
Abstract
(Z,Z’)-1,1′-(4-ortho-Caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime) intermediate 3 was synthesized by a three-step reaction with a final treatment with base to give a new series of ortho-carboranyl biphenyloxime derivatives (4–8). Compounds 7 and 8 showed high solubility and the in vitro study results revealed high levels of accumulation in HeLa cells with higher cytotoxicity and boron uptake compared to l-boronphenylalanine.

Similar content being viewed by others

Introduction
Carborane (C2B10H12, Fig. 1) is a spherical compound formed by one or more boron peaks of polyhedral boron compounds, which is formed by carbon atoms. The volume is similar to that of a benzene ring [1,2,3,4,5]. This is a special large steric skeleton with a very strong hydrophobic structure. Therefore, improvement of the chemical structure can alter the stability, water solubility, and biological activity of compatibility and allow wider applications of carborane as a BNCT agent [6,7,8,9]. Boron neutron capture therapy (BNCT) was first proposed as a potential cancer therapy in 1936, based on the thermal neutron captured by 10B atoms then produces a 4He (α-particle) and a 7Li ion [10, 11]. However, its successful application in the treatment of cancer patients still presents a challenge in medical research [12]. A major challenge in designing boron containing drugs for BNCT of cancer is the selective delivery of 10B to the tumor as well as water solubility [13]. Our synthetic strategy was to use heterocyclic alkyl chains as a boron delivery system, the target molecules being the heterocyclic alkyl oxime chains in which the boron functionality was present as a ortho-carborane. The large number of boron atoms has a clear advantage for BNCT [14]. This paper reports the hydrophilic carboranylbenzyloxime moiety, such as alkylmorpholine, alkylpiperidine, phenoxyalkyl, and pyridine, on carbon–oxygen combined with chemical bonding. These compounds have higher solubility in polar solvents and increased the boron uptake in tumor cells, highlighting the potential use of carborane as a hydrophilic carrier into the body that can pass the Blood Brain Barrier (BBB rule) to the cells within the organization for drug evaluation.

Comparison of the o-Carborane and benzene
Experimental
All manipulations were performed under a dry nitrogen atmosphere using standard Schlenk techniques. Tetrahydrofuran (THF) was purchased from Aladdin Pure Chemical Company and dried over sodium metal distillation prior use. The reactions were monitored on Merck F-254 pre-coated TLC plastic sheets using hexane as the mobile phase. All yields refer to the isolated yields of the products after column chromatography using silica gel (200–230 mesh). All glassware, syringes, magnetic stirring bars, and needles were dried overnight in a convection oven. Ortho-carborane (C2H2B10H10) was purchased from HENAN WANXIANG Fine Chemical Company and used after sublimation. The NMR spectra were recorded on a Bruker 300 spectrometer operated and the chemical shifts were measured relative to the internal residual peaks from the lock solvent (99.9% CDCl3 and CD3COCD3), and then referenced to Si(CH3)4 (0.00 ppm). The Fourier transform infrared (FTIR) spectra of the samples were recorded on an Agilent Cary 600 Series FT-IR spectrometer using KBr disks. Elemental analyses were performed using a Carlo Erba Instruments CHNS–O EA1108 analyzer (Additional file 1).
