
Abstract
Depression remains a debilitating condition with an uncertain aetiology. Recently, attention has been given to the renin–angiotensin system. In the central nervous system, angiotensin II may be important in multiple pathways related to neurodevelopment and regulation of the stress response. Studies of drugs targeting the renin–angiotensin system have yielded promising results. Here, we review the potential beneficial effects of angiotensin blockers in depression and their mechanisms of action. Drugs blocking the angiotensin system have efficacy in several animal models of depression. While no randomised clinical trials were found, case reports and observational studies showed that angiotensin-converting enzyme inhibitors or angiotensin receptor blockers had positive effects on depression, whereas other antihypertensive agents did not. Drugs targeting the renin–angiotensin system act on inflammatory pathways implicated in depression. Both preclinical and clinical data suggest that these drugs possess antidepressant properties. In light of these results, angiotensin system-blocking agents offer new horizons in mood disorder treatment.
Similar content being viewed by others


Background
The pathophysiology of depression remains elusive and current treatments, which focus on traditional pathways (monoamine alterations), are only partially effective. Remission rates in the treatment of depression are only about 30% for those treated with traditional pharmacotherapy, and multiple agents are often required to achieve an adequate level of recovery [1] Evidence points to the involvement of neuroinflammation, oxidative and nitrosative stress pathways, mitochondrial dysfunction and neurotrophic signalling in depression [2].
Recently, the renin–angiotensin system (RAS) was proposed to be implicated in depression, and that blocking this system, either with angiotensin-converting enzyme inhibitors (ACEIs) or with angiotensin II type 1 receptor (AT1R) blockers, would translate into clinical benefits for the depression treatment [3,4,5,6,7]. Here, we review the literature so far on RAS-targeting drugs in depression.
Methods
A PubMed search was conducted for literature published between January 1974 and June 2017. Search terms included were: depression OR inflammation OR anxiety OR mood AND renin–angiotensin system, angiotensin, ATR1, ATR2, angiotensin receptor blockers, angiotensin-converting enzyme inhibitors, ATR3, ATR4, Mas, and aldosterone. Systematic reviews, randomised controlled trials (RCTs), observational studies, case series and animal studies with an emphasis on the angiotensin system and its role in depression were included. Articles not in English were excluded. The PubMed search was augmented by manually searching the references of key papers and related literature. The results were presented as a narrative review.
The RAS in the brain
The RAS was discovered in the 19th Century, after the blood pressure-raising agent renin was first identified in the rabbit kidney [8]. In time, the RAS became an established and extensively studied peripheral regulator of blood pressure and renal-mediated body fluid homeostasis, and was discovered to be a central target in clinical hypertension therapy. Renin, a protein synthesised by the juxtaglomerular cells of the kidney, cleaves the polypeptide angiotensinogen to generate angiotensin I (Ang I). This peptide is metabolised to angiotensin II (Ang II) by angiotensin I-converting enzyme (ACE).
It was surprising when renin was identified in the dog brain in 1971 [9, 10]. Subsequently, intracranial Ang II was shown to elevate blood pressure and to promote fluid intake [11,12,13,14], suggesting that angiotensin receptors were present in the brain. The actions of Ang II in the central nervous system are mediated mainly by two receptor types: AT1R and AT2R [15, 16]. Other receptors, including MAS [17], the (pro)renin receptor (PRR) [18] and AT4R [19], have also recently been identified but their roles remain less well characterised. AT3R was first reported as a new binding site for Ang II in mouse neuroblastoma cell cultures [20], but a separate gene for this receptor remains to be sequenced in humans.
AT1R mediates most of the peripheral and central actions of Ang II [21] and is implicated in multiple pathways related to regulation of the stress response. Stimulating AT1R contributes to the release of inflammatory markers [22]. Ang II interacts with AT1Rs, activating the NADPH–oxidase complex [23,24,25], the microglial RhoA/Rho kinase pathway [26,27,28], NF-kappa B, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2). In turn, activated COX-2 forms an intermediate in several key aspects of central nervous system inflammation, and in oxidative and nitrosative stress (see Fig. 1). AT1R stimulation also releases tumour necrosis factor α (TNF-α) [29, 30], which is important in several neurodegenerative disorders [29, 31,32,33], and regulates activation of the hypothalamic–pituitary–adrenal axis. Stimulation of AT1R in the parvocellular hypothalamic paraventricular nucleus (PVN) by Ang II increases production of corticotrophin-releasing factor [34,35,36]. In turn, this spurs adrenocorticotropic hormone secretion in the anterior pituitary gland, starting the stress response cascade. Accordingly, in humans, AT1R blockade downregulates hypothalamic–pituitary–adrenal axis activation [37].

