9Vδ2 T Cells. Front. Immunol. 5, 657 (2014).
Peng, G. et al. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 27, 334–348 (2007).
Maniar, A. et al. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood 116, 1726–1733 (2010).
Caccamo, N. et al. Aminobisphosphonates as new weapons for gammadelta T Cell-based immunotherapy of cancer. Curr. Med. Chem. 15, 1147–1153 (2008).
Nishio, N. et al. Zoledronate sensitizes neuroblastoma-derived tumor-initiating cells to cytolysis mediated by human γδ T cells. J. Immunother. 35, 598–606 (2012).
Todaro, M. et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J. Immunol. 182, 7287–7296 (2009).
Chen, H. C. et al. Synergistic targeting of breast cancer stem-like cells by human γδ T cells and CD8(+) T cells. Immunol. Cell Biol. 95, 620–629 (2017).
Todaro, M. et al. Chemotherapy sensitizes colon cancer initiating cells to Vγ9Vδ2 T cell-mediated cytotoxicity. PLoS One 8, e65145 (2013).
Lai, D. et al. Human ovarian cancer stem-like cells can be efficiently killed by γδ T lymphocytes. Cancer Immunol. Immunother. 61, 979–989 (2012).
Gammaitoni, L. et al. Effective activity of cytokine-induced killer cells against autologous metastatic melanoma including cells with stemness features. Clin Cancer Res. 19, 4347–4358 (2013).
Sangiolo, D. et al. Cytokine-induced killer cells eradicate bone and soft-tissue sarcomas. Cancer Res. 74, 119–129 (2014).
Yang, T. et al. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC Cancer 18, 984 (2018).
Mesiano, G. et al. Cytokine Induced Killer cells are effective against sarcoma cancer stem cells spared by chemotherapy and target therapy. Oncoimmunology 7, e1465161 (2018).
Xu, Q. et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 27, 1734–1740 (2009).
Bol, K. F. et al. Dendritic cell-based immunotherapy: state of the art and beyond. Clin. Cancer Res. 22, 1897–1906 (2016).
Pellegatta, S. et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 66, 10247–10252 (2006).
Lu, L. et al. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology 4, e990767 (2015).
Hu, Y. et al. Therapeutic efficacy of cancer stem cell vaccines in the adjuvant setting. Cancer Res. 76, 4661–4672 (2016).
Vik-Mo, E. O. et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 62, 1499–1509 (2013).
Lin, M. et al. Safety and efficacy study of lung cancer stem cell vaccine. Immunol. Res. 62, 16–22 (2015).
Lin, M. et al. Prospective study of the safety and efficacy of a pancreatic cancer stem cell vaccine. J. Cancer Res. Clin. Oncol. 141, 1827–1833 (2015).
Yang, Y. et al. B7-H1 enhances proliferation ability of gastric cancer stem-like cells as a receptor. Oncol. Lett. 9, 1833–1838 (2015).
Zhang, B. et al. Potential function of CTLA-4 in the tumourigenic capacity of melanoma stem cells. Oncol. Lett. 16, 6163–6170 (2018).
Zheng, F. et al. Cancer stem cell vaccination With PD-L1 and CTLA-4 blockades enhances the eradication of melanoma stem cells in a mouse tumor model. J. Immunother. 41, 361–368 (2018).
Shi, X. et al. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int. J. Cancer 142, 2106–2117 (2018).
Powell, S. & McMillan, T. J. DNA damage and repair following treatment with ionizing radiation. Radiother. Oncol. 19, 95–108 (1990).
Vlashi, E. et al. Radiation-induced dedifferentiation of head and neck cancer cells into cancer stem cells depends on human papillomavirus status. Int. J. Radiat. Oncol. Biol. Phys. 94, 1198–1206 (2016).
Dai, W. W., Liu, S., Liu, X. J. & Peng, Z. P. Stemness-related changes of CD133- cells in nasopharyngeal carcinoma after x-ray radiation at the median lethal dose. Eur. Rev. Med. Pharmacol. Sci. 22, 2334–2342 (2018).
Gomez-Casal, R. et al. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 12, 94 (2013).
Qi, X. S. et al. Radioresistance of the breast tumor is highly correlated to its level of cancer stem cell and its clinical implication for breast irradiation. Radiother. Oncol. 124, 455–461 (2017).
Smit, J. K. et al. Prediction of response to radiotherapy in the treatment of esophageal cancer using stem cell markers. Radiother. Oncol. 107, 434–441 (2013).
Kim, S. Y. et al. Breast cancer stem cell-like cells are more sensitive to ionizing radiation than non-stem cells: role of ATM. PLoS One 7, e50423 (2012).
Sahlberg, S. H. et al. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLoS One 9, e94621 (2014).
Ghisolfi, L. et al. Ionizing radiation induces stemness in cancer cells. PLoS One 7, e43628 (2012).
Wang, S. et al. SOX2 promotes radioresistance in non-small cell lung cancer by regulating tumor cells dedifferentiation. Int. J. Med. Sci. 20, 781–796 (2023).
Lee, Y. et al. FoxM1 promotes stemness and radio-resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS One 10, e0137703 (2015).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
Phillips, T. M., McBride, W. H. & Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl Cancer Inst. 98, 1777–1785 (2006).
Lan, X. et al. CD133 silencing inhibits stemness properties and enhances chemoradiosensitivity in CD133-positive liver cancer stem cells. Int. J. Mol. Med. 31, 315–324 (2013).
Bontemps, I. et al. Loss of CD24 promotes radiation‑ and chemo‑resistance by inducing stemness properties associated with a hybrid E/M state in breast cancer cells. Oncol. Rep. 49, 4 (2023).
Falke, I. et al. Knockdown of the stem cell marker Musashi-1 inhibits endometrial cancer growth and sensitizes cells to radiation. Stem Cell Res. Ther. 13, 212 (2022).
Troschel, F. M. et al. Knockdown of the prognostic cancer stem cell marker Musashi-1 decreases radio-resistance while enhancing apoptosis in hormone receptor-positive breast cancer cells via p21(WAF1/CIP1). J. Cancer Res. Clin. Oncol. 147, 3299–3312 (2021).
Fang, X. et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 13, eabc7275 (2021).
Kang, H. et al. Downregulated CLIP3 induces radioresistance by enhancing stemness and glycolytic flux in glioblastoma. J. Exp. Clin. Cancer Res. 40, 282 (2021).
Wu, Q. et al. Downregulation of SFRP2 facilitates cancer stemness and radioresistance of glioma cells via activating Wnt/β-catenin signaling. PLoS One 16, e0260864 (2021).
Tang, T., Wang, L. X., Yang, M. L. & Zhang, R. M. lncRNA TPTEP1 inhibits stemness and radioresistance of glioma through miR‑106a‑5p‑mediated P38 MAPK signaling. Mol. Med. Rep. 22, 4857–4867 (2020).
Ramakrishnan, V. et al. Radiation-induced extracellular vesicle (EV) release of miR-603 promotes IGF1-mediated stem cell state in glioblastomas. EBioMedicine 55, 102736 (2020).
Stanzani, E. et al. Dual role of integrin Alpha-6 in glioblastoma: supporting stemness in proneural stem-like cells while inducing radioresistance in mesenchymal stem-like cells. Cancers 13, 3055 (2021).
Bai, X. et al. THOC2 and THOC5 regulate stemness and radioresistance in triple-negative breast cancer. Adv. Sci. 8, e2102658 (2021).
Zhao, X. et al. GDF15 contributes to radioresistance by mediating the EMT and stemness of breast cancer cells. Int. J. Mol. Sci. 23, 10911 (2022).
Sun, X. et al. ALG3 contributes to stemness and radioresistance through regulating glycosylation of TGF-β receptor II in breast cancer. J. Exp. Clin. Cancer Res. 40, 149 (2021).
Wang, Y. et al. NRP1 contributes to stemness and potentiates radioresistance via WTAP-mediated m6A methylation of Bcl-2 mRNA in breast cancer. Apoptosis 28, 233–246 (2023).
Li, B., Cheng, X. L., Yang, Y. P. & Li, Z. Q. GRP78 mediates radiation resistance of a stem cell-like subpopulation within the MCF-7 breast cancer cell line. Oncol. Rep. 30, 2119–2126 (2013).
Park, S. Y. et al. The JAK2/STAT3/CCND2 axis promotes colorectal cancer stem cell persistence and radioresistance. J. Exp. Clin. Cancer Res. 38, 399 (2019).
Sun, T. et al. Exosomal microRNA-19b targets FBXW7 to promote colorectal cancer stem cell stemness and induce resistance to radiotherapy. Kaohsiung J. Med. Sci. 38, 108–119 (2022).
Tanaka, H. et al. Nuclear Accumulation of β-Catenin in Cancer Stem Cell Radioresistance and Stemness in Human Colon Cancer. Anticancer Res. 39, 6575–6583 (2019).
Yang, M. et al. FOXQ1-mediated SIRT1 upregulation enhances stemness and radio-resistance of colorectal cancer cells and restores intestinal microbiota function by promoting β-catenin nuclear translocation. J. Exp. Clin. Cancer Res. 41, 70 (2022).
Fu, W. et al. LINC01224/ZNF91 promote stem cell-like properties and drive radioresistance in non-small cell lung cancer. Cancer Manag. Res. 13, 5671–5681 (2021).
Chen, W. et al. HSP27 associates with epithelial-mesenchymal transition, stemness and radioresistance of salivary adenoid cystic carcinoma. J. Cell Mol. Med. 22, 2283–2298 (2018).
Lin, C. S. et al. Silencing JARID1B suppresses oncogenicity, stemness and increases radiation sensitivity in human oral carcinoma. Cancer Lett. 368, 36–45 (2015).
Wiechec, E., Matic, N., Ali, A. & Roberg, K. Hypoxia induces radioresistance, epithelial‑mesenchymal transition, cancer stem cell‑like phenotype and changes in genes possessing multiple biological functions in head and neck squamous cell carcinoma. Oncol. Rep. 47, 58 (2022).
