
Abstract
Background
Recent mitochondrial phylogenomics studies have reported a sister-group relationship of the orders Capsalidea and Dactylogyridea, which is inconsistent with previous morphology- and molecular-based phylogenies. As Dactylogyridea mitochondrial genomes (mitogenomes) are currently represented by only one family, to improve the phylogenetic resolution, we sequenced and characterized two dactylogyridean parasites, Lamellodiscus spari and Lepidotrema longipenis, belonging to a non-represented family Diplectanidae.
Results
The L. longipenis mitogenome (15,433 bp) contains the standard 36 flatworm mitochondrial genes (atp8 is absent), whereas we failed to detect trnS1, trnC and trnG in L. spari (14,614 bp). Both mitogenomes exhibit unique gene orders (among the Monogenea), with a number of tRNA rearrangements. Both long non-coding regions contain a number of different (partially overlapping) repeat sequences. Intriguingly, these include putative tRNA pseudogenes in a tandem array (17 trnV pseudogenes in L. longipenis, 13 trnY pseudogenes in L. spari). Combined nucleotide diversity, non-synonymous/synonymous substitutions ratio and average sequence identity analyses consistently showed that nad2, nad5 and nad4 were the most variable PCGs, whereas cox1, cox2 and cytb were the most conserved. Phylogenomic analysis showed that the newly sequenced species of the family Diplectanidae formed a sister-group with the Dactylogyridae + Capsalidae clade. Thus Dactylogyridea (represented by the Diplectanidae and Dactylogyridae) was rendered paraphyletic (with high statistical support) by the nested Capsalidea (represented by the Capsalidae) clade.
Conclusions
Our results show that nad2, nad5 and nad4 (fast-evolving) would be better candidates than cox1 (slow-evolving) for species identification and population genetics studies in the Diplectanidae. The unique gene order pattern further suggests discontinuous evolution of mitogenomic gene order arrangement in the Class Monogenea. This first report of paraphyly of the Dactylogyridea highlights the need to generate more molecular data for monogenean parasites, in order to be able to clarify their relationships using large datasets, as single-gene markers appear to provide a phylogenetic resolution which is too low for the task.
Similar content being viewed by others


Background
Monogeneans of the family Diplectanidae (Dactylogyridea: Dactylogyrinea) are parasites found on the gills of (mostly) marine perciform fishes [1]. The family comprises approximately 250 species and is mainly studied for the adverse health effects these parasites cause to the hosts; the fixation of their opisthaptors on the gills causes haemorrhages and a white mucoid exudate, which often leads to secondary fungal, bacterial and/or viral infections [2, 3]. An example is Diplectanum aequans (Wagener, 1857), which can cause high mortality of juvenile European sea bass in the Mediterranean aquaculture [2].
Traditionally used phylogenetic markers, morphology and single-genes, are often not suitable for resolving evolutionary history with high confidence; morphological traits can be homoplastic, which often causes taxonomic and phylogenetic artifacts [4,5,6] and, due to the small amount of information (phylogenetic signal) they carry, single-gene molecular markers may have limited resolving power [7]. This is reflected in the unresolved phylogeny of Monogenean parasites [8,9,10,11]. Specifically, studies based on spermatozoal ultrastructural characters [12, 13], the 18S rRNA gene [9,10,11], and a combination of three unlinked nuclear genes [5], supported a phylogenetically closer relationship between monogenean orders Capsalidea and Gyrodactylidea than (either of the two) to the Dactylogyridea. However, 66 homologous series of morphological characters resolved the Gyrodactylidea and Dactylogyridea as sister groups [14]. Therefore, molecular markers carrying more powerful phylogenetic signals are needed to resolve their phylogenetic relationships with high resolution. Mitogenome is a good candidate marker, with an approximately ten times larger nucleotide alignment length than commonly used single-gene molecular markers (ITS, 18S and 28S rRNA). Although their applicability for studies of the Neodermata is still hampered by their relative scarcity, they are increasingly used in population genetics [15], phylogenetics [16, 17] and diagnostics [7, 18] of parasitic flatworms, despite this limitation. Intriguingly, recent researches [4, 19, 20] relying on the mitochondrial (mt) phylogenomics approach consistently resolved the Dactylogyridea and Capsalidea as sister-groups, thereby further complicating phylogenetic hypotheses for the three aforementioned orders.
As the resolution power of mitochondrial genomics is still limited by the very low number of sequenced monogenean mitogenomes available, where many taxonomic categories remain poorly represented or unrepresented (only one dactylogyridean family represented), we sequenced and characterized two complete mitochondrial genomes belonging to a non-represented dactylogyridean family, the Diplectanidae: Lamellodiscus spari (Zhukov, 1970) and Lepidotrema longipenis (Yamaguti, 1934), collected from the gills of two marine fish species. Their availability shall enable us to employ mitochondrial phylogenomics to investigate relationships of these three orders with improved resolution.
Methods
Specimen collection and identification
According to the records in Zhang et al. [21], we searched for diplectanid parasites by exploring fish markets in several coastal cities in the southern China. Lepidotrema longipenis was obtained from Terapon jarbua (Forsskål, 1775) (Perciformes: Terapontidae) bought at a local market in Zhanjiang city, Guangdong Province (21°15'5"-21°15'16"N, 110°23'46"-110°24'12"E), on the 18th June 2016. Lamellodiscus spari was obtained from the black sea bream Acanthopagrus schlegelii (Bleeker, 1854) (Perciformes: Sparidae) caught by fishermen in Daya Bay, Guangdong Province (22°42'58"-22°42'56"N, 114°32'16"-114°32'25"E) on the 10th July 2017. Discriminative morphological characteristics for the Diplectanidae are their opisthaptor equipped by three transversal bars connected to two pairs of central hooks, 14 marginal hooks and accessory adhesive organ (lamellodisc or squamodisc) that can be present or absent [22, 23]. Parasites were further morphologically identified to the genus level as described in Domingues & Boeger [23], and to the species level under a light microscope according to the traits described in Ogawa & Egusa [24] for L. spari, and the traits described in Zhang et al. [21] for L. longipenis. Additionally, to confirm the taxonomic identity, the 28S rRNA gene was amplified using universal primers [25] (Additional file 1: Dataset S1); both species share a very high identity of 99.6% with corresponding conspecific homologs available in GenBank: 744/747 identical bp for L. longipenis (EF100563), and (837/840) for L. spari (DQ054823). All sampled and identified parasites were first washed in 0.6% saline and then stored in 100% ethanol at 4 °C.
