
Abstract
Background
The microbial cell factory Bacillus subtilis is a popular industrial platform for high-level production of secreted technical enzymes. Nonetheless, the effective secretion of particular heterologous enzymes remains challenging. Over the past decades various studies have tackled this problem, and major improvements were achieved by optimizing signal peptides or removing proteases involved in product degradation. On the other hand, serious bottlenecks in the protein export process per se remained enigmatic, especially for protein secretion at commercially significant levels by cells grown to high density. The aim of our present study was to assess the relevance of the intramembrane protease RasP for high-level protein production in B. subtilis.
Results
Deletion of the rasP gene resulted in reduced precursor processing and extracellular levels of the overproduced α-amylases AmyE from B. subtilis and AmyL from Bacillus licheniformis. Further, secretion of the overproduced serine protease BPN’ from Bacillus amyloliquefaciens was severely impaired in the absence of RasP. Importantly, overexpression of rasP resulted in threefold increased production of a serine protease from Bacillus clausii, and 2.5- to 10-fold increased production of an AmyAc α-amylase from Paenibacillus curdlanolyticus, depending on the culture conditions. Of note, growth defects due to overproduction of the two latter enzymes were suppressed by rasP-overexpression.
Conclusion
Here we show that an intramembrane protease, RasP, sets a limit to high-level production of two secreted heterologous enzymes that are difficult to produce in the B. subtilis cell factory. This finding was unexpected and suggests that proteolytic membrane sanitation is key to effective enzyme production in Bacillus.
Similar content being viewed by others

Background
Secretory protein production is of critical importance in biotechnology, because this generally delivers high amounts of correctly folded proteins for which downstream processing from the fermentation broth is relatively easy. The Gram-positive bacterium Bacillus subtilis and related bacilli are amongst the best protein producers known today and, therefore, frequently used to produce commercially relevant enzymes. In particular, these organisms are suitable for large-scale high-density fermentation, leading to product yields in the 25 g/l range [1]. Thus, they have a long history in the production of, for example, amylases [2, 3] and proteases [2, 4] used in the food, textile and pharmaceutical industries [5]. Another advantage of B. subtilis and its close relatives is that they lack toxic by-products, such as endotoxin, which makes them suitable for the Qualified Presumption of Safety (QPS) status of the European Food Safety Authority. Accordingly, many Bacillus products have obtained the Generally Recognized As Safe (GRAS) status from the US Food and Drug Administration [6, 7].
Much effort has been made to optimize protein secretion in B. subtilis. Major improvements were achieved by optimizing signal peptides [8, 9] or removing proteases involved in product degradation [10]. For example, the deletion of multiple genes for extracellular proteolytic enzymes allowed efficient production not only of technical enzymes, such as a thermostable β-1,3-1,4-glucanase from Clostridium thermocellum [11], but also of pharmaceutical proteins, such as single-chain antibodies [12] or human interleukin-3 [13]. Further improvements in protein secretion were achieved at lab-scale by overexpression of the signal peptidase SipS [14], the peptidyl-prolyl cis/trans isomerase PrsA [15], or the staphylococcal thiol-disulphide oxidoreductase DsbA [16]. Nonetheless, serious bottlenecks in the protein export process per se have remained enigmatic, especially for protein secretion at commercially significant levels by cells grown to high density [17].
Notably, it was previously shown that deleting the gene for the intramembrane protease RasP of B. subtilis led to elevated levels of the membrane proteins FtsL [18], HtrA and HtrB, but compromised the production of various other membrane proteins [19] and processing of the α-amylase AmyQ of Bacillus amyloliquefaciens [20, 21]. This focused our attention on a possible role of RasP in secretory protein production, especially because the biogenesis of many membrane proteins relies on the general secretory pathway [22]. RasP belongs to the family of site-2 proteases (S2P), more specifically the zink-metallo proteases, which cleave their substrates within the plane of the cytoplasmic membrane [23]. These proteases are conserved in all domains of life where they have roles in regulated intramembrane proteolysis. For instance, the Escherichia coli S2P named RseP was shown to cleave signal peptides upon their signal peptidase-mediated liberation from secretory precursor proteins [24, 25]. B. subtilis RasP was shown to cleave the anti-sigma factor RsiW under conditions of oxidative- or temperature stress, causing induction of the so-called σW regulon, which is believed to support cell envelope integrity and to mitigate effects of extracellular stress [20, 21, 26, 27]. Therefore, the present study was aimed at determining whether RasP could be a bottleneck for protein production in B. subtilis. Indeed, our results show that RasP overexpression can boost protein production in this important cell factory.