Pathways involved in neuronal damage of angiotensin II through AT1 receptor agonism. Ang II, angiotensin II; AT1R, angiotensin II receptor type 1; PGE2, prostaglandin E2; Cox-2, Cyclooxygenase-2; PPAR-γ, peroxisome proliferator-activated receptor gamma; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; iNOS, inducible nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species
Ang II also stimulates the release of aldosterone via AT1R in the adrenal cortex of the kidney [38]. Thus, the acronym ‘RAAS’ (as in renin–angiotensin–aldosterone system) is often used. Besides being regulated by Ang II, aldosterone release is also stimulated by adrenocorticotropic hormone and the sympathetic nervous system. The role of aldosterone in the brain has previously been downplayed because its specific intracellular receptor, the mineralocorticoid receptor (MR), shares affinity with cortisol, which circulates at a ~1000-fold higher concentration than aldosterone [39]. For a tissue to be sensitive to aldosterone, it must express 11β-hydroxysteroid dehydrogenase type 2 (HSD-2) protein, which degrades cortisol, freeing the MR to the action of aldosterone. HSD-2 has been identified in the brain, mainly in the nucleus of the solitary tract, but also in the PVN [40]; regions that also express AT1R. Surprisingly – paralleling the history of angiotensin – aldosterone synthesis was also recognised in the amygdala, hippocampus and hypothalamus of the brain [41].
AT1R is particularly dense in the anterior pituitary; the circumventricular organs (area postrema; subfornical organ, the vascular organ of lamina terminalis and the median eminence); the lateral geniculate body; inferior olivary nucleus; the nucleus of the solitary tract and in the PVN, the preoptic and the supraoptic nuclei of the hypothalamus [42].
Modern molecular approaches have revealed that AT2R is also expressed in the adult brain [43, 44]. AT2R is involved in neurodevelopment [45,46,47,48,49] and participates in cell growth inhibition, fetal tissue development, extracellular matrix modulation, neuronal regeneration, apoptosis, cellular differentiation, and, possibly, vasodilation and left ventricular hypertrophy [50]. AT2R stimulation exerts neuroprotective effects in ischaemic stroke in rodents [51,52,53,54,55], and while the underlying mechanism remains to be fully characterised, it seems to partly involve an increase in the anti-inflammatory cytokine interleukin-10 [56]. AT2R is particularly dense in the amygdala, caudate putamen, medial geniculate body, globus pallidus, habenula, hypoglossal nucleus, inferior colliculus, inferior olivary nucleus, locus coeruleus, thalamus, and ventral tegmental area [42].
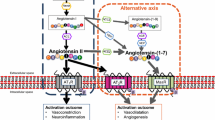
More components of the RAS such as ACE2, angiotensin-(1–7) and the Mas receptor have recently been identified in the brain. This alternative pathway is sometimes referred to as the non-classical RAS [57]. Originally identified in 1986 as an oncogene in mice [58], the tumorigenic power of Mas was later discredited and remained an orphan receptor until it was subsequently shown to bind with Ang (1–7) [17]. ACE2 can hydrolyse Ang II to produce Ang-(1–7). It can also cleave Ang I, producing Ang-(1–9) with subsequent Ang-(1-7) formation, although with much less efficiency. Mas is thus proposed to be a receptor for Ang-(1-7), with its highest expression in the brain [59]. The action of Ang-(1–7) through Mas is thought to influence arachidonic acid production and nitric oxide synthase activation [60] (see Fig. 2).

Pathway from angiotensinogen to AT1, AT2 and Mas receptors. ACE, Angiotensin-converting enzyme
The recently discovered PRR is highly expressed in the brain [18]. Its large extracellular domain binds and captures renin and its almost inactive precursor prorenin, increasing their enzymatic activities [61], but it also mimics the actions of AT1R through intracellular signalling [62].
A specific receptor for angiotensin IV (Ang IV), another less active peptide than Ang II, was first identified in a guinea pig hippocampus [19]. It is thought that the identity of AT4R was established when it was discovered that Ang IV is a strong inhibitor of insulin-regulated aminopeptidase (IRAP) [63]. IRAP is responsible for oxytocin degradation and, as demonstrated when an injection of Ang IV abolished the antidepressant effects of oxytocin in mice [64], is apparently required for its mood effects to take place. Yet recently, discrepancies between Ang IV binding site-antagonist and IRAP inhibitors [60], or the unaltered cognitive response of Ang IV in IRAP knockout mice [65], have cast doubt on whether IRAP is the only AT4R receptor. Further candidates for the role of AT4R have been proposed [42].
Ang II is also involved in cerebral blood flow regulation [21, 22]. Rising circulating Ang II is free to cross into the subfornical organ. This is a circumventricular organ lacking the blood–brain barrier, which, via AT1R, signals the paraventricular nucleus of the hypothalamus to activate the rostral ventrolateral medullary neurons and peripheral sympathetic nerves, thereby raising blood pressure [66]. Overstimulation of AT1Rs can lead to endothelial dysfunction [67] and neuronal injury and vulnerability caused by cerebrovascular remodelling [68,69,70,71,72].