DNA extraction, amplification and sequencing
Due to the small size of these parasites, we used two kinds of genomic DNA to ensure a sufficient amount for amplification and sequencing, both extracted using a TIANamp MicroDNA Kit (Tiangen Biotech, Beijing, China): mixture DNA (20 parasite specimens) and individual DNA (a single parasite specimen). Mixture DNA was first used to amplify the whole mitogenome. First, we selected 14 monogenean mitogenomes from GenBank, aligned them using ClustalX [26], and designed degenerate primer pairs (Additional file 1: Dataset S1) matching the generally conserved regions of mitochondrial genes (16S, 12S, cox1, cox2, nad1, nad4 and cytb). On the basis of these obtained fragments, specific primers were then designed using Primer Premier 5 [27], and the remaining mitogenome was amplified and sequenced in several PCR steps (Additional file 1: Dataset S1). Both mitogenomes were amplified exactly following the procedures previously described [4, 17, 19, 28]; detailed PCR conditions are provided in Additional file 1: Dataset S1. PCR products were sequenced bi-directionally using both degenerate and specific primers mentioned above on an ABI 3730 automatic sequencer (Sangon, Shanghai, China) using the Sanger method. During the sequencing we paid close attention to chromatograms, carefully examining them for double peaks, or any other sign of the existence of two different sequences. A BLAST [29] check was used to confirm that all amplicons are the actual target sequences. To address the possibility of intraspecific sequence variation present in the mixture DNA, we then used individual DNA and long-range PCR to verify the obtained sequences (primers used are listed in Additional file 1: Dataset S1). If we found two different sequences, we used the DNA extracted from a single individual to infer the mitogenomic sequence, thereby ensuring that each sequence belongs to a single specimen.
Sequence annotation and analyses
Both mitogenomes were assembled and annotated following a previously described procedure [4, 17, 19, 28] using DNAstar v.7.1 software [30], MITOS [31], ARWEN [32] and DOGMA [33] web tools, so detailed methodology is provided in Additional file 1: Dataset S1. Codon usage and relative synonymous codon usage (RSCU) for 12 protein-encoding genes (PCGs) of the two studied diplectanids, two dactylogyrids (Dactylogyrus lamellatus Achmerov, 1952 and Tetrancistrum nebulosi Young, 1967) and three capsalids (Neobenedenia melleni MacCallum, 1927, Benedenia seriolae Yamaguti, 1934 and B. hoshinai Ogawa, 1984) were computed and sorted using MitoTool [34] (an in-house GUI-based software), and finally the RSCU figure drawn using the ggplot2 [35] plugin. Non-synonymous (dN) / synonymous (dS) mutation rate ratios among the 12 PCGs of the two studied diplectanid mitogenomes were calculated with DnaSP v.5 [36]. The same software was also employed to conduct the sliding window analysis: a sliding window of 200 bp and a step size of 20 bp were implemented to estimate the nucleotide divergence Pi between the mitogenomes of L. longipenis and L. spari. Tandem Repeats Finder [37] was employed to find tandem repeats in the long non-coding regions (LNCR), and their secondary structures were predicted by Mfold software [38]. Rearrangement events in the mitogenomes and pairwise comparisons of gene orders of all 20 available monogeneans were calculated with the CREx program [39] using the common intervals measurement. Due to limitations of the CREx algorithm, and to facilitate comparative analyses of gene orders, we provisionally added the gene block trnS1-trnC-trnG to the gene order sequence of L. spari, corresponding to the position where these genes were found in L. longipenis (between nad5 and cox3). Genetic distances (identity) among mitogenomic sequences were calculated with the “DistanceCalculator” function in Biopython using the “identity” model.
Phylogenetic analyses
Phylogenetic analyses were conducted using the two newly sequenced diplectanid mitogenomes and all 18 monogenean mitogenomes available in GenBank (5th May 2018). Two species of the order Tricladida, Crenobia alpina (Dana, 1766) (KP208776) and Obama sp. (NC_026978), were used as outgroups, thus making a total of 22 mitogenomes (Additional file 2: Table S1). Two datasets were used for phylogenetic analysis: amino acid alignment of 12 protein-coding genes (PCGAA) and codon-based alignment of nucleotide sequences of 12 protein-coding genes + secondary structure alignment of 22 tRNAs and 2 rRNAs (PCGRT). As data processing was conducted as previously described [4, 17, 19, 28, 40], using MitoTool, MAFFT [41] and Gblocks [42]; details are given in Additional file 1: Dataset S1. The heterogeneity of sequence divergence within data sets was analyzed using AliGROOVE [43], wherein indels in nucleotide dataset were treated as ambiguity, and a BLOSUM62 matrix was used for amino acids. Best partitioning scheme and evolutionary models were selected using PartitionFinder2 [44], with greedy algorithm and AICc criterion. Phylogenetic analyses were conducted using two different algorithms: maximum likelihood (ML) and Bayesian inference (BI). Based on the Akaike information criterion implemented in ProtTest [45], MTART+I+G+F was chosen as the optimal evolutionary model for the downstream phylogenetic analyses. Under this optimal model and partition model, ML analysis was conducted in RAxML [46] using a ML+rapid bootstrap (BS) algorithm with 1000 replicates. Bayesian inference analyses with the empirical MTART model were conducted using PhyloBayes (PB) MPI 1.5a [47]. For each analysis, two MCMC chains were run after the removal of invariable sites from the alignment, and the analysis was stopped when the conditions considered to indicate a good run (according to the PhyloBayes manual) were reached: maxdiff < 0.1 and minimum effective size > 300. Phylograms and gene orders were visualized and annotated by iTOL [48] with the help of several dataset files generated by MitoTool, as described in our recent papers [4, 17].
Results and discussion
Genome organization and base composition
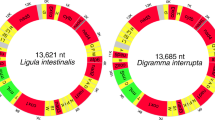
The full circular mitochondrial genome of L. longipenis (GenBank: MH328203), at 15,433 bp, is the longest among the monopisthocotylids characterized so far (Additional file 2: Table S1). The mitogenome of L. spari is 14,614 bp in size (GenBank: MH328204). The L. longipenis mitogenome contains the standard [49] 36 flatworm mitochondrial genes, including 12 protein-encoding genes (PCGs; atp8 is absent), 22 tRNA genes, and two rRNA genes, whereas trnS1, trnC and trnG genes are missing in L. spari (Table 1 and Fig. 1). The architecture, gene contents and similarity of orthologous sequences for the two studied mitogenomes are summarized in Table 1. Average sequence similarity of PCGs between the two studied mitogenomes ranged from 55.58 (nad5) to 79.45% (cox1) (Table 1). In comparison to PCGs, average sequence similarity values between the rRNAs of the two species were higher: 75.43% for rrnL and 70.87% for rrnS.