Le Grand, M. et al. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 10, 6411–6429 (2020).
Wang, C., Liu, L., Cheng, Y. & Shi, H. Combined GSK-3β and MEK inhibitors modulate the stemness and radiotherapy sensitivity of cervical cancer stem cells through the Wnt signaling pathway. Chem. Biol. Interact 380, 110515 (2023).
Liu, Z. et al. A methyltransferase-like 14/miR-99a-5p/tribble 2 positive feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2-mediated epigenetic modulation in esophageal squamous cell carcinoma. Clin. Transl. Med. 11, e545 (2021).
Terraneo, N. et al. L1 Cell Adhesion Molecule Confers Radioresistance to Ovarian Cancer and Defines a New Cancer Stem Cell Population. Cancers 12, 217 (2020).
Chi, H. C. et al. DOCK6 promotes chemo- and radioresistance of gastric cancer by modulating WNT/β-catenin signaling and cancer stem cell traits. Oncogene 39, 5933–5949 (2020).
Cahu, J., Bustany, S. & Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 3, e446 (2012).
Zhang, L. et al. Dedifferentiation process driven by radiotherapy-induced HMGB1/TLR2/YAP/HIF-1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 10, 724 (2019).
Zhao, Y. et al. K-RAS Acts as a Critical Regulator of CD44 to Promote the Invasiveness and Stemness of GBM in Response to Ionizing Radiation. Int. J. Mol. Sci. 22, 10923 (2021).
Lagadec, C. et al. Radiation-induced reprogramming of breast cancer cells. Stem Cells 30, 833–844 (2012).
Wang, W. J. et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 73, 1219–1231 (2013).
Nathansen, J. et al. Oct4 confers stemness and radioresistance to head and neck squamous cell carcinoma by regulating the homologous recombination factors PSMC3IP and RAD54L. Oncogene 40, 4214–4228 (2021).
Huang, C. et al. SOX2 regulates radioresistance in cervical cancer via the hedgehog signaling pathway. Gynecol. Oncol. 151, 533–541 (2018).
Lee, J. K. et al. USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro. Oncol. 18, 37–47 (2016).
de Araujo, P. R. et al. Musashi1 impacts radio-resistance in glioblastoma by controlling DNA-protein kinase catalytic subunit. Am. J. Pathol. 186, 2271–2278 (2016).
Zakharchenko, O., Cojoc, M., Dubrovska, A. & Souchelnytskyi, S. A role of TGFß1 dependent 14-3-3σ phosphorylation at Ser69 and Ser74 in the regulation of gene transcription, stemness and radioresistance. PLoS One 8, e65163 (2013).
Lu, Y. et al. EVI1 promotes epithelial-to-mesenchymal transition, cancer stem cell features and chemo-/radioresistance in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 38, 82 (2019).
Chang, L. et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 4, e875 (2013).
Lubanska, D. et al. The cyclin-like protein Spy1 regulates growth and division characteristics of the CD133+ population in human glioma. Cancer Cell 25, 64–76 (2014).
Ong, D. S. T. et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl Acad. Sci. USA 114, E9086–e9095 (2017).
Park, S. J. et al. Integrin β1 regulates the perineural invasion and radioresistance of oral squamous carcinoma cells by modulating cancer cell stemness. Cell Signal. 110, 110808 (2023).
Shang, Y. et al. MiR-7-5p/KLF4 signaling inhibits stemness and radioresistance in colorectal cancer. Cell Death Discov. 9, 42 (2023).
Zhu, Y. et al. miR-145 Antagonizes SNAI1-mediated stemness and radiation resistance in colorectal cancer. Mol. Ther. 26, 744–754 (2018).
Sharanek, A. et al. OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat. Commun. 11, 4116 (2020).
Jia, C. et al. Apigenin sensitizes radiotherapy of mouse subcutaneous glioma through attenuations of cell stemness and DNA damage repair by inhibiting NF-κB/HIF-1α-mediated glycolysis. J. Nutr. Biochem. 107, 109038 (2022).
Yang, W. et al. MiR-146b-5p overexpression attenuates stemness and radioresistance of glioma stem cells by targeting HuR/lincRNA-p21/β-catenin pathway. Oncotarget 7, 41505–41526 (2016).
Chen, K. et al. Silencing hTERT attenuates cancer stem cell-like characteristics and radioresistance in the radioresistant nasopharyngeal carcinoma cell line CNE-2R. Aging 12, 25599–25613 (2020).
Yang, W. et al. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 326, 22–35 (2014).
Tan, B. et al. Tumor-suppressive E3 ubiquitin ligase CHIP inhibits the PBK/ERK axis to repress stem cell properties and radioresistance in non-small cell lung cancer. Apoptosis 28, 397–413 (2023).
Lin, C. et al. Phase I trial of concurrent stereotactic body radiotherapy and nelfinavir for locally advanced borderline or unresectable pancreatic adenocarcinoma. Radiother. Oncol. 132, 55–62 (2019).
Qian, Y. et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 13, 130 (2020).
Bedard, P. L., Hyman, D. M., Davids, M. S. & Siu, L. L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 395, 1078–1088 (2020).
Jokinen, E., Laurila, N., Koivunen, P. & Koivunen, J. P. Combining targeted drugs to overcome and prevent resistance of solid cancers with some stem-like cell features. Oncotarget 5, 9295–9307 (2014).
Yao, W. et al. All-trans retinoic acid reduces cancer stem cell-like cell-mediated resistance to gefitinib in NSCLC adenocarcinoma cells. BMC Cancer 20, 315 (2020).
Tan, Q. et al. Ginsenoside Rg3 attenuates the osimertinib resistance by reducing the stemness of non-small cell lung cancer cells. Environ. Toxicol. 35, 643–651 (2020).
Corominas-Faja, B. et al. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: a novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 12, 3390–3404 (2013).
Torigoe, H. et al. Therapeutic strategies for afatinib-resistant lung cancer harboring HER2 alterations. Cancer Sci. 109, 1493–1502 (2018).
Lei, H. M. et al. Aldehyde dehydrogenase 1A1 confers erlotinib resistance via facilitating the reactive oxygen species-reactive carbonyl species metabolic pathway in lung adenocarcinomas. Theranostics 9, 7122–7139 (2019).
Tang, C. H. et al. APE1 shRNA-loaded cancer stem cell-derived extracellular vesicles reverse Erlotinib resistance in non-small cell lung cancer via the IL-6/STAT3 signalling. Clin Transl. Med. 12, e876 (2022).
Cheng, Q. et al. FGFR1 overexpression induces cancer cell stemness and enhanced Akt/Erk-ER signaling to promote palbociclib resistance in luminal a breast cancer cells. Cells 10, 3008 (2021).
Huang, W. C. et al. Novel function of THEMIS2 in the enhancement of cancer stemness and chemoresistance by releasing PTP1B from MET. Oncogene 41, 997–1010 (2022).
Chang, H. L. et al. Ovatodiolide suppresses yes-associated protein 1-modulated cancer stem cell phenotypes in highly malignant hepatocellular carcinoma and sensitizes cancer cells to chemotherapy in vitro. Toxicol In Vitro 51, 74–82 (2018).
Witt-Kehati, D. et al. Inhibition of pMAPK14 overcomes resistance to sorafenib in hepatoma cells with Hepatitis B Virus. Transl. Oncol. 11, 511–517 (2018).
Cherng, Y. G. et al. Induced mitochondrial alteration and DNA damage via IFNGR-JAK2-STAT1-PARP1 pathway facilitates viral hepatitis associated hepatocellular carcinoma aggressiveness and stemness. Cancers 13, 2755 (2021).
Ng, K. Y. et al. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 7, 24005–24017 (2016).
Wang, X. et al. Musashi2 contributes to the maintenance of CD44v6+ liver cancer stem cells via notch1 signaling pathway. J. Exp. Clin. Cancer Res. 38, 505 (2019).
Li, Y., Tang, T., Lee, H. J. & Song, K. Selective anti-cancer effects of plasma-activated medium and its high efficacy with cisplatin on hepatocellular carcinoma with cancer stem cell characteristics. Int. J. Mol. Sci. 22, 3956 (2021).
Ma, X. L. et al. CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J. Hematol. Oncol. 13, 11 (2020).
Kahraman, D. C., Kahraman, T. & Cetin-Atalay, R. Targeting PI3K/Akt/mTOR pathway identifies differential expression and functional role of IL8 in liver cancer stem cell enrichment. Mol. Cancer Ther. 18, 2146–2157 (2019).
Huang, H. et al. RAB27A-dependent release of exosomes by liver cancer stem cells induces Nanog expression in their differentiated progenies and confers regorafenib resistance. J. Gastroenterol Hepatol. 36, 3429–3437 (2021).
Zubrilov, I. et al. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett. 361, 86–96 (2015).
Jebali, A., Battistella, M., Lebbé, C. & Dumaz, N. RICTOR affects melanoma tumorigenesis and its resistance to targeted therapy. Biomedicines 9, 1498 (2021).
Hüser, L. et al. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int. J. Cancer 143, 3131–3142 (2018).
Prasetyanti, P. R. et al. ErbB-3 activation by NRG-1β sustains growth and promotes vemurafenib resistance in BRAF-V600E colon cancer stem cells (CSCs). Oncotarget 6, 16902–16911 (2015).
Tung, M. C. et al. Targeting DRD2 by the antipsychotic drug, penfluridol, retards growth of renal cell carcinoma via inducing stemness inhibition and autophagy-mediated apoptosis. Cell Death Dis. 13, 400 (2022).
He, M. et al. Sunitinib increases the cancer stem cells and vasculogenic mimicry formation via modulating the lncRNA-ECVSR/ERβ/Hif2-α signaling. Cancer Lett. 524, 15–28 (2022).
Wang, T. et al. The TGFβ-miR-499a-SHKBP1 pathway induces resistance to EGFR inhibitors in osteosarcoma cancer stem cell-like cells. J. Exp. Clin. Cancer Res. 38, 226 (2019).