Synthetic routes and experimental data
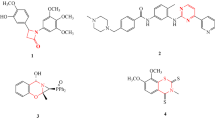
Synthesis of bis(3-methoxybenzyl)-ortho-carborane (1). A 2.5 M n-BuLi (4.0 mL, 10 mmol) solution was added via a syringe to a solution of o-carborane (1.44 g, 10 mmol) in 50 mL of THF at − 78 °C. A solution of 1-(bromomethyl)-3-methoxybenzene (4.22 g, 21 mmol) in THF 10 mL was added slowly to the reaction flask at − 78 °C, and the reaction temperature was maintained at − 78 °C for 1 h. The reaction mixture was then warmed slowly to room temperature, stirred for an additional 12 h, and quenched with distilled H2O (30 mL). The crude product was then extracted with methylene chloride (30 mL × 3). The organic layer was washed with H2O, dried with anhydrous Na2SO4, and filtered then concentrated. The residue was purified by flash column chromatography (ethyl acetate/hexane 1:10) to give compound 1 as a colorless oil: yield: 3.6 g (93%). IR(KBr pellet), cm−1, ν: (B-Ho-carborane) 2593. 1HNMR (CDCl3), δ, ppm: 3.2–0.8 (br, B-Ho-carborane, 10H), 3.61 (s, –CH2, 4H), 3.83 (s, –OCH3, 6H), 6.77 (s, 1-Hbenzene, 2H), 6.84–6.82 (d, J = 6.9 Hz, 2-Hbenzene, 2H), 6.90–6.88 (d, J = 6.9 Hz, 3-Hbenzene, 2H), 7.32–7.29 (m, 4-Hbenzene, 2H). Found, %: C 56.31; H 7.65. C18H28B10O2. Calculated, %: C 56.23; H 7.34.
Synthesis of 1,1′-(4-caboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one) (2). Acetyl chloride (1.4 mL, 20 mmol) was added via a syringe to a solution of aluminum chloride (2.6 g, 20 mmol) in 50 mL of methylene chloride at 0 °C and stirred for 30 min. A solution of compound 1 (3.5 g, 10 mmol) in methylene chloride 10 mL was added slowly to the reaction flask at 0 °C, and the reaction temperature was maintained at 0 °C for 30 min. The reaction mixture was then warmed slowly to room temperature, stirred for an additional 3 h, and quenched with a saturated NaHCO3 (30 mL) solution. The crude product was then extracted, and the organic layer was washed with H2O, dried with anhydrous Na2SO4, and filtered then concentrated. The residue was purified by flash column chromatography (ethyl acetate/hexane 1:8) to give compound 2 as a colorless oil: yield: 4.1 g (97%). IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2602. 1HNMR(CDCl3), δ, ppm: 3.2–0.8 (br, B-Ho-carborane, 10H), 3.64 (s, –CH3, 6H), 3.66 (s, –CH2, 4H), 3.95 (s, –OCH3, 6H), 6.82 (s, 1-Hbenzene, 2H), 6.89–6.86 (d, J = 7.8 Hz, 2-Hbenzene, 2H), 7.77–7.74 (d, J = 7.8 Hz, 3-Hbenzene, 2H). Found, %: C 56.42; H 6.67. C22H32B10O4. Calculated, %: C 56.39; H 6.88.
Synthesis of (Z,Z′)-1,1′-(4-caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime) (3). A solution of compound 2 (3.8 g, 8.1 mmol) and hydroxylamine (1.2 g, 17.8 mmol) in 40 mL of methanol was heated under reflux for 2 h. The reaction mixture was then cooled to room temperature, and the crude product was concentrated. The residue was purified by flash column chromatography (ethyl acetate/hexane 1:4) to give compound 3 as a colorless oil: Yield: 3.7 g (92%). IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2586. 1H NMR (CD3COCD3), δ, ppm: 3.16 (s, –CH3, 6H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.88 (s, –OCH3, 6H), 3.93 (s, –CH2, 4H), 6.97–6.95 (d, J = 7.5 Hz, 2-Hbenzene, 2H), 7.05 (s, 1-Hbenzene, 2H), 7.30–7.28 (d, J = 7.5 Hz, 3-Hbenzene, 2H). Found, %: C 52.68; H 6.81; N 5.69. C22H34B10N2O4. Calculated, %: C 52.99; H 6.87; N 5.62.