Methods
Bacterial strains and growth conditions
The bacterial strains used in this study are listed in Table 1. B. subtilis strains were grown at 37 °C, 280 rpm in Lysogeny Broth (LB; Oxoid Limited), MBU medium or 5SM12 medium. The MBU medium is similar to the MBD medium as described by Vogtentanz et al. [28], but lacks soytone and instead of 7.5% glucose it contains 2.1% glucose and 3.5% maltodextrin DE13-17. The 5SM12 medium consists of 75 mM K2HPO4, 25 mM NaH2PO4, 12% maltodextrin, 5% Difco Bacto Soytone, 2 mM sodium citrate, 0.5 mM MgSO4, 0.2 mM MnCl2, 0.03 mM calcium chloride, and 0.0053% ferric ammonium citrate. Growth media were supplemented with neomycin 15 µg/ml or phleomycin 4 µg/ml to select for particular gene deletions. Chloramphenicol was added to 5 or 25 µg/ml for, respectively, the selection of chromosomally integrated amylase or protease expression cassettes and their amplification. In pulse-chase labeling experiments with cells grown on MBU medium, 2.5 µg/ml chloramphenicol was used.
Construction of strains and plasmids
Ex Taq polymerase, dNTPs and buffers used for the construction of mutant strains were purchased from Takara Bio Inc. Phusion High Fidelity DNA polymerase (New England Biolabs) was used for the construction of plasmids. Primers were obtained from Eurogentec. Construction of deletion mutants in a B. subtilis Δupp::neo R strain was performed using the modified mutation delivery method described by Fabret et al. [29]. To delete a particular gene of interest (i.e. rasP or tepA), its 5′ and 3′ flanking regions were amplified using primer pairs designated P1/P2 and P3/P4 (Table 2). The amplified fragments were fused to a cassette containing a phleomycin resistance marker, the upp gene and the cI gene [29]. The resulting fusion product was used to transform B. subtilis Δupp::neo R, where competence was induced with 0.3% xylose due to the presence of a xylose-inducible comK gene. This resulted in phleomycin resistant and neomycin sensitive strains lacking the target gene. PCRs using primer combinations P0/P4 and P0/CI2.rev (Table 2) were performed to verify the correct deletion. To achieve overproduction and secretion of AmyE [30], AmyL [31] or BPNʹ-Y217L (in short BPNʹ) [32, 33], the promoter of aprE (600 bp upstream of the GTG start codon) [34] and the signal sequence of aprE were fused to the 7-codon pro-sequence of amyE, the amyL gene lacking its signal sequence, or the eighth codon of the BPNʹ signal sequence, respectively. For AmyE, the C-terminal starch-binding module was removed by the introduction of a stop codon after the codon for residue 425 in amyE and the complete gene expression cassette was produced synthetically (GeneOracle, Santa Clara, CA). Transcription of amyE or bpnʹ was terminated using the native BPNʹ terminator, and the native terminator in case of amyL. To accomplish the expression and secretion of Properase® (i.e. the subtilisin variant of Bacillus clausii) or AmyAc [i.e. an engineered α-amylase from Paenibacillus curdlanolyticus that belongs to the AmyAc family (NCBI reference sequence: WP_040711139)], the respective genes were ordered synthetically (GeneArt, Thermofisher Scientific) and fused to the promoter and signal sequence of aprE as described above. For transcription termination, the native BPNʹ terminator was used. Genes encoding the five afore-mentioned secretory proteins were integrated into the aprE locus by single cross-over recombination using a vector based on plasmid pJH101 [35]. Lastly, gene amplification was achieved by growing transformants at increasing chloramphenicol concentrations up to 25 µg/ml.
To complement the ΔrasP mutation, plasmid pHTK315, a derivative of pHT315-Pspac [36] containing the kan gene for kanamycin resistance was constructed using the CPEC strategy described by Quan et al. [37]. Plasmid pHT315-Pspac was amplified by PCR using the primers pHT315CPEC.fw and pHT315CPEC.rev. In parallel, the kan gene was amplified from the vector pGDL48 [38], using the primers kanaCPEC.fw and kanaCPEC.rev both containing approximately 30 bp overlap with the pHT315-Pspac vector. The two resulting PCR fragments were fused and amplified by PCR, and the resulting amplicon was used to transform competent cells of E. coli strain TG1. The plasmid thus obtained was named pHTK315. Next, plasmid pHTK315-rasP was constructed following the same strategy. For amplification of pHTK315, the primer combination pHT315rasP.fw/pHT315rasP.rev was used, and rasP was amplified using the primer combinations rasP.fw/rasP.rev. Both fragments were fused by PCR based on the 30 bp overlaps. The resulting amplicon was used to transform competent cells of E. coli TG1, and the plasmid thus obtained was named pHTK315-rasP. The correctness of pHTK315 and pHTK315-rasP was verified by sequencing and, subsequently, these plasmids were introduced into B. subtilis using xylose-induced competence, as described above. After the transformation, mutant strains containing amplified amyE, amyL or bpnʹ expression cassettes were selected by repeated transfers to fresh LB plates containing 25 μg/ml chloramphenicol.
