
Abstract
Incidence of hepatocellular carcinoma (HCC) is on the rise due to the prevalence of chronic hepatitis and cirrhosis. Although there are surgical and chemotherapy treatment avenues the mortality rate of HCC remains high. Immunotherapy is currently the new frontier of cancer treatment and the immunobiology of HCC is emerging as an area for further exploration. The tumor microenvironment coexists and interacts with various immune cells to sustain the growth of HCC. Thus, immunosuppressive cells play an important role in the anti-tumor immune response. This review will discuss the current concepts of immunosuppressive cells, including tumor-associated macrophages, marrow-derived suppressor cells, tumor-associated neutrophils, cancer-associated fibroblasts, and regulatory T cell interactions to actively promote tumorigenesis. It further elaborates on current treatment modalities and future areas of exploration.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths worldwide, with approximately 800,000 cases per year [1]. Most cases appear in the context of cirrhosis and are most often associated with chronic hepatitis B and hepatitis C virus infection. In the case of localized HCC, surgical resection, liver transplantation, and tumor ablation are potential cures. Advancements such as laparoscopic liver resection and living donor transplantation continue to develop and influence treatment options [2, 3]. For patients with locally advanced disease, interventional techniques such as transarterial chemoembolization or transradial radioembolization can provide disease control or lead to tumor regression and hypertrophy in future liver remnants [4, 5]. Systemic sorafenib has been shown to be effective in patients with severe cirrhosis who are not suitable for liver-directed therapy and patients with metastatic HCC who have slow disease progression. However, it only exerts a weak therapeutic effect [6, 7]. Therefore, a novel treatment strategy with different mechanisms from those of conventional treatments is needed to improve the prognosis of HCC.
Since the approval of cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) inhibitors for the treatment of melanoma, immunotherapy has emerged as a potential alternative treatment option in clinical practice. Get widespread attention. Malignant tumors [8]. HCC is an inflammation-driven disease with potentially chronic liver inflammation and cirrhosis, and a quarter of HCC cases express markers of inflammatory response. However, these tumors also have fewer chromosomal aberrations, suggesting a combination of immunological interventions may be more effective with conventional treatment of this disease [9]. Major immunotherapeutic strategies for HCC can be classified into five categories: adoptive cell therapies; cytokines; vaccines; immune checkpoint inhibitors; and oncolytic viruses. A phase II clinical trial using tremelimumab included 21 patients with advanced HCC, with partial response rates and disease control rates of 17.6 and 76.4%, respectively [10]. PD-1 is expressed on B cells, T cells, natural killer (NK) cells, and dendritic cells (DCs) [11]. The PD-1 monoclonal antibody (mAb) blocks receptor binding of PD-L1 and PD-L2 to activate immune cells [12]. The researchers found that the PD-1 inhibitor nivolumab activates a sustained tumor-specific immune response and that side effects are controllable [13]. Treatment with PD-1 and CTLA-4 can stimulate T cell activation to enhance tumor eradication.
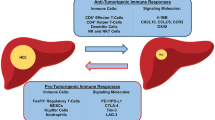
In the tumor microenvironment, non-malignant cells can help tumor cells to proliferate, invade and metastasize. The immunosuppressive features of tumor lesions participate not only as one of the major players inducing cancer progression but also a big challenge for effective immunotherapy. It has been found that immunosuppression associated with chronic inflammatory factors, such as growth factors, cytokines, and chemokines is generated by stroma and tumor cells [14, 15]. Multiple immune cells coexist and interact in a complex series of pathways that ultimately lead to tumor carcinogenesis. In the review, we will document some immunosuppressive cells, including tumor-associated macrophages (TAMs), marrow-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), cancer-associated fibroblasts (CAFs) and regulatory T cells (Tregs) and their roles in cancer formation, which can lead to HCC.
Tumor-associated macrophages
TAMs inhibit anti-tumor immunity and promote tumor progression by expressing cytokines and chemokines. Preclinical studies have identified key pathways for TAMs recruitment, polarization and metabolism during tumor progression, and new therapies for these pathways can indirectly stimulate cytotoxic T-cell activation and recruitment [16,17,18]. Clinical trials of therapeutic agents currently promoting phagocytosis or inhibiting the survival, proliferation, transport or polarization of TAMs have shown improvement of cancer outcomes (Fig. 1).

Macrophage targeting strategy in HCC therapy. Preclinical studies have identified key pathways that regulate the recruitment, polarization, survival, and autophagy of TAMs during tumor progression. In addition, inhibition of macrophage-derived VEGF can inhibit tumor angiogenesis and progression. Targeting key receptors or signaling proteins can inhibit these macrophage properties and inhibit tumor progression. These molecular targets form the basis of several therapeutic HCC strategies currently being clinically developed to promote an effective anti-tumor immune response. Abbreviations: TAM, tumor-associated macrophages; Treg cells: regulatory T cells;VEGF, vascular endothelial growth factor
TAMs are significant components of the microenvironment in HCC and associated with a poor prognosis of HCC patients. TAMs expression and density have been assessed by immunohistochemical staining from 253 HCC patients and results showed that CD68+ TAMs were not associated with clinicopathologic features and prognosis in HCC. However, low presence of CD86+ TAMs and high presence of CD206+ TAMs were significantly associated with invasive tumor phenotypes and with poorer overall survival (OS) as well as reduced time to recurrence [19]. TAMs develop from monocytes to functional macrophages and acquire various immunosuppressive functions at each stage of its differentiation to maintain the tumor microenvironment [20]. Repolarization of TAMs towards an antitumor phenotype was one approach to contributing to tumor regression [21]. Yang Y et al. first demonstrated that tumor cell-derived Wnt ligand stimulates M2 to transduce the polarization of TAMs via classical Wnt/β-catenin signaling, which results in immunosuppression in HCC [22]. Therefore, blocking Wnt secretion in tumor cells and/or activation of Wnt/β-catenin signaling in TAMs may be a potential strategy for future HCC treatment.
