
Abstract
Background
Vertebrate mitochondrial genomes (mitogenomes) are 16–18 kbp double-stranded circular DNAs that encode a set of 37 genes. The arrangement of these genes and the major noncoding region is relatively conserved through evolution although gene rearrangements have been described for diverse lineages. The tandem duplication-random loss model has been invoked to explain the mechanisms of most mitochondrial gene rearrangements. Previously reported mitogenomic sequences for geckos rarely included gene rearrangements, which we explore in the present study.
Results
We determined seven new mitogenomic sequences from Gekkonidae using a high-throughput sequencing method. The Tropiocolotes tripolitanus mitogenome involves a tandem duplication of the gene block: tRNAArg, NADH dehydrogenase subunit 4L, and NADH dehydrogenase subunit 4. One of the duplicate copies for each protein-coding gene may be pseudogenized. A duplicate copy of the tRNAArg gene appears to have been converted to a tRNAGln gene by a C to T base substitution at the second anticodon position, although this gene may not be fully functional in protein synthesis. The Stenodactylus petrii mitogenome includes several tandem duplications of tRNALeu genes, as well as a translocation of the tRNAAla gene and a putative origin of light-strand replication within a tRNA gene cluster. Finally, the Uroplatus fimbriatus and U. ebenaui mitogenomes feature the apparent loss of the tRNAGlu gene from its original position. Uroplatus fimbriatus appears to retain a translocated tRNAGlu gene adjacent to the 5’ end of the major noncoding region.
Conclusions
The present study describes several new mitochondrial gene rearrangements from Gekkonidae. The loss and reassignment of tRNA genes is not very common in vertebrate mitogenomes and our findings raise new questions as to how missing tRNAs are supplied and if the reassigned tRNA gene is fully functional. These new examples of mitochondrial gene rearrangements in geckos should broaden our understanding of the evolution of mitochondrial gene arrangements.
Similar content being viewed by others
Background
Metazoan mitochondrial genomes (mitogenomes) are double-stranded circular DNAs typically 16–18 kbp in size (reviewed in [1–3]). They are maternally inherited as haploid genomes with multiple copy numbers in a cell. Metazoan mitogenomes encode a set of 37 genes for two ribosomal RNAs, 22 tRNAs, and 13 respiratory protein subunits [1, 4] and also possess a major noncoding region or control region that contains signals for the initiation of replication and transcription (reviewed in [5, 6]). In addition, most vertebrate mitogenomes conserve a characteristic stem-and-loop structure between tRNAAsn and tRNACys genes that acts as the putative origin of light-strand replication (OL) [5].
The organization of the 37 genes and the major noncoding region varies considerably between metazoan classes but is relatively conserved within Vertebrata [2]. The typical vertebrate gene organization (Figure 1A), which was first revealed for the human mitogenome [4], is shared by many species of fishes, amphibians, reptiles, and mammals. However, deviation from this typical organization has been found in species of all these vertebrate groups and birds ([2, 3, 7, 8] and refs. therein). The majority of gene rearrangement cases in vertebrate mitochondrial genomes involve shuffling of some neighboring genes (most typically clustered tRNA genes) or the translocation of genes across duplicated control regions, for example in snake mitogenomes [2, 9]. Gene inversions are quite rare in vertebrate mitogenomes though not unknown [10].

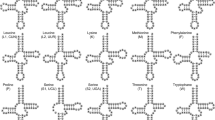
Gene organizations of mitogenomes for (A) many vertebrates including Tropiocolotes steudneri , Lepidodactylus lugubris, and Phelsuma guimbeaui (the typical vertebrate gene arrangement), (B) Tropiocolotes tripolitanus , (C) Stenodactylus petrii , (D) Uroplatus fimbriatus , and (E) Uroplatus ebenaui . Circular mitogenomes are represented linearly as bars and genes encoded by the H-strand and L-strand are shown, respectively, above and below the bar. Genes with an asterisk are probable pseudogenes. Several genes relevant to our discussions on gene rearrangements are highlighted with colors. For gene names, ND1-6 and 4L represent NADH dehydrogenase subunits 1-6 and 4L. CO1-3 stand for cytochrome oxidase subunit 1-3. cytb, A6 and A8 represent cytochrome b, ATPase subunit 6 and 8, respectively. 12S and 16S stand for 12S rRNA and 16S rRNA, respectively. Transfer RNA genes are depicted with the corresponding single-letter amino acid and, in T. tripolitanus, two glutamine tRNA genes are discriminated by Q1 and Q2. L1 and L2 represent tRNALeu(UUR) and tRNALeu(CUN) genes, respectively, and S1 and S2 represent tRNASer(UCN) and tRNASer(AGY) genes, respectively. OL represents the putative L-strand replication origin. IGS stands for an intergenic sequence in the ND6/cytb gene boundary (see text). The position and orientation of several PCR primers (see Additional file 1: Table S1 for their sequences) that were used to amplify and sequence the rearrangement-related regions are also shown.
Recent technical advances in sequencing have led to the rapid accumulation of complete or nearly complete mitogenomic sequences and the mitogenomic sequences of over 1800 vertebrate species are currently known [3]. Approximately half of them are from fishes with the remainder from tetrapods, where mammals dominate over reptiles. To the best of our knowledge, only 13 species have been sequenced from the Gekkota, which consists of more than 1500 species of geckos and their allies [11]. These known gekkotan mitogenomes share the typical vertebrate gene organization although polymorphic tandem duplications with varying sizes (6–9 kbp) have been reported for the parthenogenetic Heteronotia binoei[12] and potential pseudogenization of the tRNAGln gene was found in Hemitheconyx caudicinctus[13].
Previous studies proposed that some vertebrate groups may be more susceptible to mitogenomic gene rearrangements than the others. For example, ranoid frogs include a variety of gene rearrangements in their mitogenomes while most non-neobatrachian frogs conserve the typical gene organization ([8, 14] and refs. therein). Amongst lizards, Agamidae contain several different types of gene rearrangement [7, 10, 15, 16] but no mitogenomic rearrangement has been reported from the closely related family, Iguanidae [17]. This heterogeneity in the occurrence of gene rearrangements among different vertebrate groups has been examined in relation to loss of the light-strand replication origin [15, 18], duplication of the control region [2, 19], or changes of the rate of molecular evolution [20], although none of these causal hypotheses have been fully examined across diverse vertebrate groups.
Here, we report seven new mitogenomic sequences from Gekkonidae (Reptilia; Squamata) and describe several new gene rearrangements that involve shuffling, loss, and reassignment of tRNA genes. We discuss evolutionary mechanisms for the gene rearrangements and their effects on the mitochondrial translational system.
Results
Gene arrangement in the Tropiocolotes tripolitanusmitogenome
We used high-throughput sequencing to determine nucleotide sequences of seven new mitochondrial genomes from Gekkonidae (see Table 1 for scientific names and accompanying information of the seven geckos). Although mitogenomic sequences of Tropiocolotes steudneri, Lepidodactylus lugubris, and Phelsuma guimbeaui were found to possess the typical vertebrate gene organization (Figure 1A), deviations from this organization were seen in the other gekkonid mitogenomes.