It is well established that angiotensin receptors are present in the brain, yet the origin of active angiotensin peptides in the brain remains somewhat controversial. Researchers are puzzled because while Ang II is too hydrophilic to cross the blood–brain barrier [73], expression of renin in the brain is too low to account for its local synthesis [74]. Among the hypotheses advanced to solve this apparent paradox are renin-independent synthesis of angiotensin peptides [75]; impaired blood–brain barrier in hypertension leading to Ang II leaking into the cerebrospinal fluid [73]; an intracellular form of renin in the brain [76] or undetectable renin caused by its sequestration by PRR [62]. Although uncertainties persist, targeting the brain RAS or the peripheral RAS cannot be equal because ACEIs that penetrate the blood–brain barrier are superior to non-centrally acting ones in preventing cognitive decline [77, 78].
Major depressive disorder (MDD) and neuroinflammation: pre-clinical data
Inflammation is essential for restoring homeostasis in stress, infection and injury [79]. Hormones and circulating pro-inflammatory cytokines, products of neuronal injury and bacterial endotoxins, activate transcription factors. Activated inflammatory cascades with brain parenchymal microglia and blood-derived infiltrating macrophages also participate [80]. A well-regulated central inflammatory chain is fundamental to restore homeostasis, but an exaggerated response can be responsible for chronic inflammation, neuronal damage and a decrease in brain-derived neurotrophic factor [81,82,83,84,85,86]. Thus, excess or sustained activation of immune responses augments the risk of disease in vulnerable individuals, and can be important in the pathophysiology of many neurological and psychiatric disorders [2, 81, 87,88,89,90,91,92,93,94,95,96].
The inflammatory hypothesis [97, 98] postulates that depression is the result of altered immune-inflammatory pathways. This leads to increased immune activation, inflammation, nitro-oxidative stress and alteration of the kynurenine pathway, which ultimately causes changes in monoamine levels. MDD is characterised by a low-grade inflammatory state with increased peripheral levels of inflammatory cytokines, and microglial activation [98,99,100,101,102,103]. Normalised levels of inflammatory markers are associated with remission of clinical depression [104], while persistently elevated levels are associated with a lack of response to antidepressants [105]. Elevated levels of inflammatory markers such as C-reactive protein (CRP) may increase the risk of a first episode of depression [106, 107]. However, a large Mendelian randomisation study found no causal association between increased CRP levels and depression in people with genetically elevated CRP [108], and also that inflammation may better stratify those who will or will not benefit from anti-inflammatory treatments [109]. More compelling is the strong observation of depressive symptoms induced by interferon-α treatment, both in humans and in animal models [110,111,112,113].
Consequently, it has been hypothesised that drugs with anti-inflammatory properties might also demonstrate antidepressant potential. Nonsteroidal anti-inflammatory drugs have shown benefits [114, 115], although no influence was observed in association with antidepressants [116]. Cytokine inhibitors were found to improve depression [117,118,119] and specific depressive symptoms, such as anxiety [120] and fatigue [117], among patients with psoriasis [117, 118, 120] or ankylosing spondylitis [119]. This finding is supported by evidence from animal models [121]. In an open-label report, aspirin exhibited antidepressive effects, even at low doses [122], and may have a more favourable benefit/risk ratio compared with selective COX-2 inhibitors [123, 124]. Epidemiological reports also support antidepressant effects of aspirin [106, 125]. N-acetylcysteine may also be useful in treating MDD [126,127,128]. Statins, which apart from their antiatherosclerotic and cardioprotective effects also display neuroprotective and anti-inflammatory effects [129,130,131], showed the potential to produce mood-related benefits [132] and are associated with a reduced risk of depression [133]. Clinical trials of statins seem to show antidepressant effects in aggregate [134]. In a meta-analysis, supplementing the treatment of severe MDD with polyunsaturated fatty acids (PUFAs) was found to be beneficial, even though its role in mild-to-moderate depression or prevention seems limited [135].
Studies attempting to link depression with genetic variations in the RAS provide additional evidence. Initial reports for the most studied ACE polymorphism (I/D) – the presence or absence of a 287-bp fragment in intron 16 related to ACE serum levels [136] – were inconsistent and a meta-analysis showed no significance [137, 138]. However, other single nucleotide polymorphisms have been associated with depression [139, 140], including the GG genotype of ACE A2350G, which also correlated with higher ACE serum activity [141]. Recently, seven single nucleotide polymorphisms were significantly tied to late-life depression and cortisol levels under stressful circumstances [142]. The AT1R genotype (A1166C) CC is also associated with depression and increased responsiveness to Ang II [6], as well as clinical response [143, 144]. Epigenetic mechanisms also appear to be important, as altered methylation of the regulatory region of the ACE gene has been associated with depression [145]. ACE polymorphisms even seem able to influence antidepressant response [145,146,147], cognitive function after a depression episode in the elderly [148, 149], or suicide behaviour [150, 151].