Previous literature has demonstrated that TAMs can produce a variety of chemokines, such as CCL17, CCL18 and CCL22, which attract Tregs cells to cancer sites, thereby impeding cytotoxic T cell activation [23, 24]. This may lead to a positive feedback loop between TAMs and Tregs, providing a new dimension to the immunosuppressive effects of cancer. TAMs in HCC can also attract Tregs to regulate the tumor microenvironment. This phenomenon was found by Zhou J that the intratumoral distribution of FoxP3+ Tregs were associated with high density TAMs. The anti-interleukin-10 (IL-10) antibody partially blocked this increase, suggesting that TAMs may trigger an increase in the population of FoxP3+ Treg cells in the tumor, thereby promoting the progression of HCC [25]. Mechanistically, Wu Q et al. recently confirmed that TREM-1+ TAMs can respond to hypoxia and tumor metabolites via the ERK/NF-κβ pathway leading to accumulation of CCR6+ Foxp3+ Tregs [26]. This study highlights hypoxia-induced tumor immunosuppression by TREM-1+ TAMs that attracts CCR6 + Foxp3 + Tregs, and TREM-1+ TAMs confers HCC resistance to Programmed cell death 1 ligand 1 (PD-L1) treatment.
TAMs are able to produce angiogenic factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor and transforming growth factor β. TAMS also induce angiogenesis by expressing matrix metalloproteinase (MMPs) [27,28,29]. Angiogenesis inhibition therapy has recently become a promising therapeutic strategy for HCC. Lipid-based nanoparticles (NP) targeting CXCR4 were developed to deliver VEGF siRNA specifically to HCC as an anti-angiogenic substance. The researchers demonstrated that AMD-modified NPs (AMD-NPs) can effectively transfer VEGF siRNAs into HCC and down-regulate VEGF expression in vitro and in vivo. Although SDF1α/CXCR4 axis is upregulation in hypoxia induction after anti-angiogenesis therapy, AMD-NPs combined with conventional sorafenib therapy or VEGF siRNA inhibition of CXCR4 can prevent the infiltration of TAMs [30]. However, has there been any association between TAMs and VEGF in HCC? One study has investigated the role of macrophages in HCC progression under sorafenib treatment and explored whether combination of drugs that deplete macrophages improved the antitumor effect of sorafenib. Results showed that although sorafenib significantly inhibited tumor growth and lung metastasis, it induced a significant increase in peripheral recruitment and intratumoral infiltration of F4/80- and CD11b-positive cells, which causes an increase of VEGF. Depletion of macrophages by clodrolip or zoledronic acid (ZA) in combination with sorafenib significantly inhibited tumor progression, tumor angiogenesis, and lung metastasis [31]. These results indicate macrophages may have an important role in tumor progression under sorafenib treatment.
Preclinical studies have established that autophagy may play a role in the manipulation of TAMs function and tumor-related immunity. Chang CP et al. recently found that toll-like receptor 2 (TLR2)-associated ligands derived from HCC can stimulate the differentiation of M2 macrophages by selectively autophagy to control the homeostasis of NF-κβ RELA/p65 protein. TLR2 signaling induces NF-κβ RELA cytoplasmic ubiquitination and leads to degradation by SQSTM1/p62-mediated autophagy. Inhibition of autophagy will save NF-κβ activity and shape the phenotype of HCC polarized M2 macrophages [32]. The findings reveal a new role for autophagy in TAMs, which controls cellular function. Previous studies reported that Abies georgei’s natural product, named 747, is associated with xenophenol structure and exhibits sensitivity and selectivity as a CCR2 antagonist. In animals, 747 increases the number of CD8+ T cells in tumors by blocking tumor-infiltrating macrophage-mediated immunosuppression and inhibits in situ and subcutaneous tumor growth in a CD8+ T cell-dependent manner. In addition, 747 enhances the therapeutic effect of low-dose sorafenib without significant toxicity [33]. As shown in Table 1, treatment of TAMs targets in HCC has been documented and could be a new approach for treating HCC [34,35,36,37,38].
