
Abstract
Plasmodium knowlesi is a zoonotic malaria parasite normally residing in long-tailed and pig-tailed macaques (Macaca fascicularis and Macaca nemestrina, respectively) found throughout Southeast Asia. Recently, knowlesi malaria has become the predominant malaria affecting humans in Malaysian Borneo, being responsible for approximately 70% of reported cases. Largely as a result of anthropogenic land use changes in Borneo, vectors which transmit the parasite, along with macaque hosts, are both now frequently found in disturbed forest habitats, or at the forest fringes, thus having more frequent contact with humans. Having access to human hosts provides the parasite with the opportunity to further its adaption to the human immune system. The ecological drivers of the transmission and spread of P. knowlesi are operating over many different spatial (and, therefore, temporal) scales, from the molecular to the continental. Strategies to prevent and manage zoonoses, such as P. knowlesi malaria require interdisciplinary research exploring the impact of land use change and biodiversity loss on the evolving relationship between parasite, reservoir hosts, vectors, and humans over multiple spatial scales.
Similar content being viewed by others


Background
Numerous ecosystems across the globe have been altered anthropogenically by deforestation, pollution, agricultural expansion, road construction, dam building, mining, eutrophication, increased nitrogen fixation, urbanization and other activities, thereby creating conditions suitable for the emergence of infectious diseases [1, 2]. Changing patterns of land use may instigate contact between humans and both domestic and wild animals, and disturb population distribution and abundance [3]. As a result, many of the current emerging infectious diseases (EIDs) are zoonotic in origin and present serious public health concerns. The zoonotic parasite, Plasmodium knowlesi, is one such disease which threatens Malaysia’s malaria eradication policy [4]. Eradication policies for the human malarias focus on behavioural prevention and treatment practices based around insecticide-treated bed nets, indoor spraying, and drug therapy. The challenge with zoonotic P. knowlesi malaria is that the mosquito vectors do not enter dwellings to rest and feed. Traditional behavioural methods offer no easy options for breaking the transmission cycle. Furthermore, accurate diagnosis of the parasite requires molecular methods, and ways to reduce this time-consuming process are being investigated [5].
Infectious disease emergence has been described as a complex interplay between social and ecological systems over many scales, ranging from the molecular to the continental [6,7,8]. Ecological drivers operating at multiple spatial scales are altering transmission dynamics between pathogens, vectors and hosts [7]. Anthropogenic land use change reduces biodiversity, increases the contact between natural hosts, vectors, and humans, and alters the ecology in ways that may be favouring pathogen transmission to humans [9, 10]. A detailed examination of ecological changes at the local landscape scale in relation to the transmission and spread of P. knowlesi has been presented in the relevant paper [11]. However, this local landscape scale is nested within a complex array of interactions between social and biological systems ranging from the global scale, where pathogen phylogenetic history plays a role, down to the molecular level, where genetic factors influencing pathogen, vector, and host dynamics are crucial to emerging infection rates [7, 8, 12, 13]. For this reason, multi-scaled socio-ecological models of infectious disease may be effectively applied to P. knowlesi emergence in order to determining the complex drivers of its emergence [13].
Drivers of the transmission and spread of Plasmodium knowlesi at different ecological scales
Evolutionary drivers of Plasmodium knowlesi
Non-human primates (NHPs) share malaria parasites with humans and host-switching is an ongoing evolutionary process. Plasmodium knowlesi contrasts with many other parasite species because it has the capability of infecting both immature and mature stage erythrocytes in humans, and infects both monkey and human hosts [14]. This unique capacity relates to differences in the multigene families and the proteins they encode for in the human and NHP Plasmodium species/strains. The genetic differences appear to be critical in determining current and potential hosts and host-switching capabilities [14]. The evolution of these differences has arisen over millennia, in parallel with the evolution of mammals. It is suggested that a single host-switch event originally brought the Plasmodium parasite across into mammalian hosts from sauropsid-infecting ancestors [15]. This leap occurred at least 64 million years ago (Ma) [16]. Alongside this host-switching came the specialization into Anopheles mosquitoes as the specific vector for mammalian hosts [17].
There are now six species of human malaria parasites; Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale (Plasmodium ovale wallikeri and P. o. curtisi), Plasmodium malariae and Plasmodium knowlesi [14, 18]. At least 30 Plasmodium species cause infection in the NHPs [19, 20] infecting 53 host species over 25 genera [21]. Most Plasmodium parasites are constrained to phylogenetically related hosts that belong to the same family. The exceptions to this rule are Plasmodium brasilianum and P. knowlesi, which infect hosts from three and two families, respectively [21]. The natural hosts for P. knowlesi are macaques (Macaca fascicularis and Macaca nemestrina) as well as leaf monkeys (Presbytis melalophos) [21, 22] and now humans. Thirteen genes found in P. falciparum and P. vivax are not found in P. knowlesi and may be limiting the parasite’s transmission success in human hosts for now [23].
In Africa, the Laverania ‘subgenus’ [15] are seven tightly host-specific parasites found only in the great apes (such as chimpanzees and bonobos) but were still able to give rise to the recent (between 40 and 60,000 years ago) host-switch event of P. falciparum into humans [24,25,26,27,28,29,30]. In Southeast Asia, five NHP species predominate—P. knowlesi, Plasmodium inui, Plasmodium cynomolgi, Plasmodium fieldi, and Plasmodium coatneyi [31]. Globally, spillover from NHP malaria parasites into humans (albeit in low numbers), has been reported from Costa Rica [32] and Venezuela [33] with P. brasilianum/P. malariae, Brazil with Plasmodium simium [34, 35], Northeast Brazil with P. brasilianum/P. malariae [36], Malaysia with P. cynomolgi [37], and the Central African Republic with the P. vivax-like strain from the great apes [38].
The P. brasilianum genome is 99.7% identical to human P. malariae [39] and similarly, P. simium is considered genetically indistinguishable from P. vivax [40]. Plasmodium brasilianum/P. malariae is being considered as an anthropozoonoses, equally able to infect monkeys and humans given the opportunity [41]. It is hypothesized that P. brasilianum and P. simium are most likely the result of host-switching from P. malariae and P. vivax respectively, brought to the New World by European colonizers [42,43,44].
For P. knowlesi, it is still the subject of speculation whether it emerged along with P. vivax and other closely related species from an African origin as opposed to an archaic Asian origin [31, 42, 45, 46]. What is understood is that P. vivax and P. knowlesi (which both possess Duffy Binding Proteins for erythrocyte invasion) diverged somewhere between 18 and 34 Ma which approximates the split between their respective hosts, the apes (Hominoidea) and Old World Monkeys (Cercopithecidae) [16]. Phylogenetic analysis of CSP and MSP-9 gene sequences reveal that P. coatneyi and P. knowlesi have a very close relationship, forming one monophyletic group, all descending from one ancestor, even though they differ in many important ways biologically [47, 48]. This clade relationship between P. coatneyi and P. knowlesi has been corroborated using SSU rRNA genes by Chua and colleagues [49]. Another monophyletic group is comprised of P. cynomolgi, Plasmodium simiovale, P. simium and P. vivax and together both these groups belong to a well-defined and distant clade [47]. It has been suggested that P. vivax diverged from P. cynomolgi when it host-switched to humans [50, 51].
The Old World macaques brought P. knowlesi across into Southeast Asia when land bridges became available during glacial periods over the past several million years [52, 53]. A divergence occurred between P. knowlesi and its closest ancestor, P. coatneyi, around 98,000–478,000 years ago in this region [54] indicating that the parasite predates human settlement in SE Asia [55]. Plasmodium knowlesi parasite numbers increased significantly 30,000–40,000 years ago [54], but whether this is connected to an increase in primate hosts or mosquito vectors is still undetermined [56]. According to data on population growth and stabilization of the long-tailed macaques on Borneo, their population increase occurred prior to the population expansion of P. knowlesi on the island [57].
Given the geographical separation of the archipelagic part of Southeast Asia, it is hypothesized that different locations will host different genetic variants of the P. knowlesi parasite [58, 59]. Most human infections of P. knowlesi are dominated by single genotypes from one of three main genomic clusters that show evidence of recent strong selection [60, 61], two clusters from each of the macaque hosts in Borneo (Macaca fascicularis and Macaca nemestrina Borneo) and a third cluster from Peninsular Malaysia found in humans [60, 62,63,64,65]. Geographical separation of the island of Borneo from the peninsular by sea-level rise at the end of the last ice age would have prevented macaque movement and been responsible for any allopatric divergence [66].