The role of aldosterone in depression is an emerging area of research, thus regulation of aldosterone by the RAS is another point to take into account. Patients with primary hyperaldosteronism have depressive symptoms [152, 153]. In animal models, administering aldosterone leads to depressive behaviour [154], anxiety [155] and anhedonia [156]. Eplerone, an aldosterone antagonist, had anxiolytic properties in rats [157]. Poorer clinical outcome in MDD is predicted by higher salivary aldosterone [158, 159]. Conversely, MDD patients with suicidal behaviour had lower concentrations of aldosterone compared to suicidal patients without MDD and non-suicidal depressive patients [160]. Spironolactone, another MR antagonist, induces a sleep pattern characteristic of melancholic depression and reduces the efficacy of amitriptyline [40]. This hints at a non-linear dynamic of aldosterone throughout the MDD episode, prompting its exploration as a biomarker that is able to differentiate depression duration. Indeed, at least in women, higher aldosterone levels are associated with a shorter duration of a depressive episode [159], and in an animal model were used to mark the onset of depression [161].
Taking the above evidence in aggregate, current understanding of the pathophysiology of depression supports the search for novel therapeutics affecting the pathways of inflammation, oxidative biology, apoptosis and neurogenesis. Besides their anti-inflammatory effects, angiotensin receptor blockers (ARBs) and ACEIs have good tolerability, limited side effects and are already widely used drugs approved by the US Food and Drug Administration [162, 163]. Their neuroprotective, anti-inflammatory, vasodilatory [164] and microglia activation inhibitory effects [29] make them candidates for novel therapeutic targets for inflammatory brain diseases and cognitive disorders [21, 29, 30, 165, 166]. In this regard, interesting data is emerging from animal models.
The body of evidence supporting the antidepressant and antianxiety effects of drugs targeting the RAS in animal models is increasing. Mutant mice lacking the angiotensin gene have less depressive-like behaviour in the forced swim test [167]. Pharmacologically decreasing the production of Ang II by administering captopril (an ACEI) produces an analogous result [168].
Blockage of Ang II also leads to antidepressant-like activity in the learned helplessness [169] and chronic mild stress paradigms [170, 171], both more valid models than the forced swim test. Preclinical data also suggests a link between the antidepressant effect and a decrease in Ang II activity; AT1R antagonism by its specific blockers losartan [3], valsartan [171], irbesartan [170] and telmisartan [172] has similar actions to that caused by ACEIs. As with most antidepressants, use of these blockers also seems to have antianxiety properties. Candesartan [21, 173], losartan [174, 175] and captopril [176] reduced anxiety behaviour (promoting exploration) in the elevated plus maze test. Nevertheless, enalapril (a non-centrally acting ACEI) was not effective in normotensive rats [175].
Remarkably, different phenotypes of anxiolytic response to ARBs across different mice strains may be explained by differences in AT1R expression levels [177]. Curiously, mood effects were also apparent in an amphetamine-induced model of mania in mice, which candesartan was able to prevent and treat with comparable efficacy to lithium [30]. Transgenic rats overexpressing Ang-(1-7) [178] or ACE2 [179] showed a reduced anxiety phenotype that is seemingly dependent on Mas signalling, since antagonism of Mas reversed the phenotype. Administering Ang-(1-7) was associated with decreased oxidative stress markers in the amygdala [180]. The same Mas antagonism also prevented the anxiolytic/antidepressant effect of enalapril in transgenic hypertensive rats [181, 182].
These agents seem to influence mood disorders independently of their blood pressure-lowering activity. A study exploring the effect of valsartan in a chronic mild stress model found no change in average blood pressure after a month of treatment, while at the same time registering antianxiety and antidepressant effects [171].
Animal experiments also support the anti-inflammatory and oxidative stress-reducing effects of these drugs as part of their mechanisms of action. Both irbesartan and fluoxetine decreased levels of thiobarbituric-reactive substances – oxidative stress markers – while increasing catalase and glutathione (antioxidants) and serotonin (5-HT) levels in the brain [170]. Valsartan also increased neurogenesis in mice [171]. Captopril and perindopril (both centrally acting ACEIs) [183], telmisartan [183, 184] and candesartan [21, 185, 186] all show anti-inflammatory effects by reducing microglial activation and levels of inflammatory markers such as nitric oxide and TNF-α.
Clinical data
To date, no RCT has assessed the effects of ACEIs or ARBs in depression. However, observational studies have established a bidirectional link between cardiovascular disorders and depression. Antihypertensive sympatholytic drugs such as reserpine or clonidine can induce depression [187,188,189], prompting some to propose that sympathetic nervous system hyperreactivity is a common substrate [190, 191]. It was unclear whether this association was caused by hypertension itself, its treatment, or both [192, 193].
A meta-analysis of prospective cohort studies [194] found no evidence that hypertension is a risk factor for depression. However, the contrary – that depression increases the risk of developing hypertension – has been suggested [195] and confirmed by a meta-analysis [196]. In light of all the evidence, the RAS now emerges as a major link between mood and the cardiovascular system.
In the early 1980s, several cases reported that captopril might promote mood elevation in patients with MDD [197,198,199]. Mood benefits were reported in 9 patients with MDD, and one with bipolar disorder, who were treated with lisinopril (an ACEI) [200]. In each case, patients were being treated for hypertension or cardiac heart failure (see Table 1).