Marrow-derived suppressor cells
In cancer, the differentiation of myeloid cells often changes, producing a group of immature myeloid cells, which have strong immunosuppressive activity and impaired function as antigen-presenting cells (APCs) [39]. These cells are now known as MDSCs, a heterogeneous population of immature myeloid cell. MDSCs are also plastic and respond to microenvironment signals [40,41,42]. MDSCs can differentiate into macrophages, granulocytes and dendritic cells (DCs) in vitro. Therefore, MDSCs have significant diversity and plasticity, and are capable of changing their functional status in response to a variety of cytokines and growth factors in the tumor microenvironment. It has been reported that the immunosuppressive activity of MDSCs in the tumor microenvironment mainly includes (a) inducing differentiation and expansion of Tregs; (b) inhibiting the polarization of DCs, NKs and macrophages to the M2 phenotype; (c) depriving T cells of essential amino acids; (d) inducing oxidative stress (Fig. 2) [40,41,42,43]. We mainly reviewed the MDSCs in the tumor growth, angiogenesis, and metastasis of HCC in this context (Table 1) [44,45,46,47,48,49,50,51,52]. These evidence suggest that MDSCs contribute to the immunosuppressive network through multiple mechanisms and are potential immunotherapy targets for HCC.

The mechanism of immunosuppressive activity of MDSCs in the tumor microenvironment. MDSCs induce differentiation and expansion of Tregs during tumorigenesis; inhibit DC, NK and macrophage polarization to the M2 phenotype; deprive T cells from essential amino acids; and produce oxidative stress to mediate cancer progress. Abbreviations:MDSCs: myeloid-derived suppressor cells; Treg cells: regulatory T cells;NK cells: natural killer cells; CCL2: CC-chemokine ligand 2; DCs: dendritic cell; FOXP3: forkhead box P3; IL-10: interleukin-10; iNOS: inducible nitric oxide synthase; TCR: T-cell receptors; TGF-β: transforming growth factor β
MDSCs from HCC patients are unable to stimulate an allogeneic T-cell response, suppress T-cell proliferation, and have high arginase activity. Liu YT et al. used a hydrodynamic jet and transposon system to create a model that introduces a protein kinase and an open reading frames (ORFs) chromosomal encoding agent for tumor antigens. The transposon-based Akt/N-Ras-induced HCC mouse model allows researchers to monitor tumor growth non-invasively, quantify and characterize endogenous or over-transferred CD8+ T cell responses [53]. These characteristics make it a convenient preclinical model for the evaluation of immunological checkpoint inhibitors and cellular immunotherapy HCC. MDSCs was found to cause inhibition of CD8+ T-cell responses. In addition, recent studies have reported that inhibition of tumor CCRK or liver IL-6 increases interferon γ+ tumor necrosis factor-α+CD8+ T-cell infiltration and impaired tumorigenicity and is restored by recovery of MDSCs. It is worth noting that the tumor CCRK depletion up-regulated the expression of PD-L1 and increased the expression of intratumoral CD8+ T cells, thereby enhancing the effect of PD-L1 blocking HCC [54].
Additionally, these MDSCs cocultured with autologous T cells induce Treg expansion, which mitigates effector T-cell function. Kalathil S et al. recorded an increase in the number of Tregs, MDSC, PD-1+-exhausted T cells, and an increase in immunosuppressive cytokine levels in HCC patients, revealing the potential mechanistic network for immune disorders in HCC patients compared with the normal control group [55]. Other mechanisms have been described, in which MDSCs affect T-cell function, survival, and trafficking. Similar to TAMs, MDSCs express galectin-9 that binds to TIM-3 on T cells, inducing T-cell apoptosis [56]. MDSCs can also impair natural killer (NK) cell function. In HCC, MDSCs inhibit NK cell cytotoxicity and cytokine release, which is mediated by the NKp30 receptor [57]. Regarding the interaction between MDSCs and DC cells, it has been reported that MDSC inhibits TLR-ligand-induced IL-12 by IL-10 production and inhibits T-cell stimulating activity of DCs in HCC [58].
Because of the tumor-promoting and immunosuppressive effects of bone marrow cells, it is of great interest to target them to enhance the efficacy of conventional cancer therapies. The recently approved chemotherapeutic agent, trabectedin, not only targets tumor cells, but induces rapid apoptosis of bone marrow cells [59]. Clinical trials have shown that trabectedin also has a strong cytotoxic effect on liver cancer cells [60]. Therefore, trabectedin may be a potential treatment for HCC. Another potential target is estrogen, which inhibits myeloid cell function in HCC [61]. Estrogen inhibits IL-6 exposure to macrophages exposed to necrotic hepatocytes and reduces the risk of liver cancer in DEN-treated female mice [62]. Estrogen inhibits tumor necrotic cell-STAT6 activation by inhibiting Janus kinase, resulting in a reduced HCC model in mice [63]. Thus, estrogen therapy may help disrupt the development and function of HCC bone marrow cells. Therefore, targeting bone marrow cells represents a point of further research as a possible adjuvant therapy to attenuate HCC progression.
Tumor-associated neutrophils
It is clear that bone marrow-derived cells, including TAMs, TANs, and MDSCs, promote tumor progression [64,65,66]. In recent years, many studies have shown that TANs not only promote tumor growth, but also anti-tumor effects on tumors, and can regulate their different phenotypes through tumor signal transduction. TANs can be divided into two major subtypes, N1 (anti-tumor) and N2 (pro-tumor) and the plasticity of these subtypes depends on the presence of TGF-β [67, 68]. Neutrophils can polarize TGF-β to the N2 phenotype while TGF-β together with increased inhibition of IFN-β induces N1phenotype [69].