Lu, Y. J. et al. Lysyl oxidase-like 2 promotes stemness and enhances antitumor effects of gefitinib in head and neck cancer via IFIT1 and IFIT3. Cancer Sci. 114, 3957–3971 (2023).
Kim, H. M. et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann. Surg. Oncol. 19, S539–548 (2012).
Arabi, L., Badiee, A., Mosaffa, F. & Jaafari, M. R. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J. Control Release 220, 275–286 (2015).
Gao, Y. et al. Emodin is a potential drug targeting CD44-positive hepatocellular cancer. Curr. Cancer Drug Targets, 24, 510–518 (2023).
Han, Y., Sun, B., Cai, H. & Xuan, Y. Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and anti-CD133 CAR-T combination therapy. Cancer Immunol. Immunother. 70, 2795–2803 (2021).
Hu, C. et al. Lentivirus-mediated shRNA targeting Nanog inhibits cell proliferation and attenuates cancer stem cell activities in breast cancer. J. Drug Target 24, 422–432 (2016).
Liang, L. et al. The Wee1 kinase inhibitor MK1775 suppresses cell growth, attenuates stemness and synergises with bortezomib in multiple myeloma. Br. J. Haematol. 191, 62–76 (2020).
Chen, C. L. et al. Profiling of circulating tumor cells for screening of selective inhibitors of tumor-initiating stem-like cells. Adv. Sci. 10, e2206812 (2023).
Nguyen, P. H. et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene 35, 5619–5628 (2016).
Choi, J. H. et al. The Small-Molecule Wnt Inhibitor ICG-001 efficiently inhibits colorectal cancer stemness and metastasis by suppressing MEIS1 expression. Int. J. Mol. Sci. 22, 13413 (2021).
Karakas, D. et al. Addition of niclosamide to palladium(II) saccharinate complex of terpyridine results in enhanced cytotoxic activity inducing apoptosis on cancer stem cells of breast cancer. Bioorg. Med. Chem. 23, 5580–5586 (2015).
Rodova, M. et al. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One 7, e46083 (2012).
Hayashi, T. et al. Not all NOTCH is created equal: the oncogenic role of NOTCH2 in bladder cancer and its implications for targeted therapy. Clin. Cancer Res. 22, 2981–2992 (2016).
Moon, C. M. et al. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 134, 519–529 (2014).
Abubaker, K. et al. Targeted disruption of the JAK2/STAT3 pathway in combination with systemic administration of paclitaxel inhibits the priming of ovarian cancer stem cells leading to a reduced tumor burden. Front. Oncol. 4, 75 (2014).
Chung, S. S. & Vadgama, J. V. Curcumin and epigallocatechin gallate inhibit the cancer stem cell phenotype via down-regulation of STAT3-NFκB signaling. Anticancer Res. 35, 39–46 (2015).
Ni, T. et al. Celastrus orbiculatus extract suppresses gastric cancer stem cells through the TGF-β/Smad signaling pathway. J. Nat. Med. 78, 100–113 (2024).
Hongwiangchan, N. et al. Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc. Pharmaceuticals 14, 1112 (2021).
Surowiec, R. K. et al. Transcriptomic Analysis of Diffuse Intrinsic Pontine Glioma (DIPG) Identifies a Targetable ALDH-Positive Subset of Highly Tumorigenic Cancer Stem-like Cells. Mol. Cancer Res. 19, 223–239 (2021).
Hu, F. et al. Degree of stemness predicts micro-environmental response and clinical outcomes of diffuse large B-cell lymphoma and identifies a potential targeted therapy. Front. Immunol. 13, 1012242 (2022).
Bai, J. et al. HIF-2α regulates CD44 to promote cancer stem cell activation in triple-negative breast cancer via PI3K/AKT/mTOR signaling. World J. Stem Cells 12, 87–99 (2020).
Andreucci, E. et al. The acidic tumor microenvironment drives a stem-like phenotype in melanoma cells. J. Mol. Med. 98, 1431–1446 (2020).
Doherty, M. R. & Jackson, M. W. The critical, clinical role of interferon-beta in regulating cancer stem cell properties in triple-negative breast cancer. DNA Cell Biol. 37, 513–516 (2018).
Doherty, M. R. et al. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl Acad. Sci. USA 114, 13792–13797 (2017).
Huang, C. S. et al. Galectin-3 promotes CXCR2 to augment the stem-like property of renal cell carcinoma. J. Cell Mol. Med. 22, 5909–5918 (2018).
Singh, J. K. et al. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin. Cancer Res. 19, 643–656 (2013).
Chen, J. et al. Co-targeting FAK and Gli1 inhibits the tumor-associated macrophages-released CCL22-mediated esophageal squamous cell carcinoma malignancy. MedComm 4, e381 (2023).
You, Y. et al. M1-like tumor-associated macrophages cascade a mesenchymal/stem-like phenotype of oral squamous cell carcinoma via the IL6/Stat3/THBS1 feedback loop. J. Exp. Clin. Cancer Res. 41, 10 (2022).
Wang, H. et al. Rab13 sustains breast cancer stem cells by supporting tumor-stroma cross-talk. Cancer Res. 82, 2124–2140 (2022).
Kim, B. et al. IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol. Carcinog. 60, 188–200 (2021).
McAndrews, K. M. et al. αSMA(+) fibroblasts suppress Lgr5(+) cancer stem cells and restrain colorectal cancer progression. Oncogene 40, 4440–4452 (2021).
Ning, J. et al. The protein arginine methyltransferase family (PRMTs) regulates metastases in various tumors: from experimental study to clinical application. Biomed. Pharmacother. 167, 115456 (2023).
Wu, Q., Schapira, M., Arrowsmith, C. H. & Barsyte-Lovejoy, D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 20, 509–530 (2021).
Feng, X. et al. Chromatin target of protein arginine methyltransferase regulates invasion, chemoresistance, and stemness in epithelial ovarian cancer. Biosci. Rep. 39, BSR20190016 (2019).
Katsura, Y. et al. A novel combination cancer therapy with iron chelator targeting cancer stem cells via suppressing stemness. Cancers 11, 177 (2019).
Mana, M. D. et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 35, 109212 (2021).
Song, I. S. et al. Peroxiredoxin 3 maintains the survival of endometrial cancer stem cells by regulating oxidative stress. Oncotarget 8, 92788–92800 (2017).
Song, I. S., Jeong, Y. J. & Han, J. Mitochondrial metabolism in cancer stem cells: a therapeutic target for colon cancer. BMB Rep. 48, 539–540 (2015).
Ning, J. et al. CircRNAs and lung cancer: Insight into their roles in metastasis. Biomed. Pharmacother. 166, 115260 (2023).
Feng, Z. et al. Functions and Potential Applications of Circular RNAs in Cancer Stem Cells. Front. Oncol. 9, 500 (2019).
Lin, X., Chen, W., Wei, F. & Xie, X. TV-circRGPD6 nanoparticle suppresses breast cancer stem cell-mediated metastasis via the miR-26b/YAF2 Axis. Mol. Ther. 29, 244–262 (2021).
Ding, D., Yang, F., Chen, Z. & Ying, J. Circ_0007385 regulates cell proliferation, apoptosis and stemness via targeting miR-493-3p/RAB22A axis in non-small cell lung cancer. Thorac. Cancer 13, 571–581 (2022).
Shi, L. et al. Exosomal lncRNA Mir100hg derived from cancer stem cells enhance glycolysis and promote metastasis of lung adenocarcinoma through mircroRNA-15a-5p/31-5p. Cell Commun. Signal. 21, 248 (2023).
Tan, Y. et al. miR-148a regulates the stem cell-like side populations distribution by affecting the expression of ACVR1 in Esophageal squamous cell carcinoma. Oncol. Targets Ther. 13, 8079–8094 (2020).
Lv, L., Shi, Y., Wu, J. & Li, G. Nanosized drug delivery systems for breast cancer stem cell targeting. Int. J. Nanomed. 16, 1487–1508 (2021).
Li, L., Ni, R., Zheng, D. & Chen, L. Eradicating the tumor “seeds”: nanomedicines-based therapies against cancer stem cells. Daru 31, 83–94 (2023).
Cao, J. et al. Cancer stem cells and strategies for targeted drug delivery. Drug Deliv. Transl. Res. 11, 1779–1805 (2021).
Kola, P. et al. Innovative nanotheranostics: smart nanoparticles based approach to overcome breast cancer stem cells mediated chemo- and radioresistances. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 15, e1876 (2023).
Shen, S. et al. A nanotherapeutic strategy to overcome chemotherapeutic resistance of cancer stem-like cells. Nat. Nanotechnol. 16, 104–113 (2021).
Li, Y. et al. In vivo β-catenin attenuation by the integrin α5-targeting nano-delivery strategy suppresses triple negative breast cancer stemness and metastasis. Biomaterials 188, 160–172 (2019).
Samson, A. A. S. et al. Liposomal co-delivery-based quantitative evaluation of chemosensitivity enhancement in breast cancer stem cells by knockdown of GRP78/CLU. J. Liposome Res. 29, 44–52 (2019).
Li, W. et al. Construction and in vitro evaluation of pH-sensitive nanoparticles to reverse drug resistance of breast cancer stem cells. Discov. Oncol. 15, 21 (2024).
Reda, A., Hosseiny, S. & El-Sherbiny, I. M. Next-generation nanotheranostics targeting cancer stem cells. Nanomedicine 14, 2487–2514 (2019).
Liufu, C. et al. Echogenic PEGylated PEI-loaded microbubble as efficient gene delivery system. Int. J. Nanomed. 14, 8923–8941 (2019).
Su, W. et al. Red-emissive carbon quantum dots for nuclear drug delivery in cancer stem cells. J. Phys. Chem. Lett. 11, 1357–1363 (2020).
Oswald, J. T. et al. Drug delivery systems using surface markers for targeting cancer stem cells. Curr. Pharm. Des. 26, 2057–2071 (2020).
Eskandari, A., Kundu, A., Ghosh, S. & Suntharalingam, K. A Triangular Platinum(II) multinuclear complex with cytotoxicity towards breast cancer stem cells. Angew. Chem. Int. Ed. Engl. 58, 12059–12064 (2019).