However, recent genomic analysis has uncovered chromosomal-segment exchanges between these subpopulations [61]. Benavente and colleagues [61] investigated the P. knowlesi genotype from Cluster 1 (Mf-Pk Borneo) and found two geographic subgroups corresponding to different geographical regions: the Kapit and Betong regions in Sarawak. Chromosome 8 of the genome presented an anomaly where distinct genetic differences were noted for this chromosome between the two geographical locations. Chromosome 8 contains information relating to interaction with the vector during the parasite life cycle. The researchers found that many of the Macaca fascicularis/P. knowlesi Betong genotypes had segments on chromosome 8 almost identically matching the chromosome 8 segments from the Macaca nemestrina/P. knowlesi Cluster 2 genotype [61]. This finding suggests a recent genetic exchange between these two macaque clusters which appear to be under strong selective pressure, given that 73% of the Betong subgroup share this new haplotype and 33/60 genetic isolates examined showed evidence of genetic exchange between these two clusters [61]. The parasite is, therefore, adapting to the two macaque hosts with enough genetic differentiation to be considered as subgroups, but still capable of recombining when contact arises. Benavente et al. [61] suggest that the parasites genetic diversity and the changing environment in Borneo may assist P. knowlesi with further host transitions in the future.
Bioregional/regional drivers—distribution of vectors, hosts, and the influence of land use
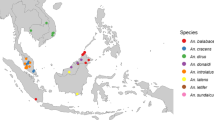
For now, transmission of P. knowlesi malaria is restricted to the Leucosphyrus Group of Anopheles mosquitoes because the sporozoites of the parasite are unable to invade the salivary glands of other mosquito species [67]. This vector group restricts P. knowlesi to the Southeast Asian biogeographic region [68]. The Leucosphyrus group consists of two different complexes made up of many different species, some of which have been identified as important for carrying P. knowlesi: the Dirus Complex of Anopheles dirus and Anopheles cracens, and the Leucosphyrus Complex of Anopheles latens, Anopheles balabacensis and Anopheles introlatus [59]. The Leucosphyrus Complex vectors are found in Malaysia, Indonesia, Singapore, Brunei and parts of the Philippines while the Dirus Complex vectors are widespread in the northern mainland countries of Myanmar, Thailand, Cambodia and Vietnam [59]. The Leucosphyrus group of mosquitos feed on both monkeys and humans, with species such as An. cracens, showing a clear preference for humans [69,70,71,72].
Anopheles cracens, although from the Dirus Complex, is the primary vector of P. knowlesi in Peninsular Malaysia. It shares ten olfactory genes with Anopheles maculatus, the species responsible for human malaria in this region, but additionally carries a further five olfactory genes thought to be responsible for its ‘monkey-seeking’ biting behaviour [72, 73]. This mosquito is more commonly found in forest and farms than in larger settlements [74], but will enter villages, although not into dwellings. It prefers biting humans over monkeys at a ratio of 2:1 with peak feeding between 7 and 9 p.m. [69,70,71,72]. Anopheles latens, the vector of P. knowlesi in Kapit, Sarawak, is predominately detected in farming plots and forest locations, with highest infection rates occurring in the forest in humans engaged in forest-based activities. However, its propensity to transmit the parasite is highest in farms [75].
Anopheles balabacensis is the principal mosquito species transmitting the P. knowlesi parasite in Sabah [76,77,78]. In Kudat district, Sabah, for example, this vector comprised 94.4% of the total mosquito collection with highest densities found in villages over forest or farms [77]. This species was five times more commonly collected from the peri-domestic environment than from inside the dwelling [79] indicating its exophilic nature. However, the highest infection rates still occur in the forest and farming plots, possibly because of the proximity to macaques [79].
Chua and colleagues [49] examined all captured An. balabacensis from the Kudat district, Sabah, for Plasmodium infection and found that although this species comprised 89.87% (n = 1586) of the total catch, only 23 An. balabacensis mosquitos carried simian Plasmodium infections. Plasmodium knowlesi was found in only two mosquitoes (in combination with P. inui in one and with P. cynomolgi in the other), almost 9% of all Plasmodium infections. The close genetic relationship between the P. knowlesi parasite found in the human cases from Kudat, previous macaque isolates and the An. balabacensis mosquito suggest that infection is largely contingent on the mosquito having access to a reservoir of hosts [49].
The natural hosts for P. knowlesi are the cynomolgus monkeys, the long-tailed and pig-tailed macaques (Macaca fascicularis and Macaca nemestrina, respectively) found throughout Southeast Asia. These two species coexist throughout much of this region, however, at a more local level they show preferences for wetter alluvial riverine terrain compared to a drier hilly terrain respectively [80]. As Macaca fascicularis is a tree-traveller and Macaca nemestrina travels along the ground, attributes of the different habitats may explain their segregation within their natural habitats. Macaca fascicularis has a preference for denser canopy foliage with fewer canopy gaps [80]. The pig-tailed macaque has an upper distribution boundary around the Surat Thani-Krabi depression in Thailand [81]. North of this boundary, this macaque is considered as a different species, Macaca leonina [81]. There is evidence that Macaca leonina may also be a putative host species for the parasite given that positive knowlesi infection has been confirmed from the Yunnan province of China and coincides with the northward range of Macaca leonina into Myanmar, well beyond the range of the two main host species [82, 83]. This species also shares a close genetic relationship to Macaca nemestrina [59].
Plasmodium knowlesi infections have been detected in macaques by molecular methods from both Borneo and Peninsular Malaysia as well as Singapore and Thailand [54, 70, 84]. The highest prevalence of malaria positive macaques has been reported in the Kapit Division of Sarawak, with 87% of 83 long-tailed macaques and 50% of 26 pig-tailed macaques found to be P. knowlesi positive [84]. In Selangor, Malaysia, 60% of the wild macaques were found to be infected with the parasite [85]. Monkeys infected with P. knowlesi are naturally asymptomatic with low level parasitaemias [22, 86].
Risk-assessment maps for the parasite have been produced based upon ranges of the host species, vectors, land use patterns and confirmed infections in humans, macaques and mosquitos [58, 59, 82]. Plasmodium knowlesi risk exists for an arc of habitats reaching from Taiwan in the north, down through the archipelago of the Philippine and Indonesian islands, up the Malay peninsula and into the mainland of Southeast Asia onto Myanmar [67]. Taiwan has no recorded cases, although the same vector species does reach this far north and various macaque species also inhabit the forest and forest fringes of the island [67].
Plasmodium knowlesi became noted as a parasite of concern after Singh and colleagues [87] published their findings in 2004 on a cluster of cases from Kapit, Sarawak between 2000 and 2002. They identified P. knowlesi as the malaria parasite responsible for infection in 58% (n = 208) of the cases, as opposed to P. malariae, as had been previously diagnosed. Similar findings have been reported in other countries of the region. In Thailand, blood samples from as far back as 1996 found P. knowlesi to be responsible for malaria (largely as a co-infection with the human malarias) in 0.48% (n = 210) of the human malaria cases [88] and more recently in 0.6% (n = 1874) of cases in 2006–2007, [89], and in 0.67% (n = 3770) of cases from 2008–2009 [88]. In the Philippines, two cases were detected in 2006 [90] and in southern Myanmar, P. knowlesi comprised 21.9% (n = 146) of cases in 2008, also mainly as a co-infection with the human malarias [91]. Further confirmed human cases of the disease have now been recorded from Singapore [92, 93], Vietnam [94, 95], Cambodia, southern Thailand [96], Indonesia [97,98,99], Indonesian Borneo [100,101,102], Brunei, Myanmar [84, 91, 96, 103], the Nicobar and Andaman Islands of India [104], and quite recently from Laos [105].
The reality is however, that the true prevalence of P. knowlesi in the Southeast Asian region is difficult to estimate, and when or where this parasite first caused a natural infection in a human is unknown. The first reported infection in a human was 1965 from peninsular Malaysia [106] and presumptively again in 1971 from the same region [107]. Confirmed human cases are increasing, especially in Malaysian Borneo, although this may simply relate to increased awareness of, and testing for, this pathogen. There is an urgent need for cross-sectional studies that can provide information on patterns of infection and exposure at the community level to assess the true threat of this parasite [82, 108].
With reference to shifts in vector and host habitats, the Southeast Asian region has undergone dramatic changes in land use patterns. The economic pressures behind such transitions relate in part to global commodity markets driving consumption of palm oil and wood which are central to Malaysia’s economic growth [109,110,111]. The demand for palm oil is increasing by an estimated 15% per year [112] with Malaysia’s production expected to rise to 23.4 million tonnes by 2020 up from current production of 15 million tonnes [110]. Oil palm is considered a threat to biodiversity in Southeast Asia because these plantations are almost totally devoid of forest-dwelling animal species [113]. Malaysian Borneo has been severely impacted by deforestation over the past several decades and estimates suggest that 80% of the country had been logged and cleared for agriculture by 2009 [114]. Since this time, deforestation has continued at a rate of 350,000 ha each year [110]. Deforestation and land-use change may be influencing the distribution of P. knowlesi host and vector populations.