The complete mitochondrial genome sequence of Tropiocolotes tripolitanus is 20,248 bp (Table 1) and encodes all 37 mitochondrial genes in addition to containing the major noncoding region (2,923 bp) located between tRNAPro and tRNAPhe genes (Figure 1B). At the 5’ end of the major noncoding region, there are four tandem repeats of a 74-bp sequence, while at its 3’ end there are 10 tandem repeats of a 100-bp sequence, followed by a second weakly repetitive sequence. In the middle of the major noncoding region, three conserved sequence block (CSB) motifs (CSB-1, CSB-2, and CSB-3) [21] are found. The central part of the major noncoding region is therefore regarded as the control region that regulates replication and transcription of the mitochondrial genome [5].
The T. tripolitanus mitogenome has the typical vertebrate gene organization, except for a region between NADH dehydrogenase subunit 3 (ND3) and tRNAHis genes (Figure 1B). This region usually contains an array of genes (ND3, tRNAArg, ND4L, ND4, and tRNAHis) in the typical vertebrate organization (Figure 1A). However, the corresponding region in T. tripolitanus contains a rearranged set of genes (ND3, tRNAGln, ND4L, ND4*, tRNAArg, ND4L*, ND4, and tRNAHis) where genes with an asterisk may be pseudogenes (see below for details). To exclude the possibility that this gene arrangement resulted from erroneous high-throughput DNA sequencing or assembly, we carefully amplified and re-sequenced this region using various combinations of 10 species-specific primers (Ttri-4L to Ttri-8L and Ttri-4H to Ttri-8H; see Additional file 1: Table S1 and Figure S1). The resultant sequence was identical to the one determined by high-throughput DNA sequencing.
The tRNAGln2 gene next to the ND3 gene (denoted Q2 in Figure 1B) is encoded by the heavy strand, while another tRNAGln1 gene (Q1 in Figure 1B) encoded by the light strand occurs between tRNAIle and tRNAMet genes. The position and orientation of the latter tRNAGln1 gene matches the typical vertebrate gene organization (Figure 1A). Figure 2 illustrates the secondary structures of these two tRNAGln genes. Both the tRNAGln genes can assume stable clover-leaf structures. The tRNAGln2 gene has a clear sequence similarity with the tRNAArg gene (Figure 3); there are only four base differences between them with one at the second anticodon position (T for the tRNAGln2 gene and C for the tRNAArg gene). These two tRNA genes are thus paralogs and one of them (the tRNAGln2 gene; see below) was created by gene duplication and subsequent base substitution at the second anticodon position.

Secondary structures of two tRNAGlngenes encoded in the T. tripolitanus mitogenome. tRNAGln1 and tRNAGln2 genes correspond to Q1 and Q2 genes of Figure 1B, respectively. Watson-Crick and wobble base pairs are shown with a bar and a dot, respectively. Anticodon sequence for the glutamine tRNA gene (TTG) is highlighted.

Sequence similarity of T. tripolitanus tRNAArgand tRNAGln 2 genes. Sense-strand sequences for the T. tripolitanus tRNAArg and tRNAGln2 genes are aligned. The tRNA gene sequences are divided into structural elements, such as the D loop and acceptor stem, and three nucleotides corresponding to the anticodon are highlighted.
Regions adjacent to the tRNAGln2 and tRNAArg genes are homologous, sharing sequences related to ND4L genes (see Additional file 2: Figure S2). The former has a complete ND4L coding region, whereas the latter has frequent indels and severely reduced sequence similarity to ND4L genes from other geckos (Additional file 2: Figure S2). In-frame translation of this ND4L pseudogene does not show any detectable level of sequence similarity with ND4L amino acid sequences of other geckos due to frameshift indels (Additional file 2: Figure S3).
These two ND4L-related regions are followed by another pair of homologous sequences with high sequence similarity to ND4 genes from other geckos (Additional file 2: Figure S4). These two ND4-related sequences do not have in-frame stop codons and are easily aligned to each other without indels (Additional file 2: Figure S4). The second ND4-related sequence preceding the tRNAHis gene is shorter than the first one preceding the tRNAArg gene but has a slightly increased sequence similarity to ND4 sequences from other geckos, especially at amino acids 291–330 and 403–426 (Additional file 2: Figure S5). We therefore tentatively assume the second sequence to be a legitimate ND4 gene and regard the first one as a possible pseudogene. However, it also seems possible that both copies are functional genes in T. tripolitanus mitochondria.
Gene arrangement in the Stenodactylus petriimitogenome
The S. petrii mitogenome is 18,672 bp in length (Table 1) and includes all 37 mitochondrial genes (Figure 1C). It shows two changes from the typical vertebrate gene organization. First, there are four tandemly duplicated copies of tRNALeu (UUR) between 16S rRNA and ND1 genes. These four genes have high sequence similarity to each other (Additional file 1: Figure S6) and it is evident that they have been created by recent tandem duplications. The first gene located at the 5’ end of this tandem duplication lacks some basic tRNA secondary structures and may now be a pseudogene. The fourth copy seems to have the most stable secondary structure but the second and third copies may also be functional tRNALeu (UUR) genes in light of the structural criterion of mitochondrial tRNA genes [22]. We have samples of two more S. petrii individuals. Sequencing the corresponding region of these individuals showed a single tRNALeu (UUR) gene between 16S rRNA and ND1 genes (data not shown).
Second, there is a shuffling of tRNA genes and the OL contained in the WAN (OL) CY tRNA gene cluster. Four tandem copies of tRNAAla and the OL were found at the 5’ end of the remaining WNCY genes (Figure 1C). All four tRNAAla and OL copies have an identical sequence (data not shown), suggesting that the tandem duplications were very recent. We amplified and sequenced a mitogenomic region between the ND2 and cytochrome oxidase subunit 1 (CO1) genes from the two additional S. petrii individuals to show that one has only two tandem repeats of tRNAAla and OL while the other has four tandem repeats (see Additional file 3: Figure S7 for sizes of the amplified products and Additional file 4: Table S2 for accession numbers of these sequences with which annotation details can be referred to). Repeat number of this region is polymorphic within species.
Occurrence of gene rearrangements in other Tropiocolotes and Stenodactylusspecies
The genus Tropiocolotes includes 10 species distributed in Saharo-Arabian regions [11]. Another genus, Stenodactylus, with a similar distribution, is closely related to Tropiocolotes according to recent molecular phylogenetic studies [23–26], although precise phylogeny at the species level has not been established. We examined the occurrence of the gene rearrangements found in T. tripolitanus and S. petrii mitogenomes among other species of these genera. First, the Tropiocolotes steudneri mitogenomic sequence is 15,863 bp in length (Table 1). The major noncoding region of this species contains rather long arrays of repetitive sequences that were not completely sequenced. The T. steudneri mitogenome includes all sets of 37 mitochondrial genes with the typical gene organization of vertebrates (Figure 1A). Neither of the two types of gene rearrangements found in T. tripolitanus and S. petrii occur in this species.