Analysis of secreted protein production by LDS-PAGE
Cultures were inoculated from LB plates with 25 μg/ml chloramphenicol and grown for approximately 8 h in LB broth with 25 µg/ml chloramphenicol. These cultures were diluted 1000-fold in MBU medium with 2.5 µg/ml chloramphenicol in Ultra Yield Flasks™ (Thomson Instrument Company) and incubated for approximately 16 h at 37 °C, 280 rpm in a Multitron orbital shaker (Infors) at high humidity. After measuring and correcting for the optical density at 600 nm (OD600), equal amounts of cells were separated from the culture medium by centrifugation. For the analysis of extracellular proteins, proteins in the culture medium were precipitated with trichloroacetic acid (TCA; 10% w/v final concentration), dissolved in LDS buffer (Life Technologies) and heated for 10 min at 95 °C. Next, proteins were separated by LDS-PAGE on 10% NuPage gels (Life Technologies). Gels were stained with SimplyBlue™ SafeStain (Life Technologies). Gel images were quantified with the ImageJ software (http://imagej.nih.gov/ij/).
Pulse-chase protein labeling experiments
Pulse-chase labeling of B. subtilis proteins was performed using Easy tag [35S]-methionine (PerkinElmer Inc.). Immunoprecipitation and LDS-PAGE were performed as described previously [39] using the following adaptations. Cells were grown for 16 h in MBU with 2.5 µg/ml chloramphenicol as described before and diluted 1 h prior to the actual labeling to OD600 ~0.7 in fresh MBU with 2.5 µg/ml chloramphenicol. Labeling was performed with 25 µCi [35S]-methionine for 30 s before adding an excess amount of unlabeled methionine (chase; 0.6 mg/ml final concentration). Samples were collected at several time points, followed by direct precipitation of the proteins with 10% TCA (w/v) on ice. Precipitates were re-suspended in lysis buffer (10 mM Tris pH 8, 25 mM MgCl2, 200 mM NaCl and 5 mg/ml lysozyme). After 10–15 min incubation at 37 °C, lysis was achieved by adding 1% (w/v) SDS and heating for 10 min at 100 °C. Specific polyclonal antibodies against AmyE or AmyL were used for immunoprecipitation of the respective labeled proteins in STD-Tris buffer (10 mM Tris pH 8.2, 0.9% (w/v) NaCl, 1.0% (v/v) triton X-100, 0.5% (w/v) sodium deoxycholate) with the help of Protein A affinity medium (Mabselect Sule, GE Healthcare Life Sciences).
Because of the high proteolytic activity of BPNʹ, which also degrades antibodies, the immunoprecipitation of BPNʹ was performed in the presence of a specific serine protease inhibitor (4 mM, Pefablock SC, Roche). Due to aspecific binding of the BPNʹ antibodies to unidentified cellular proteins of B. subtilis, the immunoprecipitation of BPNʹ was only performed to assay secreted BPNʹ in TCA-precipitated culture medium samples. Labeled proteins were separated by LDS-PAGE using 10% NuPage gels (Life Technologies) and visualized using a Cyclon Plus Phosphor Imager (Perkin Elmer). Quantification of the obtained data was achieved by making use of the ImageJ software.
For pulse-chase labeling studies on the complementation of the ΔrasP mutation, cells containing pHTK315-rasP or control cells containing the empty vector pHTK315 were pre-cultured for 20 h in MBU with 2.5 µg/ml chloramphenicol, because these cells grow slightly slower than cells without such plasmids. One hour prior to labeling with [35S]-methionine, the cells were diluted to an OD600 of ~0.7 in fresh MBU with 2.5 µg/ml chloramphenicol containing 25 µM isopropyl β-d-1-thiogalactopyranoside (IPTG) necessary for the induction of the Pspac promoter. The pulse-chase protein labeling was performed as described above.
Assays for Properase or AmyAc production
To analyze Properase secretion by cells overexpressing rasP or wt control cells, the respective strains were pre-cultured for 5 h in LB at 37 °C. From these pre-cultures 1.5 OD units were used to inoculate 25 ml of MBU medium in Ultra Yield Flasks™, and culturing was continued at 37 °C (250 rpm, 70% humidity). Samples were withdrawn from the cultures at 18, 25, 41, 48 and 65 h of growth for OD600 readings and protease activity measurements. OD600 was determined using a SpectraMax spectrophotometer (Molecular Devices, Downington, PA, USA). Protease activity in the samples was determined by incubating sample aliquots with the synthetic substrate N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide (Sigma Chemical Co) and absorbance readings at 405 nm using a SpectraMax spectrophotometer, as described previously in WO 2010/144283.
