
Abstract
Background
Until 2009, the Laverania subgenus counted only two representatives: Plasmodium falciparum and Plasmodium reichenowi. The recent development of non-invasive methods allowed re-exploration of plasmodial diversity in African apes. Although a large number of great ape populations have now been studied regarding Plasmodium infections in Africa, there are still vast areas of their distribution that remained unexplored. Gabon constitutes an important part of the range of western central African great ape subspecies (Pan troglodytes troglodytes and Gorilla gorilla gorilla), but has not been studied so far. In the present study, the diversity of Plasmodium species circulating in great apes in Gabon was analysed.
Methods
The analysis of 1,261 faecal samples from 791 chimpanzees and 470 gorillas collected from 24 sites all over Gabon was performed. Plasmodium infections were characterized by amplification and sequencing of a portion of the Plasmodium cytochrome b gene.
Results
The analysis of the 1,261 samples revealed that at least six Plasmodium species circulate in great apes in Gabon (Plasmodium praefalciparum, Plasmodium gorA (syn Plasmodium adleri), Plasmodium gorB (syn Plasmodium blacklocki) in gorillas and Plasmodium gaboni, P. reichenowi and Plasmodium billcollinsi in chimpanzees). No new phylogenetic lineages were discovered. The average infection rate was 21.3% for gorillas and 15.4% for chimpanzees. A logistic regression showed that the probability of infection was significantly dependent on the freshness of the droppings but not of the host species or of the average pluviometry of the months of collection.
Similar content being viewed by others


Background
Plasmodium falciparum is a protozoan parasite responsible for malaria in humans. Among the five parasites infecting humans (Plasmodium falciparum, Plasmodium malariae, Plasmodium ovale, Plasmodium vivax and Plasmodium knowlesi), P. falciparum is by far the most virulent, responsible every year for approximately 207 million clinical cases and 627,000 deaths in the world [1], of which 98% are in sub-Saharan Africa [2-5]. Malaria is proving to be an obstacle that can slow down economic prosperity in many tropical countries, particularly in Africa [3].
Plasmodium falciparum belongs to the subgenus Laverania, which up to 2009 included only two known representatives: P. falciparum and Plasmodium reichenowi, a parasite from chimpanzees. Since 2009, thanks to the use of molecular tools for species identification and the development of non-invasive methods, several studies re-explored the diversity of Plasmodium species circulating in non-human primates in Africa, especially great apes (gorillas and chimpanzees) [6-8]. These studies revealed the existence of several lineages/species related to P. falciparum, deeply modifying the comprehension of the evolution of this parasite and of Laverania more generally. Four Laverania species are now recognized to infect chimpanzees: P. reichenowi, Plasmodium billcollinsi, Plasmodium gaboni and Plasmodium billbrayi [9-11]. For gorillas, there are three species: Plasmodium praefalciparum (the closest relative of P. falciparum), Plasmodium gorB (syn-Plasmodium blacklocki) and Plasmodium gorA (syn-Plasmodium adleri) [2,12]. Great apes have also been shown to be infected with species of the subgenus Plasmodium (non-Laverania): P. malariae-like, P. ovale-like and P. vivax-like parasites [8,13,14].
Was the entire diversity of Plasmodium species circulating in great apes in Africa discovered? Although a large number of great ape populations in Africa have now been studied regarding Plasmodium infections, there are still vast areas of their geographic distribution that remain unexplored. This is the case, for instance, for the western, central African populations of chimpanzees and gorillas (Pan troglodytes troglodytes and Gorilla gorilla gorilla). Although the range of both species covers all Gabon, half the surface of the Republic of the Congo, the south of Cameroon (south of the Sanaga river) and south of the Central African Republic, Plasmodium infections were almost only studied in populations from Cameroon, making about two-thirds of their range still unexplored [2,3,12].