Visual representation of the circular mitochondrial genomes of Lepidotrema longipenis and Lamellodiscus spari. Key: Red, protein-coding genes; yellow, tRNAs; green, rRNAs; grey, non-coding regions
Protein-coding genes and codon usage
Eleven out of 12 PCGs of the two studied mitogenomes used ATG or GTG as the initial codons. However, it proved difficult to determine the initial codon of the nad2 gene in both species. On the basis of results reported for other related species, as a working hypothesis we proposed TTG and ATT as the initial codon of nad2 for L. longipenis and L. spari, respectively. Similarly, ATT was proposed as the start codon for nad2 in B. hoshinai (Ogawa, 1984) and Aglaiogyrodactylus forficulatus (Kritsky, Vianna & Boeger, 2007) [20, 50], and TTG as the start codon for cox2 in Paragyrodactylus variegatus (You, King, Ye & Cone, 2014) [51] (also see Additional file 3: Table S2). Canonical stop codons for the genetic code 9 (echinoderm and flatworm mitochondrion), TAA and TAG, were found in all 12 PCGs (Table 1 and Additional file 3: Table S2). Codon usage, RSCU, and codon family (corresponding to the amino acids) proportions were investigated among the seven available capsalids (three species) and dactylogyrids (four species) (Additional file 4: Figure S1). The third codon position exhibited the highest A+T bias (Table 2). Amino acids encoded by adenosine and thymine-rich codon families (such as Phe, Leu2 and Ile) were strongly preferred, whereas amino acids encoded by guanine and cytosine-rich codon families (such as Arg, Pro and Ala) appear to be selected against (Additional file 4: Figure S1).
Loss of trnS1, trnC and trnG from the L. spari mitogenome
Both ARWEN and MITOS algorithms failed to detect trnS1, trnC and trnG in the L. spari mitogenome. We made several attempts to corroborate that this is not an artifact. First, we carefully checked (via alignments with monogenean trnS1, trnC and trnG homologs) all intergenic sequences (including the LNCR). As these missing tRNA genes are located between nad5 and LNCR in L. longipenis (Table 1, Fig. 2), and as trnC is located between two rRNA genes in many other monogeneans (Fig. 2), we focused specifically on these two fragments. None of these sequences showed appreciable similarity with the queried homologs. Secondly, we re-sequenced the fragment between nad5 and cox3 using both mixture DNA and individual DNA, and checked the chromatograms carefully [52]. We did not find any evidence for sequence variability. Thirdly, referring to L. longipenis and other closely related monogeneans, as well as cestodes and trematodes in most of which trnG is located between nad5 and cox3 (Fig. 2, Additional file 5: Figure S2), we designed two primer pairs (Additional file 1: Dataset S1) to assess whether trnG is located between these two genes (or in their vicinity) in L. spari: (i) one forward primer (LS-GlyF) matching the most conserved region of trnG and two reverse primers (LSR1-6 and LSR1-8) matching the conserved regions of cox3 to amplify the fragment between trnG and cox3; and (ii) two forward primers (LSF17-0 and LSF17) matching nad5 and one reverse primer (LS-GlyR) matching trnG to amplify the fragment between nad5 and trnG. None of these primer pairs could generate a PCR product, which indicates that trnG is either not in the vicinity of these two genes in L. spari, or that its sequence is highly divergent, or that it is completely missing from the mitogenome. On the basis of these tests and high quality of chromatograms of the fragment between nad5 and cox3 [52], we suspect that all three tRNAs (trnS1-trnC-trnG) might be missing from the mitogenome of L. spari. These results would have to be corroborated either by resequencing of this mitogenome, or by sequencing of other closely related species. Loss of tRNA genes was also reported in many other metazoan taxa [53,54,55,56,57]. Given that the amino acid usage frequency of serine (AGN), glycine and cysteine is analogous between L. spari and other monogeneans (Additional file 4: Figure S1), there are at least three possible explanations for the missing tRNAs: (i) they are imported from the nucleus, as is common in mitochondria [58]; (ii) they are encoded in the mitogenome, but undergo extensive post-transcriptional RNA editing [54], so they could not be identified from their coding sequences; and (iii) they are encoded on a separate minicircle of mtDNA.

Phylogeny and mitogenomic architecture of the class Monogenea. The phylogram was constructed using MTART model on the basis of concatenated amino acid sequences of 20 monogenean mitogenomes. Crenobia alpina and Obama sp. are outgroups. The scale-bar corresponds to the estimated number of substitutions per site. Statistical support values are shown above the nodes, except for nodes with maximum support. Monogenean families are shown in different colors. Gene order is displayed to the right of the tree
Non-coding regions
A putative control region, or long non-coding region (LNCR), was found between nad5 and cox3 genes in both mitogenomes (disregarding the three missing tRNAs; Table 1 and Fig. 1). The LNCR of L. longipenis (1993 bp) was the longest among the monogeneans characterized so far [4, 7, 16, 19, 20, 50, 51, 59,60,61,62,63,64,65,66,67]. The A+T content of both LNCRs (L. longipenis = 93.8%, L. spari = 92.5%) was much higher than in other parts of the mitogenomes (Table 2). Both LNCRs contained a highly repetitive region (HRR): the HRR of L. longipenis was composed of 18 tandem repeats (TRs), where repeat units 2–18 were identical (91 bp) and unit 1 was 1 bp longer with a nucleotide insertion at the 16th position; the HRR of L. spari contained 20 TRs with the consensus size of 87 bp, but sizes and sequences of the repeat units were variable, exhibiting nucleotide mutations, deletions and insertions. This is the second report of TRs with high repeat numbers and large size in the subclass Monopisthocotylea; the first report was also in a dactylogyridean species, D. lamellatus [19]. These findings consistently reject the hypothesis that monopisthocotylids possess fewer and smaller (in size) TRs in the LNCR than polyopisthocotylids [61]. As in other monogeneans [19, 64] and cestodes [68, 69], both consensus repeat patterns of the HRRs in L. longipenis and L. spari are capable of forming stem-loop structures (Additional file 6: Figure S3). Since the presence of tandem repeats forming stable secondary structure is often associated with replication origin in mitochondria [64, 70, 71], it appears likely that these repeat regions are embedded within the control region.