Sun, S., Yang, Q., Jiang, D. & Zhang, Y. Nanobiotechnology augmented cancer stem cell guided management of cancer: liquid-biopsy, imaging, and treatment. J. Nanobiotechnol.22, 176 (2024).
Ahmad, G. & Amiji, M. M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin. Drug Deliv. 14, 997–1008 (2017).
Gao, J., Li, W., Guo, Y. & Feng, S. S. Nanomedicine strategies for sustained, controlled and targeted treatment of cancer stem cells. Nanomedicine 11, 3261–3282 (2016).
Wang, Y. et al. Hyaluronic acid mediated Fe(3)O(4) nanocubes reversing the EMT through targeted cancer stem cell. Colloids Surf B Biointerfaces 222, 113071 (2023).
Wang, Q. et al. Cancer stem-like cells-oriented surface self-assembly to conquer radioresistance. Adv. Mater 35, e2302916 (2023).
Jia, Y. et al. Tumor microenvironment-responsive nanoherb delivery system for synergistically inhibition of cancer stem cells. ACS Appl. Mater. Interfaces 15, 16329–16342 (2023).
Toshiyama, R. et al. Poly(ethylene glycol)-poly(lysine) block copolymer-ubenimex conjugate targets aminopeptidase N and exerts an antitumor effect in hepatocellular carcinoma stem cells. Oncogene 38, 244–260 (2019).
Miyano, K. et al. cRGD peptide installation on cisplatin-loaded nanomedicines enhances efficacy against locally advanced head and neck squamous cell carcinoma bearing cancer stem-like cells. J. Control Release 261, 275–286 (2017).
Xu, X. et al. Application of mPEG-CS-cRGD/Bmi-1RNAi-PTX nanoparticles in suppression of laryngeal cancer by targeting cancer stem cells. Drug Deliv. 30, 2180112 (2023).
Sun, L. et al. Activated carbon nanoparticles loaded with metformin for effective against hepatocellular cancer stem cells. Int. J. Nanomed. 18, 2891–2910 (2023).
Liu, B. et al. Recent advances in aptamer-based therapeutic strategies for targeting cancer stem cells. Mater. Today Biol. 19, 100605 (2023).
Shen, S. et al. Combating Cancer Stem-Like Cell-Derived Resistance to Anticancer Protein by Liposome-Mediated Acclimatization Strategy. Nano Lett. 22, 2419–2428 (2022).
Andrade, F. et al. Polymeric micelles targeted against CD44v6 receptor increase niclosamide efficacy against colorectal cancer stem cells and reduce circulating tumor cells in vivo. J. Control Release 331, 198–212 (2021).
Yuan, H. et al. Albumin Nanoparticle of Paclitaxel (Abraxane) Decreases while Taxol Increases Breast Cancer Stem Cells in Treatment of Triple Negative Breast Cancer. Mol. Pharm. 17, 2275–2286 (2020).
Jang, E. et al. Nanovesicle-mediated systemic delivery of microRNA-34a for CD44 overexpressing gastric cancer stem cell therapy. Biomaterials 105, 12–24 (2016).
Liu, J. et al. MicroRNA-200c delivered by solid lipid nanoparticles enhances the effect of paclitaxel on breast cancer stem cell. Int. J. Nanomed. 11, 6713–6725 (2016).
Qin, J., Zhu, Y., Zheng, D. & Zhao, Q. pH-sensitive polymeric nanocarriers for antitumor biotherapeutic molecules targeting delivery. Bio-Des. Manufact. 4, 612–626 (2021).
Xu, H. L. et al. Ratiometric delivery of two therapeutic candidates with inherently dissimilar physicochemical property through pH-sensitive core-shell nanoparticles targeting the heterogeneous tumor cells of glioma. Drug Deliv. 25, 1302–1318 (2018).
Duan, H., Liu, Y., Gao, Z. & Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin B 11, 55–70 (2021).
Li, H. et al. Nucleus-targeted nano delivery system eradicates cancer stem cells by combined thermotherapy and hypoxia-activated chemotherapy. Biomaterials 200, 1–14 (2019).
Zhu, X. et al. Cascade-responsive nano-assembly for efficient photothermal-chemo synergistic inhibition of tumor metastasis by targeting cancer stem cells. Biomaterials 280, 121305 (2022).
Fernandes, S. et al. Magnetic Nanoparticle-Based Hyperthermia Mediates Drug Delivery and Impairs the Tumorigenic Capacity of Quiescent Colorectal Cancer Stem Cells. ACS Appl. Mater. Interfaces 13, 15959–15972 (2021).
Ye, W. et al. A cisplatin and disulphiram co-loaded inclusion complex overcomes drug resistance by inhibiting cancer cell stemness in non-small cell lung cancer. J. Drug Target 32, 159–171 (2024).
Gao, Y. et al. Liposome-enabled bufalin and doxorubicin combination therapy for trastuzumab-resistant breast cancer with a focus on cancer stem cells. J. Liposome Res. 34, 489–506 (2024).
Chen, L. et al. Dual Targeted Nanoparticles for the Codelivery of Doxorubicin and siRNA Cocktails to Overcome Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 24, 11575 (2023).
Zhang, Y. et al. Construction of a target MSNs drugcarrier loaded with siRNA(GLI1) and siRNA(SMO) aim at hedgehog signal pathway and the pharmacodynamic study of drug-carriers in the treatment of leukemia stem cells. Drug Deliv. Transl. Res. 12, 2463–2473 (2022).
Wang, Q. et al. Combined delivery of salinomycin and docetaxel by dual-targeting gelatinase nanoparticles effectively inhibits cervical cancer cells and cancer stem cells. Drug Deliv. 28, 510–519 (2021).
Wang, D. et al. An assembly-inducing PDC enabling the efficient nuclear delivery of nucleic acid for cancer stem-like cell suppression. Nanoscale 14, 15384–15392 (2022).
Fernandes, Q. et al. Shrinking the battlefield in cancer therapy: nanotechnology against cancer stem cells. Eur. J. Pharm. Sci. 191, 106586 (2023).
Yin, H. et al. Delivery of Anti-miRNA for Triple-negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 27, 1252–1261 (2019).
Davodabadi, F. et al. Cancer chemotherapy resistance: mechanisms and recent breakthrough in targeted drug delivery. Eur. J. Pharmacol. 958, 176013 (2023).
Narayana, R. V. L. et al. Carboplatin- and Etoposide-Loaded Lactoferrin Protein Nanoparticles for Targeting Cancer Stem Cells in Retinoblastoma In Vitro. Investig. Ophthalmol. Vis. Sci. 62, 13 (2021).
Pesarrodona, M. et al. Engineering a Nanostructured Nucleolin-binding Peptide For Intracellular Drug Delivery In Triple-negative Breast Cancer Stem Cells. ACS Appl. Mater. Interfaces 12, 5381–5388 (2020).
Kim, D. M. et al. Anti-MUC1/CD44 dual-aptamer-conjugated liposomes for cotargeting breast cancer cells and cancer stem cells. ACS Appl. Biol. Mater. 2, 4622–4633 (2019).
Chen, H., Lin, J., Shan, Y. & Zhengmao, L. The promotion of nanoparticle delivery to two populations of gastric cancer stem cells by CD133 and CD44 antibodies. Biomed. Pharmacother. 115, 108857 (2019).
Qiao, S. et al. A novel double-targeted nondrug delivery system for targeting cancer stem cells. Int. J. Nanomed. 11, 6667–6678 (2016).
Zhang, Z. et al. Rational design of nanotherapeutics based on the five features principle for potent elimination of cancer stem cells. Acc. Chem. Res. 55, 526–536 (2022).
Oliveira, B. S. A. et al. Nanotherapeutic approach to tackle chemotherapeutic resistance of cancer stem cells. Life Sci. 279, 119667 (2021).
Bozzato, E., Bastiancich, C. & Préat, V. Nanomedicine: a useful tool against glioma stem cells. Cancers 13, 9 (2020).
Smiley, S. B. et al. Novel therapeutics and drug-delivery approaches in the modulation of glioblastoma stem cell resistance. Ther. Deliv. 13, 249–273 (2022).
Knauer, N. et al. In vitro validation of the therapeutic potential of dendrimer-based nanoformulations against tumor stem cells. Int. J. Mol. Sci. 23, 5691 (2022).
Behrooz, A. B. et al. Tailoring drug co-delivery nanosystem for mitigating U-87 stem cells drug resistance. Drug Deliv. Transl. Res. 12, 1253–1269 (2022).
Smiley, S. B. et al. Development of CD133 targeting multi-drug polymer micellar nanoparticles for glioblastoma - in vitro evaluation in glioblastoma stem cells. Pharm. Res. 38, 1067–1079 (2021).
Fadera, S., Chen, P. Y., Liu, H. L. & Lee, I. C. Induction therapy of retinoic acid with a temozolomide-loaded gold nanoparticle-associated ultrasound effect on glioblastoma cancer stem-like colonies. ACS Appl. Mater. Interfaces 13, 32845–32855 (2021).
Chang, L. et al. Nanostructured lipid carrier co-delivering paclitaxel and doxorubicin restrains the proliferation and promotes apoptosis of glioma stem cells via regulating PI3K/Akt/mTOR signaling. Nanotechnology 32, 225101 (2021).
Lépinoux-Chambaud, C. & Eyer, J. The NFL-TBS.40-63 peptide targets and kills glioblastoma stem cells derived from human patients and also targets nanocapsules into these cells. Int. J. Pharm. 566, 218–228 (2019).
Wang, Z. et al. Etoposide loaded layered double hydroxide nanoparticles reversing chemoresistance and eradicating human glioma stem cells in vitro and in vivo. Nanoscale 10, 13106–13121 (2018).
Kim, J. et al. Engineered biomimetic nanoparticle for dual targeting of the cancer stem-like cell population in sonic hedgehog medulloblastoma. Proc. Natl Acad. Sci. USA 117, 24205–24212 (2020).