In this study, using the second largest bank of faecal samples studied so far (more than 1,200 faecal samples), the diversity of Plasmodium species circulating in the great ape populations of Gabon was analysed. An investigation of the ecological factors susceptible to influence the detection of Plasmodium from these non-invasive samples was also performed.
Methods
Origin of faecal samples
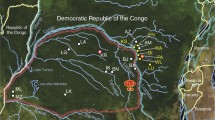
Faecal samples of chimpanzees and gorillas were collected in 24 sites in Gabon from 2010 to 2014 (Figure 1a and Table 1). In the field, the origin of the faeces (chimpanzee or gorilla) was deduced according to cues such as the type of nest near which they were found, footprints, texture, and odours. Freshness of the faeces (>24or <24 hours post excretion) was also estimated based on the freshness of surrounding nests (when present), texture, colours, humidity, and level of degradation. All samples were preserved in RNAlater® (Life technologies, USA) and conserved at the CIRMF at -80°C. Their origin (chimpanzee or gorilla) was confirmed by mitochondrial DNA analysis as previously described [15,16]. This investigation was approved by the Government of the Republic of Gabon and with the authorization of the Agence Nationale des Parcs Nationaux (ANPN). In total, 791 faecal samples of chimpanzees and 470 of gorillas were collected and analysed.

Sampling sites and variations of Plasmodium prevalences in Gabon. (a) Distribution of the sampling sites and amount of gorilla and chimpanzee samples collected and analysed in each site. Figure also shows the variations of prevalence (frequency of PCR-positives) and relative frequencies among positives of the different parasite species within the various populations of gorillas (b) and chimpanzees (c) sampled.
Extraction of DNA and PCR
Faecal DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Courteboeuf, France) as previously described [17] and Plasmodium infections were determined after amplification of a portion of Plasmodium mitochondrial genome (cytochrome b: cyt-b) as described in Prugnolle et al. [2]. All amplified products (10 μl) were run on 1.5% agarose gels in TAE buffer. The PCR-amplified products (956 bp) were used as templates for sequencing. DNA sequencing was performed by Eurofin MWG [18].
Species identification in mixed infections
When sequence chromatograms showed multiple peaks (heterozygous base calling), the program Mixed Sequences Reader (MSR) was used to determine if the isolates were mixed infected and by which species [19]. This program can directly analyse heterozygous base-calling fluorescence chromatograms and identify species in presence from a list of reference sequences (Table 2).
Phylogenetic analyses
Phylogenetic analyses were performed using only cyt-b sequences derived from chromatograms with no ambiguous base calls. To examine the relationship of the cyt-b sequences obtained with the different Plasmodium species known so far, a phylogenetic tree was constructed using a set of reference sequences belonging to different Plasmodium species. Hosts and GenBank accession numbers for these reference sequences are given in Table 3. The multiple alignment of all partial cyt-b sequences (686 nucleotides) was done using ClustalW (v 1.8.1 in BioEdit v.7.0.9.0. software) [20]. Maximum likelihood (ML) tree construction was based on the cyt-b sequences. The best-fitting ML model under the Akaike Information Criterion was GTR (general time reversible) + ModelTest. [21] The highest-likelihood DNA tree and corresponding bootstrap support values were obtained by PhyML (freely available at the ATGC bioinformatics platform [22,23]) using NNI (nearest neighbour interchange) + SPR (sub-tree pruning regrafting) branch swapping and 100 bootstrap replicates [24].

Phylogenetic relationships between the Cytochrome b sequences obtained in the study and those of known Plasmodium species (represented by their accession number). The tree was built based on cytochrome b (cyt-b) sequences of 686 bp. Red indicates sequences obtained from chimpanzees and green from gorillas. Bootstrap values are given at each node. More details on the different reference sequences can be found in Table 3.