Depending on the microenvironment of the tumor, TANs can promote or inhibit tumor progression by releasing cytokines. Zhou SL and his colleagues found that CCL2 and CCL17 are the most highly expressed cytokines in peripheral blood neutrophils (PBNs) activated by TANs and HCC cells. The number of CCL2+ or CCL17+ TANs is related to tumor size, microvascular infiltration, tumor embedding, tumor differentiation and staging. Compared to PBN-conditioned medium, TAN-conditioned media and recombinant CCL2 and CCL17 increased the migration activity of HCC cells or mouse macrophages and Tregs [70]. Taken together, this evidence suggests that TANs recruit macrophages and Treg cells into HCCs to promote their growth, progression, and resistance to sorafenib. Surprisingly, the same team reported that the secretion secreted BMP2 and TGF-β2 and triggered the expression of miR-301b-3p in HCC cells, followed by inhibition of the limbal gene expression membrane protein (LSAMP) and CYLD Lysine deubiquitinase and increased stem cell characteristics in HCC cells. These TAN-induced hepatoma stem cell-like cells are active in NF-κB signaling, a higher level of secretion of C-X-C-thematic chemokine 5 (CXCL5) and recruitment of more TANs infiltration, suggesting a positive feedback loop [71]. Additionally, we have literature some important molecules which might target TANs in HCC (Table 1) [72,73,74,75,76].
Cancer-associated fibroblasts
Recent studies have shown that communication between cancer cells and fibroblasts is very important, and these fibroblasts are called CAFs [77, 78]. CAFs promote the proliferation, invasion and metastasis of tumors by secreting various growth factors and cytokines [79, 80]. In HCC, Mano Y et al. established that primary CAFs and non-cancerous liver fibroblasts from 15 patients undergoing HCC resection. They found that endogenous and exogenous BMP4 activate hepatic fibroblasts to gain the ability to secrete cytokines and enhance the invasiveness of cancer cells. BMP4 is one of the regulators of CAFs function in the HCC microenvironment [81].
CAFs train NK cells to obtain an inactive phenotype and produce an unresponsive state in the tumor. Li T et al. found that HCC-derived fibroblasts induced NK cell dysfunction significantly better than foreskin-derived fibroblasts, which were characterized by low expression of cytotoxic molecules and cell surface markers, and impaired production of cytokines. They also noted that PGE2 and IDO from activated fibroblasts inhibit the activation of NK cells, thereby creating favorable conditions for tumor progression [82]. In addition, CAF can recruit regulatory DCs through IL-6-mediated STAT3 activation and educate them to obtain a tolerogenic phenotype and up-regulate Treg production by secreting TGF-β in the tumor microenvironment [83]. In summary, CAFs play an important role in the development and progression of cancer cells and targeting CAFs may be effective in treating fibrosis and preventing HCC progression (Tables 1, [84,85,86,87,88,89,90].
Anticancer therapies targeting CAFs or cytokine inhibitors secreted by CAFs have recently been actively studied. Inhibitors of TGF-beta signaling have been shown to block HCC growth and progression by modulating EMT in different experimental models [91]. Several studies have explored the active targeting of CAFs to provide therapeutic compounds. A recent report presented anti-fibrotic drugs such as PRI-724, conophylline, follistatin, salvianolic acid, Gliotoxin. Curcumin, sulfasalazine, and tanshinone I, which inhibit activated HSC and/or induce apoptosis [92,93,94,95,96]. It is believed that these drugs can not only control fibrosis, but also inhibit HCC by controlling the function of CAFs. It has been reported that fibroblast growth factor receptor 1 is overexpressed in the fiber layer HCC [97]. CAFs stimulate tumor cells through FGF and produces fibrosis [98, 99]. Treatments that target CAFs may be effective in fibrolamellar HCC. CAFs are one of the most important components in the tumor microenvironment and promote cancer cell growth and invasion through various mechanisms. CAFs are important for the initiation and progression of cancer cells and for the treatment of CAFs to effectively treat fibrosis and prevent HCC progression.
Tumor-infiltrating Tregs
Tregs cells are the subset of CD4+ T cells and identified with the CD25 marker. CD4+ CD25+ compartment of cells is approximately 1–2% of peripheral CD4+cells. Tregs cells do not only limit autoimmune responses, they also dampen responses against microbial and viral antigens, allergens, tumors and allografts. Tumor-associated Tregs directly promote tumor evasion by a number of contact-dependent and contact-independent mechanisms [100, 101].
The number of Tregs has been reported to increase in patients with HCC [102]. Yang et al. found that the proportion and absolute number of CD4+CD25+ T cells in the surrounding area of the tumor were significantly increased [103]. The other group showed a higher frequency of Tregs in peripheral blood from HCC patients compared to HCV patients and healthy subjects. Mechanistically, Huh7 culture supernatants appear to promote CD4+CD25+ T-cell proliferation and inhibit CD4+CD25− T-cell proliferation [104]. In addition, the frequency of circulating Tregs was linked to the disease progression and had a potential of serving as a significant biomarker in HCC patients [105, 106]. Sorafenib is a multikinase inhibitor that could reduce the frequency of hepatic infiltrating Tregs by suppressing TGF-β signaling [107]. As a result,these evidence indicates suppressing Tregs might be one of the significant targets for the induction of an immune response for HCC.