To test the effect of rasP overexpression on the production of AmyAc, four colonies from the rasP overexpressing strain or the wt control strain were used to inoculate LB with 25 µg/ml chloramphenicol and the resulting pre-cultures were grown for 4 h at 37 °C. Next, 0.075 OD units from a pre-culture were used to inoculate 2 ml of medium (5SM12 or MBU) in 24 deep well microtiter plates and cultures were grown for 48 h at 37 °C under vigorous shaking. Samples were withdrawn at 18, 25, 41 and 48 h of culturing for OD600 readings and amylase activity measurements. Amylase activity in whole-broth samples was assayed with the Ceralpha HR kit (Megazyme, Wicklow, Ireland) and absorbance readings at 400 nm according to the manufacturer’s instructions.
Results and discussion
As a first approach to assess the possible function of RasP in protein secretion under fermentation-mimicking conditions, the rasP gene was deleted from the B. subtilis genome and the secretion of three representative model proteins was assessed in the resulting ΔrasP strain. Specifically, the secreted model proteins were the α-amylase AmyE from B. subtilis, the α-amylase AmyL from Bacillus licheniformis and the serine protease BPNʹ from B. amyloliquefaciens. The respective genes were expressed to high levels using the aprE promoter, which is a preferred promoter for enzyme production at industrial scale [40]. As shown in Fig. 1, ΔrasP cells grown to stationary phase in about 16–20 h of culturing secreted less AmyE, AmyL or and BPNʹ than wild-type cells. This was clearly not the case for control cells lacking the tepA gene, which encodes an unrelated cytoplasmic protease [41]. Furthermore, the rates of AmyE and AmyL precursor processing as determined by pulse-chase labeling with [35S]-methionine after 16 h of growth were substantially reduced in ΔrasP cells compared to wild-type cells (Fig. 2a). In case of BPNʹ we were unable to detect cell-associated precursor forms of this protein but, nonetheless, we showed that the rate of appearance of the mature [35S]-methionine-labeled BPNʹ in the growth medium was strongly slowed down in cells lacking rasP (Fig. 2b). Together, these findings clearly demonstrate that RasP is needed for efficient processing and secretion of mature AmyE, AmyL and BPNʹ. As illustrated with AmyE, the wild-type rate of precursor processing and secretion of the mature protein were restored when rasP was expressed from a plasmid in the ΔrasP cells (Figs. 3, 4). This shows that the ΔrasP mutation can be complemented with rasP expressed in trans. Of note, the wild-type cells grew to higher optical densities at 600 nm (OD600 ~ 25) than the ΔrasP cells (OD600 ~ 15), but this effect was corrected for in the loading of gels shown in Figs. 1 and 3, and in the pulse-chase labelling experiments in Figs. 2 and 4 comparable amounts of cells were used.

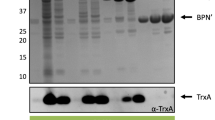
Reduced secretion levels of AmyE, AmyL and BPN’ in rasP mutant cells. ΔrasP mutant cells and wild-type (wt) or ΔtepA mutant control cells expressing AmyE, AmyL or BPNʹ were grown for 16 h in MBU medium. Next, cells and growth media were separated by centrifugation. Proteins in growth medium fractions were precipitated with TCA and analyzed by LDS-PAGE. Protein bands were visualized with the SimplyBlue SafeStain. Molecular weights of marker proteins are indicated (in kDa) on the left side of each gel segment. Secreted amounts of AmyE, AmyL and BPNʹ in the growth medium fractions from three independent cultures were assessed by ImageJ analysis of the gels, and the ratios of each of these proteins in the medium fractions of the ΔrasP or ΔtepA strains relative to the wt strain are indicated below each gel segment together with the standard deviation

Reduced rates of AmyE, AmyL and BPNʹ secretion in rasP mutant cells. a Processing of the precursor proteins of AmyE or AmyL by signal peptidase was analyzed by pulse-chase protein labeling with [35S]-methionine, immunoprecipitation of AmyE or AmyL from culture samples with specific antibodies, LDS-PAGE and phosphorimaging as described in “Methods”. The positions of precursor (p) and mature (m) forms of AmyE and AmyL are indicated. Data from three independent experiments were analyzed with ImageJ to assess the kinetics of precursor processing, and the results are plotted below the autoradiographs. The plot shows the relative amounts (%) of the precursor forms of AmyE (black symbols) or AmyL (white symbols) in the ΔrasP (triangle) or wt (square) strains at different time points after the chase with non-radioactive methionine (t = 0). Error bars show the standard deviation. b Secretion of BPNʹ was analyzed by pulse-chase labeling with [35S]-methionine, immunoprecipitation from growth medium fractions devoid of cells with specific antibodies, LDS-PAGE and phosphorimaging as described in “Methods”. The position of mature BPNʹ is indicated (m). Data from three independent experiments were analyzed with ImageJ to determine the kinetics BPN’ appearance in the growth medium, and the results are plotted below the autoradiographs. The plot shows the average of the calculated ratio of secreted BPNʹ in the ΔrasP (triangle) or wt (square) strains relative to the amount of BPNʹ secreted immediately after the chase with non-radioactive methionine (t = 0) in the wt strain. Error bars indicate the standard deviation