Statistical analyses
All statistical analyses were performed using R [31]. A logistic regression was used to analyse the variations among individuals in the infection status. In these models, the variable to be predicted was the presence/absence of a Plasmodium infection. The predictive variables were: (i) the site of collection (random effect); (ii) freshness of the faeces; (iii) host species; and, (iv) average pluviometry during months of collection (fixed effects). For the second predictive variable, faeces were subdivided into two groups: the faecal samples deposited less than 24 hours before collection and those collected after 24 hours. The host species corresponded to gorilla and chimpanzee. Finally, for each month of collection, the average Gabonese pluviometry (estimated from data collected from1960 to 1990) was retrieved from [32], which data were produced by the Climatic Research Unit (CRU) of University of East Anglia (UEA). Pluviometry was considered as a possible predictive variable because it is known to influence levels of infection in human foci [33,34].
GeneBank accession numbers published in this study
The sequences reported in this study were deposited in GenBank under the following accession numbers KP875428 to KP875480
Results
Plasmodium species infecting great apes in Gabon
Some 1,261 faecal samples from wild chimpanzees (n = 791) and gorillas (n = 470) from 24 sites were analysed (Figure 1a). Among them, 122 samples of chimpanzees (15.42%) and 100 of gorillas (21.28%) were detected positive to a Plasmodium infection by Cyt-b PCR. Sequences of quality (of sufficient size (>600 bp) and with a clear chromatogram) were obtained for 31% (n = 69) of the Cyt-b amplicons. Among them, sixteen showed multiple peaks and were identify as clear mixed infections by the program MSR. The frequency of mixed infections observed in chimpanzees and gorillas as estimated by the analysis of the chromatograms is given in Table 2. Phylogenetic analyses (Figure 2) revealed the presence of three Plasmodium species in chimpanzees (P. gaboni, P. reichenowi and P. billcollinsi) and three in gorillas (P. praefalciparum, P. gorA and P. gorB). Neither species of the subgenus Plasmodium (P. vivax-like, P. malariae-like and P. ovale-like) nor new phylogenetic lineages were found in these samples. Relative frequencies of each Plasmodium species in each site among positives are given in Figure 1a-c.
Over the entire dataset, logistic regressions revealed that the probability of infection was only significantly dependent on the variable ‘freshness of the stool’. Pluviometry as well as host species did not significantly explain the probability of infection (Table 4). As shown in Table 4, the probability of infection was higher in stools collected less than 24 hours after dropping than in the older ones. Overall, freshness of the stools did not significantly differ between chimpanzees and gorillas (p-value = 0.07).
Discussion
In the last few years, several new Plasmodium species were discovered in African non-human primates, especially great apes [2,3,6,10,26]. These discoveries were made possible by the development of a non-invasive method allowing detection of Plasmodium infections from faecal samples [2,6], despite inherent problems of DNA degradation with this type of biological material. This issue was overcome by the use of mitochondrial sequences to amplify the parasite, which presents several advantages: 1) Mitochondrial DNA is in multiple copy inside parasites (unlike nuclear DNA) and 2) if properly chosen, small portions of the mitochondrial genome (as small as 200 bp), can contain enough phylogenetic information to identify the different Plasmodium species. This method is now one of the main methods used to analyse Plasmodium from wild non-human primates [2,7,12,25].
In the present study, analyses were performed on a set of 1,261 faecal samples collected all over Gabon from chimpanzees and gorillas. All Plasmodium species found belonged to the subgenus Laverania and were all previously identified in Pan troglodytes troglodytes and Gorilla gorilla gorilla, respectively [2,12]. No new phylogenetic lineage or species were identified. Surprisingly, no species of the subgenus Plasmodium (non-Laverania) were identified either. This is at odds with recent observations made from ape blood samples or infected sylvatic anopheline mosquitoes collected in Gabon showing the circulation of P. vivax-like parasites in the area. [13] These results are nevertheless congruent with those from Liu et al. [12]. Although they analysed 3,000 ape faecal samples from west and central Africa, they only obtained seven sequences of Plasmodium belonging to the subgenus Plasmodium (non-Laverania).