We also examined gene organizations of a few other species by PCR amplification and sequencing. Figure S8 in Additional file 3 shows 1% agarose gel electrophoresis of PCR products amplified using rND3-1L and rCUN-3H primers (see Additional file 1: Table S1 for primer sequences and Figure 1B for their locations). As expected, a large product was amplified from T. tripolitanus (~4.2 kbp: lane 1) owing to the gene rearrangements described above. In contrast, Microgecko (recently moved from Tropiocolotes) persicus, Tropiocolotes steudneri, and Stenodactylus petrii gave rise to shorter products (~2.2 kbp: lanes 2–4), supporting that the mitogenomes of these species do not have the above gene rearrangements.
These results suggest that the rearranged gene arrangements shown in Figure 1B are not widely distributed among Tropiocolotes and Stenodactylus geckos. The gene rearrangements may have occurred relatively recently on a lineage leading to T. tripolitanus after its divergence from the other examined species. This view is supported by an observation that two paralogous ND4 amino acid sequences of T. tripolitanus are much more similar to each other than they are to counterparts in other gecko species (Additional file 2: Figure S5). These two paralogous sequences also share an insertion at sites 253–261 (Additional file 2: Figure S5), suggesting that this insertion event took place after the divergence from a lineage leading to T. steudneri but before the duplication of the ND4 gene.
The mitogenomic region between the ND2 and CO1 genes amplified from Stenodactylus doriae was somewhat longer than that from Stenodactylus slevini (Additional file 3: Figure S7). Stenodactylus slevini turned out to have the typical WAN(OL)CY gene organization but S. doriae had another unique gene arrangement: WAN*(OL)CNY, where N* represents a possible pseudogene of the tRNAAsn gene (see Additional file 4: Table S2 for accession numbers of nucleotide sequences deposited with complete annotation). The N* gene has a considerably weaker acceptor-stem secondary structure than the N gene (data not shown). Together with the information derived from T. tripolitanus and T. steudneri (Figure 1), these results suggest that the gene rearrangement found in S. petrii, in which tRNAAla and OL are translocated to the 5’ end of WNCY genes (Figure 1C), is not widely distributed among Tropiocolotes and Stenodactylus geckos. This translocation and the possible translocation of the tRNAAsn gene found in S. doriae probably took place independently in each lineage.
Loss of the tRNAGlu gene from the Uroplatus ebenauimitogenome
Complete mitogenomic sequences obtained for Uroplatus fimbriatus and U. ebenaui are 16,780 and 16,830 bp in length, respectively (Table 1). These mitogenomes possess the typical vertebrate gene organization, except for the disappearance of tRNAGlu, which is usually located between ND6 and cytochrome b (cytb) genes (Figures 1D and E). The corresponding intergenic region retains 54- and 62-bp sequences in each species (Figure 4A). However, these sequences do not show detectable sequence similarity to tRNAGlu genes from five non-Uroplatus geckos (Figure 4B). The tRNAGlu genes of these non-Uroplatus geckos are 68–72 bp in length, somewhat longer than the Uroplatus intergenic sequences.

Heavy-strand nucleotide sequences between ND6 and cytb genes for Uroplatus (A) and other geckos (B). Sequences in this region for non-Uroplatus geckos represent tRNAGlu genes, which are aligned based on clover-leaf secondary structure [22]. In B, tRNAGlu gene sequences found at the 5’ end of the major noncoding region for U. fimbriatus and U. sikorae are also shown. Alignment of the Uroplatus intergenic sequences in A was made with the aid of ClustalX [27]. Uroplatus gecko sequences were given the following abbreviations: Ufim, U. fimbriatus; Usik, U. sikorae; Ulin. U. lineatus; Upie, U. pietschmanni; Uebe, U. ebenaui; Upha, U. phantasticus; and Ugue, U. guentheri (see Additional file 4: Table S2 for accession numbers). Sequence data of tRNAGlu gene sequences for non-Uroplatus geckos are taken from Tropiocolotes tripolitanus (Ttri; this study), Tropiocolotes steudneri (Tste; this study), Stenodactylus petrii (Spet; this study), Gekko vittatus (Gvit; accession No. AB178897), and Coleonyx variegatus (Cvar; AB114446).
We sequenced this intergenic region for five more Uroplatus species (U. pietschmanni, U. sikorae, U. guentheri, U. phantasticus, and U. lineatus) with rND6-3L (or uND5-2L) and ucytb-1H primers (see Figure 1D and Additional file 1: Table S1 for primer positions and sequences, respectively). Intergenic sequences of 55–64 bp in length were found in each species but they do not show appreciable sequence similarity with each other (Figure 4A). Thus, it is unlikely that these intergenic sequences in Uroplatus species encode any conserved gene sequence. There is also no evidence to suggest that these intergenic sequences were evolutionarily derived from tRNAGlu genes.
Functional tRNAGlu gene sequences were carefully searched for over the complete mitogenomic sequences of the two Uroplatus taxa. It was found that the U. fimbriatus mitogenome encodes a tRNAGlu gene adjacent to the 5’ end of the major noncoding region (Figure 1D). However, no tRNAGlu-like sequence was found in the U. ebenaui mitogenome (Figure 1E). Coding regions in both taxa between tRNAPhe and tRNAPro genes do not have a notable intergenic region >50 bp in length, except for the ND6-cytb intergenic region described above.
The major noncoding regions of U. fimbriatus and U. ebenaui are 1,344 bp and 1,456 bp, respectively. These noncoding regions include tandem repeat sequences and the CSB1-3 sequences but tRNAGlu-like structures were not found, either by the COVE program as implemented in DOGMA [28] or by visual inspection for a standard mitochondrial tRNA gene structure [22]. We therefore conclude that the tRNAGlu gene is lacking from the mitogenome of U. ebenaui.
The major noncoding region was amplified and sequenced for the other five Uroplatus species using uThr-2L and r12S-1H (or rPhe-3H) primers (see Figure 1D and Additional file 1: Table S1 for primer positions and sequences, respectively). As a result, only one of them (U. sikorae) has the tRNAGlu gene at the 5’ end of the major noncoding region, similar to U. fimbriatus, whereas U. pietschmanni, U. guentheri, U. phantasticus, and U. lineatus do not have the tRNAGlu gene located near the major noncoding region, as in U. ebenaui (see Additional file 4: Table S2 for accession numbers of determined sequences). These results show that the disappearance of the tRNAGlu gene from the ND6/cytb junction is a common feature among the Uroplatus mitogenomes but that translocation to the 5’ end of the major noncoding region only occurs in some Uroplatus species.
Discussion
Mechanism of gene rearrangements
In the T. tripolitanus mitogenome (Figure 1B), a region from the tRNAGln2 gene to the ND4* gene has a sequence similarity with the region from the tRNAArg gene to the ND4 gene. This gene rearrangement originated from the tandem duplication of three genes: tRNAArg, ND4L and ND4 (Figure 5). One duplicate copy of the ND4L and ND4 genes has subsequently been pseudogenized, while in a duplicate copy of tRNAArg a base substitution (C to T) at the second anticodon position converted the identity of the tRNA gene from tRNAArg to tRNAGln. Three accompanying base substitutions, at positions between the acceptor and D stems, in the extra arm and in the T loop, have also occurred (Figure 3).