Aside from these TRs, the LNCR of L. longipenis also harbored 17 identical trnV pseudogenes, whereas the LNCR of L. spari contained 13 trnY pseudogenes (identified using ARWEN and DOGMA algorithms), all of which were located on the minus strand. These pseudogenes and HRR repeat patterns were two separate features, although the pseudogenes partially overlapped with the repeat patterns of HRR. Among the 13 trnY pseudogenes, six were 80 bases-long, and seven were 82 bases-long, with two bases inserted in the TΨC stem. trnV pseudogene had a standard TAC anticodon, whereas trnY contained modified standard anticodon (ATA). The cloverleaf structures of the two pseudogenes and the alignment with the corresponding functional monogenean tRNA homologs is shown in Fig. 3. Average sequence similarity values of the alignment for trnV and trnY were 40.78 ± 3.87% and 39.91 ± 4.13%, respectively. As the amino acid usage frequencies of valine and tyrosine for L. longipenis and L. spari were analogous with other monogeneans (Additional file 4: Figure S1), we hypothesise that the presence of these pseudogenes is non-adaptive, and that they may not be functional. A tRNA pseudogene was also found in the LNCR of Paratetraonchoides inermis (Bychowsky, Gussev & Nagibina, 1965) [4], but such a large accumulation of tRNA pseudogenes in tandem arrays is much more common in plastid genomes [72] and prokaryotes [73] than in mitochondrial genomes of metazoans. It might be of interest to sequence mitogenomes of other closely related species to infer whether they also harbor this feature, and study its evolutionary history and mutational rate.

Alignment of trnV and trnY pseudogenes with the corresponding functional monogenean tRNA homologs. Cloverleaf structures of the two pseudogenes are shown on the right of alignment. Two red bases in trnY indicate nucleotide insertions in the TΨC stem
Nucleotide diversity and evolutionary rate analysis
The sliding window analysis was conducted using concatenated alignments of 12 PCGs, two rRNAs and 19 coalescent tRNAs of the two diplectanids (trnS1, trnC and trnG were removed due to their absence from L. spari). The plot of sequence variation ratio exhibited highly variable nucleotide diversity between the two diplectanids, with Pi values for the 200 bp windows ranging from 0.201 to 0.411 (Fig. 4a). cox1 (0.201), tRNAs (0.215), rrnS (0.221), rrnL (0.224) and cytb (0.251) exhibited a comparatively low sequence variability, whereas nad2 (0.411), nad5 (0.392), nad4 (0.381) and nad6 (0.354) had a comparatively high sequence variability. This was corroborated by the non-synonymous/synonymous (dN/dS) ratio (omega) analysis, which showed that cox1 (0.163), cytb (0.167), nad4L (0.213) and cox2 (0.229) are evolving comparatively slowly, whereas nad2 (0.69), nad5 (0.49), nad6 (0.462) and nad4 (0.451) are evolving comparatively fast (Fig. 4b). Therefore, these analyses consistently indicate that cox1, which is often used as a universal barcode for species identification [74], as well as population genetics in monogeneans [75,76,77,78], is the slowest evolving and least variable gene. As rapidly evolving genes are more suitable for analyzing relationships among closely related species [79], we propose that the fast-evolving nad2, nad5, nad6 and nad4 would be better molecular markers than cox1 for diplectanids.

Sliding window and selection pressure analyses of the mitogenomes of Lepidotrema longipenis and Lamellodiscus spari. a Sliding window analysis was conducted on concatenated alignments of 12 PCGs, 2 rRNAs and 19 coalescent tRNAs (missing tRNAs, trnS1, trnC and trnG, were removed). The black line represents the value of nucleotide diversity (window size = 200 bp, step size = 20 bp, with the value inserted at its mid-point). Gene names, boundaries/direction (colored arrows) and average nucleotide diversity values are indicated above the graph. b Ratios of non-synonymous (dN) to synonymous (dS) substitution rates calculated for protein-coding genes
Phylogeny
Regardless of the dataset and model used, all analyses produced phylograms with concordant branch topologies (Fig. 2 and Additional file 7: Figure S4 and S5). As the AliGROOVE analysis indicated that the PCGAA dataset exhibits lower heterogeneity than PCGRT (Additional file 7: Figure S6), we displayed only the results of PCGAA in Fig. 2. As expected, the two diplectanids, L. longipenis and L. spari, constituted a monophyletic group with maximum support. In accordance with previous results based on mitochondrial phylogenomics [4, 19, 20], the two dactylogyrids (D. lamellatus and T. nebulosi; Dactylogyridea: Dactylogyridae) formed a sister-group with the three capsalids (N. melleni, B. hoshinai and B. seriolae; Capsalidea: Capsalidae). The two newly-sequenced diplectanids (Diplectanidae) formed a sister-group with this (Dactylogyridae + Capsalidae) clade. As the family Diplectanidae was classified into the order Dactylogyridea in the classifications proposed by Bychowsky [80], Lebedev [81] and Boeger & Kritsky [82], this topology rendered Dactylogyridea paraphyletic by the nested Capsalidea clade. In these taxonomic systems, the Dactylogyridae and Diplectanidae were assigned to the same order Dactylogyridea (or suborder Dactylogyrinea within the order Dactylogyridea in Bychowsky [80]), whereas the Capsalidae was classified into the order Capsalidea (or suborder Monopisthocotylinea within the order Dactylogyridea in Bychowsky [80]). A number of subsequent morphology- and molecular data-based studies further supported the closer phylogenetic relationship between the Dactylogyridae and Diplectanidae than either of the two with Capsalidae: chaetotaxy and ciliated cells of the oncomiracidium [83], spermatozoon ultrastructure [12, 13], comprehensive morphological characters [14, 82], 28S rRNA [8] and 18S rRNA [9,10,11]. Regarding the phylogenetic position of the Capsalidae, it was either resolved as phylogenetically closely related to the Gyrodactylidea (families Gyrodactylidae and/or Udonellidae) based on the evidence of spermatozoon ultrastructure [12, 13], 28S rRNA gene [8], 18S rRNA gene [9,10,11], and a combination of three unlinked nuclear genes (28S rRNA, Histone 3 and Elongation Factor 1α) [5], or was resolved as basal to the Gyrodactylidea and Dactylogyridea on the basis of comprehensive morphological characters [14, 82]. However, as argued before, morphological traits are liable to cause taxonomic and phylogenetic artifacts in (parasitic) microscopic animals [4,5,6] and single gene-based molecular markers may not provide sufficient resolution power to infer the relationships among these species with high precision [7, 84]. These discrepancies could also be a result of discrepant evolutionary rates between mitochondrial and nuclear sequences, which can produce differing evolutionary signals [85, 86]. Although our phylogenetic analysis was based on relatively limited mitogenomic data (four species belonging to two families for Dactylogyridea, and three species/one family for Capsalidea), the paraphyly of the Dactylogyridea with reasonably high support values (BS = 85, PB = 0.99) suggests that their relationships should be further explored using a larger number of monogenean mitogenomes and large nuclear datasets.