One main factor could explain why parasites of the subgenus Plasmodium were not detected and this is most likely linked to the nature of the primers used to perform PCRs. Indeed, as in the study of Liu et al. [12], the primers used were specifically designed to amplify sequences of Laverania parasites. As a consequence, several nucleotides of differences separated them from the homologous sequences in P. vivax, P. malariae and P. ovale, thus very likely reducing the sensitivity of this PCR to detect non-Laverania species. In addition, such problem might have been amplified by the presence of co-infections with Laverania species. Indeed, it has been demonstrated that in case of co-infection, the PCR tends to favour the amplification of the parasite with the best matching sequence to the primers [35,36]. Recently, Liu et al. [14] solved this problem by designing primers specific to P. vivax. Out of the 3,000 samples previously analysed and re-analysed with other samples, they finally detected more than 87 P. vivax infections.
In this study, no Laverania species were found to infect both hosts (gorillas and chimpanzees). This reinforces the hypothesis that Laverania lineages infect specific hosts [10], a specificity that could be associated to specific ligand/receptor interactions occurring in the vertebrate host, as suggested by several studies [37], or by ecological factors such as the trophic preferences of the vectors [38] or the fact that gorillas’ and chimpanzees’ home ranges might not overlap in space and time. Additional studies would be needed to disentangle these different possibilities.
No human Plasmodium species were found. This result is congruent with other studies performed so far on wild populations of apes [8,12,39] thus confirming that, contrarily to what some authors have feared [7,40], great apes do not (and will certainly never) constitute reservoirs of Plasmodium, in particular P. falciparum, for humans. The fact that their populations are rapidly declining [41,42] is unfortunately another element in support of this prediction. Finally, no evidence of ape-to-human transfers of Laverania species was ever recorded despite efforts to find them [37]. The only documented record of this kind of transfer, in a natural context, was for a P. vivax-like (non-Laverania) parasite [13].
Regarding the prevalence of infections, more than 15% of the chimpanzee and 21% of the gorilla samples were positive to Plasmodium. Infections were detected in 17out of 23sites for chimpanzees and 16 out of 24 for gorillas. Global rates of infection found in this study are similar to those found by Prugnolle et al. [2], Kaiser et al. [8], and Liu et al. [12] in other areas. As previously discussed [2,12], it is very likely that the accurate rates of infection are higher, because the detection of Plasmodium in this kind of biological material (faecal) is expected to be less sensitive than in blood, as it is the case for urine and saliva [12,43,44], due to sample degradation or repeated sampling (faecal samples from the same individual may have been collected several times). The effect of sample degradation (and hence DNA degradation) is evident here when comparing the rates of infection detected in the faeces that were collected before and after 24 hours post excretion. The freshest (and so the less degraded) faecal samples significantly present more Plasmodium infections than the other ones (odds ratio = 2.038).
References
WHO. World malaria report 2013. Geneva: World Health Organization; 2014. p. 178.
Prugnolle F, Ollomo B, Durand P, Yalcindag E, Arnathau C, Elguero E, et al. African monkeys are infected by Plasmodium falciparum nonhuman primate-specific strains. Proc Natl Acad Sci U S A. 2011;108:11948–53.
Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, et al. The origin of malignant malaria. Proc Natl Acad Sci U S A. 2009;106:14902–7.
Jeffares DC, Pain A, Berry A, Cox AV, Stalker J, Ingle CE, et al. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007;39:120–5.
Rich SM, Ayala FJ. Population structure and recent evolution of Plasmodium falciparum. Proc Natl Acad Sci U S A. 2000;97:6994–7001.
Prugnolle F, Ayala F, Ollomo B, Arnathau C, Durand P, Renaud F. Plasmodium falciparum is not as lonely as previously considered. Virulence. 2011;2:71–6.