Plausible pathway of the gene rearrangements found for the T. tripolitanus mitogenome based on the tandem duplication-random loss model. Mitochondrial genes are illustrated as in Figure 1 and thick horizontal bars show a unit for tandem duplication. From the typical gene arrangement (state 1), three genes were tandemly duplicated (state 2). Reassignment of a tRNA gene (R to Q) and pseudogenization of duplicate protein genes gave rise to the T. tripolitanus gene arrangement (state 3). In future, deletion of redundant genes or pseudogenes may lead to a rearranged organization shown as state 4.
This plausible mechanism for the gene rearrangement that gave rise to the T. tripolitanus mitogenome (Figure 5) is consistent with the tandem duplication-random loss (TDRL) model [29] that has been postulated to explain most vertebrate mitochondrial gene rearrangements [2]. The TDRL model assumes a tandem duplication of a mitochondrial DNA segment and subsequent deletion of one of the duplicate gene copies, leading to a rearranged gene organization or reversal to the original organization. Deletion of the redundant gene copy may happen rapidly as it is free from functional constraint and therefore base changes can readily occur, facilitating its pseudogenization or complete deletion. This may also be driven by strong pressure for size reduction of metazoan mitochondrial genomes [1, 30]. The finding of duplicate genes between tRNAGln2 and ND4 genes in the T. tripolitanus mitogenome, but not in any other mitogenomes of closely related species (Additional file 3: Figure S8), is in agreement with this reasoning if the duplication is recent. Thus, we consider that the gene organization found in the T. tripolitanus mitogenome is not stable and may soon lead to the complete deletion of redundant pseudogenes, as shown in Figure 5. Alternatively, the apparently redundant ND4L and ND4 pseudogenes may not be deleted easily if they play roles in translating mRNAs for overlapping protein genes (i.e., ND4L preceding ND4* and ND4 next to ND4L*). It is well known that reading frames for vertebrate ND4L and ND4 genes partly overlap and that mature mRNAs for these genes occur as a di-cistronic mRNA [4].
With respect to the mechanisms underlying the gene rearrangements in the Stenodactylus petrii mitogenome (Figure 1C), tandem duplications of the tRNALeu(UUR) gene can occur by slipped-strand mispairing during mitogenome replication [31]. In the WAN (OL)CY tRNA gene cluster, translocation of the tRNAAla gene and OL (i.e., from WAN (OL)CY to A(OL)WNCY) probably occurred first by a process consistent with the TDRL model [29]. Then, the slipped-strand mispairing could have resulted in 4-fold copying of A(OL) to generate the S. petrii gene arrangement (Figure 1C). The intraspecific occurrence of 2- and 4-fold copies of the A(OL), as described in Results, are consistent with this mechanism.
We also inferred the process of loss and translocation of the tRNAGlu gene for Uroplatus geckos. Because all Uroplatus taxa examined in this study lack a tRNAGlu gene at the ND6/cytb gene boundary and because all non-Uroplatus geckos examined to date have this gene at this location, the disappearance of the tRNAGlu gene from this boundary likely occurred in the common ancestor of Uroplatus. The most straightforward explanation is that the tRNAGlu gene was translocated to the 5’ end of the major noncoding region by the TDRL of a four gene block: tRNAGlu, cytb, tRNAThr, and tRNAPro (the status seen for U. fimbriatus and U. sikorae mitogenomes; Figure 1D). This translocated tRNAGlu gene was later lost from all other Uroplatus species, giving rise to the gene arrangement shown in Figure 1E.
Previous molecular phylogenetic studies of Uroplatus[32, 33] suggested a sister relationship between U. fimbriatus and U. sikorae; however, they are not sister species but are nested in a clade of other Uroplatus species (i.e., U. guentheri, U. ebenaui, U. phantasticus, U. pietschmanni, and U. lineatus). If true, this raises the possibility that the mitogenome of the most recent common ancestor of all Uroplatus taxa had the tRNAGlu gene at the 5’ end of the major noncoding region and that it has disappeared from descendant Uroplatus lineages multiple times. An alternative possibility is that the tRNAGlu gene was already lost from the most recent common ancestor but that a tRNAGlu gene was newly created in the common ancestor of U. fimbriatus and U. sikorae by a mechanism such as tandem duplication of another tRNA gene and reassignment of a duplicate gene copy to tRNAGlu via anticodon mutation. However, the tRNAGlu genes of U. fimbriatus and U. sikorae retain high sequence similarity to those of other geckos (Figure 4B), which is not consistent with the latter possibility.
Two functional tRNAGln genes in the T. tripolitanusmitogenome?
An intriguing question is whether the T. tripolitanus mitogenome encodes two functional tRNAGln genes whose products can function in mitochondrial protein synthesis. CAA and CAG are two glutamine codons in the genetic code of vertebrate mitochondria and these codons are usually decoded by a single tRNAGln encoded in a mitogenome [1, 4]. There is no need to duplicate the tRNAGln gene for mitochondrial protein synthesis. Table S3 in Additional file 4 provides evidence for no genetic code change at these codons in T. tripolitanus mitochondria. There is also no evidence for codon use change. Glutamine codons (CAA + CAG) appear in protein-coding genes of the T. tripolitanus mitogenome as frequently as in the mitogenomes of 17 other geckos (Table 2). The relative frequency of CAA vs. CAG codons is not significantly different from that averaged among the 17 geckos (Table 2).
The secondary structures of the tRNAGln1 and tRNAGln2 genes (Figure 2) conserve several features of functional mitochondrial tRNA genes [22]. Briefly, both the tRNAGln genes retain many base pairings in the stem regions and share an identical anticodon sequence (TTG) in the middle of a canonical 7-nucleotide anticodon-loop. The 5’ and 3’ nucleotides of the anticodon are, respectively, T and a purine (G) for both genes. Two intervening nucleotides occur between the acceptor-stem and D-stem, whereas a single extra nucleotide occurs between the D-stem and anticodon-stem. The extra arm between the anticodon-stem and T-stem has four nucleotides in both tRNAGln genes, the typical number for vertebrate mitochondrial tRNA genes (3–5 nucleotides) [22]. Finally, no intervening nucleotide occurs between the T-stem and acceptor-stem. These tRNAGln genes appear to comply with the basic structural requirements of mitochondrial tRNA genes.
However, an extra requirement should be considered for tRNAGln genes encoded by vertebrate mitogenomes. Eukaryotic mitochondria, as well as all known archaea and most bacteria, lack a glutaminyl-tRNA synthetase (GlnRS), which is responsible for charging tRNAGln with glutamine [34, 35]. Instead, they use the non-discriminating glutamyl-tRNA synthetase (GluRS) to charge both tRNAGlu and tRNAGln with glutamic acid, thus forming Glu-tRNAGlu and Glu-tRNAGln, respectively. Glu-tRNAGln is then converted to Gln-tRNAGln by an amidotransferase (AdT) (reviewed in [36]). The crystal structure of an archaeal non-discriminating GluRS in comparison with that of an E. coli GlnRS-tRNAGln complex [37] indicated that the non-discriminating GluRS recognizes anticodon nucleotides at positions 34 (C or U at a wobble position) and 35 (U) but not at position 36 (G for tRNAGln and C for tRNAGlu). It therefore seems possible that, in T. tripolitanus mitochondria, tRNAs expressed from both the tRNAGln1 and tRNAGln2 genes could be charged with glutamic acid by the mitochondrial non-discriminating GluRS.