These land use changes have contributed to a different relationship between disease risk and environmental variables for Malaysia in comparison to continental Asia [59, 82]. Moyes and others [59] found that in the southern countries of Malaysia, Indonesia, Singapore, Brunei and part of the Philippines, there is a risk to humans for P. knowlesi from vectors of the Leucosphyrus complex being found together with macaque hosts within disturbed forest areas, including plantations, vegetable gardens and logging concessions. When members of the Dirus complex overlap with the habitat of the long-tailed macaques (Macaca fascicularis) in the northern endemic countries, the possibility for co-occurrence exists for savannah, vegetation mosaics, and cropland as well as forested areas [59]. In Malaysia, deforestation and changes in land use have induced macaques to move away from forest to farms and semi-urban areas, with the Leucosphyrus mosquitos therefore also extending their range to become more predominant in farms, villages and areas of logged forest [74, 115].
Current knowledge suggests that the parasite may be adapting to these changes in the distribution and vectorial capacity of the mosquitos from the Leucosphyrus Complex. Benavente et al. [61] reported on the strong selective pressure on the parasite being imposed by the current vectors within the Malaysian region. For example, one recombinant gene in chromosome 12 of P. knowlesi (PKH_120710) is orthologous to the gene (Pfs47-like) in P. falciparum which is known for its role in infecting Anopheles gambiae without activating its immune system [116]. A haplotype alteration in Pfs47 in P. falciparum renders it compatible with a different species of mosquito [116]. Benavente et al. [61] found that close to half of the P. knowlesi isolates taken from the Betong region of their study in Sarawak presented a recombinant profile in this gene indicating the potential for these isolates to adapt to other mosquito vectors. Thus, other more ecologically widespread mosquitoes may be only ‘one mutation event away’ from being able to act as vectors, potentially increasing the range of the parasite considerably [67].
Individual-level drivers
Individual level drivers of emergence relate to the human characteristics and behaviours that bring humans into contact with the natural hosts and vectors of the parasite [7]. Gaining an understanding of the breadth and clinical severity of infection from P. knowlesi is hampered by the need for molecular methods for accurate diagnosis. Polymerase chain reaction (PCR) methods are accurate but not feasible in most malaria endemic regions because of the cost of the equipment and the training required. Also, they do not provide the rapid results needed for treatment selection. However, a novel non-PCR method called loop-mediated isothermal amplification (LAMP) is showing promise for accurate and fast diagnostic results with low technology requirements [117]. Microscopy of stained blood smears and rapid diagnostic immunochromatographic tests (RDTs) are used widely and successfully for the detection of the other four human malarias [118], but are inadequate when attempting to diagnose P. knowlesi at the necessary levels of sensitivity and specificity [117, 119, 120]. For now, in the P. knowlesi endemic areas, any malaria infection which appears as P. malariae through microscopy is considered as P. knowlesi and treatment provided accordingly [121].
In Malaysian Borneo, those most at risk of contracting P. knowlesi infection have been noted to be males and either traditional subsistence farmers or those working in jobs relating to agriculture and forestry [108, 122, 123]. Data from 2016 for Sabah and Sarawak show that P. knowlesi infected more men than women (80%) with most cases occurring in adults, especially those aged over 55 years [78]. In contrast to these findings, a recent prospective study from Sabah reported a median age for P. knowlesi cases to be 33 years (IQR, 21–49 years) yet still predominately male [124]. The presence of macaques in proximity to human populations increases the risk of infection [49]. High levels of forest clearing together with remaining forest cover near houses is associated with P. knowlesi exposure, with the creation of such forest fringes known to encourage macaques [108]. Males may be more prone to clinical infection due to a higher number of infective bites resulting from proximity to macaques in these work-related areas [108].
The emerging possibility that P. knowlesi is being transmitted from human to human certainly exists, but to date no evidence has been found [125]. For human to human infection, the parasite would need to differentiate into the sexual gametocyte stage within the host red blood cell [126]. Plasmodium knowlesi gametocytes have been detected in human infections in Malaysia using microscopy [127] and reverse transcription (RT-PCR) methods to measure the level of pks25, an mRNA gene that is expressed in mature gametocytes [94, 128].
Although studies have generally shown a lack of case clustering in human settlements [129], Fornace et al. [130] and Barber et al. [131] both found family case clusters in districts in Sabah with a wide age distribution of infection (children < 15 years as well as women), including one infected family that had not travelled outside of Kudat township. In Aceh, Indonesia, a family cluster of cases (mother and two teenagers) occurred in a residential location nearby to the forest, indicating infection from the peri-domestic area [97]. Manin et al. [79] emphasize the importance of peri-domestic exposure from their research in Sabah where An. balabacensis was found to be 5.5-fold more abundant outside of dwellings in the early evenings than inside them. The lack of clustering of parasite genotypes in humans or macaques is further suggestive that transmission is zoonotic [54, 63, 64] However, according to Yakob et al. [132], this scenario could also be a result of human outbreaks that remained relatively small and localized, with transmission chains that did not extend far beyond the immediate population.
To estimate community prevalence of the knowlesi parasite in endemic areas, serological investigations were undertaken in three communities: two from northern Sabah and another from the Philippines. Results showed that women were similarly exposed to P. knowlesi as men, but with a higher proportion of subclinical outcomes. Fornace et al. [108] also found P. knowlesi exposure in children under 5 years of age at all three study sites, suggestive of low-level ongoing transmission. In Kudat province, Borneo, P. knowlesi is the most common cause of childhood malaria [133]. In general, individuals exposed to P. knowlesi were younger than those with sero-prevalence for the non-zoonotic malarias. Subclinical infections and exposure have therefore been found in populations in Sabah [108, 130], Northern Sumatra [98], Sarawak [134], and the Philippines [108] and these individuals may be contributing to the transmission of P. knowlesi malaria to others.
Co-infection with P. knowlesi and the human malarias is relatively common [70, 87, 89, 98, 108, 135] and may indicate human to human transmission where the vectors pick up P. knowlesi from humans [94]. In the border region between China and Myanmar, Jiang and colleagues [91] found that P. knowlesi was most commonly found as a co-infection with either P. falciparum (13%) or P. vivax (13%) rather than as a mono-infection (4%). In Khanh Phu, southern Vietnam, sampling over 2009–2010 found P. knowlesi infections in 26% of people sampled (32/125) and always as a co-infection predominantly with P. vivax followed to a much lesser degree by P. falciparum [136]. In a more recent study from Khan Phu, Vietnam, 70% of mosquitoes tested that had P. knowlesi in their salivary glands also carried the human malaria parasites, P. falciparum and/or P. vivax [94].
Agencies in Malaysia have been efficient in reducing the incidence of the two main human malarias in the region: P. falciparum and P. vivax. However this situation may be assisting P. knowlesi in the evolutionary transition into humans [137]. It is believed that P. falciparum and P. vivax may be outcompeting P. knowlesi within the human host as these parasites are so well adapted now to humans [137]. In areas such as Sabah and Sarawak, Borneo, where the prevalence of human malaria parasite is in decline, P. knowlesi may have a better chance of infecting human hosts, therefore, resulting in increasing incidence in this region.
Imai and colleagues [138] argue that there is a very low chance of sustained human to human transmission in the absence of macaques, with an estimated Ro = 1.04 in one of their models. However, continuing and closer contact between humans and macaques may alter mosquito preference in the future and lead to increased human to human transmission [67]. According to the Ross-McDonald mathematical model for malarial transmission, even a small change in preference for humans exerts a sizeable impact upon malaria transmission because of the exponential nature of this particular variable within the model [139]. Yakob et al. [140] used mathematical modelling to demonstrate that as humans increase their contact with the knowlesi parasite, more anthropophilic vectors will be selected for, as will a parasite that has a preference for human blood meals. These conditions may occur if the population density of humans in any given endemic area exceeds that of the macaques and a tipping point is reached.
Drivers at the cellular and genetic level
Drivers of P. knowlesi transmission operating at these micro-scales relate to pathogen exposure, immunity, and health status of the human hosts. Together with these cellular factors, the different genotypes of P. knowlesi and invasion mechanisms have an important role to play in infection outcomes and continuing adaptation to humans as hosts [7]. Coevolution of malaria parasites and humans has resulted in a delicate balance between the levels of parasitaemia developed and the immune system of the host, with natural selection favouring maximum parasite transmission and limited host damage [141]. An important goal of malaria research is to better understand how the genomic differences in the Plasmodium parasites confer adaption for specific hosts and the different erythrocyte developmental stages [14].