IPTG-dependent complementation of the ΔrasP mutation in AmyE-producing cells containing pHTK315-rasP. ΔrasP mutant bacteria overproducing AmyE and carrying either pHTK315-rasP or the empty vector pHTK315 were grown for 16 h in MBU with 2.5 µg/ml chloramphenicol. Next rasP expression in cells containing pHTK315-rasP, where rasP is transcribed from the IPTG-dependent Pspac promoter, was induced for 4 h by the addition of IPTG to different end concentrations as indicated. ΔrasP mutant bacteria carrying the empty vector pHTK315 were also treated with IPTG as a negative control. Proteins in the growth medium were precipitated with TCA and separated by LDS-PAGE. Protein bands were visualized with the SimplyBlue SafeStain. The AmyE band is indicated with an arrow, and the molecular weights of marker proteins are indicated on the left of the gel (in kDa)

Complementation of pre-AmyE processing in ΔrasP mutant cells. Processing of pre-AmyE (p) to mature AmyE (m) was analyzed by pulse-chase protein labeling with [35S]-methionine in IPTG-induced ΔrasP mutant or wt cells containing either pHTK315-rasP or the empty vector pHTK315. Pre-AmyE and mature AmyE were immunoprecipitated with specific antibodies, separated by LDS-PAGE, and visualized by a phosporimaging. Data from two independent experiments were analyzed with ImageJ to assess the kinetics of pre-AmyE processing to the mature form, and the results are plotted below the autoradiographs. Specifically, the plot shows the relative amounts (%) of the precursor form of AmyE in ΔrasP mutant (triangle) or wt (square) cells carrying pHTK315-rasP (white symbols) or pHTK315 (black symbols) at different time points after the chase with non-radioactive methionine (t = 0)
Because the removal of RasP had significant influence on the processing and secretion of AmyL, AmyE and BPNʹ, we wanted to know whether rasP overexpression could be beneficial for protein secretion in B. subtilis. Notably, in ‘wild-type’ B. subtilis the secretion of AmyL, AmyE and BPNʹ is already very efficient. Moreover, these enzymes are produced really well in other industrial Bacillus species, which gives the optimization of their production in B. subtilis a lower commercial impact. Therefore, we focused our attention on two other enzymes, namely a protease (‘Properase’) from B. clausii and AmyAc from P. curdlanolyticus, which are both commercially valuable but hard to produce. Especially the large-scale production of enzymes of the AmyAc family is very challenging in Bacillus species. To overexpress rasP, this gene was placed under control of the very strong spoVG promoter (PspoVG). Importantly, the PspoVG promoter is a constitutive promoter and its strength is comparable to that of the promoter of aprE, which was used to express the secreted proteins employed in this study (data not shown). Next, the capability of the resulting strain to secrete the highly active Properase, which is even capable of degrading prions, was tested. A potential problem caused by high-level Properase production is a negative effect on the viability of B. subtilis. As shown in Fig. 5a, PspoVG-driven expression of rasP enhanced both cell viability and the secretion of Properase in the production phase when cells were grown in MBU medium. Of note, high-level rasP expression led to an increase in Properase production of about threefold. To further obtain proof-of-principle that PspoVG-driven rasP expression may be beneficial for secretory protein production, we investigated the effect on production of a bacterial α-amylase belonging to the AmyAc family. Similar to Properase, the expression of the AmyAc enzyme had a negative impact on growth in MBU medium and, in this case, both growth and amylase production remained relatively low unless rasP was overexpressed (Fig. 5b). In fact, AmyAc production by cells grown in MBU was up to tenfold increased upon rasP overexpression. Interestingly, expression of the AmyAc enzyme does not impact on growth in 5SM12 medium, which allowed us to distinguish between growth effects and effects of rasP overexpression on production of the secreted AmyAc enzyme. As shown in Fig. 5c, the yield of this enzyme in the 5SM12 growth medium was about 2.5-fold increased, which implies that the improved productivity was mostly directly related to rasP overexpression rather than an enhanced cell density of the culture. The main difference between the 5SM12 and MBU media is that the 5SM12 medium contains soytone and 3.4-fold more maltodextrin. This suggests that AmyAc production may have a negative impact on nutrient acquisition by cells grown in MBU, which can be bypassed either by rasP overexpression or the provision of soytone and/or additional maltodextrin. Altogether, our results imply that rasP overexpression gives significant benefits for producing secretory proteins commercially.