Recently, the crystal structure of the bacterial ‘glutamine transamidosome complex’, consisting of tRNAGln, GluRS, and AdT, indicated that glutamylation and transamidation may be consecutive reactions and that GluRS and AdT may take on conformational changes to compete for the acceptor stem of tRNAGln as their reaction target [38]. The same study showed that the bacterial AdT does not interact with the anticodon nucleotides of its substrate tRNAGln but recognizes the tRNAGln-specific tertiary structure at an outer corner of the L-shaped tRNAGln, especially in the D loop side.
It is well known that nonmitochondrial tRNAs conserve several nucleotides that are involved in forming the rigid L-shaped structure by tertiary hydrogen bondings, such as G18-Ψ56 (T56 at the DNA level) and G19-C57. Sequence comparison of mitochondrial tRNAGln genes from various vertebrates indicated that G18G19 in the D loop and T55T56C57R58A59 in the T loop are well conserved (Figure 6) while many other mitochondrial tRNA genes do not conserve these bases [22]. This observation is consistent with a view that formation of a standard L-shaped tertiary structure is necessary for a tRNAGln to be catalyzed by the mitochondrial AdT. Tropiocolotes tripolitanus tRNAGln1 conserves these bases for the tertiary interactions but tRNAGln2 does not (Figure 6). The latter even truncates nucleotides in the D and T loops considerably. Because mitochondrial tRNAs lacking the D loop/T loop interactions take on severely loosened tertiary structures [39, 40], they may not be a good substrate for the transamidation reaction catalyzed by the mitochondrial AdT.

Mitochondrial tRNAGlngene sequences for geckos and other vertebrates. tRNAGln gene sequences are aligned based on the standard clover-leaf structures. Asterisks indicate positions corresponding to conserved nucleotides for G18G19 in the D loop and T55T56C57R58A59 in the T loop (see text). Abbreviations and data sources are: tRNAGln1 (Ttri_1; this study) and tRNAGln2 (Ttri_2; this study) genes of Tropiocolotes tripolitanus, Tropiocolotes steudneri (Tste; this study), Stenodactylus petrii (Spet; this study), Gekko vittatus (Gvit; accession No. AB178897), Coleonyx variegatus (Cvar; AB114446), chicken (X52392), human (J01415), coelacanth (U82228), and trout (L29771).
Taken together, these results suggest that both tRNAGln1 and tRNAGln2 are possibly glutamylated in T. tripolitanus mitochondria but that only Glu-tRNAGln1 may be efficiently converted to Gln-tRNAGln1 for the protein synthesis. This implies that Glu-tRNAGln2 possibly remains as an inactive form or has become a harmful reagent that can decode CAR codons as glutamic acid, rather than glutamine. In this regard, Nagao et al. [41] found that human mitochondria do not allow Glu-tRNAGln to participate in protein synthesis because it is not efficiently recognized by mitochondrial elongation factor Tu. Thus, Glu-tRNAGln2 might simply be a harmless byproduct in T. tripolitanus mitochondria. Alternatively, though less likely, the mitochondrial AdTs may be able to recognize both tRNAGln1 and tRNAGln2 by a different mechanism from that of the bacterial AdTs. Metazoan mitochondrial aminoacyl-tRNA synthetases were suggested to have simplified recognition mechanisms towards substrate tRNAs in response to the decrease of structural constraints on mitochondrial tRNAs [39]. We therefore cannot rule out the possibility that the tRNAGln2 gene serves as the second functional tRNAGln gene in T. tripolitanus mitochondria.
Source of tRNAGlu for protein synthesis in U. ebenauimitochondria
We found that the Uroplatus ebenaui mitogenome apparently lacks the tRNAGlu gene. If so, how could protein synthesis be performed in U. ebenaui mitochondria? GAA and GAG are two codons for glutamic acid in the vertebrate mitochondrial genetic code [4] and no genetic code change is suggested for these codons in U. ebenaui mitochondria (Additional file 4: Table S3). Because glutamic acid codons (GAA + GAG) appear in protein-coding genes of the U. ebenaui mitogenome as frequently as in those of the mitogenomes of 17 geckos (Table 2), there must be a tRNAGlu that is responsible for decoding these codons.
The most straightforward explanation is an import of nuclear-encoded cytosolic tRNAGlu into the mitochondrion. The import of cytosolic tRNAs into vertebrate mitochondria has been suspected for some taxa. For example, marsupial mitogenomes do not appear to encode functional tRNALys genes [42]. Biochemical experiments later supported the import of cytosolic tRNALys into marsupial mitochondria [43]. The import of cytosolic tRNAs into mitochondria is more common in non-vertebrates (reviewed in [44]). The majority of tRNAs necessary for mitochondrial protein synthesis are not encoded by the Tetrahymena mitogenome and are imported from the cytosol [45, 46].
It seems noteworthy in this regard that the relative frequency of GAA vs. GAG codons in the U. ebenaui mitogenome is very strongly deviated (p < 0.001) from that averaged among 17 geckos, whereas there is no significant (p < 0.05) deviation of this relative frequency for U. fimbriatus and two Tropiocolotes species (Table 2). In general, codon usage in an organism reflects various factors, such as GC content of the genome and the relative abundance and translational efficiency of tRNAs that decode different codons (e.g., [47, 48]). In animal mitochondria, strand-specific base composition bias was also proposed as a major factor [49]. If U. ebenaui mitochondria do use tRNAGlu(s) imported from the cytosol, it seems possible that the usage of GAA and GAG codons is adapted to the different codon-decoding abilities of the imported cytosolic tRNAGlu(s) with regard to GAA and GAG.
An alternative explanation is that post-transcriptional modifications may create a tRNAGlu from other tRNA genes. Enzymatic modification of anticodon bases of tRNAs could change decoding specificity from one amino acid to another (reviewed in [50]). RNA editing could also change the decoding specificity of tRNAs. RNA editing is known to occur in various metazoan mitochondrial tRNAs ([51, 52] and refs. therein). In marsupial mitochondria, a C to U editing at the second anticodon position switches a tRNAGly (anticodon GCC) to tRNAAsp (anticodon GUC) [53]. Similar RNA editing of a G to C change at the third anticodon base of tRNAGln might supply the missing tRNAGlu for Uroplatus mitochondria. However, this RNA editing would need to occur concomitantly with other modifications in, e.g., the D arm, in order not to be converted from Glu-tRNAGlu to Gln-tRNAGlu by transamidation (see the preceding section for the recognition sites by AdT).
Conclusions
In the present study, seven new mitogenomic sequences were determined from Gekkonidae with the aid of high-throughput sequencing. Several new gene rearrangements were found and Gekkonidae can no longer be considered a group in which mitochondrial gene rearrangements rarely occur. Although the high-throughput sequencing has a weak point in assembling repeat sequences, we were able to demonstrate that moderately repetitive sequences, as found in the T. tripolitanus mitogenome, can be reliably assembled by this method. In future, high-throughput sequencing will further contribute to efficient and accurate mitogenomic sequencing from numerous metazoan taxa.