In a case-control study of 972 patients from primary care practices, who had both diabetes and a new diagnosis of depression, those exposed to ACEIs in the last 6 months showed a lower odds ratio for depression (OR 1.3, 95% CI: 0.8–2.2) compared to those exposed to beta-blockers (BBs) (OR 2.6, 95% CI: 1.1–7.0) and calcium channel blockers (CCBs) (OR 2.2, 95% CI: 1.2–4.2) [201]. In a recent population cohort study, ACEIs decreased the incidence of MDD [202]. These results were replicated by Boal et al. [203], who examined mood-related hospital admissions of 144,660 patients treated with antihypertensive monotherapy for a five-year follow-up. Interestingly, ACEIs and ARBs were associated with the lowest risk of mood disorder admissions (log-rank P = 0.006), while CCBs (hazard ratio (HR) = 2.28, [95% CI 1.13–4.58]; P = 0.02) and BBs (HR = 2.11, [95% CI 1.12 –3.98]; P = 0.02) were associated with increased risk compared to ACEIs and ARBs. There was no significant difference in patients receiving no antihypertensive medication (HR = 1.63 [95% CI 0.94–2.82]; P = 0.08), or those taking thiazide diuretics (HR = 1.56 [95% CI 0.65–3.73]; P = 0.32).
However, in the CREATE trial, a randomised placebo-controlled trial of citalopram in 284 coronary heart disease patients with MDD, the use of ACEIs predicted a worse response to citalopram [204]. A possible caveat is that the use of ACEIs may cause bias towards more severe coronary disease, and thus a possible vascular, more refractory type of depression. Another interesting possibility, considering the antidepressant properties of ACEIs, is that their use may have prevented or even treated milder episodes of depression, creating a selection bias for more severe depression. Indeed, we know that an increasingly smaller percentage of patients respond or remit after trying a second or third drug after failing previous treatments [205], and that antidepressant-naïve patients improve their Hamilton Depression Rating Scale score more than those taking antidepressants in response to treatment [206].
The antidepressant effects of ACEIs can be further inferred both by mood effects in the population without a formal diagnosis of MDD, and in studies looking at quality of life. Mood elation was reported in healthy volunteers taking enalapril [207]. One RCT found a higher quality of life score was attained in patients taking captopril compared to other classes of antihypertensive drugs, despite similar blood pressure control [208]. A head-to-head comparison of captopril (a centrally acting ACEI) and enalapril (a non-centrally acting ACEI) reported no difference in antihypertensive efficacy, but that captopril had a superior effect on quality of life measurements [209].
In the Norwegian HUNT study [192], the depressive symptoms of a large population of 55,472 patients with systemic hypertension taking an ACEI were compared with those of patients with untreated systemic hypertension. Results showed an important trend in favour of the depressive symptom-reducing effects of ACEIs, as assessed by the Hospital Anxiety and Depression Rating Scale (OR 0.54, 95% CI 0.28–1.08). Interestingly, those on BBs (OR 1.20, 95% CI 0.78–1.83) or on CCBs (OR 1.04, 95% CI 0.70–1.53) showed no reduction in depressive symptoms compared to the untreated systemic hypertension group. Again, this suggests that the pharmacological benefits of ACEIs and ARBs in depression are independent of their antihypertensive effects. A small open-label trial of 17 type 2 diabetic patients taking candesartan for at least 3 months found that depression scores were improved [210].
Nonetheless, there are a few negative reports of the effects of RAS drugs on mood. A small (n = 8), 6-week, double-blind crossover trial found captopril to have no positive effects on mood [211]. Another study found the BB atenolol superior to captopril for self-reported anxiety [212]. However, BBs are known to affect somatic anxiety, so measuring anxiety might not be an appropriate proxy for mood in this case. In a double-blinded trial of 451 hypertensive patients taking either enalapril or the CCB amlodipine for 38 weeks, no differences were found between the two drugs in terms of quality of life measures [213]. Another 6-month double-blind trial with 540 hypertensive patients showed no superiority of cilazapril (an ACEI) over atenolol (a BB) [214]. Losartan was also not superior to nifedepine (a CCB) in a 12-week randomised double-blind trial with 223 hypertensive patients [215].
Conclusions
A growing body of evidence suggests a role for the angiotensin system in the pathophysiology of MDD. Drugs targeting the RAS reduce oxidative and inflammatory stress and enhance neurogenesis; all documented pathological markers in depression. Despite the heavy burden of depression, new drug development has been underwhelming. While RCTs providing definitive proof are yet to come, available preclinical and clinical data suggest the potential antidepressant properties of ACEIs and ARBs. The search for novel, effective, safe anti-inflammatory drugs that act centrally in the brain are of fundamental interest. Future clinical trials targeting the brain angiotensin system are necessary to verify the usefulness of these agents in treating depression.