Lu, L. et al. The nanoparticle-facilitated autophagy inhibition of cancer stem cells for improved chemotherapeutic effects on glioblastomas. J. Mater. Chem. B 7, 2054–2062 (2019).
Kuo, Y. C., Wang, L. J. & Rajesh, R. Targeting human brain cancer stem cells by curcumin-loaded nanoparticles grafted with anti-aldehyde dehydrogenase and sialic acid: Colocalization of ALDH and CD44. Mater. Sci. Eng. C Mater. Biol. Appl. 102, 362–372 (2019).
Mu, L. M. et al. Lipid vesicles containing transferrin receptor binding peptide TfR-T(12) and octa-arginine conjugate stearyl-R(8) efficiently treat brain glioma along with glioma stem cells. Sci. Rep. 7, 3487 (2017).
Séhédic, D. et al. Locoregional Confinement and Major Clinical Benefit of (188)Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 7, 4517–4536 (2017).
Miao, Y. B. et al. Customizing delivery nano-vehicles for precise brain tumor therapy. J. Nanobiotechnol. 21, 32 (2023).
Morad, G. et al. Tumor-derived extracellular vesicles breach the intact blood-brain barrier via transcytosis. ACS Nano 13, 13853–13865 (2019).
Kim, S. S., Harford, J. B., Pirollo, K. F. & Chang, E. H. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochem. Biophys. Res. Commun. 468, 485–489 (2015).
Dupoiron, D. et al. Intrathecal catheter for chemotherapy in leptomeningeal carcinomatosis from HER2-negative metastatic breast cancer. J. Breast Cancer 26, 572–581 (2023).
Rothwell, W. T. et al. Intrathecal viral vector delivery of trastuzumab prevents or inhibits tumor growth of human HER2-Positive Xenografts in Mice. Cancer Res. 78, 6171–6182 (2018).
Wang, J. et al. Clinical effect of intrathecal injection of medicine combined with continuous lumbar cistern drainage on intracranial infection after intracranial tumor surgery. Pak. J. Pharm. Sci. 34, 65–67 (2021).
Ding, X. W., Wu, J. H. & Jiang, C. P. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci 86, 631–637 (2010).
Gisina, A., Kim, Y., Yarygin, K. & Lupatov, A. Can CD133 be regarded as a prognostic biomarker in oncology: pros and cons. Int. J. Mol. Sci. 24, 17398 (2023).
Erisik, D. et al. Differences and similarities between colorectal cancer cells and colorectal cancer stem cells: molecular insights and implications. ACS Omega 8, 30145–30157 (2023).
Liu, C. et al. ALDH1A1 activity in tumor-initiating cells remodels myeloid-derived suppressor cells to promote breast cancer progression. Cancer Res. 81, 5919–5934 (2021).
Xu, J. et al. The Crucial Roles of Bmi-1 in Cancer: Implications in Pathogenesis, Metastasis, Drug Resistance, and Targeted Therapies. Int. J. Mol. Sci. 23, 8231 (2022).
Elbadawy, M., Usui, T., Yamawaki, H. & Sasaki, K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 20, 2340 (2019).
Huang, T. et al. A positive feedback between PDIA3P1 and OCT4 promotes the cancer stem cell properties of esophageal squamous cell carcinoma. Cell Commun. Signal 22, 60 (2024).
Zhu, Y. et al. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by β-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 12, 449 (2021).
Shmelkov, S. V. et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Investig. 118, 2111–2120 (2008).
Shimokawa, M. et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 545, 187–192 (2017).
Ni, Y. et al. The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front. Cell Dev. Biol. 9, 637675 (2021).
Li, Y., Atkinson, K. & Zhang, T. Combination of chemotherapy and cancer stem cell targeting agents: Preclinical and clinical studies. Cancer Lett. 396, 103–109 (2017).
Lee, H., Kim, J. W., Lee, D. S. & Min, S. H. Combined Poziotinib with manidipine treatment suppresses ovarian cancer stem-cell proliferation and stemness. Int. J. Mol. Sci. 21, 7379 (2020).
Raghav, P. K. & Mann, Z. Cancer stem cells targets and combined therapies to prevent cancer recurrence. Life Sci. 277, 119465 (2021).
Wang, A., Qu, L. & Wang, L. At the crossroads of cancer stem cells and targeted therapy resistance. Cancer Lett. 385, 87–96 (2017).
Montazersaheb, P. et al. Magnetic nanoparticle-based hyperthermia: a prospect in cancer stem cell tracking and therapy. Life Sci. 323, 121714 (2023).
Wang, W. D. et al. Sniping cancer stem cells with nanomaterials. ACS Nano 17, 23262–23298 (2023).
Bo, Y. et al. Leveraging intracellular ALDH1A1 activity for selective cancer stem-like cell labeling and targeted treatment via in vivo click reaction. Proc. Natl Acad. Sci. USA 120, e2302342120 (2023).
Corn, K. C., Windham, M. A. & Rafat, M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 80, 101055 (2020).
Lu, G. et al. Two-step tumor-targeting therapy via integrating metabolic lipid-engineering with in situ click chemistry. Biomater. Sci .8, 2283–2288 (2020).
Best, M. D., Zhang, H. & Prestwich, G. D. Inositol polyphosphates, diphosphoinositol polyphosphates and phosphatidylinositol polyphosphate lipids: structure, synthesis, and development of probes for studying biological activity. Nat. Prod. Rep. 27, 1403–1430 (2010).
Zhou, T. et al. ESE3/EHF, a promising target of rosiglitazone, suppresses pancreatic cancer stemness by downregulating CXCR4. Gut 71, 357–371 (2022).
Zhao, H. et al. The regulatory role of cancer stem cell marker gene CXCR4 in the growth and metastasis of gastric cancer. NPJ Precis. Oncol. 7, 86 (2023).
Zhu, T. et al. ETV4 promotes breast cancer cell stemness by activating glycolysis and CXCR4-mediated sonic Hedgehog signaling. Cell Death Discov. 7, 126 (2021).
Cheng, C. W. et al. MiR-139 modulates cancer stem cell function of human breast cancer through targeting CXCR4. Cancers 13, 2582 (2021).
Dubrovska, A. et al. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br. J. Cancer 107, 43–52 (2012).
Liu, B. Q. et al. BAG3 promotes stem cell-like phenotype in breast cancer by upregulation of CXCR4 via interaction with its transcript. Cell Death Dis. 8, e2933 (2017).
DeCastro, A. J., Cherukuri, P., Balboni, A. & DiRenzo, J. ΔNP63α transcriptionally activates chemokine receptor 4 (CXCR4) expression to regulate breast cancer stem cell activity and chemotaxis. Mol. Cancer Ther. 14, 225–235 (2015).
Peng, H. et al. Activin and hepatocyte growth factor promotes colorectal cancer stemness and metastasis through FOXM1/SOX2/CXCR4 signaling. Gut Liver 18, 476-488 (2024).
Yue, D. et al. Chloroquine inhibits stemness of esophageal squamous cell carcinoma cells through targeting CXCR4-STAT3 pathway. Front. Oncol. 10, 311 (2020).
Kang, N. et al. Hypoxia-induced cancer stemness acquisition is associated with CXCR4 activation by its aberrant promoter demethylation. BMC Cancer 19, 148 (2019).
Nian, W. Q., Chen, F. L., Ao, X. J. & Chen, Z. T. CXCR4 positive cells from Lewis lung carcinoma cell line have cancer metastatic stem cell characteristics. Mol. Cell Biochem. 355, 241–248 (2011).
Ping, Y. F. et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 224, 344–354 (2011).
Schulte, A. et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 59, 590–602 (2011).
Rasti, A. et al. Reduced expression of CXCR4, a novel renal cancer stem cell marker, is associated with high-grade renal cell carcinoma. J. Cancer Res. Clin. Oncol. 143, 95–104 (2017).
Fatehullah, A. et al. A tumour-resident Lgr5(+) stem-cell-like pool drives the establishment and progression of advanced gastric cancers. Nat. Cell Biol. 23, 1299–1313 (2021).
Wang, C. et al. Rspondin-1 contributes to the progression and stemness of gastric cancer by LGR5. Biochem. Biophys. Res. Commun. 627, 91–96 (2022).
Zhang, J. et al. LGR5, a novel functional glioma stem cell marker, promotes EMT by activating the Wnt/β-catenin pathway and predicts poor survival of glioma patients. J. Exp. Clin. Cancer Res. 37, 225 (2018).
Cao, H. Z. et al. LGR5 promotes cancer stem cell traits and chemoresistance in cervical cancer. Cell Death Dis. 8, e3039 (2017).
Alharbi, S. A., Ovchinnikov, D. A. & Wolvetang, E. Leucine-rich repeat-containing G protein-coupled receptor 5 marks different cancer stem cell compartments in human Caco-2 and LoVo colon cancer lines. World J. Gastroenterol. 27, 1578–1594 (2021).
Akbari, S. et al. LGR5/R-Spo1/Wnt3a axis promotes stemness and aggressive phenotype in hepatoblast-like hepatocellular carcinoma cell lines. Cell Signal. 82, 109972 (2021).
Amsterdam, A. et al. LGR5 and Nanog identify stem cell signature of pancreas beta cells which initiate pancreatic cancer. Biochem. Biophys. Res. Commun. 433, 157–162 (2013).
Mani, S. K. et al. EpCAM-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis B virus-infected hepatocytes. J. Hepatol. 65, 888–898 (2016).
Wang, M. H. et al. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 9, 2 (2018).
Zhang, D. et al. Hypoxia modulates stem cell properties and induces EMT through N-glycosylation of EpCAM in breast cancer cells. J. Cell Physiol. 235, 3626–3633 (2020).
Shi, R. et al. Downregulation of cytokeratin 18 induces cellular partial EMT and stemness through increasing EpCAM expression in breast cancer. Cell Signal. 76, 109810 (2020).
Zhang, P. et al. Protein C receptor maintains cancer stem cell properties via activating lipid synthesis in nasopharyngeal carcinoma. Signal. Transduct Target Ther. 7, 46 (2022).