Gene order
The order of tRNA genes of the two studied diplectanids exhibits notable rearrangements in comparison to all other sequenced monogenean mitogenomes (Fig. 2, Additional file 8: Table S4). Disregarding the three missing tRNAs, the gene order of the two diplectanids was very similar, with only two transposition events: position interchanges of trnF and trnQ, and trnP and trnI (Fig. 2 and Additional file 8: Table S4). This unique gene order pattern of the two diplectanids is further manifested by low pairwise similarity values in comparison with those observed among other monogeneans: the highest, between L. spari and B. hoshinai, is only 354 over 1254 (Additional file 8: Table S4). The transformational pathway from L. spari to the most similar gene arrangement, belonging to B. hoshinai, required one transposition, one TDRL (tandem-duplication-random-loss) and two coupled transposition events (Additional file 9: Figure S7). The transformational pathway from L. longipenis to the most similar gene arrangement found in T. nebulosi and P. variegatus (similarity value: 322 over 1254; gene orders of T. nebulosi and P. variegatus were identical) required two transpositions, one TDRL and two coupled transposition events (Additional file 9: Figure S7). In addition to the patterns summarized in our recent paper [4], we assigned pattern 1c to the gene order of the two diplectanids, which seems to be synapomorphic to the family Diplectanidae (Additional file 5: Figure S2).
Conserved gene arrangement is considered to be a typical feature of mitochondrial genomes [87,88,89], but our results suggest that extensive gene order rearrangements are not rare events in the class Monogenea. As five out of six proposed main patterns are found in the Monogenea (1a, 1b, 3, 4 and the new pattern 1c; Additional file 5: Figure S2), this further confirms the hypothesis [4] that gene order in monogeneans is evolving at a relatively rapid rate. However, evidence is emerging that the evolution of mitogenomic gene order arrangements is discontinuous in monogeneans, as some taxonomic categories appear to be particularly prone to mitogenomic rearrangements (diplectanids, tetraonchids and A. forficulatus), whereas others exhibit relatively conserved gene orders. Of course, our conclusions should be interpreted within the context of the limited number of monogenean mitogenomes currently available. Discontinuity in mitogenomic architecture evolution was also found in nematodes [28], snails [90], insects [91] and vertebrates [92]. Although gene order is sometimes used as a tool for inferring phylogenetic relationships [93, 94], this discontinuity in gene order rearrangements in monogeneans might produce misleading evolutionary signals, such as disproportionately long branches, which in turn might cause long branch attraction artifacts. Thus, gene order may only be used for phylogenetic analyses in this group of animals with this limitation in mind. The provisional addition of three missing tRNAs certainly affected the similarity values and transformational pathways between L. spari and other monogeneans, but it would not affect the assignment of a new gene order pattern to diplectanids, nor our conclusion that diplectanids are prone to mitogenomic rearrangements.
Conclusions
The present study reports four findings worthy of emphasis. First, on the basis of nucleotide diversity, dN/dS and average sequence identity, we propose that nad2, nad5 and nad4 genes are better-suited as molecular markers for species identification and population genetics studies of diplectanids than the commonly used cox1. Secondly, the long non-coding region of both mitogenomes contains two interesting features: (i) a highly repetitive region, which is often associated with replication origin in mitochondria; and (ii) tRNA pseudogenes in tandem arrays, which is common in plastid genomes and prokaryotes, but rare in metazoan mitogenomes. Thirdly, phylogenetic analysis showed that the two new diplectanids (Dactylogyridea) formed a sister group with a clade comprised of two other dactylogyrids (Dactylogyridea) and three capsalids (Capsalidea). Thus, Dactylogyridea was rendered paraphyletic by the nested Capsalidea clade. Fourthly, due to the extensive tRNA gene rearrangements in the two diplectanids, we assigned them a new gene order pattern, and concluded that the evolution of mitogenomic gene order arrangements is discontinuous in monogeneans. However, our confidence in the unorthodox phylogeny produced here, and understanding of genomic architecture evolution, is curbed by the scarcity of available mitogenomes (only four dactylogyrideans and 20 monogeneans), so we encourage researchers to accumulate more samples and molecular data (especially large molecular data) for monogenean parasites in order to infer their evolutionary history with confidence.
Abbreviations
- ORF:
-
open reading frame
- RSCU:
-
relative synonymous codon usage
- PCGs:
-
protein-encoding genes
- ML:
-
maximum likelihood
- BI:
-
Bayesian inference
- dN:
-
non-synonymous mutation rate
- dS:
-
synonymous mutation rate
- LNCR:
-
long non-coding regions
- BS:
-
bootstrap
- PB:
-
PhyloBayes
- HRR:
-
highly repetitive region
- TRs:
-
tandem repeats
- TDRL:
-
tandem-duplication-random-loss
References
Oliver G. Les Diplectanidae Bychowsky, 1957 (Monogenea, Monopisthocotylea, Dactylogyridea). Systématique. Biologie. Ontogénie. Écologie. Essai de phylogénèse. Thesis. France: Université des Sciences et Techniques du Languedoc; 1987.
Dezfuli BS, Giari L, Simoni E, Menegatti R, Shinn AP, Manera M. Gill histopathology of cultured European sea bass, Dicentrarchus labrax (L.), infected with Diplectanum aequans (Wagener, 1857) Diesing 1958 (Diplectanidae: Monogenea). Parasitol Res. 2007;100:707–13.
Cone DK. Monogenea (Phylum Platyhelminthes). In: Woo PTK, editor. Fish Diseases and Disorders: Protozoan and Metazoan Infections. Wallingford: CAB International; 1995. p. 289–327.