Improved production of Properase and AmyAc upon overexpression of rasP. a Growth (left panel) and extracellular Properase activity (right panel) of cells cultured in MBU medium. Measurements on cells that overexpress rasP from the strong PspoVG promoter are indicated with black lines and measurements on wt cells are indicated with grey lines. b Growth (left panel) and extracellular AmyAc activity (right panel) of cells that overexpress rasP from the PspoVG promoter (black lines) or wt cells (grey lines) cultured in MBU medium. c Growth (left panel) and extracellular AmyAc activity (right panel) of cells that overexpress rasP from the PspoVG promoter (black lines) or wt cells (grey lines) cultured in 5SM12 medium. All plots in a–c show average values from three independent experiments, and the error bars represent the standard deviations of the respective measurements
Conclusions
In conclusion, our present study shows that the S2P intramembrane protease RasP sets the limit to efficient extracellular production of two proteins in B. subtilis, namely Properase and an AmyAc type amylase. These proteins are difficult to produce, which is partly due to effects on cell growth and/or viability in late stages of the fermentation process. Enhanced expression of rasP can overcome these negative effects, and seems even capable of boosting the secretion of the AmyAc type amylase up to tenfold. Our present findings are unprecedented, giving the first proof-of-principle that overexpression of a protease that cleaves within the plane of the cytoplasmic membrane of a bacterium can lead to improved protein production. The precise mechanism by which RasP exerts this effect in Bacillus is not yet known but, based on knowledge from other studies on S2P proteases, we envisage at least three possible scenarios. Firstly, RasP may facilitate the removal of cleaved signal peptides from the membrane [25], secondly, RasP may clear the membrane of mislocalized secretory precursor proteins that may interfere with essential membrane processes [20], or thirdly, overproduced RasP may modulate expression of σw-dependent genes that somehow influence productivity [26]. In these three scenarios, rasP overexpression could increase the fitness of the producing cells through the prevention of membrane and cell envelope perturbations. This would then lead to enhanced growth and productivity. A fourth possible explanation would be that RasP activity precludes potentially inhibitory effects of accumulating signal peptides on the SecA preprotein translocation motor [42] and/or on signal peptidases that convert translocated precursors of secretory proteins to the mature form [43]. Of course, combinations of these four scenarios are also conceivable. Irrespective of the precise mechanisms, we conclude that RasP can be applied to boost protein secretion in Bacillus, and that the overexpression of this and other S2P proteases represents a promising avenue for future cell factory engineering.
Abbreviations
- GRAS:
-
Generally Recognized As Safe
- IPTG:
-
isopropyl β-d-1-thiogalactopyranoside
- LDS:
-
lithium dodecyl sulphate
- OD600 :
-
optical density at 600 nm
- PAGE:
-
polyacrylamide gel electrophoresis
- PCR:
-
polymerase chain reaction
- QPS:
-
Qualified Presumption of Safety
- S2P:
-
site-2 protease
- TCA:
-
trichloroacetic acid
References
Van Dijl JM, Hecker M. Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb Cell Fact. 2013;12(1):3.
Sarvas M, Harwood CR, Bron S, Van Dijl JM. Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim Biophys Acta. 2004;1694:311–27.
Palva I. Molecular cloning of alpha-amylase gene from Bacillus amyloliquefaciens and its expression in B. subtilis. Gene. 1982;19(1):81–7.
Krishnappa L, Dreisbach A, Otto A, Goosens VJ, Cranenburgh R, Harwood CR, Becher D, Van Dijl JM. Extracytoplasmic proteases determining the cleavage and release of secreted proteins, lipoproteins, and membrane proteins in Bacillus subtilis. J Proteome Res. 2013;12:4101–10.
Westers L, Westers H, Quax WJ. Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta. 2004;1694:299–310.
Olempska-Beer ZS, Merker RI, Ditto MD, DiNovi MJ. Food-processing enzymes from recombinant microorganisms—a review. Regul Toxicol Pharmacol. 2006;45(2):144–58.
Earl AM, Losick R, Kolter R. Ecology and genomics of Bacillus subtilis. Trends Microbiol. 2008;16(6):269–75.