References
Jakubovski E, Bloch MH. Prognostic subgroups for citalopram response in the STAR*D trial. J Clin Psychiatry. 2014;75:738–47.
Moylan S, Berk M, Dean OM, Samuni Y, Williams LJ, O’Neil A, et al. Oxidative & nitrosative stress in depression: why so much stress? Neurosci Biobehav Rev. 2014;45:46–62.
Gard PR, Mandy A, Sutcliffe MA. Evidence of a possible role of altered angiotensin function in the treatment, but not etiology, of depression. Biol Psychiatry. 1999;45:1030–4.
Gard PR. The role of angiotensin II in cognition and behaviour. Eur J Pharmacol. 2002;438:1–14.
Gard PR. The brain renin-angiotensin system: a target for novel antidepressants and anxiolytics. Drug Dev Res. 2005;65:270–7.
Saab YB, Gard PR, Yeoman MS, Mfarrej B, El-Moalem H, Ingram MJ. Renin-angiotensin-system gene polymorphisms and depression. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1113–8.
Saavedra JM, Pavel J. Angiotensin II AT1 receptor antagonists inhibit the angiotensin-CRF-AVP axis and are potentially useful for the treatment of stress-related and mood disorders. Drug Dev Res. 2005;65:237–69.
Phillips MI, Schmidt-Ott KM. The discovery of renin 100 years ago. News Physiol Sci. 1999;14:271–4.
Ganten D, Boucher R, Genest J. Renin activity in brain tissue of puppies and adult dogs. Brain Res. 1971;33:557–9.
Ganten D, Marquez-Julio A, Granger P, Hayduk K, Karsunky KP, Boucher R, et al. Renin in dog brain. Am J Physiol. 1971;221:1733–7.
Bickerton RK, Buckley JP. Evidence for a central mechanism in angiotensin induced hypertension. Exp Biol Med. 1961;106:834–6.
Buggy J, Johnson AK. Angiotensin-induced thirst: effects of third ventricle obstruction and periventricular ablation. Brain Res. 1978;149:117–28.
Phillips MI, Felix D. Specific angiotensin II receptive neurons in the cat subfornical organ. Brain Res. 1976;109:531–40.
Johnson AK, Epstein AN. The cerebral ventricles as the avenue for the dipsogenic action of intracranial angiotensin. Brain Res. 1975;86:399–418.
Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther. 2008;120:292–316.
Oro C, Qian H, Thomas WG. Type 1 angiotensin receptor pharmacology: signaling beyond G proteins. Pharmacol Ther. 2007;113:210–26.
Santos RA, Silva AC S e, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor. Mas Proc Natl Acad Sci U S A. 2003;100:8258–63.
Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27.
Harding JW, Cook VI, Miller-Wing AV, Hanesworth JM, Sardinia MF, Hall KL, et al. Identification of an AII(3–8) [AIV] binding site in guinea pig hippocampus. Brain Res. 1992;583:340–3.
Chaki S, Inagami T. Identification and characterization of a new binding site for angiotensin II in mouse neuroblastoma neuro-2A cells. Biochem Biophys Res Commun. 1992;182:388–94.
Benicky J, Sánchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–70.
Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36:1–18.
Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J Neurochem. 2009;109:656–69.
Rodriguez-Pallares J, Rey P, Parga JA, Muñoz A, Guerra MJ, Labandeira-Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol Dis. 2008;31:58–73.
Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinski AL, et al. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J Neuroinflammation. 2011;8:129.
Rodriguez-Perez AI, Dominguez-Meijide A, Lanciego JL, Guerra MJ, Labandeira-Garcia JL. Inhibition of Rho kinase mediates the neuroprotective effects of estrogen in the MPTP model of Parkinson’s disease. Neurobiol Dis. 2013;58:209–19.
Tönges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego ÉM, et al. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain. 2012;135:3355–70.
Villar-Cheda B, Valenzuela R, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol Aging. 2012;33:e1–e11.
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Microglial TNF-α mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia. 2014;62:145–57.
de Souza Gomes JA, de Souza GC, Berk M, Cavalcante LM, de Sousa FC, Budni J, et al. Antimanic-like activity of candesartan in mice: possible involvement of antioxidant, anti-inflammatory and neurotrophic mechanisms. Eur Neuropsychopharmacol. 2015;25:2086–97.
Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129:318–20.
Hofman FM, Hinton DR, Johnson K, Merrill JE. Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med. 1989;170:607–12.
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–10.
Aguilera G, Scott Young W, Kiss A, Bathia A. Direct regulation of hypothalamic corticotropin-releasing-hormone neurons by angiotensin II. Neuroendocrinology. 1995;61:437–44.
Sumitomo T, Suda T, Nakano Y, Tozawa F, Yamada M, Demura H. Angiotensin II increases the corticotropin-releasing factor messenger ribonucleic acid level in the rat hypothalamus. Endocrinology. 1991;128:2248–52.