CTLA-4 is member of immunoglobulin superfamily homologous to CD28 with higher affinity for B7. CTLA-4 inhibits early T cell immune response, mainly in lymphoid tissues [108]. In naive T cells, CTLA-4 localized in the intracellular space and expressed on the cell surface upon stimulatory signals. Whereas in Tregs, CTLA-4 is constitutively expressed and is involved in Treg suppressive functions [109]. Several studies investigated PD-1 and PD-L1 expression in the context of HCC and found that PD-L1 expression was localized mainly in neoplastic or intratumoral inflammatory cells [110, 111]. Moreover, PD-1/PD-L1 interaction was demonstrated to contribute to immune suppression in HCC [112]. High level of PD-L1 expression was shown in tumors with a high number of tumor infiltrating lymphocytes (TILs) and shorter overall survival [113]. There are several clinical trials investigating the efficacy of monoclonal antibodies (mAb) against CTLA-4 and PD-1/PD-L1 as single agent treatment. In this review, we have extensively studied the efficacy of the current proposed immunotherapy for Tregs in patients with HCC. (Fig. 3) (Tables 1,2) [114,115,116,117,118,119,120,121,122,123,124,125,126,127].

Summary of potential candidates for Treg-targeted anti-tumor immunotherapy in HCC. Targets such as CCR4, PD-1, LAG3, TIM3 and GITR are primarily expressed on the membrane surface of Treg cells. Checkpoint inhibitors overcome T cell failure in HCC progression and restore the immunosuppressive state of the HCC microenvironment by blocking immunological checkpoint molecules. Abbreviations:CTLA4:cytotoxic T-lymphocyte-associated protein 4;PD-1:programmed cell death 1; TIM3: T cell immunoglobulin domain and mucin domain-3;LAG3: lymphocyte-activation gene 3;GITR:tumor necrosis factor receptor superfamily, member 18;ICOS: inducible T-cell co-stimulator;OX40: tumor necrosis factor receptor superfamily, member 4;CCR8:chemokine (C-C motif) receptor 8;CCR4:chemokine (C-C motif) receptor 4;GARP:glycoprotein-A repetitions predominant
Conclusion and perspectives
Due to high mortality, limited treatment and poor prognosis of HCC, new immunotherapy treatments are urgently needed. For example, blocking the activation of immunosuppressive receptors on T-cells has become a new focus of therapy. Immunosuppressive cells, including TAMs, MDSCs, TANs, CAFs, and Tregs are key components of the tumor microenvironment that promote HCC growth and invasion. There is an interaction between these types of immune cells leading to tumor immune escape. These cells often have both anti-cancer and cancer-promoting effects, and their specific regulation and mechanism of action are not well understood. The differentiation, maturation and function of immune cells require the participation and regulation of cytokines and chemical factors, as well as the interaction of receptors and related ligands. These factors create a tumour microenviroment that inhibits the anti-tumor activity of immune cells, promoting the occurrence of HCC. Further research involving paired tumor biopsy will inform immunotherapy treatments and improve the prognosis of patients with HCC. In addition, advances in DNA and RNA sequencing technologies provide insights into the mechanisms of HCC progression to identify other targets. Altogether, these approaches to treatment brings new hope to HCC patients.
Availability of data and materials
Not applicable.
Abbreviations
- AMD-NPs:
-
AMD-modified NPs
- APCs:
-
Antigen-Presenting Cells
- CAFs:
-
Cancer-associated fibroblasts
- CSF:
-
Colony stimulating factor-1 receptor
- CTLA-4:
-
Cytotoxic T lymphocyte-associated protein 4
- CXCL5:
-
C-X-C-thematic chemokine 5
- DCs:
-
Dendritic Cells
- HCC:
-
Hepatocellular carcinoma
- IL-10:
-
Interleukin-10
- LSAMP:
-
Limbal gene expression membrane protein
- mAb:
-
Monoclonal antibodies
- MDSCs:
-
Marrow-derived suppressor cells
- MMPs:
-
Matrix metalloproteinase
- NK:
-
Natural killer
- NP:
-
Nanoparticles
- ORFs:
-
Open Reading Frames
- PBNs:
-
Peripheral blood neutrophils
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death ligand 1
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- TILs:
-
Tumor infiltrating lymphocytes
- TLR2:
-
Toll-like Receptor 2
- Tregs:
-
Regulatory T cells
- VEGF:
-
Vascular Endothelial Growth Factor
- ZA:
-
Zoledronic Acid
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Guro H, Cho JY, Han HS, et al. Current status of laparoscopic liver resection for hepatocellular carcinoma. Clin Mol Hepatol. 2016;22(2):212–8.
Yoon YI, Lee SG. Living Donor Liver Transplantation for Hepatocellular Carcinoma: An Asian Perspective. Dig Dis Sci. 2019;64(4):993–1000.
Sun JY, Yin T, Zhang XY, et al. Therapeutic advances for patients with intermediate hepatocellular carcinoma. J Cell Physiol. 2019;234(8):12116–21.
Bishay VL, Biederman DM, Ward TJ, et al. Transradial Approach for Hepatic Radioembolization: Initial Results and Technique. AJR. 2016;207(5):1112–21.
Galun D, Srdic-Rajic T, Bogdanovic A, et al. Targeted therapy and personalized medicine in hepatocellular carcinoma: drug resistance, mechanisms, and treatment strategies. J Hepatocell Carcinoma. 2017;4:93–103.
Saffo S, Taddei TH. Systemic Management for Advanced Hepatocellular Carcinoma: A Review of the Molecular Pathways of Carcinogenesis, Current and Emerging Therapies, and Neovel Tratment Strategies. Dig Dis Sci. 2019;64(4):1016–29.