Brockmeier U, Caspers M, Freudl R, Jockwer A, Noll T, Eggert T. Systematic screening of all signal peptides from Bacillus subtilis: a powerful strategy in optimizing heterologous protein secretion in Gram-positive bacteria. J Mol Biol. 2006;362(3):393–402.
Caspers M, Brockmeier U, Degering C, Eggert T, Freudl R. Improvement of Sec-dependent secretion of a heterologous model protein in Bacillus subtilis by saturation mutagenesis of the N-domain of the AmyE signal peptide. Appl Microbiol Biotechnol. 2010;86(6):1877–85.
Pohl S, Bhavsar G, Hulme J, Bloor AE, Misirli G, Leckenby MW, Radford DS, Smith W, Wipat A, Williamson ED, Harwood CR, Cranenburgh RM. Proteomic analysis of Bacillus subtilis strains engineered for improved production of heterologous proteins. Proteomics. 2013;13(22):3298–308.
Luo Z, Gao Q, Li X, Bao J. Cloning of LicB from Clostridium thermocellum and its efficient secretive expression of thermostable beta-1,3-1,4-glucanase. Appl Biochem Biotechnol. 2014;173(2):562–70.
Wu XC, Ng SC, Near RI, Wong SL. Efficient production of a functional single-chain antidigoxin antibody via an engineered Bacillus subtilis expression-secretion system. Biotechnology (N Y). 1993;11(1):71–6.
Westers L, Dijkstra DS, Westers H, van Dijl JM, Quax WJ. Secretion of functional human interleukin-3 from Bacillus subtilis. J Biotechnol. 2006;123(2):211–24.
Bolhuis A, Sorokin A, Azevedo V, Ehrlich SD, Braun PG, De Jong A, Venema G, Bron S, Van Dijl JM. Bacillus subtilis can modulate its capacity and specificity for protein secretion through temporally controlled expression of the sipS gene for signal peptidase I. Mol Microbiol. 1996;22(4):605–18.
Kontinen VP, Sarvas M. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol Microbiol. 1993;8(4):727–37.
Kouwen TR, van der Goot A, Dorenbos R, Winter T, Antelmann H, Plaisier MC, Quax WJ, van Dijl JM, Dubois JY. Thiol-disulphide oxidoreductase modules in the low-GC Gram-positive bacteria. Mol Microbiol. 2007;64(4):984–99.
Chen J, Fu G, Gai Y, Zheng P, Zhang D, Wen J. Combinatorial Sec pathway analysis for improved heterologous protein secretion in Bacillus subtilis: identification of bottlenecks by systematic gene overexpression. Microb Cell Fact. 2015;14:92. doi:10.1186/s12934-015-0282-9.
Bramkamp M, Weston L, Daniel RA, Errington J. Regulated intramembrane proteolysis of FtsL protein and the control of cell division in Bacillus subtilis. Mol Microbiol. 2006;62(2):580–91.
Zweers JC, Wiegert T, van Dijl JM. Stress-responsive systems set specific limits to the overproduction of membrane proteins in Bacillus subtilis. Appl Environ Microbiol. 2009;75(23):7356–64.
Heinrich J, Hein K, Wiegert T. Two proteolytic modules are involved in regulated intramembrane proteolysis of Bacillus subtilis RsiW. Mol Microbiol. 2009;74(6):1412–26.
Heinrich J, Lundén T, Kontinen VP, Wiegert T. The Bacillus subtilis ABC transporter EcsAB influences intramembrane proteolysis through RasP. Microbiology. 2008;154:1989–97.
Yuan J, Zweers JC, van Dijl JM, Dalbey RE. Protein transport across and into cell membranes in bacteria and archaea. Cell Mol Life Sci. 2010;67(2):179–99.
Dalbey RE, Wang P, van Dijl JM. Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol Mol Biol Rev. 2012;76(2):311–30.
Akiyama Y, Kanehara K, Ito K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 2004;23:4434–42.
Saito A, Hizukuri Y, Matsuo E, Chiba S, Mori H, Nishimura O, Ito K, Akiyama Y. Post-liberation cleavage of signal peptides is catalyzed by the site-2 protease (S2P) in bacteria. Proc Natl Acad Sci USA. 2011;108(33):13740–5.
Zweers JC, Nicolas P, Wiegert T, Van Dijl JM, Denham EL. Definition of the σ (W) regulon of Bacillus subtilis in the absence of stress. PLoS ONE. 2012;7(11):e48471.
Helmann JD. Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr Opin Microbiol. 2016;30:122–32.