Wang, D. et al. Protein C receptor is a therapeutic stem cell target in a distinct group of breast cancers. Cell Res. 29, 832–845 (2019).
Wang, D. et al. Identification of multipotent mammary stem cells by protein C receptor expression. Nature 517, 81–84 (2015).
Jo, J. H. et al. Novel gastric cancer stem cell-related marker LINGO2 is associated with cancer cell phenotype and patient outcome. Int. J. Mol. Sci. 20, 555 (2019).
Belle, N. M. et al. TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun. 10, 4408 (2019).
Weng, C. C. et al. Mutant Kras-induced upregulation of CD24 enhances prostate cancer stemness and bone metastasis. Oncogene 38, 2005–2019 (2019).
Yang, C. H. et al. Identification of CD24 as a cancer stem cell marker in human nasopharyngeal carcinoma. PLoS One 9, e99412 (2014).
Ghuwalewala, S. et al. MiRNA-146a/AKT/β-Catenin activation regulates cancer stem cell phenotype in oral squamous cell carcinoma by targeting CD24. Front. Oncol. 11, 651692 (2021).
Wang, T. W. et al. SIRT1-mediated expression of CD24 and epigenetic suppression of novel tumor suppressor miR-1185-1 Increases colorectal cancer stemness. Cancer Res. 80, 5257–5269 (2020).
Ke, J. et al. A subpopulation of CD24+ cells in colon cancer cell lines possess stem cell characteristics. Neoplasma 59, 282–288 (2012).
Fujikuni, N. et al. Hypoxia-mediated CD24 expression is correlated with gastric cancer aggressiveness by promoting cell migration and invasion. Cancer Sci. 105, 1411–1420 (2014).
Ooki, A. et al. CD24 regulates cancer stem cell (CSC)-like traits and a panel of CSC-related molecules serves as a non-invasive urinary biomarker for the detection of bladder cancer. Br. J. Cancer 119, 961–970 (2018).
Liu, H. et al. CD44+/CD24+ cervical cancer cells resist radiotherapy and exhibit properties of cancer stem cells. Eur. Rev. Med. Pharmacol. Sci. 20, 1745–1754 (2016).
Lin et al. STAT3 as a potential therapeutic target in ALDH+ and CD44+/CD24+ stem cell-like pancreatic cancer cells. Int. J. Oncol. 49, 2265–2274 (2016).
Zhang, L. et al. miR-205/RunX2 axis negatively regulates CD44(+)/CD24(-) breast cancer stem cell activity. Am. J. Cancer Res. 10, 1871–1887 (2020).
Koh, M. Z. et al. Regulation of cellular and cancer stem cell-related putative gene expression of parental and CD44(+)CD24(-) sorted MDA-MB-231 cells by cisplatin. Pharmaceuticals 14, 391 (2021).
Hurt, E. M. et al. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 98, 756–765 (2008).
Chiu, C. C. et al. Grp78 as a therapeutic target for refractory head-neck cancer with CD24(-)CD44(+) stemness phenotype. Cancer Gene Ther. 20, 606–615 (2013).
Ghuwalewala, S. et al. CD44(high)CD24(low) molecular signature determines the Cancer Stem Cell and EMT phenotype in Oral Squamous Cell Carcinoma. Stem Cell Res. 16, 405–417 (2016).
Meng, E. et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 29, 939–948 (2012).
Zhang, J. et al. CD44+/CD24+-expressing cervical cancer cells and radioresistant cervical cancer cells exhibit cancer stem cell characteristics. Gynecol. Obstet. Investig. 84, 174–182 (2019).
Nallasamy, P. et al. Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1-CD44 Axis. Gastroenterology 161, 1998–2013.e1997 (2021).
Wei, C. Y. et al. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. J. Hematol. Oncol. 12, 21 (2019).
Ding, K. et al. JWA inhibits nicotine-induced lung cancer stemness and progression through CHRNA5/AKT-mediated JWA/SP1/CD44 axis. Ecotoxicol. Environ. Saf. 259, 115043 (2023).
Liu, Y. et al. A novel EHD1/CD44/Hippo/SP1 positive feedback loop potentiates stemness and metastasis in lung adenocarcinoma. Clin. Transl. Med. 12, e836 (2022).
Bishnupuri, K. S. et al. Reg4 interacts with CD44 to regulate proliferation and stemness of colorectal and pancreatic cancer cells. Mol. Cancer Res. 20, 387–399 (2022).
Su, Y. J. et al. Direct reprogramming of stem cell properties in colon cancer cells by CD44. Embo J. 30, 3186–3199 (2011).
Kamarajan, P. et al. ADAM17-mediated CD44 cleavage promotes orasphere formation or stemness and tumorigenesis in HNSCC. Cancer Med. 2, 793–802 (2013).
Janisiewicz, A. M. et al. CD44(+) cells have cancer stem cell-like properties in nasopharyngeal carcinoma. Int. Forum Allergy Rhinol. 2, 465–470 (2012).
Lu, Y., Wang, W. & Tan, S. EHD1 promotes the cancer stem cell (CSC)-like traits of glioma cells via interacting with CD44 and suppressing CD44 degradation. Environ. Toxicol. 37, 2259–2268 (2022).
Wang, J. et al. EMP1 regulates cell proliferation, migration, and stemness in gliomas through PI3K-AKT signaling and CD44. J. Cell Biochem. 120, 17142–17150 (2019).
Zhang, M. et al. ITPR3 facilitates tumor growth, metastasis and stemness by inducing the NF-ĸB/CD44 pathway in urinary bladder carcinoma. J. Exp. Clin. Cancer Res. 40, 65 (2021).
Matsumoto, Y., Itou, J., Sato, F. & Toi, M. SALL4 - KHDRBS3 network enhances stemness by modulating CD44 splicing in basal-like breast cancer. Cancer Med. 7, 454–462 (2018).
Chen, Q. et al. TGF-β1 promotes epithelial-to-mesenchymal transition and stemness of prostate cancer cells by inducing PCBP1 degradation and alternative splicing of CD44. Cell Mol. Life Sci. 78, 949–962 (2021).
Jiang, Y. X. et al. Ascites-derived ALDH+CD44+ tumour cell subsets endow stemness, metastasis and metabolic switch via PDK4-mediated STAT3/AKT/NF-κB/IL-8 signalling in ovarian cancer. Br. J. Cancer 123, 275–287 (2020).
Park, J. et al. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front. Onco.l 11, 808300 (2021).
Wang, J. et al. A non-metabolic function of hexokinase 2 in small cell lung cancer: promotes cancer cell stemness by increasing USP11-mediated CD133 stability. Cancer Commun. 42, 1008–1027 (2022).
Liu, K. et al. Hypoxia-induced GLT8D1 promotes glioma stem cell maintenance by inhibiting CD133 degradation through N-linked glycosylation. Cell Death Differ. 29, 1834–1849 (2022).
Brescia, P. et al. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 31, 857–869 (2013).
Xu, W. W. et al. IGF2 induces CD133 expression in esophageal cancer cells to promote cancer stemness. Cancer Lett. 425, 88–100 (2018).
Wang, Y. et al. Aquaporin 3 maintains the stemness of CD133+ hepatocellular carcinoma cells by activating STAT3. Cell Death Dis. 10, 465 (2019).
Attia, S., Atwan, N., Arafa, M. & Shahin, R. A. Expression of CD133 as a cancer stem cell marker in invasive gastric carcinoma. Pathologica 111, 18–23 (2019).
Jamal, S. M. E. et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 39, 5468–5478 (2020).
Leung, C. O. N. et al. MicroRNA-135a-induced formation of CD133+ subpopulation with cancer stem cell properties in cervical cancer. Carcinogenesis 41, 1592–1604 (2020).
Liu, T. J. et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 32, 544–553 (2013).
Dang, S. C. et al. G-protein-signaling modulator 2 expression and role in a CD133(+) pancreatic cancer stem cell subset. OncoL. Targets Ther. 12, 785–794 (2019).
Kanwal, R., Shukla, S., Walker, E. & Gupta, S. Acquisition of tumorigenic potential and therapeutic resistance in CD133+ subpopulation of prostate cancer cells exhibiting stem-cell like characteristics. Cancer Lett. 430, 25–33 (2018).
Wang, R. et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl Acad. Sci. USA 115, E10127–e10136 (2018).
Levin, T. G. et al. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 139, 2072–2082.e2075 (2010).
Chen, X. et al. CD166 promotes cancer stem cell-like phenotype via the EGFR/ERK1/2 pathway in the nasopharyngeal carcinoma cell line CNE-2R. Life Sci. 267, 118983 (2021).
Qiu, X. et al. Characterization of sphere-forming cells with stem-like properties from the small cell lung cancer cell line H446. Cancer Lett. 323, 161–170 (2012).
Gopinath, S. et al. Cathepsin B and uPAR regulate self-renewal of glioma-initiating cells through GLI-regulated Sox2 and Bmi1 expression. Carcinogenesis 34, 550–559 (2013).
Asuthkar, S. et al. Urokinase-type plasminogen activator receptor (uPAR)-mediated regulation of WNT/β-catenin signaling is enhanced in irradiated medulloblastoma cells. J. Biol. Chem. 287, 20576–20589 (2012).
Shi, J. et al. CD90 highly expressed population harbors a stemness signature and creates an immunosuppressive niche in pancreatic cancer. Cancer Lett. 453, 158–169 (2019).
Buishand, F. O. et al. Identification of CD90 as putative cancer stem cell marker and therapeutic target in insulinomas. Stem Cells Dev. 25, 826–835 (2016).
Lobba, A. R., Forni, M. F., Carreira, A. C. & Sogayar, M. C. Differential expression of CD90 and CD14 stem cell markers in malignant breast cancer cell lines. Cytometry A 81, 1084–1091 (2012).
Zhang, K. et al. The SHH/Gli axis regulates CD90-mediated liver cancer stem cell function by activating the IL6/JAK2 pathway. J. Cell Mol. Med. 22, 3679–3690 (2018).