Synthesis of (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-dipyridin-2-ylmethyldioxime (4). A solution of compound 3 (0.7 g, 1.4 mmol) and potassium carbonate (0.4 g, 3.0 mmol) in 10 mL of acetonitrile was stirred at room temperature for 30 min. Subsequently, (2-bromomethyl)pyridine (0.5 g, 3.0 mmol) was added at room temperature, and then heated under reflux for 5 h. The crude product was then concentrated, and the residue was purified by flash column chromatography (ethyl acetate/hexane 1:4) to give compound 4 as a yellow oil: Yield: 0.8 g (88%). IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2607. 1HNMR (CD3Cl), δ, ppm: 2.31 (s, –CH2, 6H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.63 (s, –CH3, 4H), 3.84 (s, –OCH3, 6H), 5.37 (s, –CH2, 2H), 6.73 (s, 1-Hbenzene, 2H), 6.80–6.77 (d, J = 7.8 Hz, 2-Hbenzene, 2H), 7.29–7.24 (m, 3-Hbenzene and pyridine, 4H), 7.47–7.44 (d, J = 7.8 Hz, 3-Hpyridine, 2H), 7.76–7.70 (t, J = 7.8 Hz, 2-Hpyridine, 2H), 8.61–8.59 (d, J = 4.8 Hz, 1-Hpyridine, 2H). Found, %: C 59.36; H 6.63; N 8.35. C34H44B10N4O4. Calculated, %: C 59.98; H 6.51; N 8.23.
Synthesis of (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-phenoxyethyl)dioxime (5). A procedure analogous to the preparation of 4 was used and a colorless oil was obtained. Yield: 0.9 g (89%). IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2577. 1H NMR (CD3Cl) δ, ppm: 2.22 (s, –CH3, 6H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.64 (s, –CH2, 4H), 3.85 (s, –OCH3, 6H), 4.31–4.28 (t, J = 4.8 Hz, –CH2 alkyl-1, 4H), 4.56–4.52 (t, J = 5.1 Hz, –CH2 alkyl-2 4H), 6.75 (s, 1-Hbenzene-1 2H), 6.83–6.80 (d, J = 7.5 Hz, 2-Hbenzene-1, 2H), 7.00–6.95 (m, 1-Hbenzene-2, 6H), 7.34–7.29 (m, 2-Hbenzene-1 and 2, 6H). Found, %: C 61.47; H 6.92; N 3.84. C38H50B10N2O6. Calculated, %: C 61.77; H 6.82; N 3.79.
Synthesis of (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(3-phenoxypropyl)dioxime (6). A procedure analogous to the preparation of 4 was used and a colorless oil was obtained. Yield: 0.9 g (86%). IR (KBr pellet), cm−1, ν: (B–H) 2589. 1H NMR(CD3Cl), δ, ppm: 2.25–2.17 (m, –CH3 and -CH2 alkyl-1, 10H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.64 (s, –CH2, 4H), 3.85 (s, –OCH3, 6H), 4.16–4.12 (t, J = 6.0 Hz, –CH2 alkyl-2, 4H), 4.40–4.36 (t, J = 6.0 Hz, –CH2 alkyl-3, 4H), 6.74 (s, 1-Hbenzene-1, 2H), 6.82–6.79 (d, J = 7.8 Hz, 2-Hbenzene-1, 2H), 6.96–6.93 (m, 1-Hbenzene-2, 6H), 7.33–7.30 (m, 2-Hbenzene-1 and 2, 6H). Found, %: C 62.52; H 7.12; N 3.77. C40H54B10N2O6. Calculated, %: C 62.64; H 7.10; N 3.65.
Synthesis of (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-piperidin-1-ylethyl)dioxime (7). A procedure analogous to the preparation of 4 was used and a colorless oil was obtained. Yield: 0.8 g (82%) colorless oil. IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2591. 1H NMR (CD3Cl), δ, ppm: 1.47–1.45 (m, 1-Hpiperidine, 4H), 1.64–1.60 (m, 2-Hpiperidine, 4H), 1.88–1.86 (m, 3-Hpiperidine, 4H), 2.19 (s, –CH3, 6H), 2.53–2.51 (m, 8H), 2.76–2.72 (t, J = 6.0 Hz, –CH2 alkyl-1, 4H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.63 (s, –CH2, 4H), 3.85 (s, –OCH3, 6H), 4.36–4.32 (t, J = 6.0 Hz, –CH2 alkyl-2, 4H), 6.74 (s, 1-Hbenzene, 2H), 6.82–6.79 (d, J = 7.8 Hz, 2-Hbenzene, 2H), 7.31–7.29 (d, J = 7.8 Hz, 3-Hbenzene, 2H). Found, %: C 59.65; H 8.34; N 7.68. C36H60B10N4O4. C 59.97; H 8.39; N 7.77.