Vogtentanz G, Collier KD, Bodo M, Chang JH, Day AG, Estell DA, Falcon BC, Ganshaw G, Jarnagin AS, Kellis JT Jr, Kolkman MA, Lai CS, Meneses R, Miller JV, de Nobel H, Power S, Weyler W, Wong DL, Schmidt BF. A Bacillus subtilis fusion protein system to produce soybean Bowman–Birk protease inhibitor. Protein Expr Purif. 2007;55(1):40–52.
Fabret C, Ehrlich SD, Noirot P. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol. 2002;46(1):25–36.
Yang M, Galizzi A, Henner D. Nucleotide sequence of the amylase gene from Bacillus subtilis. Nucleic Acids Res. 1983;11(2):237–49.
Yuuki T, Nomura T, Tezuka H, Tsuboi A, Yamagata H, Tsukagoshi N, Udaka S. Complete nucleotide sequence of a gene coding for heat- and pH-stable alpha-amylase of Bacillus licheniformis: comparison of the amino acid sequences of three bacterial liquefying alpha-amylases deduced from the DNA sequences. J Biochem. 1985;98(5):1147–56.
Wells JA, Ferrari E, Henner DJ, Estell DA, Chen EY. Cloning, sequencing, and secretion of Bacillus amyloliquefaciens subtilisin in Bacillus subtilis. Nucleic Acids Res. 1983;11(22):7911–25.
Wells JA, Cunningham BC, Graycar TP, Estell DA. Recruitment of substrate-specificity properties from one enzyme into a related one by protein engineering. Proc Natl Acad Sci USA. 1987;84(15):5167–71.
Valle F, Ferrari E. Subtilisin: a redundantly temporally regulated gene. In: Smith I, Slepecky AR, Selow P, editors. Regulation of prokaryotic development. Washington: American Society for Microbiology; 1989. p. 131–46.
Perego M. Integrational vectors for genetic manipulation in Bacillus subtilis. In: Sonenshein AL, Hoch JA, Losick R, editors. Bacillus subtilis and other Gram-positive bacteria: biochemistry, physiology, and molecular genetics. Washington: American Society for Microbiology; 1993. p. 615–24.
Fukushima T, Furihata I, Emmins R, Daniel RA, Hoch JA, Szurmant H. A role for the essential YycG sensor histidine kinase in sensing cell division. Mol Microbiol. 2011;79(2):503–22.
Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS ONE. 2009;4(7):e6441.
Meijer WJ, de Jong A, Bea G, Wisman A, Tjalsma H, Venema G, Bron S, van Dijl JM. The endogenous Bacillus subtilis (natto) plasmids pTA1015 and pTA1040 contain signal peptidase-encoding genes: identification of a new structural module on cryptic plasmids. Mol Microbiol. 1995;17(4):621–31.
Van Dijl JM, De Jong A, Smith H, Bron S, Venema G. Non-functional expression of Escherichia coli signal peptidase I in Bacillus subtilis. J Gen Microbiol. 1991;137:2073–83.
Ferrari E, Valle F. Mutant aprE promoter. Genencor. 2000; EP1244794 B1.
Bolhuis A, Matzen A, Hyyryläinen HL, Kontinen VP, Meima R, Chapuis J, Venema G, Van Dijl JM. Singal peptide peptidase- and ClpP-like proteins of Bacillus subtilis required for efficient translocation and processing of secretory proteins. J Biol Chem. 1999;274(35):24585–92.
Cunningham K, Wickner W. Specific recognition of the leader region of precursor proteins is required for the activation of translocation ATPase of Escherichia coli. Proc Natl Acad Sci USA. 1989;86:8630–4.
Wickner W, Moore K, Dibb N, Geissert D, Rice M. Inhibition of purified Escherichia coli leader peptidase by the leader (signal) peptide of bacteriophage M13 procoat. J Bacteriol. 1987;169:3821–2.
Worner K, Szurmant H, Chiang C, Hoch JA. Phosphorylation and functional analysis of the sporulation initiation factor Spo0A from Clostridium botulinum. Mol Microbiol. 2006;59(3):1000–12.
Authors’ contributions
JN, CB, BS and JMvD conceived the study and analyzed the data; JN, CB, VJG and JMvD performed the experiments; and JN, CB and JMvD wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
JN, VJG and JMvD declare no competing financial interests. CB and BS are employees of DuPont Industrial Biosciences.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Consent for publication
The present manuscript has been seen and approved by all authors. They agree with its content and submission to Microbial Cell Factories.
Funding
This work was supported by funding from Genencor/Dupont.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Neef, J., Bongiorni, C., Goosens, V.J. et al. Intramembrane protease RasP boosts protein production in Bacillus . Microb Cell Fact 16, 57 (2017). https://doi.org/10.1186/s12934-017-0673-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0673-1