Leyton, L. et al. Thy-1/CD90 a Bidirectional and Lateral Signaling Scaffold. Front. Cell Dev. Biol. 7, 132 (2019).
Huang, Y. H. et al. EXOSC5 maintains cancer stem cell activity in endometrial cancer by regulating the NTN4/integrin β1 signalling axis. Int. J. Biol. Sci. 20, 265–279 (2024).
Barnawi, R. et al. β1 Integrin is essential for fascin-mediated breast cancer stem cell function and disease progression. Int. J. Cancer 145, 830–841 (2019).
Gardelli, C. et al. Differential glycosylation of collagen modulates lung cancer stem cell subsets through β1 integrin-mediated interactions. Cancer Sci. 112, 217–230 (2021).
Moon, J. H. et al. Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 96, 34–41 (2019).
Seguin, L. et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 16, 457–468 (2014).
Cheng, S. et al. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 20, 328–337 (2019).
Seyfrid, M. et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. J. Immunother. Cancer 10, e003289 (2022).
Liu, L. et al. Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer 25, 706–716 (2018).
Velázquez-Quesada, I. et al. Pranlukast Antagonizes CD49f and reduces stemness in triple-negative breast cancer cells. Drug Des Dev. Ther. 14, 1799–1811 (2020).
Vieira, A. F. et al. P-cadherin is coexpressed with CD44 and CD49f and mediates stem cell properties in basal-like breast cancer. Stem Cells 30, 854–864 (2012).
Herrmann, A. et al. Integrin α6 signaling induces STAT3-TET3-mediated hydroxymethylation of genes critical for maintenance of glioma stem cells. Oncogene 39, 2156–2169 (2020).
Kowalski-Chauvel, A. et al. Alpha6-Integrin Regulates FGFR1 Expression through the ZEB1/YAP1 Transcription Complex in Glioblastoma Stem Cells Resulting in Enhanced Proliferation and Stemness. Cancers 11, 406 (2019).
Fisher, M. L. et al. Transglutaminase Interaction with α6/β4-Integrin Stimulates YAP1-Dependent ΔNp63α Stabilization and Leads to enhanced cancer stem cell survival and tumor formation. Cancer Res. 76, 7265–7276 (2016).
Yu, J. M. et al. TRIB3 supports breast cancer stemness by suppressing FOXO1 degradation and enhancing SOX2 transcription. Nat. Commun. 10, 5720 (2019).
An, C. et al. LINC00662 enhances cell progression and stemness in breast cancer by MiR-144-3p/SOX2 axis. Cancer Cell Int. 22, 184 (2022).
Wang, X. et al. Stem Cell Factor SOX2 Confers Ferroptosis Resistance in Lung Cancer via Upregulation of SLC7A11. Cancer Res. 81, 5217–5229 (2021).
Huang, X. et al. A Self-Propagating c-Met-SOX2 Axis Drives Cancer-Derived IgG Signaling That Promotes Lung Cancer Cell Stemness. Cancer Res. 83, 1866–1882 (2023).
Jiang, X. et al. LncRNA GSCAR promotes glioma stem cell maintenance via stabilizing SOX2 expression. Int. J. Biol. Sci. 19, 1681–1697 (2023).
Lopez-Bertoni, H. et al. Sox2 induces glioblastoma cell stemness and tumor propagation by repressing TET2 and deregulating 5hmC and 5mC DNA modifications. Signal. Transduct Target Ther. 7, 37 (2022).
Zheng, Q. et al. Inhibiting NR5A2 targets stemness in pancreatic cancer by disrupting SOX2/MYC signaling and restoring chemosensitivity. J. Exp. Clin. Cancer Res. 42, 323 (2023).
Herreros-Villanueva, M. et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2, e61 (2013).
Zhao, N. et al. SOX2 maintains the stemness of retinoblastoma stem-like cells through Hippo/YAP signaling pathway. Exp. Eye Res. 214, 108887 (2022).
Praharaj, P. P. et al. CLU (clusterin) promotes mitophagic degradation of MSX2 through an AKT-DNM1L/Drp1 axis to maintain SOX2-mediated stemness in oral cancer stem cells. Autophagy 19, 2196–2216 (2023).
Yao, Z. et al. MTA3-SOX2 module regulates cancer stemness and contributes to clinical outcomes of tongue carcinoma. Front. Oncol. 9, 816 (2019).
Wang, Z. et al. AKT drives SOX2 overexpression and cancer cell stemness in esophageal cancer by protecting SOX2 from UBR5-mediated degradation. Oncogene 38, 5250–5264 (2019).
Forghanifard, M. M., Kasebi, P. & Abbaszadegan, M. R. SOX2/SALL4 stemness axis modulates Notch signaling genes to maintain self-renewal capacity of esophageal squamous cell carcinoma. Mol. Cell Biochem. 476, 921–929 (2021).
Guo, C. et al. Hypoxia increases RCC stem cell phenotype via altering the androgen receptor (AR)-lncTCFL5-2-YBX1-SOX2 signaling axis. Cell Biosci. 12, 185 (2022).
Gao, Z. et al. Exosomal lncRNA UCA1 modulates cervical cancer stem cell self-renewal and differentiation through microRNA-122-5p/SOX2 axis. J. Transl. Med. 19, 229 (2021).
Sarkar Bhattacharya, S. et al. PFKFB3 works on the FAK-STAT3-SOX2 axis to regulate the stemness in MPM. Br. J. Cancer 127, 1352–1364 (2022).
Yang, L. et al. Predictive Value of Stemness Factor Sox2 in Gastric Cancer Is Associated with Tumor Location and Stage. PLoS One 12, e0169124 (2017).
Ye, P. et al. Alanine-Glyoxylate Aminotransferase Sustains Cancer Stemness Properties through the Upregulation of SOX2 and OCT4 in Hepatocellular Carcinoma Cells. Biomolecules 12, 668 (2022).
Zhang, L. et al. KLF8 promotes cancer stem cell-like phenotypes in osteosarcoma through miR-429-SOX2 signaling. Neoplasma 67, 519–527 (2020).
Yang, Z. et al. LncRNA WAC-AS1 promotes osteosarcoma Metastasis and stemness by sponging miR-5047 to upregulate SOX2. Biol. Direct 18, 74 (2023).
Bareiss, P. M. et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 73, 5544–5555 (2013).
Long, W. et al. PHF20 collaborates with PARP1 to promote stemness and aggressiveness of neuroblastoma cells through activation of SOX2 and OCT4. J. Mol. Cell Biol. 10, 147–160 (2018).
Hui, K. et al. RASAL2, a RAS GTPase-activating protein, inhibits stemness and epithelial-mesenchymal transition via MAPK/SOX2 pathway in bladder cancer. Cell Death Dis. 8, e2600 (2017).
Zhan, Y. et al. Long non-coding RNA SOX2OT promotes the stemness phenotype of bladder cancer cells by modulating SOX2. Mol. Cancer 19, 25 (2020).
Kim, Y., Yeon, M. & Jeoung, D. DDX53 Regulates Cancer Stem Cell-Like Properties by Binding to SOX-2. Mol. Cells 40, 322–330 (2017).
Bhagat, M. et al. HIF-2α mediates a marked increase in migration and stemness characteristics in a subset of glioma cells under hypoxia by activating an Oct-4/Sox-2-Mena (INV) axis. Int. J. Biochem. Cell Biol. 74, 60–71 (2016).
Cheng, C. C. et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 53, 339–348 (2018).
Reers, S. et al. Stem cell profiling in head and neck cancer reveals an Oct-4 expressing subpopulation with properties of chemoresistance. Oral Oncol. 50, 155–162 (2014).
Li, L. et al. siRNA-mediated knockdown of ID1 disrupts Nanog- and Oct-4-mediated cancer stem cell-likeness and resistance to chemotherapy in gastric cancer cells. Oncol. Lett. 13, 3014–3024 (2017).
Liu, X. et al. Niche stiffness sustains cancer stemness via TAZ and NANOG phase separation. Nat. Commun. 14, 238 (2023).
Zhang, C. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. USA 113, E2047–2056 (2016).
Tong, X. et al. Nanog maintains stemness of Lkb1-deficient lung adenocarcinoma and prevents gastric differentiation. EMBO Mol. Med. 13, e12627 (2021).
Ye, T. et al. Nr5a2 promotes cancer stem cell properties and tumorigenesis in nonsmall cell lung cancer by regulating Nanog. Cancer Med. 8, 1232–1245 (2019).
Chen, J. et al. ABI2-mediated MEOX2/KLF4-NANOG axis promotes liver cancer stem cell and drives tumour recurrence. Liver Int. 42, 2562–2576 (2022).
Chen, W. et al. CD44v6+ hepatocellular carcinoma cells maintain stemness properties through Met/cJun/Nanog Signaling. Stem Cells Int. 2022, 5853707 (2022).
Kang, K. T. et al. TRRAP enhances cancer stem cell characteristics by regulating NANOG protein stability in colon cancer cells. Int. J. Mol. Sci. 24, 6260 (2023).
Zhang, J. et al. NANOG modulates stemness in human colorectal cancer. Oncogene 32, 4397–4405 (2013).
Siu, M. K. Y. et al. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers 11, 813 (2019).
Sun, X. et al. Nanog-mediated stem cell properties are critical for MBNL3-associated paclitaxel resistance of ovarian cancer. J. Biochem. 169, 747–756 (2021).
Deng, L. et al. NANOG promotes cell proliferation, invasion, and stemness via IL-6/STAT3 signaling in esophageal squamous carcinoma. Technol. Cancer Res. Treat. 20, 15330338211038492 (2021).
Yong, X. et al. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 374, 292–303 (2016).
Wang, X. et al. AMPK promotes SPOP-Mediated NANOG degradation to regulate prostate cancer cell stemness. Dev. Cell 48, 345–360.e347 (2019).
Zhang, T. et al. Nanog mediates tobacco smoke-induced enhancement of renal cancer stem cell properties. Environ. Toxicol. 35, 1274–1283 (2020).