Although mitochondrial gene rearrangements have been described in various taxa, an intermediate type of gene arrangement that is indicative of molecular evolutionary mechanisms is rarely found. The unique gene arrangement found in the T. tripolitanus mitogenome provides an opportunity to study relatively new gene rearrangements in which the duplicate state of genes is maintained. Based on the characterization of duplicated genes (Additional file 2), the order of genes for tRNAArg, ND4L, and ND4 (as in the typical gene organization) may be changed to ND4L, tRNAArg, and ND4 after complete deletion of redundant genes or pseudogenes (Figure 5). However, if the duplicate pseudogenes retain a functional role in translating overlapping genes, they may not be deleted from the mitogenome, as seen in parrotfish mitochondrial tRNA pseudogenes that are retained as punctuation marks for mRNA processing [54].
In addition, the T. tripolitanus mitogenome may have gone through tRNA gene reassignment from tRNAArg to tRNAGln by a point mutation at the second anticodon position (Figure 5), although the novel tRNAGln gene may not be fully functional in translation. Mitochondrial tRNA gene reassignment has been reported in some invertebrates (e.g., [55]) but, to the best of our knowledge, it is uncommon in vertebrates. Together with the finding of tRNAGlu gene loss in the U. ebenaui mitogenome, these new features should broaden our understanding of the evolution of mitochondrial gene arrangements.
Methods
Samples and general experimental procedures
Lepidodactylus lugubris sample was collected at Chichi-jima Island of the Bonin Islands, Japan. Other animal samples of either dead or live individuals were obtained from local shops or animal dealers in Japan. All experiments in which live animals are handled were conducted carefully under the guideline of the Animal Experiment Committee of Nagoya City University with permission (No. H21N-02).
A small amount of tissue from the tail muscle was used for crude DNA extraction with a DNeasy Tissue Kit (Qiagen). PCR amplifications were conducted with SpeedSTAR HS DNA polymerase (Takara) or PrimeSTAR GXL DNA polymerase (Takara) according to the manufacturer’s instructions. The former was routinely used for <2 kbp amplifications and the latter was selected for longer amplifications. Short amplified products were purified with a High Pure PCR Cleanup Micro Kit (Roche), followed by the dye termination sequencing reaction using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies). The resultant reaction mixture was ethanol precipitated and applied to the 8-capillary 3500 Genetic Analyzer (Life Technologies) in the standard run mode.
Complete mitochondrial genome sequencing
Crude extracted DNA from a tiny amount of animal tissue was used as template for long PCR amplification that nearly covered an entire mitochondrial genome (see Additional file 1: Table S1 for primer sequences used for the long PCR amplifications for each taxon). Approximately 2 μg of amplified products were pooled and sonicated using a Bioruptor UCD-250 (Cosmo Bio) into shorter fragments (~600 bp on average). After exclusion of short DNAs (<200 bp) by binding to Solid-Phase Reversible Immobilization beads (Agencourt AMPure XP: Beckman Coulter) [56, 57], recovered DNAs were end-repaired with T4 polynucleotide kinase (Takara) and T4 DNA polymerase (Takara) using the manufacturer’s protocol.
Using the parallel tagged sequencing method described by Meyer et al. [57], palindromic 20-bp DNAs, whose sequences differ from species to species, were ligated to both 5’ and 3’ ends of the repaired products. Ligated products from multiple species were quantified simultaneously with a Quant-iT Picogreen dsDNA Assay Kit and the Qubit Fluorometer (Life Technologies) to combine them at a nearly equal molar ratio. The pooled DNAs were digested with an 8-bp recognizing restriction enzyme, SrfI (Stratagene), at the central position of the ligated tag sequences. The final product (>500 ng) was sent to Hokkaido System Science Co. for Roche GS FLX Titanium high-throughput DNA sequencing.
Roche GS FLX Titanium sequencing produced short reads of 300–400 bp in length, which were then sorted into individual species based on the attached index tag sequences. After tags were removed, reads were assembled by the GS De Novo Assembler (Roche) into one to several contigs. Highly repetitive sequences inside the control region of mitochondrial genomes are not usually assembled together. In addition, short regions between the two long PCR primers (see above) are absent. These gap regions were amplified with reptile-oriented primers [58] or species-specific primers (data not shown), sequenced with the 8-capillary 3500 Genetic Analyzer (Life Technologies), and finally assembled into a contiguous circular mitogenome sequence with Sequencher 4.8 (Gene Codes).
The pyrosequencing chemistry adopted by Roche GS FLX Titanium sequencing has a weak point with homopolymer sequences (repeats of a single base) that tend to be miscalled. Majority-rule consensus sequence was trusted only when each site is covered by many reads (typically >20 reads) and ambiguous regions, if any, were independently amplified and sequenced by the Sanger method to confirm their sequences. Note that the accuracy of the above-mentioned method for sequencing a mitogenome was confirmed using several animal species in which their complete mitogenomic sequences for the same individuals had been determined in our laboratory by Sanger sequencing alone (Kumazawa, Y., unpublished data). The determined mitogenomic sequences (Table 1) did not include any unexpected frameshifts or stop codons inside protein-coding genes, except for the potential pseudogenes of T. tripolitanus.
Analysis of gene arrangement and codon usage
Genes encoded in the determined mitogenomic sequences were identified by initial characterization with DOGMA [28] and subsequent manual inspections of gene structure, especially in light of secondary structures for mitochondrial tRNA genes [22]. In addition, we used the software Getmitogenome [59] to excise nucleotide sequences of 37 encoded genes, as well as amino acid sequences of 13 protein genes, which were added to a pre-existing alignment dataset for >150 vertebrates (data not shown). This procedure helped us to evaluate gene boundaries more carefully and to identify possible pseudogenes. Tandem repeats and CSB motifs [21] in the control region were identified with DNASIS-Mac ver. 3.5 (Hitachi Software Engineering).
Codon usage at codons for glutamine and glutamic acid was calculated using MEGA 5 [60]. Statistical significance was evaluated using the chi-square test with 5% significance level. First, the relative frequency of glutamine codons (CAA + CAG) vs. glutamic acid codons (GAA + GAG) was assumed to be equal from species to species as a null hypothesis. Deviation of the relative frequency in a species from that averaged among 17 gecko species was evaluated with the chi-square test. Second, the relative frequency of CAA vs. CAG codons was assumed to be equal from species to species as a null hypothesis and deviation of the relative frequency in a species from that averaged among 17 gecko species was evaluated in the same way. Finally, the same test was conducted to evaluate deviation of the relative frequency of GAA vs. GAG codons.
Availability of supporting data
All nucleotide sequences and annotations reported in this work will be publicly available in the DDBJ nucleotide sequence database with accession numbers shown in Table 1 and Additional file 4: Table S2.
Abbreviations
- AdT:
-
Amidotransferase
- AT6 and AT8:
-
ATPase subunit 6 and 8
- CO1-3:
-
Cytochrome oxidase subunit 1-3
- CSB:
-
Conserved sequence block
- cytb:
-
Cytochrome b
- GlnRS:
-
Glutaminyl-tRNA synthetase
- GluRS:
-
Glutamyl-tRNA synthetase
- mitogenome:
-
Mitochondrial genome
- ND1-6:
-
NADH dehydrogenase subunit 1-6
- OL:
-
Light-strand replication origin
- TDRL:
-
Tandem duplication-random loss.
References
Wolstenholme DR: Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992, 141: 173-216.