Zhang D, Zou H, Wu SG, Li M, Jakovlić I, Zhang J, et al. Sequencing of the complete mitochondrial genome of a fish-parasitic flatworm Paratetraonchoides inermis (Platyhelminthes: Monogenea): tRNA gene arrangement reshuffling and implications for phylogeny. Parasit Vectors. 2017;10:462.
Perkins EM, Donnellan SC, Bertozzi T, Chisholm LA, Whittington ID. Looks can deceive: molecular phylogeny of a family of flatworm ectoparasites (Monogenea: Capsalidae) does not reflect current morphological classification. Mol Phylogenet Evol. 2009;52:705–14.
Poulin R, Morand S. The diversity of parasites. Q Rev Biol. 2000;75:277–93.
Huyse T, Buchmann K, Littlewood DT. The mitochondrial genome of Gyrodactylus derjavinoides (Platyhelminthes: Monogenea) - a mitogenomic approach for Gyrodactylus species and strain identification. Gene. 2008;417:27–34.
Mollaret I, Jamieson BG, Justine J-L. Phylogeny of the Monopisthocotylea and Polyopisthocotylea (Platyhelminthes) inferred from 28S rDNA sequences. Int J Parasitol. 2000;30:171–85.
Olson P, Littlewood D. Phylogenetics of the Monogenea - evidence from a medley of molecules. Int J Parasitol. 2002;32:233–44.
Simkova A, Plaisance L, Matejusova I, Morand S, Verneau O. Phylogenetic relationships of the Dactylogyridae Bychowsky, 1933 (Monogenea: Dactylogyridea): the need for the systematic revision of the Ancyrocephalinae Bychowsky, 1937. Syst Parasitol. 2003;54:1–11.
Plaisance L, Littlewood DTJ, Olson PD, Morand S. Molecular phylogeny of gill monogeneans (Platyhelminthes, Monogenea, Dactylogyridae) and colonization of Indo-West Pacific butterflyfish hosts (Perciformes, Chaetodontidae). Zool Scr. 2005;34:425–36.
Justine J-L. Cladistic study in the Monogenea (Platyhelminthes), based upon a parsimony analysis of spermiogenetic and spermatozoal ultrastructural characters. Int J Parasitol. 1991;21:821–38.
Justine J-L, Lambert A, Mattei X. Spermatozoon ultrastructure and phylogenetic relationships in the monogeneans (Platyhelminthes). Int J Parasitol. 1985;15:601–8.
Boeger WA, Kritsky DC. Phylogenetic relationships of the Monogenoidea. In: Littlewood DTJ, Bray RA, editors. Interrelationships of the Platyhelminthes. London: Taylor & Francis; 2001. p. 92–102.
Yin M, Zheng HX, Su J, Feng Z, McManus DP, Zhou XN, et al. Co-dispersal of the blood fluke Schistosoma japonicum and Homo sapiens in the Neolithic Age. Sci Rep. 2015;5:18058.
Perkins EM, Donnellan SC, Bertozzi T, Whittington ID. Closing the mitochondrial circle on paraphyly of the Monogenea (Platyhelminthes) infers evolution in the diet of parasitic flatworms. Int J Parasitol. 2010;40:1237–45.
Li WX, Zhang D, Boyce K, Xi BW, Zou H, Wu SG, et al. The complete mitochondrial DNA of three monozoic tapeworms in the Caryophyllidea: a mitogenomic perspective on the phylogeny of eucestodes. Parasit Vectors. 2017;10:314.
Jia W-Z, Yan H-B, Guo A-J, Zhu X-Q, Wang Y-C, Shi W-G, et al. Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics. 2010;11:447.
Zhang D, Zou H, Wu SG, Li M, Jakovlic I, Zhang J, et al. Sequencing, characterization and phylogenomics of the complete mitochondrial genome of Dactylogyrus lamellatus (Monogenea: Dactylogyridae). J Helminthol. 2018;92:455–66.
Bachmann L, Fromm B, de Azambuja LP, Boeger WA. The mitochondrial genome of the egg-laying flatworm Aglaiogyrodactylus forficulatus (Platyhelminthes: Monogenoidea). Parasit Vectors. 2016;9:285.
Zhang JY, Yang TB, Liu L. Monogeneans of Chinese Marine Fishes. Beijing: Agriculture Press; 2001.
Sánchez-García N, Padrós F, Raga JA, Montero FE. Comparative study of the three attachment mechanisms of diplectanid monogeneans. Aquaculture. 2011;318:290–9.
Domingues MV, Boeger WA. Phylogeny and revision of Diplectanidae Monticelli, 1903 (Platyhelminthes: Monogenoidea). Zootaxa. 2008;1698:1–40.
Ogawa K, Egusa S. Three species of Lamellodiscus (Monogenea: diplectanidae) from the gills of the Japanese black sea bream, Acanthopagrus schlegeli (Bleeker). Bull Jap Soc Sci Fish. 1978;44:607–12.
Wu XY, Li AX, Zhu XQ, Xie MQ. Description of Pseudorhabdosynochus seabassi sp. n. (Monogenea: Diplectanidae) from Lates calcarifer and revision of the phylogenetic position of Diplectanum grouperi (Monogenea: Diplectanidae) based on rDNA sequence data. Folia Parasitol. 2005;52:231.
Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2:Unit 2.3.
Lalitha S. Primer Premier 5. Biot Soft Int Rep. 2000;1:270–2.
Zou H, Jakovlic I, Chen R, Zhang D, Zhang J, Li WX, et al. The complete mitochondrial genome of parasitic nematode Camallanus cotti: extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genomics. 2017;18:840.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Burland TG. DNASTAR’s Lasergene sequence analysis software. Methods Mol Biol. 2000;132:71–91.
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9.
Laslett D, Canback BARWEN. a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5.
Zhang D. MitoTool software. 2016. https://github.com/dongzhang0725/MitoTool. Accessed 22 July 2018.
Hadley W. ggplot2: Elegant graphics for data analysis. New York: Springer; 2009.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573.
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15.
Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D, et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23:2957–8.
Liu F-F, Li Y-P, Jakovlic I, Yuan X-Q. Tandem duplication of two tRNA genes in the mitochondrial genome of Tagiades vajuna (Lepidoptera: Hesperiidae). Eur J Entomol. 2017;114:407–15.