Gun SY, Lee SWL, Sieow JL, et al. Targeting immune cells for cancer therapy. Redox Biol. 2019;101:–174.
Okusaka T, Ikeda M. Immunotherapy for hepatocellular carcinoma: current status and future perspectives. ESMO Open. 2018;3:e000455.
Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8.
Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front Immunol. 2016;7:550.
Hida T. Nivolumab for the treatment of Japanese patients with advanced metastatic non-small cell lung cancer: a review of clinical trial evidence for efficacy and safety. Ther Adv Respir Dis. 2018;12:1753466618801167.
Liu B, Salgado OC, Singh S, et al. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat Commun. 2019;10(1):354.
Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):10.
Pathria P, Louis TL, Varner JA. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019;40(4):310–27.
Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76(3):513–6.
Jeong H, Hwang I, Kang SH, et al. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J Breast Cancer. 2019;22(1):38–51.
Dong P, Ma L, Liu L, et al. CD86/CD206, Diametrically Polarized Tumor-Associated Macrophages, Predict Hepatocellular Carcinoma Patient Prognosis. Int J Mol Sci. 2016;17(3):320.
Galdiero MR, Bonavita E, Barajon I, et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95.
Yang Y, Ye YC, Chen Y, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9(8):793.
Wang D, Yang L, Yue D, et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019;452:244–53.
Mamrot J, Balachandran S, Steele EJ, et al. Molecular model linking Th2 polarized M2 tumour-associated macrophages with deaminase-mediated cancer progression mutation signatures. Scand J Immunol. 2019:e12760.
Zhou J, Ding T, Pan W, et al. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer. 2009;125(7):1640–8.
Wu Q, Zhou W, Yin S, et al. Blocking TREM-1+ Tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-PD-L1 resistance in liver cancer. Hepatology. 2019. https://doi.org/10.1002/hep.30593.
Zhang D, Qiu X, Li J, et al. TGF-βsecreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle. 2018;17(24):2766–78.
Darvishi B, Majidzadeh-A K, Ghadirian R, et al. Recruited bone marrow derived cells, local stromal cells and IL-17 at the front line of resistance development to anti-VEGF targeted therapies. Life Sci. 2019;217:34–40.
Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015;(44-46):94–112.
Liu JY, Chiang T, Liu CH, et al. Delivery of siRNA Using CXCR4-targeted Nanoparticles Modulates Tumor Microenvironment and Achieves a Potent Antitumor Response in Liver Cancer, Mol Ther. 2015;23(11):1772–82.
Zhang W, Zhu XD, Sun HC, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16(13):3420–30.
Chang CP, Su YC, Lee PH, et al. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy. 2013;9(4):619–21.
Yao W, Ba Q, Li X, et al. A Natural CCR2 Antagonist Relieves Tumor-associated Macrophage-mediated Immunosuppression to Produce a Therapeutic Effect for Liver Cancer. EBioMedicine. 2017;22:58–67.
Li X, Yao W, Yuan Y, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157–67.
Li H, Li H, Li XP, et al. C‑C chemokine receptor type 2 promotes epithelial‑to‑mesenchymal transition by upregulating matrix metalloproteinase‑2 in human liver cancer. Oncol Rep. 2018;40:2734–41.
Teng KY, Han J, Zhang X, et al. Blocking the Ccl2-Ccr2 axis using Ccl2 neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther. 2017;16:312–22.
Ao JY, Zhu XD, Chai ZT, et al. Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular carcinoma. Mol Cancer Ther. 2017;16:1544–54.
Wan S, Zhao E, Kryczek I, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–404.
Rahma OE, Hodi FS. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin Cancer Res. 2019. https://doi.org/10.1158/1078-0432.
Salmaninejad A, Valilou SF, Soltani A, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (Dordr). 2019. https://doi.org/10.1007/s13402-019-00453-z.
Consonni FM, Porta C, Marino A, et al. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front Immunol. 2019;10:949.
Guilbaud E, Gautier EL, Yvan-Charvet L. Macrophage Origin, Metabolic Reprogramming and IL-1 Signaling: Promises and Pitfalls in Lung Cancer. Cancers (Basel). 2019;11(3):E298.
Bruno A, Mortara L, Baci D, Noonan DM, et al. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front Immunol. 2019;10:771.
Xu M, Zhao Z, Song J, et al. Interactions between interleukin-6 and myeloid-derived suppressor cells drive the chemoresistant phenotype of hepatocellular cancer. Exp Cell Res. 2017;351(2):142–9.
Chiu DK, Xu IM, Lai RK, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64(3):797–813.
Li R, Li H, Luo HJ, et al. SSAO inhibitors suppress hepatocellular tumor growth in mice. Cell Immunol. 2013;283(1-2):61–9.
Guha P, Gardell J, Darpolor J, et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene. 2019;38(4):533–48.
Sun H, Yang W, Tian Y, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018;9(1):5214.
Li B, Zhang S, Huang N, et al. CCL9/CCR1 induces myeloid‑derived suppressor cell recruitment to the spleen in a murine H22 orthotopic hepatoma model. Oncol Rep. 2019;41(1):608–18.
Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517.
Iwata T, Kondo Y, Kimura O, et al. PD-L1+ MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep. 2016;6:39296.