Lim et al. [142] has established that primate-adapted P. knowlesi multiplies poorly in human blood because of its affinity for reticulocytes. In vertebrate blood, reticulocytes (1% of blood cells) are immature red blood cells still undergoing active protein synthesis of haemoglobin which takes place over several days until the cells mature and are classified as normocytes (99% of blood cells) [143]. In vitro testing of P. knowlesi in human blood found that over time the parasite quickly adapts to invade normocytes as well [142]. Plasmodium knowlesi contains proteins referred to as NBPXa and NBPXb (normocyte binding-like proteins-NBPs) which have been found essential for the invasion of human erythrocytes [144, 145]. The parasite’s short multiplication time (quotidian cycle) and its ability to infect both immature and mature erythrocytes results in hyper-parasitaemia developing quickly (within several days) if the disease goes undetected [146,147,148]. Respiratory distress along with jaundice, hypotension, acute renal failure, and organ dysfunction are typical of the severe complications manifested, with thrombocytopaenia nearly universal in cases of infection [146, 149, 150].
Disease virulence appears to relate to the NBPXa allele where patients in Sarawak infected with NBPXa had different disease markers (i.e. renal dysfunction) to those infected with NBPXb [151]. The allele is dimorphic and one of these forms (KH195) is more virulent than the other form (KH273) and responsible for the high levels of parasitaemia found in some cases [151, 152]. Investigations in Sabah and Peninsular Malaysia to compare all three geographical areas found that three distinct types of P. knowlesi exist: NBPXa (Type-1 and Type-2) throughout Malaysia with the existence of a third type (Type-3) detected only in Peninsular Malaysia [153]. All three types showed dimorphism (with the virulent and less virulent forms) indicating that virulent disease progression is also possible in Peninsular Malaysia [153], although far less common [154]. Whether these different types represent sub-species would require further genetic analysis [153]. Disease severity may also relate to a decreased capacity of the P. knowlesi-infected erythrocytes to deform (to pass through a capillary wall) thereby impeding microvascular flow and leading to severe symptoms, such as impaired organ perfusion, as occurs with P. falciparum [155].
Plasmodium knowlesi employs antigenic variation similarly to P. falciparum, to systematically alter antigens presented to the host’s immune system, thereby ‘hiding’ and leading to chronic infection [156,157,158]. A further conserved erythrocyte invasion function of P. knowlesi, equivalent to the tether structure of P. falciparum, involves protein trafficking where the knowlesi parasite generates membranous structures to modify the host erythrocyte for improved protein transport and parasite nutrition [159]. Three tryptophan-rich antigens (PkTRAg) also show binding to human erythrocytes and demonstrate a sharing of human erythrocyte receptors with P. falciparum in in vitro cultures [125].
A pathway that may act to currently restrict the P. knowlesi parasite in humans is the sialic acid pathway [51]. Sialic acid is a product released by mammals from hydrolysis of brain glycolipids or salivary mucins [160]. The most common forms are N-acetyl-neuraminic acid (Neu5Ac) and N-glycolyl-neuraminic acid (Neu5Gc) [160, 161]. Humans are unable to produce Neu5Gc from Neu5Ac due to a mutation in the required gene, CMAH, and therefore human erythrocytes express only Neu5Ac. In contrast, this gene remains active in the Old World monkeys, who have retained their ability to express Neu5Gc [160, 161]. Plasmodium knowlesi is unique in its ability to infect Neu5Gc positive macaques as well as Neu5Ac positive humans [84]. Two ligands of the Duffy binding proteins (similarly found in P. vivax), PkDBPβ and PkDBPγ, invade macaque erythrocytes using Neu5Gc, but a laboratory-developed line invades human erythrocytes using PkDBPα independently of Neu5Gc receptors, demonstrating a tolerance for the Neu5Ac variant in humans.
Discussion and implications
Factors that influence the emergence of zoonotic P. knowlesi are parasite-host evolutionary dynamics, diversity, abundance and range of host and vector species, and the spatial and temporal overlap of contact between them [8]. These factors are in turn influenced by socio-ecological processes such as population growth, anthropogenic land use change, globalization of agricultural markets, and climate change [8]. These influences impact upon natural habitats and ecosystem dynamics at multiple scales in ways that alter microclimatic conditions, contact between parasite, host, and vector, and, sometimes, create new parasite-host dynamics with humans [6].
This connectivity with eco-social processes requires an interdisciplinary approach to enhance our understanding of the feedback loops involved with infectious disease emergence and spread [8]. In the case of P. knowlesi, there are many gaps in knowledge that need addressing, as outlined in Table 1. At the level of global phylogenetic analysis of malaria parasites, the ecological and evolutionary events that have led to host-switching and species diversification are not well understood, but are important to unravel for a greater knowledge of the disease in humans [15, 17]. The low occurrence of spill-over into humans from the NHP malaria parasites seems to show a tight host-specificity where significant ecological and molecular hurdles must be overcome to cross species [51]. For example, even though chimpanzees share > 99% genetic similarity with humans, the main malarial parasite infecting chimpanzees, Plasmodium reichenowi, does not infect humans and conversely P. falciparum only causes subclinical infection in them [162]. For P. knowlesi, more information is required on its phylogenetic history, life-history traits and host usage [15] to determine how it has evolved the capacity to spill-over into humans so readily now.
More information is needed on the distribution and abundance of reservoir hosts and vectors of the parasite and how these relate to the rapid landscape-scale changes occurring in many parts of Southeast Asia, and notably in Borneo [163]. The first case of P. knowlesi infecting a human occurred during the 1960s in West Malaysia when most of the interior of the country was virgin forest and the mosquito vectors and humans had little contact [164]. As emphasized using this multiscale approach, the ecological situation in Malaysia today is very different. The vector species, along with the macaques can be found at the forest fringes, and have begun to encroach into farms and peri-domestic areas around villages [74, 79, 122]. Landscape scale ecological changes and potential links to P. knowlesi transmission in Malaysian Borneo have been described in the relevant paper [11]. There is a shortage of information on community exposure patterns as most data on P. knowlesi comes from reported cases of clinical infection [163]. In order to build models of transmission and spread for the parasite, wide geographical data is needed on community exposure levels, distribution of hosts and vectors, and associated ecological factors [108, 129, 163]. This information would assist with determining where hotspots of transmission are occurring and inform strategies for prevention and treatment [163].
Currently, human infections appear to be a result of human–macaque proximity [49, 165]. However, the potential exists for P. knowlesi to be transmitted from human to human, or to adapt to utilizing other NHPs as hosts, or other mosquito species as vectors [61, 138, 166]. As subclinical infection and exposure in humans is being more readily detected [108, 130], there is a need to better understand and monitor which subpopulation of the parasite is responsible and any genetic changes that are occurring [65]. The parasite has evolved into different strains adapted to the two different macaque hosts on Borneo and which have also diverged from the isolates in Peninsular Malaysia found in humans [60]. Historically, this emergence of three distinct genetic subpopulations in Malaysia occurred through genetic isolation, but currently recombination is occurring between the different macaque isolates. This process may be accelerated as their habitats are lost to deforestation and competition for resources forces them together [61]. This genetic recombination provides compelling evidence that rapid ecological change occurring on the island of Borneo may be furthering the parasites ability to host switch. Increased contact with humans also provides evolutionary opportunity for erythrocyte invasion pathways such as the sialic acid-independent pathway using of the Duffy-binding protein PkDBPα, or the tryptophan-rich antigens (utilized similarly by P. vivax and P. falciparum) to be exploited by P. knowlesi [125, 161].
This review had demonstrated how the evolution and biology of vectors, hosts, and pathogen, the changing landscape and ecology, and human behaviour, all interact at multiple ecological scales to produce the emerging disease risk that is now apparent for P. knowlesi [167]. The transmission and spread of P. knowlesi and its potential for host-switching needs to be considered with an interdisciplinary approach that incorporates the multi-scaled influences upon its emergence [7, 8, 10]. If any of the ‘host-switching’ events were to occur, there could be an increase in transmission and spread of this parasite, and a future in which P. knowlesi poses an even greater threat to population health.
Abbreviations
- NHPs:
-
non-human primates
- Ma:
-
million years ago
- PCR:
-
polymerase chain reaction
- NBPs:
-
normocyte binding-like proteins
References
Patz JA, Daszak P, Tabor GM, Aguirre AA, Pearl M, Epstein J, et al. Unhealthy landscapes: policy recommendations on land use change and infectious disease emergence. Environ Health Perspect. 2004;112:1092–8.
Patz JA, Graczyk TK, Geller N, Vittor AY. Effects of environmental change on emerging parasitic diseases. Int J Parasitol. 2000;30:1395–405.
Murray KA, Daszak P. Human ecology in pathogenic landscapes: two hypotheses on how land use change drives viral emergence. Curr Opin Virol. 2013;3:79–83.
Barber BE, Rajahram GS, Grigg MJ, William T, Anstey NM. World Malaria Report: time to acknowledge Plasmodium knowlesi malaria. Malar J. 2017;16:135.
Komaki-Yasuda K, Vincent JP, Nakatsu M, Kato Y, Ohmagari N, Kano S. A novel PCR-based system for the detection of four species of human malaria parasites and Plasmodium knowlesi. PLoS ONE. 2018;13:e0191886.