Katoh K. Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Kück P, Meid SA, Groß C, Wägele JW, Misof B. AliGROOVE - visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinformatics. 2014;15:294.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34:772–3.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62:611–5.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Wey-Fabrizius AR, Podsiadlowski L, Herlyn H, Hankeln T. Platyzoan mitochondrial genomes. Mol Phylogenet Evol. 2013;69:365–75.
Kang S, Kim J, Lee J, Kim S, Min GS, Park JK. The complete mitochondrial genome of an ectoparasitic monopisthocotylean fluke Benedenia hoshinai (Monogenea: Platyhelminthes). Mitochondrial DNA. 2012;23:176–8.
Ye F, King SD, Cone DK, You P. The mitochondrial genome of Paragyrodactylus variegatus (Platyhelminthes: Monogenea): differences in major non-coding region and gene order compared to Gyrodactylus. Parasit Vectors. 2014;7:377.
Zhang D. The chromatograms of the fragments between nad5 and cox3. 2018. https://figshare.com/articles/The_chromatograms_of_the_fragments_between_nad5_and_cox3/6363965. Accessed 26 May 2018.
Doublet V, Ubrig E, Alioua A, Bouchon D, Marcade I, Marechal-Drouard L. Large gene overlaps and tRNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biol. 2015;12:1159–68.
Kilpert F, Podsiadlowski L. The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genomics. 2006;7:241.
Kumazawa Y, Miura S, Yamada C, Hashiguchi Y. Gene rearrangements in gekkonid mitochondrial genomes with shuffling, lossand reassignment of tRNA genes. BMC Genomics. 2014;15:930.
Wu X, Xu X, Yu Z, Kong X. Comparative mitogenomic analyses of three scallops (Bivalvia: Pectinidae) reveal high level variation of genomic organization and a diversity of transfer RNA gene sets. BMC Res Notes. 2009;2:69.
Domes K, Maraun M, Scheu S, Cameron SL. The complete mitochondrial genome of the sexual oribatid mite Steganacarus magnus: genome rearrangements and loss of tRNAs. BMC Genomics. 2008;9:532.
Huot JL, Enkler L, Megel C, Karim L, Laporte D, Becker HD, et al. Idiosyncrasies in decoding mitochondrial genomes. Biochimie. 2014;100:95–106.
Huyse T, Plaisance L, Webster BL, Mo TA, Bakke TA, Bachmann L, et al. The mitochondrial genome of Gyrodactylus salaris (Platyhelminthes: Monogenea), a pathogen of Atlantic salmon (Salmo salar). Parasitology. 2007;134:739–47.
Plaisance L, Huyse T, Littlewood DT, Bakke TA, Bachmann L. The complete mitochondrial DNA sequence of the monogenean Gyrodactylus thymalli (Platyhelminthes: Monogenea), a parasite of grayling (Thymallus thymallus). Mol Biochem Parasitol. 2007;154:190–4.
Zhang J, Wu X, Xie M, Li A. The complete mitochondrial genome of Pseudochauhanea macrorchis (Monogenea: Chauhaneidae) revealed a highly repetitive region and a gene rearrangement hot spot in Polyopisthocotylea. Mol Biol Rep. 2012;39:8115–25.
Zhang J, Wu X, Xie M, Xu X, Li A. The mitochondrial genome of Polylabris halichoeres (Monogenea: Microcotylidae). Mitochondrial DNA. 2011;22:3–5.
Zhang J, Wu X, Li Y, Zhao M, Xie M, Li A. The complete mitochondrial genome of Neobenedenia melleni (Platyhelminthes: Monogenea): mitochondrial gene content, arrangement and composition compared with two Benedenia species. Mol Biol Rep. 2014;41:6583–9.
Park JK, Kim KH, Kang S, Kim W, Eom KS, Littlewood DT. A common origin of complex life cycles in parasitic flatworms: evidence from the complete mitochondrial genome of Microcotyle sebastis (Monogenea: Platyhelminthes). BMC Evol Biol. 2007;7:11.
Zhang D, Zou H, Zhou S, Wu SG, Li WX, Wang GT. The complete mitochondrial genome of Gyrodactylus kobayashii (Platyhelminthes: Monogenea). Mitochondrial DNA Part B. 2016;1:146–7.
Zhang J, Wu X, Li Y, Xie M, Li A. The complete mitochondrial genome of Tetrancistrum nebulosi (Monogenea: Ancyrocephalidae). Mitochondrial DNA Part B. 2016;27:22–3.
Zou H, Zhang D, Li W, Zhou S, Wu S, Wang G. The complete mitochondrial genome of Gyrodactylus gurleyi (Platyhelminthes: Monogenea). Mitochondrial DNA Part B. 2016;1:383–5.
Kim K, Jeon H, Kang S, Sultana T, Kim GJ, Eom KS, et al. Characterization of the complete mitochondrial genome of Diphyllobothrium nihonkaiense (Diphyllobothriidae: Cestoda), and development of molecular markers for differentiating fish tapeworms. Mol Cells. 2007;23:379.
von Nickisch-Rosenegk M, Brown WM, Boore JL. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that platyhelminths are eutrochozoans. Mol Biol Evol. 2001;18:721–30.
Le TH, Blair D, McManus DP. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002;18:206–13.
Fumagalli L, Taberlet P, Favre L, Hausser J. Origin and evolution of homologous repeated sequences in the mitochondrial DNA control region of shrews. Mol Biol Evol. 1996;13:31–46.
Amiryousefi A, Hyvonen J, Poczai P. The chloroplast genome sequence of bittersweet (Solanum dulcamara): plastid genome structure evolution in Solanaceae. PLoS One. 2018;13:e0196069.
Tran TT, Belahbib H, Bonnefoy V. Talla E. A comprehensive tRNA genomic survey unravels the evolutionary history of tRNA arrays in prokaryotes. Genome Biol Evol. 2015;8:282–95.
Hebert PD, Cywinska A, Ball SL. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–21.
Hansen H, Bachmann L, Bakke TA. Mitochondrial DNA variation of Gyrodactylus spp. (Monogenea, Gyrodactylidae) populations infecting Atlantic salmon, grayling, and rainbow trout in Norway and Sweden. Int J Parasitol. 2003;33:1471–8.
Hansen H, Martinsen L, Bakke T, Bachmann L. The incongruence of nuclear and mitochondrial DNA variation supports conspecificity of the monogenean parasites Gyrodactylus salaris and G. thymalli. Parasitology. 2006;133:639–50.