Li S, Sun R, Chen Y, et al. TLR2 limits development of hepatocellular carcinoma by reducing IL18-mediated immunosuppression. Cancer Res. 2015;75(6):986–95.
Liu YT, Tseng TC, Soong RS, et al. A novel spontaneous hepatocellular carcinoma mouse model for studying T-cell exhaustion in the tumor microenvironment. J Immunother Cancer. 2018;6(1):144.
Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):931–44.
Kalathil S, Lugade AA, Miller A, et al. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73(8):2435–44.
Limagne E, Richard C, Thibaudin M, et al. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology. 2019;8(4):e1564505.
Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807.
Hu CE, Gan J, Zhang RD, et al. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46(2):156–64.
Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–62.
Beumer JH, Schellens JH, Beijnen JH. Hepatotoxicity and metabolism of trabectedin: a literature review. Pharmacol Res. 2005;51:391–8.
Shi L, Feng Y, Lin H, et al. Role of estrogen in hepatocellular carcinoma: is inflammation the key? J Transl Med. 2014;12:93.
Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4.
Yang W, Lu Y, Xu Y, et al. Estrogen represses hepatocellular carcinoma (HCC) growth via inhibiting alternative activation of tumor-associated macrophages (TAMs). J Biol Chem. 2012;287:40140–9.
Murdoch C, Muthana M, Coffelt SB, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86.
Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94.
Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol. 2017;102:343–9.
Andzinski L, Kasnitz N, Stahnke S, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. 2016;138:1982–93.
Zhou SL, Zhou ZJ, Hu ZQ, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646–1658.e17.
Zhou SL, Yin D, Hu ZQ, et al. A Positive Feedback Loop between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology. 2019. https://doi.org/10.1002/hep.30630.
Haider C, Hnat J, Wagner R, et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology. 2019;69(1):222–36.
Yan C, Yang Q, Gong Z. Tumor-Associated Neutrophils and Macrophages Promote Gender Disparity in Hepatocellular Carcinoma in Zebrafish. Cancer Res. 2017;77(6):1395–407.
Li L, Xu L, Yan J, et al. CXCR2-CXCL1 axis is correlated with neutrophil infiltration and predicts a poor prognosis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:129.
Li L, Yan J, Xu J, et al. CXCL17 expression predicts poor prognosis and correlates with adverse immune infiltration in hepatocellular carcinoma. PLoS One. 2014;9(10):e110064.
Gao Q, Zhao YJ, Wang XY, et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. 2012;72(14):3546–56.
De Wever O, Demetter P, Mareel M, et al. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123:2229–38.
Sukowati CH, Anfuso B, Crocé LS, et al. The role of multipotent cancer associated fibroblasts in hepatocarcinogenesis. BMC Cancer. 2015;15:188.
Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63.
Terada T, Makimoto K, Terayama N, et al. Alpha-smooth muscle actin-positive stromal cells in cholangiocarcinomas, hepatocellular carcinomas and metastatic liver carcinomas. J Hepatol. 1996;24:706–12.
Mano Y, Yoshio S, Shoji H, et al. Bone morphogenetic protein 4 provides cancer-supportive phenotypes to liver fibroblasts in patients with hepatocellular carcinoma. J Gastroenterol. 2019. https://doi.org/10.1007/s00535-019-01579-5.
Li T, Yang Y, Hua X, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318(2):154–61.
Cheng JT, Deng YN, Yi HM, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis .2016;5:e198.
Zhou Y, Ren H, Dai B, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Exp Clin Cancer Res. 2018;37(1):324.
Li Y, Wang R, Xiong S, et al. Cancer-associated fibroblasts promote the stemness of CD24+ liver cells via paracrine signaling. J Mol Med. 2019;97(2):243–55.
Wu S, Zheng Q, Xing X, et al. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res. 2018;37(1):99.
Cheng Y, Li H, Deng Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9(4):422.
Xiong S, Wang R, Chen Q, et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018;8(2):302–16.
Rhee H, Kim HY, Choi JH, et al. Keratin 19 Expression in Hepatocellular Carcinoma Is Regulated by Fibroblast-Derived HGF via a MET-ERK1/2-AP1 and SP1 Axis. Cancer Res. 2018;78(7):1619–31.
Liu C, Liu L, Chen X, et al. LSD1 Stimulates Cancer-Associated Fibroblasts to Drive Notch3-Dependent Self-Renewal of Liver Cancer Stem-like Cells. Cancer Res. 2018;78(4):938–49.
Giannelli G, Villa E, Lahn M. Transforming growth factor-β as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014;74:1890–4.
Osawa Y, Oboki K, Imamura J, et al. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/β-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine. 2015;2:1751–8.
Kubo N, Saito R, Hamano K, et al. Conophylline suppresses hepatic stellate cells and attenuates thioacetamide-induced liver fbrosis in rats. Liver Int. 2014;34:1057–67.
Wright MC, Issa R, Smart DE, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fbrosis in rats. Gastroenterology. 2001;121:685–98.
Wahl C, Liptay S, Adler G, et al. Sulfasalazine: a potent and specifc inhibitor of nuclear factor kappa B. J Clin Invest. 1998;101:1163–74.
Tsai MK, Lin YL, Huang YT. Effects of salvianolic acids on oxidative stress and hepatic fibrosis in rats. Toxicol Appl Pharmacol. 2010;242:155–64.