Zhao, M. et al. A direct negative feedback loop of miR-4721/FOXA1/Nanog promotes nasopharyngeal cell stem cell enrichment and metastasis. J. Transl. Med. 19, 387 (2021).
Huang, W. et al. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics 9, 5497–5516 (2019).
Ding, Y. et al. Forced expression of Nanog with mRNA synthesized in vitro to evaluate the malignancy of HeLa cells through acquiring cancer stem cell phenotypes. Oncol. Rep. 35, 2643–2650 (2016).
Oikawa, T. et al. Sal-like protein 4 (SALL4), a stem cell biomarker in liver cancers. Hepatology 57, 1469–1483 (2013).
Zhao, B. et al. Inflammatory micro-environment contributes to stemness properties and metastatic potential of HCC via the NF-κB/miR-497/SALL4 Axis. Mol. Ther. Oncolytics 15, 79–90 (2019).
Diener, J. et al. Epigenetic control of melanoma cell invasiveness by the stem cell factor SALL4. Nat. Commun. 12, 5056 (2021).
Peng, Z. et al. miR-497-5p/SALL4 axis promotes stemness phenotype of choriocarcinoma and forms a feedback loop with DNMT-mediated epigenetic regulation. Cell Death Dis. 12, 1046 (2021).
Sharbatoghli, M. et al. Co-expression of cancer stem cell markers, SALL4/ALDH1A1, is associated with tumor aggressiveness and poor survival in patients with serous ovarian carcinoma. J. Ovarian Res. 15, 17 (2022).
Zhang, J. et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat. Cell Biol. 8, 1114–1123 (2006).
Zhou, F. et al. Aldehyde dehydrogenase 1: a specific cancer stem cell marker for human colorectal carcinoma. Mol. Med. Rep. 11, 3894–3899 (2015).
Zou, B., Sun, S., Qi, X. & Ji, P. Aldehyde dehydrogenase activity is a cancer stem cell marker of tongue squamous cell carcinoma. Mol. Med. Rep. 5, 1116–1120 (2012).
Jiang, F. et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 7, 330–338 (2009).
Rasper, M. et al. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro. Oncol. 12, 1024–1033 (2010).
Mori, Y. et al. ALDH-dependent glycolytic activation mediates stemness and paclitaxel resistance in patient-derived spheroid models of uterine endometrial cancer. Stem Cell Rep. 13, 730–746 (2019).
Hartomo, T. B. et al. Involvement of aldehyde dehydrogenase 1A2 in the regulation of cancer stem cell properties in neuroblastoma. Int. J. Oncol. 46, 1089–1098 (2015).
Ueda, K. et al. Aldehyde dehydrogenase 1 identifies cells with cancer stem cell-like properties in a human renal cell carcinoma cell line. PLoS One 8, e75463 (2013).
Sun, S. & Wang, Z. ALDH high adenoid cystic carcinoma cells display cancer stem cell properties and are responsible for mediating metastasis. Biochem. Biophys. Res. Commun. 396, 843–848 (2010).
Kim, R. J. et al. High aldehyde dehydrogenase activity enhances stem cell features in breast cancer cells by activating hypoxia-inducible factor-2α. Cancer Lett. 333, 18–31 (2013).
Shuang, Z. Y. et al. Transforming growth factor-β1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 354, 320–328 (2014).
Yarmishyn, A. A. et al. Musashi-1 promotes cancer stem cell properties of glioblastoma cells via upregulation of YTHDF1. Cancer Cell Int 20, 597 (2020).
Wang, X. Y. et al. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol. Cancer 9, 221 (2010).
Chiou, G. Y. et al. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 7, 2172 (2017).
Qin, G. et al. Musashi1, a potential prognostic marker in esophageal squamous cell carcinoma. Oncol. Rep. 38, 1724–1732 (2017).
Kudinov, A. E. et al. Musashi-2 (MSI2) supports TGF-β signaling and inhibits claudins to promote non-small cell lung cancer (NSCLC) metastasis. Proc. Natl Acad. Sci. USA 113, 6955–6960 (2016).
Wang, X. et al. Bmi-1 regulates stem cell-like properties of gastric cancer cells via modulating miRNAs. J. Hematol. Oncol. 9, 90 (2016).
Zhu, M. et al. BMI1 silencing liposomes suppress postradiotherapy cancer stemness against radioresistant hepatocellular carcinoma. ACS Nano 17, 23405–23421 (2023).
Kim, M. et al. Silencing Bmi1 expression suppresses cancer stemness and enhances chemosensitivity in endometrial cancer cells. Biomed. Pharmacother. 108, 584–589 (2018).
Ni, Y. L. et al. Disulfiram/Copper suppresses cancer stem cell activity in differentiated thyroid cancer cells by inhibiting BMI1 expression. Int. J. Mol. Sci. 23, 13276 (2022).
Lukacs, R. U., Memarzadeh, S., Wu, H. & Witte, O. N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 7, 682–693 (2010).
Lei, Y. et al. Hairy gene homolog increases nasopharyngeal carcinoma cell stemness by upregulating Bmi-1. Aging 15, 4391–4410 (2023).
Chen, J. et al. Fas signaling induces stemness properties in colorectal cancer by regulation of Bmi1. Mol. Carcinog. 56, 2267–2278 (2017).
Qiu, G. Z. et al. Hypoxia-induced USP22-BMI1 axis promotes the stemness and malignancy of glioma stem cells via regulation of HIF-1α. Life Sci. 247, 117438 (2020).
Proctor, E. et al. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PLoS One 8, e55820 (2013).
Zhao, Y. et al. The role of BMI1 in endometrial cancer and other cancers. Gene 856, 147129 (2023).
Cox, C. V. et al. Expression of CD133 on leukemia-initiating cells in childhood ALL. Blood 113, 3287–3296 (2009).
Heo, S. K. et al. CD45(dim)CD34(+)CD38(-)CD133(+) cells have the potential as leukemic stem cells in acute myeloid leukemia. BMC Cancer 20, 285 (2020).
Herrmann, H. et al. CD34(+)/CD38(-) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 97, 219–226 (2012).
Zeijlemaker, W. et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 33, 1102–1112 (2019).
Guedes, A. G. et al. Role of CD38/cADPR signaling in obstructive pulmonary diseases. Curr. Opin. Pharmacol. 51, 29–33 (2020).
Herrmann, H. et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 123, 3951–3962 (2014).
Enz, N., Vliegen, G., De Meester, I. & Jungraithmayr, W. CD26/DPP4 - a potential biomarker and target for cancer therapy. Pharmacol. Ther. 198, 135–159 (2019).
Landberg, N. et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica 103, 447–455 (2018).
van Rhenen, A. et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 110, 2659–2666 (2007).
Kikushige, Y. et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 7, 708–717 (2010).
Cai, B. et al. A truncated derivative of FGFR1 kinase cooperates with FLT3 and KIT to transform hematopoietic stem cells in syndromic and de novo AML. Mol. Cancer 21, 156 (2022).
Das, B. et al. MYC Regulates the HIF2α Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 79, 4015–4025 (2019).
Anorma, C. et al. Surveillance of cancer stem cell plasticity using an isoform-selective fluorescent probe for aldehyde dehydrogenase 1A1. ACS Cent. Sci. 4, 1045–1055 (2018).
Gerber, J. M. et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood 119, 3571–3577 (2012).
Jin, N., Zhu, X., Cheng, F. & Zhang, L. Disulfiram/copper targets stem cell-like ALDH(+) population of multiple myeloma by inhibition of ALDH1A1 and Hedgehog pathway. J. Cell. Biochem. 119, 6882–6893 (2018).
Xu, D. D. et al. The IGF2/IGF1R/Nanog signaling pathway regulates the proliferation of acute myeloid leukemia stem cells. Front. Pharmacol. 9, 687 (2018).
Picot, T. et al. Potential role of OCT4 in Leukemogenesis. Stem Cells Dev. 26, 1637–1647 (2017).
Yang, L. et al. The stem cell factor SALL4 is an essential transcriptional regulator in mixed lineage leukemia-rearranged leukemogenesis. J. Hematol. Oncol. 10, 159 (2017).
Lu, J. et al. Dissecting the role of SALL4, a newly identified stem cell factor, in chronic myelogenous leukemia. Leukemia 25, 1211–1213 (2011).
Mariani, S. A. et al. CDKN2A-independent role of BMI1 in promoting growth and survival of Ph+ acute lymphoblastic leukemia. Leukemia 30, 1682–1690 (2016).
Galimberti, S. et al. The Polycomb BMI1 Protein Is Co-expressed With CD26+ in Leukemic Stem Cells of Chronic Myeloid Leukemia. Front. Oncol. 8, 555 (2018).
Zhang, Y. et al. miR-203 inhibits proliferation and self-renewal of leukemia stem cells by targeting survivin and Bmi-1. Sci. Rep. 6, 19995 (2016).
Acknowledgements
This work was supported by Advanced Lung Cancer Targeted Therapy Research Foundation of China (CTONG-YC20210303), Chen Xiao-Ping Foundation for the Development of Science and Technology of Hubei Province(CXPJJH121005-01), National Multidisciplinary Cooperative Diagnosis and Treatment Capacity (lung cancer z027002), Health Research Project of Hunan Provincial Health Commission (W20242005) and Postdoctoral Fellowship Program of CPSF (GZC20233187).
Author information
Authors and Affiliations
Contributions
X.C., W.T. and JN reviewed the literatures, developed the concept, and wrote the manuscript. G.T., G.X. and Y.Z. were responsible for discussing the manuscript and making critical revisions to the logic and grammar. Z.W., Z.Z. drafted and polished the figures and tables. J.Y. and R.Z. provided financial support. X.C., W.T. and J.N. contributed equally to the first author while J.Y., G.T. and RZ were equated to the corresponding author. All authors have reviewed and approved the publication of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The author declares no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chu, X., Tian, W., Ning, J. et al. Cancer stem cells: advances in knowledge and implications for cancer therapy. Sig Transduct Target Ther 9, 170 (2024). https://doi.org/10.1038/s41392-024-01851-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01851-y
- Springer Nature Limited