Aguilera G, Kiss A, Luo X. Increased expression of type 1 angiotensin II receptors in the hypothalamic paraventricular nucleus following stress and glucocorticoid administration. J Neuroendocrinol. 1995;7:775–83.
Wincewicz D, Juchniewicz A, Waszkiewicz N, Braszko JJ. Angiotensin II type 1 receptor blockade by telmisartan prevents stress-induced impairment of memory via HPA axis deactivation and up-regulation of brain-derived neurotrophic factor gene expression. Pharmacol Biochem Behav. 2016;148:108–18.
Balla T, Baukal AJ, Eng S, Catt KJ. Angiotensin II receptor subtypes and biological responses in the adrenal cortex and medulla. Mol Pharmacol. 1991;40:401–6.
Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Ren Physiol. 2009;297:F559–76.
Murck H, Schüssler P, Steiger A. Renin-angiotensin-aldosterone system: the forgotten stress hormone system: relationship to depression and sleep. Pharmacopsychiatry. 2012;45:83–95.
Gomez-Sanchez CE, Zhou MY, Cozza EN, Morita H, Foecking MF, Gomez-Sanchez EP. Aldosterone biosynthesis in the rat brain. Endocrinology. 1997;138:3369–73.
Wright JW, Harding JW. Brain renin-angiotensin - a new look at an old system. Prog Neurobiol. 2011;95:49–67.
de Kloet AD, Wang L, Ludin JA, Smith JA, Pioquinto DJ, Hiller H, et al. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct. 2016;221:891–912.
Yu L, Shao C, Gao L. Developmental expression patterns for angiotensin receptors in mouse skin and brain. J Renin Angiotensin Aldosterone Syst. 2014;15:139–49.
Mao C, Shi L, Xu F, Zhang L, Xu Z. Development of fetal brain renin-angiotensin system and hypertension programmed in fetal origins. Prog Neurobiol. 2009;87:252–63.
Meffert S, Stoll M, Steckelings UM, Bottari SP, Unger T. The angiotensin II AT2 receptor inhibits proliferation and promotes differentiation in PC12W cells. Mol Cell Endocrinol. 1996;122:59–67.
Li JM, Mogi M, Tsukuda K, Tomochika H, Iwanami J, Min LJ, et al. Angiotensin II-induced neural differentiation via angiotensin II type 2 (AT2) receptor-MMS2 cascade involving interaction between AT2 receptor-interacting protein and Src homology 2 domain-containing protein-tyrosine phosphatase 1. Mol Endocrinol. 2007;21:499–511.
Stroth U, Meffert S, Gallinat S, Unger T. Angiotensin II and NGF differentially influence microtubule proteins in PC12W cells: role of the AT2 receptor. Mol Brain Res. 1998;53:187–95.
Gendron L, Laflamme L, Rivard N, Asselin C, Payet MD, Gallo-Payet N. Signals from the AT2 (angiotensin type 2) receptor of angiotensin II inhibit p21ras and activate MAPK (mitogen-activated protein kinase) to induce morphological neuronal differentiation in NG108-15 cells. Mol Endocrinol. 1999;13:1615–26.
Cernes R, Mashavi M, Zimlichman R. Differential clinical profile of candesartan compared to other angiotensin receptor blockers. Vasc Health Risk Man. 2011;7:749–59.
Alhusban A, Fouda AY, Bindu P, Ishrat T, Soliman S, Fagan SC. Compound 21 is pro-angiogenic in the brain and results in sustained recovery after ischemic stroke. J Hypertens. 2015;33:170–80.
Joseph JP, Mecca AP, Regenhardt RW, Bennion DM, Rodríguez V, Desland F, et al. The angiotensin type 2 receptor agonist Compound 21 elicits cerebroprotection in endothelin-1 induced ischemic stroke. Neuropharmacology. 2014;81:134–41.
McCarthy CA, Vinh A, Miller AA, Hallberg A, Alterman M, Callaway JK, et al. Direct angiotensin AT2 receptor stimulation using a novel AT2 receptor agonist, compound 21, evokes neuroprotection in conscious hypertensive rats. PLoS One. 2014;9, e95762.
Min LJ, Mogi M, Tsukuda K, Jing F, Ohshima K, Nakaoka H, et al. Direct stimulation of angiotensin II type 2 receptor initiated after stroke ameliorates ischemic brain damage. Am J Hypertens. 2014;27:1036–44.
Schwengel K, Namsolleck P, Lucht K, Clausen BH, Lambertsen KL, Valero-Esquitino V, et al. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J Mol Med (Berl). 2016;94:957–66.
Fouda AY, Pillai B, Dhandapani KM, Ergul A, Fagan SC. Role of interleukin-10 in the neuroprotective effect of the Angiotensin type 2 receptor agonist, Compound 21, after ischemia/reperfusion injury. Eur J Pharmacol. 2017;799:128–34.
Mascolo A, Sessa M, Scavone C, De Angelis A, Vitale C, Berrino L, et al. New and old roles of the peripheral and brain renin–angiotensin–aldosterone system (RAAS): focus on cardiovascular and neurological diseases. Int J Cardiol. 2017;227:734–42.