Riehle KJ, Yeh MM, Yu JJ, et al. mTORC1 and FGFR1 signaling in fibrolamellar hepatocellular carcinoma. Mod Pathol. 2015;28:103–10.
Spaeth EL, Dembinski JL, Sasser AK, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992.
Giulianelli S, Cerliani JP, Lamb CA, et al. Carcinoma-associated fbroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int J Cancer. 2008;123:2518–31.
Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87.
Sakaguchi S, Miyara M, Costantino CM, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500.
Ormandy LA, Hillemann T, Wedemeyer H, et al. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65:2457–64.
Yang XH, Yamagiwa S, Ichida T, et al. Increase of CD4+CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol. 2006;45:254–62.
Cao M, Cabrera R, Xu Y, et al. Hepatocellular carcinoma cell supernatants increase expansion and function of CD4+ CD25+ regulatory T cells. Lab Investig. 2007;87:582–90.
Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–39.
Fu J, Zhang Z, Zhou L, et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology. 2013;58:139–49.
Wang Q, Yu T, Yuan Y, et al. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-β signal. J Surg Oncol. 2013;107:422–7.
Chambers CA, Kuhns MS, Egen JG, et al. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–94.
Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–40.
Ishida Y, Agata Y, Shibahara K, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95.
Wang G, Liu Y, Huang J, et al. PPARα Suppresses PD-L1-Mediated Immune Escape by Down-regulating SPP1 in Human Hepatocellular Carcinoma. Cancer Res Treat. 2019. https://doi.org/10.4143/crt.2019.111.
Wu K, Kryczek I, Chen L, et al. Kupffer Cell Suppression of CD8+ T cells in Human Hepatocellular Carcinoma is Mediated by B7-H1/PD-1 Interactions. Cancer Res. 2009;69:8067–75.
Semaan A, Dietrich D, Bergheim D, et al. CXCL12 expression and PD-L1 expression serve as prognostic biomarkers in HCC and are induced by hypoxia. Virchows Arch. 2017;470:185–96.
Kalathil SG, Lugade AA, Miller A, et al. PD-1+ and Foxp3+ T cell reduction correlates with survival of HCC patients after sorafenib therapy. JCI Insight. 2016;1(11):e86182.
Li G, Liu D, Cooper TK, et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol. 2017;66(1):75–85.
Chen ML, Yan BS, Lu WC, et al. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int J Cancer. 2014;134(2):319–31.
Wei R, Hu Y, Dong F, et al. Hepatoma cell-derived leptin downregulates the immunosuppressive function of regulatory T-cells to enhance the anti-tumor activity of CD8+ T-cells. Immunol Cell Biol. 2016;94(4):388–99.
Han Y, Chen Z, Yang Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59(2):567–79.
Zhou G, Sprengers D, Boor PPC, et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology. 2017;153(4):1107–1119.e10.
Ji J, Yin Y, Ju H, et al. Long non-coding RNA Lnc-Tim3 exacerbates CD8 T cell exhaustion via binding to Tim-3 and inducing nuclear translocation of Bat3 in HCC. Cell Death Dis. 2018;9(5):478.
Li Z, Li N, Li F, et al. Genetic polymorphisms of immune checkpoint proteins PD-1 and TIM-3 are associated with survival of patients with hepatitis B virus-related hepatocellular carcinoma. Oncotarget. 2016;7(18):26168–80.
Li Z, Liu Z, Zhang G, et al. TIM3 gene polymorphisms in patients with chronic hepatitis B virus infection: impact on disease susceptibility and hepatocellular carcinoma traits. Tissue Antigens. 2012;80(2):151–7.
Van Beek AA, Zhou G, Doukas M, et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int J Cancer. 2019;145(4):1111–24.
Pedroza-Gonzalez A, Zhou G, Singh SP, et al. GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology. 2015;4(12):e1051297.
Tu JF, Ding YH, Ying XH, et al. Regulatory T cells, especially ICOS+ FOXP3+regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep. 2016;6:35056.
Xie K, Xu L, Wu H, et al. OX40 expression in hepatocellular carcinoma is associated with a distinct immune microenvironment, specific mutation signature, and poor prognosis. Oncoimmunology. 2018;7(4):e1404214.
Busato D, Mossenta M, Baboci L, et al. Novel immunotherapeutic approaches for hepatocellular carcinoma treatment. Expert Rev Clin Pharmacol. 2019;12(5):453–70.
Acknowledgements
None.
Funding
This work was supported by grants from the National Natural Science Key Foundation of China (Grant No.81530048).
Author information
Authors and Affiliations
Contributions
There are 3 first authors in this manuscript and they have equally contributed to this project. Dr. CL, DR and BZ was responsible for gathering information of the related research and designing the review. Dr. WZ was responsible for drawing the pictures. Furthermore, we have three corresponding authors in this manuscript. Dr. WT has contributed to information interpretation, editing and critical revision of the manuscript. Dr. XW and ZC have contributed to study design and critical revision of the manuscript. Dr. WT, ZC, and XW were also responsible for handling the revisions and resubmission of revised manuscripts. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lu, C., Rong, D., Zhang, B. et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer 18, 130 (2019). https://doi.org/10.1186/s12943-019-1047-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1047-6