Wilcox BA, Colwell RR. Emerging and reemerging infectious diseases: biocomplexity as an interdisciplinary paradigm. EcoHealth. 2005;2:244.
Estrada-Peña A, Ostfeld RS, Peterson AT, Poulin R, de la Fuente J. Effects of environmental change on zoonotic disease risk: an ecological primer. Trends Parasitol. 2014;30:205–14.
Jones BA, Betson M, Pfeiffer DU. Eco-social processes influencing infectious disease emergence and spread. Parasitology. 2017;144:26–36.
Hausermann H, Tschakert P, Smithwick EA, Ferring D, Amankwah R, Klutse E, et al. Contours of risk: spatializing human behaviors to understand disease dynamics in changing landscapes. EcoHealth. 2012;9:251–5.
Loh E, Murray KA, Nava A, Aguirre A, Daszak P. Evaluating the links between biodiversity, land-use change, and infectious disease emergence in tropical fragmented landscapes. In: Tropical conservation: perspectives on local and global priorities. 2016. p. 79.
Davidson G, Chua TH, Cook A, Speldewinde PC, Weinstein P. The role of ecological linkage mechanisms in Plasmodium knowlesi transmission and spread. EcoHealth. 2019 (Epub ahead of print).
Finucane ML, Fox J, Saksena S, Spencer JH. A conceptual framework for analyzing social-ecological models of emerging infectious diseases. In: Manfredo MJ, Vaske J, Rechkemmer A, Duke EA, editors. Understanding society and natural resources. Open Access: The East-West Centre; 2014.
Waltner-Toews D. Zoonoses, One Health and complexity: wicked problems and constructive conflict. Phil Trans R Soc B. 2017;372:20160171.
Carlton JM, Sullivan SA. A feast of malaria parasite genomes. Cell Host Microbe. 2017;21:310–2.
Galen SC, Borner J, Martinsen ES, Schaer J, Austin CC, West CJ, et al. The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R Soc Open Sci. 2018;5:171780.
Silva JC, Egan A, Arze C, Spouge JL, Harris DG. A new method for estimating species age supports the coexistence of malaria parasites and their mammalian hosts. Mol Biol Evol. 2015;32:1354–64.
Martinsen ES, Perkins SL, Schall JJ. A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): evolution of life-history traits and host switches. Mol Phylogenet Evol. 2008;47:261–73.
Ansari HR, Templeton TJ, Subudhi AK, Ramaprasad A, Tang J, Lu F, et al. Genome-scale comparison of expanded gene families in Plasmodium ovale wallikeri and Plasmodium ovale curtisi with Plasmodium malariae and with other Plasmodium species. Int J Parasitol. 2016;46:685–96.
Kantele A, Jokiranta TS. Review of cases with the emerging fifth human malaria parasite, Plasmodium knowlesi. Clin Infect Dis. 2011;52:1356–62.
Ramasamy R. Zoonotic malaria—global overview and research and policy needs. Front Public Health. 2014;2:123.
Garamszegi LZ. Patterns of co-speciation and host switching in primate malaria parasites. Malar J. 2009;8:110.
Baird JK. Malaria zoonoses. Travel Med Infect Dis. 2009;7:269–77.
Frech C, Chen N. Genome comparison of human and non-human malaria parasites reveals species subset-specific genes potentially linked to human disease (Plasmodium genes linked to human disease). PLoS Comput Biol. 2011;7:e1002320.
Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH. A plethora of Plasmodium species in wild apes: a source of human infection? Trends Parasitol. 2011;27:222–9.
Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, et al. Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010;467:420–5.
Liu W, Sundararaman SA, Loy DE, Learn GH, Li Y, Plenderleith LJ, et al. Multigenomic delineation of Plasmodium species of the Laverania subgenus infecting wild-living chimpanzees and gorillas. Genome Biol Evol. 2016;8:1929–39.
Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, Nkoghe D, et al. A new malaria agent in African hominids. PLoS Pathog. 2009;5:e1000446.
Boundenga L, Ollomo B, Rougeron V, Mouele LY, Mve-Ondo B, Delicat-Loembet LM, et al. Diversity of malaria parasites in great apes in Gabon. Malar J. 2015;14:111.
Sundararaman SA, Plenderleith LJ, Liu W, Loy DE, Learn GH, Li Y, et al. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat Commun. 2016;7:11078.
Otto TD, Gilabert A, Crellen T, Böhme U, Arnathau C, Sanders M, et al. Genomes of an entire Plasmodium subgenus reveal paths to virulent human malaria. bioRxiv. 2017:095679.
Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, et al. Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2015;32:422–39.
Fuentes-Ramírez A, Jiménez-Soto M, Castro R, Romero-Zuñiga JJ, Dolz G. Molecular detection of Plasmodium malariae/Plasmodium brasilianum in non-human primates in captivity in Costa Rica. PLoS ONE. 2017;12:e0170704.
Aitken EH, Bueno MG, dos Santos Ortolan L, Alvaréz JM, Pissinatti A, Kierulff MCM, et al. Survey of Plasmodium in the golden-headed lion tamarin (Leontopithecus chrysomelas) living in urban Atlantic forest in Rio de Janeiro, Brazil. Malar J. 2016;15:93.
Brasil P, Zalis MG, de Pina-Costa A, Siqueira AM, Júnior CB, Silva S, et al. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health. 2017;5:e1038–46.
Pina-Costa AD, Brasil P, Santi SMD, Araujo MPD, Suárez-Mutis MC, Oliveira-Ferreira J, et al. Malaria in Brazil: what happens outside the Amazonian endemic region. Mem Inst Oswaldo Cruz. 2014;109:618–33.
Figueiredo MAP, Di Santi SM, Manrique WG, André MR, Machado RZ. Identification of Plasmodium spp. in Neotropical primates of Maranhense Amazon in Northeast Brazil. PLoS ONE. 2017;12:e0182905.
Ta TH, Hisam S, Lanza M, Jiram AI, Ismail N, Rubio JM. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J. 2014;13:68.
Prugnolle F, Rougeron V, Becquart P, Berry A, Makanga B, Rahola N, et al. Diversity, host switching and evolution of Plasmodium vivax infecting African great apes. Proc Natl Acad Sci USA. 2013;110:8123–8.
Talundzic E, Ravishankar S, Nayak V, Patel DS, Olsen C, Sheth M, et al. First full draft genome sequence of Plasmodium brasilianum. Genome Announc. 2017;5:e01566-16.
Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995;12:616.
Lalremruata A, Magris M, Vivas-Martínez S, Koehler M, Esen M, Kempaiah P, et al. Natural infection of Plasmodium brasilianum in humans: man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine. 2015;2:1186–92.
Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, Malenke JA, et al. African origin of the malaria parasite Plasmodium vivax. Nat Commun. 2014;5:3346.
Lim CS, Tazi L, Ayala F. Plasmodium vivax: recent world expansion and genetic identity to Plasmodium simium. Proc Natl Acad Sci USA. 2005;102:15523–8.
Tazi L, Ayala FJ. Unresolved direction of host transfer of Plasmodium vivax v. P. simium and P. malariae v. P. brasilianum. Infect Genet Evol. 2011;11:209–21.
Loy DE, Liu W, Li Y, Learn GH, Plenderleith LJ, Sundararaman SA, et al. Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax. Int J Parasitol. 2017;47:87–97.
Rodrigues P, Valdivia H, de Oliveira T, Alves J, Cerutti-Junior C, Buery J, et al. Human migration and the spread of malaria parasites to the New World. Sci Rep. 2018;8:1993.
Vargas-Serrato E, Corredor V, Galinski MR. Phylogenetic analysis of CSP and MSP-9 gene sequences demonstrates the close relationship of Plasmodium coatneyi to Plasmodium knowlesi. Infect Genet Evol. 2003;3:67–73.
Sutton PL, Luo Z, Divis PCS, Friedrich VK, Conway DJ, Singh B, et al. Characterizing the genetic diversity of the monkey malaria parasite Plasmodium cynomolgi. Infect Genet Evol. 2016;40:243.
Chua TH, Manin BO, Daim S, Vythilingam I, Drakeley C. Phylogenetic analysis of simian Plasmodium spp. infecting Anopheles balabacensis Baisas in Sabah, Malaysia. PLoS Negl Trop Dis. 2017;11:e0005991.
Chatterjee S, Mukhopadhyay P, Bandyopadhyay R, Dhal P, Biswal D, Bandyopadhyay PK. Molecular characterization and phylogenetic analysis of Plasmodium vivax, Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae and Plasmodium cynomolgi. J Parasit Dis. 2017;41:230–6.
Scully EJ, Kanjee U, Duraisingh MT. Molecular interactions governing host-specificity of blood stage malaria parasites. Curr Opin Microbiol. 2017;40:21–31.
Stewart C-B, Disotell TR. Primate evolution—in and out of Africa. Curr Biol. 1998;8:R582–8.