Boore JL: Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27: 1767-1780. 10.1093/nar/27.8.1767.
Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzsch G, Golombek A, Hadrys H, Juhling F, Meusemann K, Middendorf M, Misof B, Perseke M, Podsiadlowski L, Von Reumont B, Schierwater B, Schlegel M, Schrodl M, Simon S, Stadler PF, Stoger I, Struck TH: A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 2013, 69: 352-364. 10.1016/j.ympev.2013.05.002.
Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG: Sequence and organization of the human mitochondrial genome. Nature. 1981, 290: 457-465. 10.1038/290457a0.
Clayton DA: Transcription and replication of animal mitochondrial DNAs. Int Rev Cytol. 1992, 141: 217-232.
Scarpulla RC: Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008, 88: 611-638. 10.1152/physrev.00025.2007.
Okajima Y, Kumazawa Y: Mitochondrial genomes of acrodont lizards: timing of gene rearrangements and phylogenetic and biogeographic implications. BMC Evol Biol. 2010, 10: 141-10.1186/1471-2148-10-141.
Kurabayashi A, Sumida M: Afrobatrachian mitochondrial genomes: genome reorganization, gene rearrangement mechanisms, and evolutionary trends of duplicated and rearranged genes. BMC Genomics. 2013, 14: 633-10.1186/1471-2164-14-633.
Kumazawa Y, Ota H, Nishida M, Ozawa T: Gene rearrangements in snake mitochondrial genomes: highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol Biol Evol. 1996, 13: 1242-1254. 10.1093/oxfordjournals.molbev.a025690.
Amer SAM, Kumazawa Y: The mitochondrial genome of the lizard Calotes versicolor and a novel gene inversion in South Asian draconine agamids. Mol Biol Evol. 2007, 24: 1330-1339. 10.1093/molbev/msm054.
Uetz P, Hosek J: The Reptile Database. URL: http://www.reptile-database.org
Fujita MK, Boore JL, Moritz C: Multiple origins and rapid evolution of duplicated mitochondrial genes in parthenogenetic geckos (Heteronotia binoei; Squamata, Gekkonidae). Mol Biol Evol. 2007, 24: 2775-2786. 10.1093/molbev/msm212.
Jonniaux P, Hashiguchi Y, Kumazawa Y: Mitochondrial genomes of two African geckos of genus Hemitheconyx (Squamata: Eublepharidae). Mitochondrial DNA. 2012, 23: 278-279. 10.3109/19401736.2012.668898.
Zhang P, Liang D, Mao RL, Hillis DM, Wake DB, Cannatella DC: Efficient sequencing of Anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol Biol Evol. 2013, 30: 1899-1915. 10.1093/molbev/mst091.
Macey JR, Larson A, Ananjeva NB, Fang Z, Papenfuss TJ: Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol Biol Evol. 1997, 14: 91-104. 10.1093/oxfordjournals.molbev.a025706.
Amer SAM, Kumazawa Y: Mitochondrial genome of Pogona vitticepes (Reptilia; Agamidae): control region duplication and the origin of Australasian agamids. Gene. 2005, 346: 249-256.
Okajima Y, Kumazawa Y: Mitogenomic perspectives into iguanid phylogeny and biogeography: Gondwanan vicariance for the origin of Madagascan oplurines. Gene. 2009, 441: 28-35. 10.1016/j.gene.2008.06.011.
Fonseca MM, Harris DJ: Relationship between mitochondrial gene rearrangements and stability of the origin of light strand replication. Genet Mol Biol. 2008, 30: 566-574.
Kurabayashi A, Sumida M, Yonekawa H, Glaw F, Vences M, Hasegawa M: Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol Biol Evol. 2008, 25: 874-891. 10.1093/molbev/msn031.
Xu W, Jameson D, Tang B, Higgs PG: The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006, 63: 375-392. 10.1007/s00239-005-0246-5.
Sbisá E, Tanzariello F, Reyes A, Pesole G, Saccone C: Mammalian mitochondrial D-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene. 1997, 205: 125-140. 10.1016/S0378-1119(97)00404-6.
Kumazawa Y, Nishida M: Sequence evolution of mitochondrial tRNA genes and deep-branch animal phylogenetics. J Mol Evol. 1993, 37: 380-398.
Fujita MK, Papenfuss TJ: Molecular systematics of Stenodactylus (Gekkonidae), an Afro-Arabian gecko species complex. Mol Phylogenet Evol. 2011, 58: 71-75. 10.1016/j.ympev.2010.10.014.
Gamble T, Greenbaum E, Jackman TR, Russell AP, Bauer AM: Repeated origin and loss of adhesive toepads in geckos. PLoS One. 2012, 7: e39429-10.1371/journal.pone.0039429.
Metallinou M, Arnold EN, Crochet PA, Geniez P, Brito JC, Lymberakis P, Baha El Din S, Sindaco R, Robinson M, Carranza S: Conquering the Sahara and Arabian deserts: systematics and biogeography of Stenodactylus geckos (Reptilia: Gekkonidae). BMC Evol Biol. 2012, 12: 258-10.1186/1471-2148-12-258.
Bauer AM, Masroor R, Titus-McQuillan J, Heinicke MP, Daza JD, Jackman TR: A preliminary phylogeny of the Palearctic naked-toed geckos (Reptilia: Squamata: Gekkonidae) with taxonomic implications. Zootaxa. 2013, 3599: 301-324.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Wyman SK, Jansen RK, Boore JL: Automatic annnotation of organellar genomes with DOGMA. Bioinformatics. 2004, 20: 3252-3255. 10.1093/bioinformatics/bth352.
Moritz C, Dowling TE, Brown WM: Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Ann Rev Ecol Syst. 1987, 18: 269-292. 10.1146/annurev.es.18.110187.001413.
Selosse M-A, Albert B, Godelle B: Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol Evol. 2001, 16: 135-141. 10.1016/S0169-5347(00)02084-X.
Levinson G, Gutman GA: Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987, 4: 203-221.
Glaw F, Kosuch J, Henkel F-W, Sound P, Böhme W: Genetic and morphological variation of the leaf-tailed gecko Uroplatus fimbriatus from Madagascar, with description of a new giant species. Salamandra. 2006, 42: 129-144.
Greenbaum E, Bauer AM, Jackman TR, Vences M, Glaw F: A phylogeny of the enigmatic Madagascan geckos of the genus Uroplatus (Squamata: Gekkonidae). Zootaxa. 2007, 1493: 41-51.
Schön A, Kannangara CG, Gough S, Söll D: Protein biosynthesis in organelles requires misaminoacylation of tRNA. Nature. 1988, 331: 187-190. 10.1038/331187a0.
Frechin M, Duchene AM, Becker HD: Translating organellar glutamine codons: a case by case scenario?. RNA Biol. 2009, 6: 31-34. 10.4161/rna.6.1.7564.
Sheppard K, Yuan J, Hohn MJ, Jester B, Devine KM, Söll D: From one amino acid to another: tRNA-dependent amino acid biosynthesis. Nucleic Acids Res. 2008, 36: 1813-1825. 10.1093/nar/gkn015.