Synthesis of (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-morpholinoethyl)dioxime (8). A procedure analogous to the preparation of 4 was used and a colorless oil was obtained. Yield: 0.9 g (84%). IR (KBr pellet), cm−1, ν: (B-Ho-carborane) 2596. 1HNMR (CD3Cl), δ, ppm: 2.52 (s, –CH3, 6H), 2.55–2.54 (m, –CH2 alkyl-1, 4H), 2.77–2.72 (t, J = 6.9 Hz, –CH2 alkyl-2, 4H), 3.2–0.8 (br, B-Ho-carborane, 10H), 3.64–3.59 (m, 1-Hmorpholine, 8H), 3.76–3.73 (m, 2-Hmorpholine, 8H), 3.85 (s, –OCH3, 6H), 6.83–6.76 (m, 2-Hbenzene, 4H), 7.31 (s, 2-Hbenzene, 2H). Found, %: C 56.38; H 7.83; N 7.64. C34H56B10N4O6. C 56.33; H 7.79; N 7.73.
Cell viability assay (MTT assay)
HeLa cells in a 3 × 104/mL cell suspension per hole in 96 well plates were digested by adding 100 μL of a cell suspension and culturing for 24 h to absorb the original culture medium followed by the addition of 200 μL configured compounds-4, 5, 6, 7, 8 and BPA (l-boronphenylalanine). Each concentration was made from 4 compound holes, and the holes around the 96 well plates were sealed with PBS, the negative control. The blank control group lacked the compounds. After 24 h, 20 μL of a MTT solution was added to each hole, and cultured for 4 h. Subsequently, DMSO 150 μL was added to the medium through a suction hole and shaken for 10 min. The OD of each hole was determined at 490 nM, and the sample inhibition rate in different concentrations was calculated: inhibition rate = (Control OD value/Delivery OD value)/Control OD value × 100%. Finally, the IC50 value of the sample was calculated using the related software.
Boron uptake
HeLa cells (5 × 103) were incubated for 48 h in the presence of various concentrations of compounds 4, 5, 6, 7, 8, and BPA. After washing three times, the cumulative boron concentration was determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) [15, 16]. (± is the average value).
Results and discussion
This paper reports the hydrophilic function of the ortho-carboranylbenzyloxime moiety, such as alkylmorpholine, alkylpiperidine, phenoxyalkyl and pyridine, on carbon–oxygen combined with chemical bonding. These compounds have higher solubility in polar solvents and increasing boron uptake in tumor cells within the organization for a drug evaluation.
A general procedure for the preparation for 4-ortho-caboranyldimethyl-bis(phenyloxime) consisted of a serial reaction, such as Grignard, Friedel–Crafts, amination, and electrophilic substitution under basic conditions. A series of carborane intermediates 1–3 were prepared using the optimized procedure from the starting material. Ortho-Carborane was dissolved in dry tetrahydrofuran at − 78 °C, and treated with a Grignard reagent carbanion, and then substituted with an aromatic halide. Subsequently, aluminum chloride was used in the Friedel–Craft reaction to afford 1,1′-(4-ortho-caboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one), which was followed by the addition of hydroxylamine-hydrochloride salt to give the (Z,Z′)-1,1′-(4-ortho-Caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime) form in the presence of compound-3 (Scheme 1) [17,18,19,20,21].