Ziegler T, Abegg C, Meijaard E, Perwitasari-Farajallah D, Walter L, Hodges JK, et al. Molecular phylogeny and evolutionary history of Southeast Asian macaques forming the M. silenus group. Mol Phylogenet Evol. 2007;42:807–16.
Lee K-S, Divis PC, Zakaria SK, Matusop A, Julin RA, Conway DJ, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7:e1002015.
Macaulay V, Hill C, Achilli A, Rengo C, Clarke D, Meehan W, et al. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science. 2005;308:1034.
Chong ETJ, Neoh JWF, Lau TY, Lim YA-L, Chua KH, Lee P-C. Genetic and haplotype analyses targeting cytochrome b gene of Plasmodium knowlesi isolates of Malaysian Borneo and Peninsular Malaysia. Acta Trop. 2018;181:35–9.
Raja TN, Hu TH, Zainudin R, Lee KS, Perkins SL, Singh B. Malaria parasites of long-tailed macaques in Sarawak, Malaysian Borneo: a novel species and demographic and evolutionary histories. BMC Evol Biol. 2018;18:49.
Moyes CL, Henry AJ, Golding N, Huang Z, Singh B, Baird JK, et al. Defining the geographical range of the Plasmodium knowlesi reservoir. PLoS Negl Trop Dis. 2014;8:e2780.
Moyes CL, Shearer FM, Huang Z, Wiebe A, Gibson HS, Nijman V, et al. Predicting the geographical distributions of the macaque hosts and mosquito vectors of Plasmodium knowlesi malaria in forested and non-forested areas. Parasit Vectors. 2016;9:242.
Assefa S, Lim C, Preston MD, Duffy CW, Nair MB, Adroub SA, et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci USA. 2015;112:13027–32.
Benavente ED, de Sessions PF, Moon RW, Holder AA, Blackman MJ, Roper C, et al. Analysis of nuclear and organellar genomes of Plasmodium knowlesi in humans reveals ancient population structure and recent recombination among host-specific subpopulations. PLoS Genet. 2017;13:e1007008.
Rawa MSA, Fong M-Y, Lau Y-L. Genetic diversity and natural selection in the rhoptry-associated protein 1 (RAP-1) of recent Plasmodium knowlesi clinical isolates from Malaysia. Malar J. 2016;15:62.
Divis PC, Singh B, Anderios F, Hisam S, Matusop A, Kocken CH, et al. Admixture in humans of two divergent Plasmodium knowlesi populations associated with different macaque host species. PLoS Pathog. 2015;11:e1004888.
Divis PCS, Lin LC, Rovie-Ryan JJ, Kadir KA, Anderios F, Hisam S, et al. Three divergent subpopulations of the malaria parasite Plasmodium knowlesi. Emerg Infect Dis. 2017;23:616–24.
Divis P, Duffy CW, Kadir KA, Singh B, Conway DJ. Genome-wide mosaicism in divergence between zoonotic malaria parasite subpopulations with separate sympatric transmission cycles. Mol Ecol. 2018;27:860–70.
Liedigk R, Kolleck J, Böker KO, Meijaard E, Md-Zain BM, Abdul-Latiff MAB, et al. Mitogenomic phylogeny of the common long-tailed macaque (Macaca fascicularis fascicularis). BMC Genomics. 2015;16:222.
Galinski MR, Barnwell JW. Monkey malaria kills four humans. Trends Parasitol. 2009;25:200–4.
Sallum M, Peyton E, Wilkerson R. Six new species of the Anopheles leucosphyrus group, reinterpretation of An. elegans and vector implications. Med Vet Entomol. 2005;19:158–99.
Vythilingam I. Plasmodium knowlesi in humans: a review on the role of its vectors in Malaysia. Trop Biomed. 2010;27:1–12.
Vythilingam I, NoorAzian YM, Huat TC, Jiram AI, Yusri YM, Azahari AH, et al. Plasmodium knowlesi in humans, macaques and mosquitoes in peninsular Malaysia. Parasit Vectors. 2008;1:26.
Vythilingam I. Plasmodium knowlesi and Wuchereria bancrofti: their vectors and challenges for the future. Front Physiol. 2012;3:115.
Jiram AI, Vythilingam I, NoorAzian YM, Yusof YM, Azahari AH, Fong M-Y. Entomologic investigation of Plasmodium knowlesi vectors in Kuala Lipis, Pahang, Malaysia. Malar J. 2012;11:213.
Lau Y-L, Lee W-C, Chen J, Zhong Z, Jian J, Amir A, et al. Draft genomes of Anopheles cracens and Anopheles maculatus: comparison of simian malaria and human malaria vectors in Peninsular Malaysia. PLoS ONE. 2016;11:e0157893.
Vythilingam I, Wong ML, Wan-Yussof WS. Current status of Plasmodium knowlesi vectors: a public health concern? Parasitology. 2018;145(1):32–40. https://doi.org/10.1017/S0031182016000901.
Tan CH, Vythilingam I, Matusop A, Chan ST, Singh B. Bionomics of Anopheles latens in Kapit, Sarawak, Malaysian Borneo in relation to the transmission of zoonotic simian malaria parasite Plasmodium knowlesi. Malar J. 2008;7:52.
Wong ML, Chua TH, Leong CS, Khaw LT, Fornace K, Wan-Sulaiman W-Y, et al. Seasonal and spatial dynamics of the primary vector of Plasmodium knowlesi within a major transmission focus in Sabah, Malaysia. PLoS Negl Trop Dis. 2015;9:e0004135.
Wong ML, Vythilingam I, Leong CS, Khaw LT, Chua TH, Obrain B, et al. Incrimination of Anopheles balabacensis as the vector for simian malaria in Kudat Division, Sabah, Malaysia. J Microbiol Immunol Infect. 2015;48:S47–8.
WHO. Expert consultation on Plasmodium knowlesi malaria to guide malaria elimination strategies, Kota Kinabalu, Malaysia, 1–2 March 2017: meeting report. Manila: World Health Organization Regional Office for the Western Pacific, 2017.
Manin BO, Ferguson HM, Vythilingam I, Fornace K, William T, Torr SJ, et al. Investigating the contribution of peri-domestic transmission to risk of zoonotic malaria infection in humans. PLoS Negl Trop Dis. 2016;10:e0005064.
Rodman PS. Structural differentiation of microhabitats of sympatric Macaca fascicularis and M. nemestrina in East Kalimantan, Indonesia. Int J Primatol. 1991;12:357–75.
Malaivijitnond S, Arsaithamkul V, Tanaka H, Pomchote P, Jaroenporn S, Suryobroto B, et al. Boundary zone between northern and southern pig-tailed macaques and their morphological differences. Primates. 2012;53:377–89.
Shearer FM, Huang Z, Weiss DJ, Wiebe A, Gibson HS, Battle KE, et al. Estimating geographical variation in the risk of zoonotic Plasmodium knowlesi infection in countries eliminating malaria. PLoS Negl Trop Dis. 2016;10:e0004915.
Zhu H, Li J, Zheng H. Human natural infection of Plasmodium knowlesi. Chinese J Parasitol Parasitic Dis. 2006;24:70–1 (in Chinese).
Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev. 2013;26:165–84.
Akter R, Vythilingam I, Khaw LT, Qvist R, Lim YA-L, Sitam FT, et al. Simian malaria in wild macaques: first report from Hulu Selangor district, Selangor, Malaysia. Malar J. 2015;14:386.
Vadivelan M, Dutta T. Recent advances in the management of Plasmodium knowlesi infection. Trop Parasitol. 2014;4:31.
Singh B, Sung LK, Matusop A, Radhakrishnan A, Shamsul SSG, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24.
Jongwutiwes S, Buppan P, Kosuvin R, Seethamchai S, Pattanawong U, Sirichaisinthop J, et al. Plasmodium knowlesi malaria in humans and macaques, Thailand. Emerg Infect Dis. 2011;17:1799.
Putaporntip C, Hongsrimuang T, Seethamchai S, Kobasa T, Limkittikul K, Cui L, et al. Differential prevalence of Plasmodium infections and cryptic Plasmodium knowlesi malaria in humans in Thailand. J Infect Dis. 2009;199:1143–50.
Luchavez J, Espino F, Curameng P, Espina R, Bell D, Chiodini P, et al. Human infections with Plasmodium knowlesi, the Philippines. Emerg Infect Dis. 2008;14:811.
Jiang N, Chang Q, Sun X, Lu H, Yin J, Zhang Z, et al. Co-infections with Plasmodium knowlesi and other malaria parasites, Myanmar. Emerg Infect Dis. 2010;16:1476.
Jeslyn WPS, Huat TC, Vernon L, Irene LMZ, Sung LK, Jarrod LP, et al. Molecular epidemiological investigation of Plasmodium knowlesi in humans and macaques in Singapore. Vector Borne Zoonotic Dis. 2011;11:131–5.