Meinilä M, Kuusela J, Ziętara MS, Lumme J. Initial steps of speciation by geographic isolation and host switch in salmonid pathogen Gyrodactylus salaris (Monogenea: Gyrodactylidae). Int J Parasitol. 2004;34:515–26.
Blasco-Costa I, Miguez-Lozano R, Sarabeev V, Balbuena JA. Molecular phylogeny of species of Ligophorus (Monogenea: Dactylogyridae) and their affinities within the Dactylogyridae. Parasitol Int. 2012;61:619–27.
Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW. An evaluation of genetic distances for use with microsatellite loci. Genetics. 1995;139:463–71.
Bychowsky BE. Monogenetic Trematodes, their systematics and phylogeny. Moscow-Leningrad, USSR: Academy of Sciences; 1957 (In Russian; English translation by Hargis WJ, Oustinoff PC, 1961. American Institute of Biological Sciences, Washington).
Lebedev B. Monogenea in the light of new evidence and their position among platyhelminths. Angew Parasitol. 1988;29:149–67.
Boeger WA, Kritsky DC. Phylogeny and a revised classification of the Monogenoidea Bychowsky, 1937 (Platyhelminthes). Syst Parasitol. 1993;26:1–32.
Lambert A. Oncomiracidiums et phylogénèse des Monogènes (Plathelminthes), 2ème partie: Structures argyrophiles des oncomiracidiums et phylogénèse des Monogenea. Ann Parasitol Hum Comp. 1980;55:281–326.
Delsuc F, Tsagkogeorga G, Lartillot N, Philippe H. Additional molecular support for the new chordate phylogeny. Genesis. 2008;46:592–604.
Park S, Ruhlman TA, Weng ML, Hajrah NH, Sabir JSM, Jansen RK. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol Evol. 2017;9:1766–80.
Grechko VV. The problems of molecular phylogenetics with the example of squamate reptiles: mitochondrial DNA markers. Mol Biol. 2013;47:55–74.
Boore JL. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In: Sankoff D, Nadeau JH, editors. Comparative Genomics. Dordrecht: Kluwer Academic Publishers; 2000. p. 133–47.
Black WC 4th, Roehrdanz RL. Mitochondrial gene order is not conserved in arthropods: prostriate and metastriate tick mitochondrial genomes. Mol Biol Evol. 1998;15:1772–85.
Li H, Liu H, Shi A, Stys P, Zhou X, Cai W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS One. 2012;7:e29419.
Wang JG, Zhang D, Jakovlic I, Wang WM. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). PLoS One. 2017;12:e0181699.
Xiong H, Barker SC, Burger TD, Raoult D, Shao R. Heteroplasmy in the mitochondrial genomes of human lice and ticks revealed by high throughput sequencing. PLoS One. 2013;8:e73329.
Mueller RL, Boore JL. Molecular mechanisms of extensive mitochondrial gene rearrangement in plethodontid salamanders. Mol Biol Evol. 2005;22:2104–12.
Boore J. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol Evol. 2006;21:439–46.
Sultana T, Kim J, Lee S-H, Han H, Kim S, Min G-S, et al. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol Biol. 2013;13:12.
Acknowledgements
The authors would like to thank MSc Run Q. Wang for their assistance in sampling the monogeneans. We would also like to thank the editor and the two anonymous reviewers for the time they invested in reviewing our manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (31872604, 31572658), the Earmarked Fund for China Agriculture Research System (CARS-45-15) and the Major Scientific and Technological Innovation Project of Hubei Province (2015ABA045).
Availability of data and materials
All data are fully available without restriction. The datasets supporting the conclusions of this article are included within the article and its additional files. The mitogenomic sequences of Lepidotrema longipenis and Lamellodiscus spari are available from GenBank under the accession numbers MH328203 and MH328204, respectively. Chromatograms of the fragments between nad5 and cox3 are available from Figshare: https://figshare.com/articles/The_chromatograms_of_the_fragments_between_nad5_and_cox3/6363965.
Author information
Authors and Affiliations
Contributions
GTW, WXL and DZ designed the study. DZ, HZ, JZ and RC conducted the experiments. DZ conducted the data analysis. DZ, WXL and IJ wrote the paper. All authors revised the manuscript critically for important intellectual content, read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All applicable international, national and/or institutional guidelines for the care and use of animals were followed.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Dataset S1. Supplementary methods. (DOCX 41 kb)
Additional file 2:
Table S1. Monogenean species and outgroups used for comparative mitogenomic and phylogenetic analyses. (XLSX 14 kb)
Additional file 3:
Table S2. General statistics for the protein-coding and rRNA genes of the two diplectanids and 18 other available monogeneans. (XLSX 16 kb)
Additional file 4:
Figure S1. Relative synonymous codon usage (RSCU) of six monopisthocotylid mitogenomes. (PDF 57 kb)
Additional file 5:
Figure S2. The 23 unique gene orders in neodermatan mitochondrial genomes filtered from 113 species. Representative species and corresponding taxonomic categories at the class/subclass level are shown on the left; a star symbol denotes that the gene order is shared by Monogenea and Cestoda. Pattern types used here to classify gene orders are shown on the right. In Lamellodiscus spari, the missing tRNAs are represented by the “?” symbol in the positions homologous to the closest available diplectanid relative, Lepidotrema longipenis. (PDF 1961 kb)
Additional file 6:
Figure S3. Stem-loop structures of the consensus repeat pattern in highly repetitive regions of the long non-coding regions of Lepidotrema longipenis and Lamellodiscus spari. dG denotes the structure’s free energy (ΔG in Kcal/mol at 37 °C). (PDF 297 kb)
Additional file 7:
Figure S4. Maximum likelihood tree of nucleotide dataset with partition model. Figure S5. Maximum likelihood tree of amino acid dataset with partition model. Figure S6. AliGROOVE analysis. Table S3. The best partitioning scheme. (DOCX 517 kb)
Additional file 8:
Table S4. Pairwise comparison of mitochondrial DNA gene orders among 13 monogenean species based on the order of all 36 genes. Scores correspond to the similarity between gene orders, where “1254” represents an identical gene order. N is the number identifying the taxon in the pairwise comparisons. (XLSX 11 kb)
Additional file 9:
Figure S7. Transformational pathway from the gene orders of the two diplectanids to the most similar monogenean gene arrangements. (PDF 992 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, D., Li, W.X., Zou, H. et al. Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements. Parasites Vectors 11, 601 (2018). https://doi.org/10.1186/s13071-018-3144-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3144-6