Preparation of (Z,Z’)-1, 1′-(4-Caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime)
Finally, ortho-carboranyl hydrophilic ether compounds were generated from (Z,Z′)-1,1′-(4-ortho-Caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime) and side hydrophilic alkyl or aromatic halide reagents, followed by a treatment with potassium carbonate to result in the target compounds 4–8 (Scheme 2) [22, 23]. A treatment of ortho-carborane (C2H2B10H10) with aromatic halide as a base in tetrahydrofuran produced the target compounds 1–3 in moderate yields (1 93, 2 97, and 3 92%). Compounds 1–3 showed absorption bands in the infrared (IR) spectrum at 2602 and 2593 cm−1. The diagnostic signals of compounds 1–3 were the aromatic peaks observed at δ 7.77 and 6.77 in the 1H NMR spectra and a broad signal caused by B–H peaks for the ortho-carborane units from δ 3.2–0.8.

Preparation of (Z,Z′)-1,1′-(4-Caboranyldimethyl)-bis (hydrophilic functional) derivatives(4–8)
The major requirement of a BNCT agent is a high water solubility, high boron uptake, and low cytotoxicity. The HeLa cervical carcinoma cells were treated with the candidate compounds 4–8 for 2 days, and the cell viability was determined by a MTT assay. Compounds 4–8 exhibited boron uptake in the range of 0.106–0.520 ppm (Table 1), and the cell cytotoxicity was in the range of 1.134–2.516 µM, as shown Fig. 2. In particular, compounds 7 and 8 showed high boron uptake in HeLa cells, and both compounds had higher cytotoxicity than BPA (l-boronphenylalanine). Morpholine and piperidine is a heterocyclic nitrogen and oxygen member six-ring reagent with a simple structure that improves the water solubility and bioactivity improvement. They are used in the preparation of pharmaceutical drugs for their anti-inflammation, anticancer, and antiviral activity [24,25,26,27,28].

Accumulation of compounds 4–8 into HeLa cells
Conclusion
In conclusion, we reported the series of ortho-carborane substituted bipolar-function derivatives, such as alkyl pyridine, alkyl phenoxide, alkyl morpholine, and alkyl piperidine, were synthesized. The target compounds coupling of the aryl-oxime with chain functional group proceeded successfully for introduction of an ortho-carborane moiety in the molecules, which can easily be further four-step substituted to high yield final compound. The effects of synthesized compounds on biology activity were assay in HeLa cells. Both cyclic alkyl derivatives of ortho-carborane and oxime containing compounds, 7 and 8, respectively, were exhibit high boron uptake and higher cytotoxicity than BPA (l-boronphenylalanine). This resulted in carborane compounds with improved water solubility for the BNCT agent. The knowledge gained from modified bipolar groups could facilitate both drug selection and evaluations.
References
Nicoud JF, Bolze F, Sun XH, Hayek A, Baldeck P (2011) Boron-containing two-photon-absorbing chromophores. 3.1 One- and two-photon photophysical properties of p-carborane-containing fluorescent bioprobes. Inorg Chem 50:4272–4278
Dávalos JZ, González J, Ramos R, Hnyk D, Holub J, Santaballa JA, Moisés CL, Oliva JM (2014) Acidities of closo-1-COOH-1,7‑C2B10H11 and amino acids based on icosahedral carbaboranes. J Phys Chem A 118:2788–2793
Kaszynski P, Januszko A, Glab KL (2014) Comparative analysis of fluorine-containing mesogenic derivatives of carborane, Bicyclo[2.2.2]octane, cyclohexane, and benzene using the Maier–Meier theory. J Phys Chem B 118:2238–2248