Ng OT, Ooi EE, Lee CC, Lee PJ, Ng LC, Pei SW, et al. Naturally acquired human Plasmodium knowlesi infection, Singapore. Emerg Infect Dis. 2008;14:814–6.
Maeno Y, Culleton R, Quang NT, Kawai S, Marchand RP, Nakazawa S. Plasmodium knowlesi and human malaria parasites in Khan Phu, Vietnam: gametocyte production in humans and frequent co-infection of mosquitoes. Parasitology. 2016;144:527–35.
Eede PVd, Van HN, Van Overmeir C, Vythilingam I, Duc TN, Hung LX, et al. Human Plasmodium knowlesi infections in young children in central Vietnam. Malar J. 2009;8:249.
Sermwittayawong N, Singh B, Nishibuchi M, Sawangjaroen N, Vuddhakul V. Human Plasmodium knowlesi infection in Ranong province, southwestern border of Thailand. Malar J. 2012;11:36.
Herdiana H, Irnawati I, Coutrier FN, Munthe A, Mardiati M, Yuniarti T, et al. Two clusters of Plasmodium knowlesi cases in a malaria elimination area, Sabang Municipality, Aceh, Indonesia. Malar J. 2018;17:186.
Lubis IN, Wijaya H, Lubis M, Lubis CP, Divis P, Beshir KB, et al. Contribution of Plasmodium knowlesi to multispecies human malaria infections in North Sumatera, Indonesia. J Infect Dis. 2017;215:1148–55.
Herdiana H, Cotter C, Coutrier FN, Zarlinda I, Zelman BW, Tirta YK, et al. Malaria risk factor assessment using active and passive surveillance data from Aceh Besar, Indonesia, a low endemic, malaria elimination setting with Plasmodium knowlesi, Plasmodium vivax, and Plasmodium falciparum. Malar J. 2016;15:468.
Sulistyaningsih E, Fitri LE, Löscher T, Berens-Riha N. Diagnostic difficulties with Plasmodium knowlesi infection in humans. Emerg Infect Dis. 2010;16:1033–4.
Figtree M, Lee R, Bain L, Kennedy T, Mackertich S, Urban M, et al. Plasmodium knowlesi in human, Indonesian Borneo. Emerg Infect Dis. 2010;16:672.
Setiadi W, Sudoyo H, Trimarsanto H, Sihite BA, Saragih RJ, Juliawaty R, et al. A zoonotic human infection with simian malaria, Plasmodium knowlesi, in Central Kalimantan, Indonesia. Malar J. 2016;15:218.
Ghinai I, Cook J, Hla TTW, Htet HMT, Hall T, Lubis IN, et al. Malaria epidemiology in central Myanmar: identification of a multi-species asymptomatic reservoir of infection. Malar J. 2017;16:16.
Tyagi RK, Das MK, Singh SS, Sharma YD. Discordance in drug resistance-associated mutation patterns in marker genes of Plasmodium falciparum and Plasmodium knowlesi during coinfections. J Antimicrob Chemother. 2013;68:1081–8.
Iwagami M, Nakatsu M, Khattignavong P, Soundala P, Lorphachan L, Keomalaphet S, et al. First case of human infection with Plasmodium knowlesi in Laos. PLoS Negl Trop Dis. 2018;12:e0006244.
Chin W, Contacos PG, Coatney GR, Kimball HR. A naturally acquired quotidian-type malaria in man transferable to monkeys. Science. 1965;149:865.
Fong YL, Cadigan FC, Coatney GR. A presumptive case of naturally occurring Plasmodium knowlesi malaria in man in Malaysia. Trans R Soc Trop Med Hyg. 1971;65:839–40.
Fornace KM, Herman LS, Abidin TR, Chua TH, Daim S, Lorenzo PJ, et al. Exposure and infection to Plasmodium knowlesi in case study communities in Northern Sabah, Malaysia and Palawan, The Philippines. PLoS Negl Trop Dis. 2018;12:e0006432.
Fisher B, Edwards DP, Giam X, Wilcove DS. The high costs of conserving Southeast Asia’s lowland rainforests. Front Ecol Environ. 2011;9:329–34.
Kassam Z. Considerations of development in Malaysian Borneo. EnviroLab Asia. 2017;1:5.
Padfield R, Drew S, Syayuti K, Page S, Evers S, Campos-Arceiz A, et al. Landscapes in transition: an analysis of sustainable policy initiatives and emerging corporate commitments in the palm oil industry. Landscape Res. 2016;41:744–56.
Gaveau DL, Sheil D, Salim MA, Arjasakusuma S, Ancrenaz M, Pacheco P, et al. Rapid conversions and avoided deforestation: examining four decades of industrial plantation expansion in Borneo. Sci Rep. 2016;6:32017.
Wilcove DS, Koh LP. Addressing the threats to biodiversity from oil-palm agriculture. Biodivers Conserv. 2010;19:999–1007.
Bryan JE, Shearman PL, Asner GP, Knapp DE, Aoro G, Lokes B. Extreme differences in forest degradation in Borneo: comparing practices in Sarawak, Sabah, and Brunei. PLoS ONE. 2013;8:e69679.
Brant HL, Ewers RM, Vythilingam I, Drakeley C, Benedick S, Mumford JD. Vertical stratification of adult mosquitoes (Diptera: Culicidae) within a tropical rainforest in Sabah, Malaysia. Malar J. 2016;15:370.
Molina-Cruz A, Garver LS, Alabaster A, Bangiolo L, Haile A, Winikor J, et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science. 2013;340:984–7.
Piera KA, Aziz A, William T, Bell D, González IJ, Barber BE, et al. Detection of Plasmodium knowlesi, Plasmodium falciparum and Plasmodium vivax using loop-mediated isothermal amplification (LAMP) in a co-endemic area in Malaysia. Malar J. 2017;16:29.
Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev. 2002;15:66–78.
Kawai S, Hirai M, Haruki K, Tanabe K, Chigusa Y. Cross-reactivity in rapid diagnostic tests between human malaria and zoonotic simian malaria parasite Plasmodium knowlesi infections. Parasitol Int. 2009;58:300–2.
Foster D, Cox-Singh J, Mohamad DS, Krishna S, Chin PP, Singh B. Evaluation of three rapid diagnostic tests for the detection of human infections with Plasmodium knowlesi. Malar J. 2014;13:60.
Ooi CH, Bujang MA, Tg Abu Bakar Sidik TMI, Ngui R, Lim YA-L. Over two decades of Plasmodium knowlesi infections in Sarawak: trend and forecast. Acta Trop. 2017;176:83–90.
Vythilingam I, Lim YA, Venugopalan B, Ngui R, Leong CS, Wong ML, et al. Plasmodium knowlesi malaria an emerging public health problem in Hulu Selangor, Selangor, Malaysia (2009–2013): epidemiologic and entomologic analysis. Parasit Vectors. 2014;7:436.
Grigg MJ, Cox J, William T, Jelip J, Fornace KM, Brock PM, et al. Individual-level factors associated with the risk of acquiring human Plasmodium knowlesi malaria in Malaysia: a case-control study. Lancet Planet Health. 2017;1:e97–104.
Grigg MJ, William T, Barber BE, Rajahram GS, Menon J, Schimann E, et al. Age-related clinical spectrum of Plasmodium knowlesi malaria and predictors of severity. Clin Infect Dis. 2018;67:350–9.
Tyagi K, Gupta D, Saini E, Choudhary S, Jamwal A, Alam MS, et al. Recognition of human erythrocyte receptors by the tryptophan-rich antigens of monkey malaria parasite Plasmodium knowlesi. PLoS ONE. 2015;10:e0138691.
Malaria. Centers for Disease Control and Prevention. 2016. https://www.cdc.gov/malaria/about/biology/. Accessed 4 Sept 2017.
Cox-Singh J, Singh B. Knowlesi malaria: newly emergent and of public health importance? Trends Parasitol. 2008;24:406–10.
Grigg MJ, William T, Menon J, Dhanaraj P, Barber BE, Wilkes CS, et al. Artesunate–mefloquine versus chloroquine for treatment of uncomplicated Plasmodium knowlesi malaria in Malaysia (ACT KNOW): an open-label, randomised controlled trial. Lancet Infect Dis. 2016;16:180–8.
Fornace KM, Abidin TR, Alexander N, Brock P, Grigg MJ, Murphy A, et al. Association between landscape factors and spatial patterns of Plasmodium knowlesi Infections in Sabah, Malaysia. Emerg Infect Dis. 2016;22:201–9.
Fornace KM, Nuin NA, Betson M, Grigg MJ, William T, Anstey NM, et al. Asymptomatic and submicroscopic carriage of Plasmodium knowlesi malaria in household and community members of clinical cases in Sabah, Malaysia. J Infect Dis. 2016;213:784–7.