Duval L, Fourment M, Nerrienet E, Rousset D, Sadeuh SA, Goodman SM, et al. African apes as reservoirs of Plasmodium falciparum and the origin and diversification of the Laverania subgenus. Proc Natl Acad Sci U S A. 2010;107:10561–6.
Kaiser M, Lowa A, Ulrich M, Ellerbrok H, Goffe AS, Blasse A, et al. Wild chimpanzees infected with 5 Plasmodium species. Emerg Infect Dis. 2010;16:1956–9.
Snounou G, Escalante A, Kasenene J, Renia L, Gruner AC, Krief S. [Malaria in hominids](in French). Bull Acad Natl Med. 2011;195:1945–54.
Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH. A plethora of Plasmodium species in wild apes: a source of human infection? Trends Parasitol. 2011;27:222–9.
Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, Nkoghe D, et al. A new malaria agent in African hominids. PLoS Pathog. 2009;5:e1000446.
Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, et al. Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010;467:420–5.
Prugnolle F, Rougeron V, Becquart P, Berry A, Makanga B, Rahola N, et al. Diversity, host switching and evolution of Plasmodium vivax infecting African great apes. Proc Natl Acad Sci U S A. 2013;110:8123–8.
Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, Malenke JA, et al. African origin of the malaria parasite Plasmodium vivax. Nat Commun. 2014;5:3346.
Santiago ML, Lukasik M, Kamenya S, Li Y, Bibollet-Ruche F, Bailes E, et al. Foci of endemic simian immunodeficiency virus infection in wild-living eastern chimpanzees (Pan troglodytes schweinfurthii). J Virol. 2003;77:7545–62.
Santiago ML, Range F, Keele BF, Li Y, Bailes E, Bibollet-Ruche F, et al. Simian immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Tai Forest, Cote d’Ivoire: implications for the origin of epidemic human immunodeficiency virus type 2. J Virol. 2005;79:12515–27.
Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–6.
Eurofine ME. [www.eurofinsgenomics.com]
Chang CT, Tsai CN, Tang CY, Chen CH, Lian JH, Hu CY, et al. Mixed sequence reader: a program for analyzing DNA sequences with heterozygous base calling. Sci World J. 2012;2012:365104.
Hall T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:45–98.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:465–9.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Sundararaman SA, Liu W, Keele BF, Learn GH, Bittinger K, Mouacha F, et al. Plasmodium falciparum-like parasites infecting wild apes in southern Cameroon do not represent a recurrent source of human malaria. Proc Natl Acad Sci U S A. 2013;110:7020–5.
Krief S, Escalante AA, Pacheco MA, Mugisha L, Andre C, Halbwax M, et al. On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from Bonobos. PLoS Pathog. 2010;6:e1000765.
Duval L, Nerrienet E, Rousset D, Sadeuh Mba SA, Houze S, Fourment M, et al. Chimpanzee malaria parasites related to Plasmodium ovale in Africa. PLoS One. 2009;4:e5520.
Joveen-Neoh WF, Chong KL, Wong CM, Lau TY. Incidence of malaria in the interior division of sabah, malaysian borneo, based on nested PCR. J Parasitol Res. 2011;2011:104284.
Conway DJ, Fanello C, Lloyd JM, Al-Joubori BM, Baloch AH, Somanath SD, et al. Origin of Plasmodium falciparum malaria is traced by mitochondrial DNA. Mol Biochem Parasitol. 2000;111:163–71.
Pacheco MA, Cranfield M, Cameron K, Escalante AA. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J. 2013;12:328.
R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. URL: http://www.R-project.org. ISBN 3-900051-07-0.
Group WB. Climate change Knowledge Portal: Average Monthly Rainfall 1901 to 2009 of Gabon. [www.worldbank.org/contry/gabon]. 2014.
Robert V, Dieng H, Lochouran L, Traore SF, Trape JF, Simondon F, et al. [Malaria transmission in the rural zone of Niakhar, Senegal](in French). Trop Med Int Health. 1998;3:667–77.