Kobr L, Zhao K, Shen YQ, Polívková K, Shoemaker RK, Clark NA, Price JC, Rogers CT, Michl J (2013) Inclusion compound based approach to arrays of artificial dipolar molecular rotors: bulk inclusions. J Org Chem 78:1768–1777
Otsuka M, Takita R, Kanazawa J, Miyamoto K, Muranaka A, Uchiyama M (2015) Conjugation between σ- and π-aromaticity in 1-C-arylated monocarba-closo-dodecaborate anions. J Am Chem Soc 137:15082–15085
Wang JL, Chen LF, Ye J, Li ZY, Jiang H, Yan H, Igor B, Sivaev MYS, Wang VI, Bregadze XM (2017) Carborane derivative conjugated with gold nanoclusters for targeted cancer cell imaging. Biomacromolecules 18:1466–1472
Ruan Z, Yuan P, Jing TT, Xing T, Yan LF (2018) pH-Sensitive polypeptide conjugated with carborane clusters and cyanine for NIR bioimaging and multi-therapies. Macromol Res 26:270–277
Calabrese G, Daou A, Barbu E, Tsibouklis J (2018) Towards carborane-functionalised structures for the treatment of brain cancer. Drug Discov Today 23:63–75
Kawasaki R, Sasaki Y, Akiyoshi K (2017) Self-assembled nanogels of carborane-bearing polysaccharides for boron neutron capture therapy. Drug Discov Today 46:513–515
Locher GL (1936) Biological effects and therapeutic possibilities of neutrons. Am J Roentgenol 36:1–13
Sumitani S, Nagasaki Y (2012) Boron neutron capture therapy assisted by boron-conjugated nanoparticles. Polym J 44:522–530
Wang LW, Wang LW, Liu YWH, Chou FI, Jiang SH, Chou FI (2018) Clinical trials for treating recurrent head and neck cancer with boron neutron capture therapy using the Tsing-Hua Open Pool Reactor. Cancer Commun 38:37–46
Daniela P, Angela R, Antonietta P, Saverio A, Silva B, Ian P, Nicoletta P, Cinzia F, Laura C, Maria CA (2015) Water-soluble carboranyl-phthalocyanines for BNCT. Synthesis, characterization, and in vitro tests of the Zn(II)-nido-carboranyl-hexylthiophthalocyanine. Dalton Trans 44:11021–11028
Issei T, Kensuke N, Kimiko M (2017) Hydrophobic boron compound-loaded poly(l-lactide-co-glycolide) nanoparticles for boron neutron capture therapy. Colloids Surf B 159:360–365
Li L, Zheng SH, Yang QZ, Chen SL, Huang LF (2016) Distinguishing Astragalus mongholicus and its planting soil samples from different regions by ICP-AES. Molecules 21:482–493
Haselsberger K, Radner H, Pendi G (2017) Boron neutron capture therapy: boron biodistribution and pharmacokinetics of Na2B12H, SH in Patients with Glioblastoma1. Cancer Res 40:444–457
Lee CH, Jin GF, Yoon JH, Jung YJ, Lee JD, Cho SD, Nakamura H, Kang SO (2008) Synthesis and characterization of polar functional group substituted mono- and bis-(o-carboranyl)-1,3,5-triazine derivatives. Tetrahedron Lett 49:159–164
Dijkstra PJ, Hertog HJ, Steen BJ, Zijlstra S, Skowronska-Ptasinska M, Reinhoudt DN, Eerden J, Harkemat S (1987) Use of pyrylium synthons in the synthesis of hemispherands with modified cavities. X-ray structures of the 21-hemispherand and a pyrido hemispherand. J Org Chem 52:2433–2442
Naik R, Won M, Ban HS, Bhattarai D, Xu XZ, Eo Y, Hong YS, Singh S, Choi Y, Ahn HC, Lee K (2014) Synthesis and structure–activity relationship study of chemical probes as hypoxia induced factor-1α/malate dehydrogenase 2 inhibitors. J Med Chem 57:9522–9538
Ma XP, Li K, Wu SY, Liang C, Su GF, Mo DL (2017) Construction of 2,3-quaternary fused indolines from alkynyl tethered oximes and diaryliodonium salts through A cascade strategy of N-arylation/cycloaddition/[3]-rearrangement. Green Chem 1:1–7