Nureki O, O’Donoghue P, Watanabe N, Ohmori A, Oshikane H, Araiso Y, Sheppard K, Söll D, Ishitani R: Structure of an archaeal non-discriminating glutamyl-tRNA synthetase: a missing link in the evolution of Gln-tRNAGln formations. Nucleic Acids Res. 2010, 38: 7286-7297. 10.1093/nar/gkq605.
Ito T, Yokoyama S: Two enzymes bound to one transfer RNA assume alternative conformations for consecutive reactions. Nature. 2010, 467: 612-616. 10.1038/nature09411.
Kumazawa Y, Himeno H, Miura K, Watanabe K: Unilateral aminoacylation specificity between bovine mitochondria and eubacteria. J Biochem. 1991, 109: 421-427.
Wakita K, Watanabe Y, Yokogawa T, Kumazawa Y, Nakamura S, Ueda T, Watanabe K, Nishikawa K: Higher-order structure of bovine mitochondrial tRNAPhe lacking the 'conserved' GG and TΨCG sequences as inferred by enzymatic and chemical probing. Nucleic Acids Res. 1994, 22: 347-353. 10.1093/nar/22.3.347.
Nagao A, Suzuki T, Katoh T, Sakaguchi Y: Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc Natl Acad Sci U S A. 2009, 106: 16209-16214. 10.1073/pnas.0907602106.
Janke A, Feldmaier-Fuchs G, Thomas WK, Von Haeseler A, Pääbo S: The marsupial mitochondrial genome and the evolution of placental mammals. Genetics. 1994, 137: 243-256.
Dörner M, Altmann M, Pääbo S, Mörl M: Evidence for import of a lysyl-tRNA into marsupial mitochondria. Mol Biol Cell. 2001, 12: 2688-2698. 10.1091/mbc.12.9.2688.
Schneider A: Mitochondrial tRNA import and its consequences for mitochondrial translation. Ann Rev Biochem. 2011, 80: 1033-1053. 10.1146/annurev-biochem-060109-092838.
Suyama Y: The origins of mitochondrial ribonucleic acids in Tetrahymena pyriformis. Biochemistry. 1967, 6: 2829-2839. 10.1021/bi00861a025.
Rusconi CP, Cech TR: The anticodon is the signal sequence for mitochondrial import of glutamine tRNA in Tetrahymena. Genes Dev. 1996, 10: 2870-2880. 10.1101/gad.10.22.2870.
Osawa S, Jukes TH, Watanabe K, Muto A: Recent evidence for evolution of the genetic code. Microbiol Rev. 1992, 56: 229-264.
Satoh TP, Sato Y, Masuyama N, Miya M, Nishida M: Transfer RNA gene arrangement and codon usage in vertebrate mitochondrial genomes: a new insight into gene order conservation. BMC Genomics. 2010, 11: 479-10.1186/1471-2164-11-479.
Asakawa S, Kumazawa Y, Araki T, Himeno H, Miura K, Watanabe K: Strand-specific nucleotide composition bias in echinoderm and vertebrate mitochondrial genomes. J Mol Evol. 1991, 32: 511-520. 10.1007/BF02102653.
Watanabe K, Yokobori S: tRNA modification and genetic code variations in animal mitochondria. J Nucleic Acids. 2011, 2011: 623095-
Yokobori SI, Pääbo S: tRNA editing in metazoans. Nature. 1995, 377: 490-10.1038/377490a0.
Chateigner-Boutin AL, Small I: Organellar RNA editing. Wiley Interdiscip Rev RNA. 2011, 2: 493-506. 10.1002/wrna.72.
Börner GV, Mörl M, Janke A, Pääbo S: RNA editing changes the identity of a mitochondrial tRNA in marsupials. EMBO J. 1996, 15: 5949-5957.
Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M: Gene rearrangements and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae). J Mol Evol. 2004, 59: 287-297. 10.1007/s00239-004-2621-z.
Cantatore P, Gadaleta MN, Roberti M, Saccone C, Wilson AC: Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature. 1987, 329: 853-855. 10.1038/329853a0.
De Angelis MM, Wang DG, Hawkins TL: Solid-phase immobilization for the isolation of PCR products. Nucleic Acids Res. 1995, 23: 4742-4743. 10.1093/nar/23.22.4742.
Meyer M, Stenzel U, Hofreiter M: Parallel tagged sequencing on the 454 platform. Nat Protoc. 2008, 3: 267-278. 10.1038/nprot.2007.520.
Kumazawa Y, Endo H: Mitochondrial genome of the Komodo dragon: efficient sequencing method with reptile-oriented primers and novel gene rearrangements. DNA Res. 2004, 11: 115-125. 10.1093/dnares/11.2.115.
Jonniaux P: Getmitogenome: a software for manipulating animal mitochondrial gene sequences. 2012, URL: http://www.nsc.nagoya-cu.ac.jp/~kuma/lab_homepage/getmitogenome.html
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Acknowledgements
We thank Messrs. Takeshi Ebinuma and Kosho Yagi and Ms. Yuki Dehara for providing animal samples. We also thank Dr. Hideaki Tagami for allowing us to use his laboratory equipment. We are grateful to Mr. Yao Sun for technical assistance in a part of sequencing experiments and Dr. Morihiro Notohara for advice on statistical analyses. Our gratitude is extended to Dr. Pierre Jonniaux for his bioinformatic instructions and Dr. Hyouta Himeno for invaluable comments on the functional structures of tRNAGln. This study was funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant No. 20370033) to YK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YK designed the study and obtained a fund to conduct it. YK, SM, CY, and YH collaborated in experiments for determining complete mitogenomic sequences of geckos. YK and SM carried out additional experiments to examine gene arrangements in closely related species. Gene annotations on determined sequences were done by YK, SM, and CY. An initial manuscript was written by YK and improved by YH. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2014_6628_MOESM1_ESM.pdf
Additional file 1: Table S1: Primers used in this study. Figure S1. Location of primers used to verify the gene arrangement of T. tripolitanus mitogenome. Figure S6. Four tRNALeu(UUR) genes found in the S. petrii mitogenome. (PDF 356 KB)
12864_2014_6628_MOESM2_ESM.pdf
Additional file 2: Figure S2: Alignment of ND4L gene sequences of geckos. Figure S3. Alignment of ND4L amino acid sequences of geckos. Figure S4. Alignment of ND4 gene sequences of geckos. Figure S5. Alignment of ND4 amino acid sequences of geckos. (PDF 6 MB)
12864_2014_6628_MOESM3_ESM.pdf
Additional file 3: Figure S7: 1% agarose gel electrophoresis of PCR products amplified from closely related gecko species using rND2-4L and H5934m. Figure S8. 1% agarose gel electrophoresis of PCR products amplified from closely related gecko species using rND3-1L and rCUN-3H. (PDF 141 KB)
12864_2014_6628_MOESM4_ESM.pdf
Additional file 4: Table S2: Sequences determined from closely related species in this study. Table S3. Evidence for no genetic code change in glutamine and glutamic acid codons for four gecko species. (PDF 83 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kumazawa, Y., Miura, S., Yamada, C. et al. Gene rearrangements in gekkonid mitochondrial genomes with shuffling, loss, and reassignment of tRNA genes. BMC Genomics 15, 930 (2014). https://doi.org/10.1186/1471-2164-15-930
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-930