Ouldabdallahi M, Ouldbezeid M, Diop C, Dem E, Lassana K. [Epidemiology of human schistosomiasis in Mauritania. The right bank of the Senegal River as model](in French). Bull Soc Pathol Exot. 2010;103:317–22.
Valkiunas G, Palinauskas V, Ilgunas M, Bukauskaite D, Dimitrov D, Bernotiene R, et al. Molecular characterization of five widespread avian haemosporidian parasites (Haemosporida), with perspectives on the PCR-based detection of haemosporidians in wildlife. Parasitol Res. 2014;113:2251–63.
Zehtindjiev P, Krizanauskiene A, Bensch S, Palinauskas V, Asghar M, Dimitrov D, et al. A new morphologically distinct avian malaria parasite that fails detection by established polymerase chain reaction-based protocols for amplification of the cytochrome B gene. J Parasitol. 2012;98:657–65.
Wanaguru M, Liu W, Hahn BH, Rayner JC, Wright GJ. RH5-Basigin interaction plays a major role in the host tropism of Plasmodium falciparum. Proc Natl Acad Sci U S A. 2013;110:20735–40.
Paupy C, Makanga B, Ollomo B, Rahola N, Durand P, Magnus J, et al. Anopheles moucheti and Anopheles vinckei are candidate vectors of ape Plasmodium parasites, including Plasmodium praefalciparum in Gabon. PLoS One. 2013;8:e57294.
Prugnolle F, Durand P, Neel C, Ollomo B, Ayala FJ, Arnathau C, et al. African great apes are natural hosts of multiple related malaria species, including Plasmodium falciparum. Proc Natl Acad Sci U S A. 2010;107:1458–63.
Duval L, Ariey F. Ape Plasmodium parasites as a source of human outbreaks. Clin Microbiol Infect. 2012;18:528–32.
Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–5.
Campbell G, Kuehl H, N’Goran Kouame P, Boesch C. Alarming decline of West African chimpanzees in Cote d’Ivoire. Curr Biol. 2008;18:R903–4.
Nwakanma DC, Gomez-Escobar N, Walther M, Crozier S, Dubovsky F, Malkin E, et al. Quantitative detection of Plasmodium falciparum DNA in saliva, blood, and urine. J Infect Dis. 2009;199:1567–74.
Mharakurwa S, Simoloka C, Thuma PE, Shiff CJ, Sullivan DJ. PCR detection of Plasmodium falciparum in human urine and saliva samples. Malar J. 2006;5:103.
Acknowledgements
Authors thank the three reviewers for their constructive comments. The study was funded by Centre International de Recherches Médicales de Franceville (CIRMF, Gabon), Centre National de la Recherche Scientifique (CNRS, France), Institut de Recherche pour le Développement (IRD, France) and Agence Nationale de la Recherche (ANR, France, grant ORIGIN JCJC 012). We thank all the persons that were involved in the sampling as well as the Gabonese national agency of the national parks (authorization of collection: N° AE13009/PR/ANPN/SE/CS/AEPN). We also thank Bouanga Miriame for her help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LB, BO, FP, VB, VR, APO, FL, AN, GL, PM, and CP contributed to the acquisition of samples in fieldwork; LB, BO, FP, APO, NDM, BMO, LMDL, LYM, CA, EE, PD, CTB, PM, GL, CP, and FR analysed and interpreted the data; LB, BO, CTB, FR, and FP conducted and supervised this work; LB, BO, FP, and FR wrote this paper. All authors read and approved the final manuscript.
Benjamin Ollomo and Franck Prugnolle contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Boundenga, L., Ollomo, B., Rougeron, V. et al. Diversity of malaria parasites in great apes in Gabon. Malar J 14, 111 (2015). https://doi.org/10.1186/s12936-015-0622-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0622-6