Funakoshi S, Murayama E, Guo L, Fujii N, Yajima H (1988) A modified benzhydrylamine as a handle reagent for the solid phase synthesis of peptide amides based on the fluorenylmethoxycarbonyl method. J Chem Soc Chem Commun 5:382–384
Sun L, Wu J, Luo M, Wang XL, Pan M, Gou ZP, Sun DQ (2011) Diversity oriented design of various benzophenone derivatives and their in vitro antifungal and antibacterial activities. Molecules 16:9739–9754
Chakravarti B, Akhtar T, Rai B, Yadav M, Siddiqui JA, Dwivedi SKD, Thakur R, Singh AK, Singh AK, Kumar H, Khan K, Pal S, Rath SK, Lal J, Konwar R, Trivedi AK, Mishra DP, Godbole MM, Sanyal S, Chattopadhyay N, Kumar A (2014) Thioaryl naphthylmethanone oxime ether analogs as novel anticancer agents. J Med Chem 57:8010–8025
Hofmans S, Devisscher L, Martens S, Rompaey DV, Goossens K, Divert T, Nerinckxm W, Takahashi N, Winter HD, Veken PVD, Goossens V, Vandenabeele P, Tozasertib Augustyns K (2018) Analogues as inhibitors of necroptotic cell death. J Med Chem 61:1895–1920
Sayed MTE, Sharief MAMSE, Zarie ES, Morsy NM, Elsheakh AR, Voronkov A, Berishvili V, Hassan GS (2018) Design, synthesis, anti-inflammatory activity and molecular docking of potential novel antipyrine and pyrazolone analogs as cyclooxygenase enzyme (COX) inhibitors. Bioorg Med Chem Lett 28:952–957
Barberot C, Moniot A, Allart-Simon I, Malleret L, Yegorova T, Laronze-Cochard M, Bentaher A, Médebielle M, Bouillon JP, Hénon E, Sapi J, Velard F, Gérard S (2018) Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Euro J Med Chem 18:1–5
Singh J, Saini V, Kumar A, Bansal R (2017) Synthesis, molecular docking and biological evaluation of some newer 2- substituted-4-(benzo[d][1,3]dioxol-5-yl)-6-phenylpyridazin-3(2H)-ones as potential anti-inflammatory and analgesic agents. Bioorg Chem 17:1–31
Kamble A, Kamble R, Dodamani S, Jalalpure S, Rasal V, Kumbar M, Joshi S, Dixit S (2017) Design, synthesis and pharmacological analysis of 5-[40- (substituted-methyl)[1,10-biphenyl]-2-yl]-1H-tetrazoles. Arch Pharm Res 40:444–457
Authors’ contributions
XFY designed and finalized the scheme; LRJ performed review work and JGF wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
This study was supported financially by the scientific research foundation of Jiangsu University (Grant No. 5501290005).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data are fully available without restriction at the author’s institutions.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1.
1H-NMR bis(3-methoxybenzyl)carborane (1). Figure S2. 1H-NMR1,1′-(4-caboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one) (2). Figure S3. 1H-NMR (Z,Z′)-1,1′-(4-caboranyldimethyl)-bis(2-methoxyphenylethan-1-oxime) (3). Figure S4. 1H-NMR (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-dipyridin-2-ylmethyldioxime (4). Figure S5. 1H-NMR (1Z,1′Z)-1,1′-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-phenoxyethyl)dioxime (5). Figure S6. 1H-NMR (1Z,1′Z)-1,1’-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(3-phenoxypropyl)dioxime (6). Figure S7. 1H-NMR (1Z,1′Z)-1,1’-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-piperidin-1-ylethyl)dioxime (7). Figure S8. 1H-NMR (1Z,1′Z)-1,1’-(carboranyldimethyl)-bis(2-methoxy-4,1-phenylene-ethan-1-one)-O,O-di(2-morpholinoethyl)dioxime (8).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jin, G., Xiao, F. & Liu, R. Synthesis and biological evaluation of a new series of ortho-carboranyl biphenyloxime derivatives. Chemistry Central Journal 12, 76 (2018). https://doi.org/10.1186/s13065-018-0444-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0444-z