Barber BE, William T, Dhararaj P, Anderios F, Grigg MJ, Yeo TW, et al. Epidemiology of Plasmodium knowlesi malaria in north-east Sabah, Malaysia: family clusters and wide age distribution. Malar J. 2012;11:401.
Yakob L, Lloyd AL, Kao RR, Ferguson HM, Brock PM, Drakeley C, et al. Plasmodium knowlesi invasion following spread by infected mosquitoes, macaques and humans. Parasitology. 2018;145:101–10.
Barber BE, William T, Jikal M, Jilip J, Dhararaj P, Menon J, et al. Plasmodium knowlesi malaria in children. Emerg Infect Dis. 2011;17:814.
Siner A, Liew S-T, Kadir KA, Mohamad DSA, Thomas FK, Zulkarnaen M, et al. Absence of Plasmodium inui and Plasmodium cynomolgi, but detection of Plasmodium knowlesi and Plasmodium vivax infections in asymptomatic humans in the Betong division of Sarawak, Malaysian Borneo. Malar J. 2017;16:417.
Cox-Singh J, Davis TME, Lee K-S, Shamsul SSG, Matusop A, Ratnam S, et al. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis. 2008;46:165–71.
Marchand RP, Culleton R, Maeno Y, Quang NT, Nakazawa S. Co-infections of Plasmodium knowlesi, P. falciparum, and P. vivax among humans and Anopheles dirus mosquitoes, Southern Vietnam. Emerg Infect Dis. 2011;17:1232.
Cox-Singh J. Knowlesi malaria in Vietnam. Malar J. 2009;8:269.
Imai N, White MT, Ghani AC, Drakeley CJ. Transmission and control of Plasmodium knowlesi: a mathematical modelling study. PLoS Negl Trop Dis. 2014;8:e2978.
Lyimo IN, Ferguson HM. Ecological and evolutionary determinants of host species choice in mosquito vectors. Trends Parasitol. 2009;25:189–96.
Yakob L, Bonsall MB, Yan G. Modelling knowlesi malaria transmission in humans: vector preference and host competence. Malar J. 2010;9:329.
Deroost K, Pham T-T, Opdenakker G. The immunological balance between host and parasite in malaria. Delft: Oxford University Press; 2016. p. 208–57.
Lim C, Hansen E, DeSimone TM, Moreno Y, Junker K, Bei A, et al. Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat Commun. 2013;4:1638.
Iyer J, Grüner AC, Rénia L, Snounou G, Preiser PR. Invasion of host cells by malaria parasites: a tale of two protein families. Mol Microbiol. 2007;65:231–49.
Moon RW, Sharaf H, Hastings CH, Ho YS, Nair MB, Rchiad Z, et al. Normocyte-binding protein required for human erythrocyte invasion by the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci USA. 2016;113:7231–6.
Tachibana S-I, Sullivan S, Kawai S, Nakamura S, Kim H, Goto N, et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat Genet. 2012;44:1051–5.
Jeremiah S, Janagond AB, Parija SC. Challenges in diagnosis of Plasmodium knowlesi infections. Trop Parasitol. 2014;4:25.
Lee W-C, Chin P-W, Lau Y-L, Chin L-C, Fong M-Y, Yap C-J, et al. Hyperparasitaemic human Plasmodium knowlesi infection with atypical morphology in peninsular Malaysia. Malar J. 2013;12:88.
Paul AS, Egan ES, Duraisingh MT. Host-parasite interactions that guide red blood cell invasion by malaria parasites. Curr Opin Hematol. 2015;2:220.
William T, Menon J, Rajahram G, Chan L, Ma G, Donaldson S, et al. Severe Plasmodium knowlesi malaria in a tertiary hospital, Sabah, Malaysia. Emerg Infect Dis. 2011;17:1248–55.
Barber BE, Grigg MJ, Piera KA, William T, Cooper DJ, Plewes K, et al. Intravascular haemolysis in severe Plasmodium knowlesi malaria: association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerg Microbes Infect. 2018;7:106.
Ahmed AM, Pinheiro MM, Divis PC, Siner A, Zainudin R, Wong T, et al. Disease progression in Plasmodium knowlesi malaria is linked to variation in invasion gene family members. PLoS Negl Trop Dis. 2014;8:e3086.
Pinheiro MM, Ahmed MA, Millar SB, Sanderson T, Otto TD, Lu WC, et al. Plasmodium knowlesi genome sequences from clinical isolates reveal extensive genomic dimorphism. PLoS ONE. 2015;10:e0121303.
Ahmed MA, Fong MY, Lau YL, Yusof R. Clustering and genetic differentiation of the normocyte binding protein [nbpxa] of Plasmodium knowlesi clinical isolates from Peninsular Malaysia and Malaysia Borneo. Malar J. 2016;15:241.
Daneshvar C, William T, Davis TME. Clinical features and management of Plasmodium knowlesi infections in humans. Parasitology. 2018;145:18–31.
Barber BE, Russell B, Grigg MJ, Zhang R, William T, Amir A, et al. Reduced red blood cell deformability in Plasmodium knowlesi malaria. Blood Adv. 2018;2:433–43.
Deitsch KW, Dzikowski R. Variant gene expression and antigenic variation by malaria parasites. Annu Rev Microbiol. 2017;71:625–41.
Galinski M, Lapp S, Peterson M, Ay F, Joyner C, Le Roch K, et al. Plasmodium knowlesi: a superb in vivo nonhuman primate model of antigenic variation in malaria. Parasitology. 2018;145:85–100.
Lapp S, Geraldo J, Chien J-T, Ay F, Pakala S, Batugedara G, et al. PacBio assembly of a Plasmodium knowlesi genome sequence with Hi-C correction and manual annotation of the SICAvar gene family. Parasitology. 2018;145:71–84.
Asare KK, Sakaguchi M, Lucky AB, Asada M, Miyazaki S, Katakai Y, et al. The Plasmodium knowlesi MAHRP2 ortholog localizes to structures connecting Sinton Mulligan’s clefts in the infected erythrocyte. Parasitol Int. 2018;67:481–92.
Varki A, Schauer R. Sialic acids. In: Essentials of glycobiology. Cold Spring Harbour: Cold Spring Harbour Laboratory Press; 2009. https://www.ncbi.nlm.nih.gov/books/NBK1920/.
Dankwa S, Lim C, Bei AK, Jiang RH, Abshire JR, Patel SD, et al. Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat Commun. 2016;7:11187.
Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci USA. 2005;102:12819–24.
Brock PM, Fornace KM, Parmiter M, Cox J, Drakeley CJ, Ferguson HM, et al. Plasmodium knowlesi transmission: integrating quantitative approaches from epidemiology and ecology to understand malaria as a zoonosis. Parasitology. 2016;143:389–400.
Chin W, Contacos P, Collins W, Jeter M, Alpert E. Experimental mosquito-transmission of Plasmodium knowlesi to man and monkey. Am J Trop Med Hyg. 1968;17:355–8.
Barber BE, William T, Grigg MJ, Menon J, Auburn S, Marfurt J, et al. A prospective comparative study of knowlesi, falciparum, and vivax malaria in Sabah, Malaysia: high proportion with severe disease from Plasmodium knowlesi and Plasmodium vivax but no mortality with early referral and artesunate therapy. Clin Infect Dis. 2013;56:383–97.
Fong IW. Zoonotic malaria: Plasmodium knowlesi. In: Fong IW, editor. Emerging zoonoses: a worldwide perspective. Berlin: Springer; 2017. p. 173–88.
Lambin EF, Tran A, Vanwambeke SO, Linard C, Soti V. Pathogenic landscapes: interactions between land, people, disease vectors, and their animal hosts. Int J Health Geogr. 2010;9:54.
Authors’ contributions
Not applicable. All authors read and approved the final manuscript.
Acknowledgements
With gratitude, we would like to thank David Alloysius, the manager at the Inikea field site in Sabah, for his time and patience in showing us potential sampling sites and assisting in organizing the many aspects of the field work as needed. Also, thanks must go to the Swedish Research Council and the Swedish University of Agricultural Science for providing funding for this ongoing research within the wider project ‘Balancing production and ecosystem services from degraded tropical rainforests to aid the transition to a more sustainable economy’. The University of Western Australia has provided the opportunity to investigate the effects of forest restoration on human health as a Ph.D. project and I would like to thank all my supervisors overseeing this research for their time and efforts.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
All authors have given their consent for this publication.
Ethics approval and consent to participate
Not applicable.
Funding
This research is funded by the Swedish Research Council (FORMAS:2016-20005) and the Swedish University of Agricultural Science.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Davidson, G., Chua, T.H., Cook, A. et al. Defining the ecological and evolutionary drivers of Plasmodium knowlesi transmission within a multi-scale framework. Malar J 18, 66 (2019). https://doi.org/10.1186/s12936-019-2693-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-019-2693-2