Young D, Waitches G, Birchmeier C, Fasano O, Wigler M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell. 1986;45:711–9.
Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, et al. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin receptors: interpreters of pathophysiological angiotensinergic stimuli. Pharmacol Rev. 2015;67:754–819.
Singh KD, Karnik SS. Angiotensin receptors: structure, function, signaling and clinical applications. J Cell Signal. 2016;1:1–8.
Xu Q, Jensen DD, Peng H, Feng Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol Ther. 2016;164:126–34.
Cuadra AE, Shan Z, Sumners C, Raizada MK. A current view of brain renin-angiotensin system: is the (pro)renin receptor the missing link? Pharmacol Ther. 2010;125:27–38.
Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J Neurochem. 2003;86:344–50.
Loyens E, De Bundel D, Demaegdt H, Chai SY, Vanderheyden P, Michotte Y, et al. Antidepressant-like effects of oxytocin in mice are dependent on the presence of insulin-regulated aminopeptidase. Int J Neuropsychopharmacol. 2013;16:1153–63.
Albiston AL, Fernando RN, Yeatman HR, Burns P, Ng L, Daswani D, et al. Gene knockout of insulin-regulated aminopeptidase: Loss of the specific binding site for angiotensin IV and age-related deficit in spatial memory. Neurobiol Learn Mem. 2010;93:19–30.
Tan PS, Killinger S, Horiuchi J, Dampney RA. Baroreceptor reflex modulation by circulating angiotensin II is mediated by AT1 receptors in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2267–78.
Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond). 2007;112:375–84.
Ozacmak VH, Sayan H, Cetin A, Akyildiz-Igdem A. AT1 receptor blocker candesartan-induced attenuation of brain injury of rats subjected to chronic cerebral hypoperfusion. Neurochem Res. 2007;32:1314–21.
Ando H, Zhou J, Macova M, Imboden H, Saavedra JM. Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke. 2004;35:1726–31.
Nishimura Y, Ito T, Hoe KL, Saavedra JM. Chronic peripheral administration of the angiotensin II AT1 receptor antagonist Candesartan blocks brain AT1 receptors. Brain Res. 2000;871:29–38.
Yamakawa H, Jezova M, Ando H, Saavedra JM. Normalization of endothelial and inducible nitric oxide synthase expression in brain microvessels of spontaneously hypertensive rats by angiotensin II AT1 receptor inhibition. J Cereb Blood Flow Metab. 2003;23:371–80.
Zhou J, Ando H, Macova M, Dou J, Saavedra JM. Angiotensin II AT1 receptor blockade abolishes brain microvascular inflammation and heat shock protein responses in hypertensive rats. J Cereb Blood Flow Metab. 2005;25:878–86.
Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J Physiol. 2016;594:1591–600.
Saavedra JM. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol Neurobiol. 2005;25:485–512.
van Thiel BS, Góes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, et al. Brain renin–angiotensin system: does It exist? Hypertension. 2017;69:1136–44.
Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda). 2008;23:187–93.
Ohrui T, Tomita N, Sato-Nakagawa T, Matsui T, Maruyama M, Niwa K, et al. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology. 2004;63:1324–5.
Sink KM, Leng X, Williamson J, Kritchevsky SB, Yaffe K, Kuller L, et al. Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: results from the Cardiovascular Health Study. Arch Intern Med. 2009;169:1195–202.
Yong VW, Rivest S. Taking advantage of the systemic immune system to cure brain diseases. Neuron. 2009;64:55–60.
Licinio J, Wong ML. Pathways and mechanisms for cytokine signaling of the central nervous system. J Clin Invest. 1997;100:2941–7.
Brietzke E, Stertz L, Fernandes BS, Kauer-Sant'anna M, Mascarenhas M, Escosteguy Vargas A, et al. Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord. 2009;116:214–7.
Fernandes BS, Molendijk ML, Köhler CA, Soares JC, Leite CM, Machado-Vieira R, et al. Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: a meta-analysis of 52 studies. BMC Med. 2015;13:289.
de Oliveira GS, Ceresér KM, Fernandes BS, Kauer-Sant'Anna M, Fries GR, Stertz L, et al. Decreased brain-derived neurotrophic factor in medicated and drug-free bipolar patients. J Psychiatr Res. 2009;43:1171–4.
Fernandes BS, Gama CS, Ceresér KM, Yatham LN, Fries GR, Colpo G, et al. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res. 2011;45:995–1004.
Fernandes BS, Berk M, Turck CW, Steiner J, Gonçalves C. Decreased peripheral brain-derived neurotrophic factor levels are a biomarker of disease activity in major psychiatric disorders: a comparative meta-analysis. Mol Psychiatry. 2013;19:749–51.
Fernandes BS, Steiner J, Berk M, Molendijk ML, Gonzalez-Pinto A, Turck CW, et al. Peripheral brain-derived neurotrophic factor in schizophrenia and the role of antipsychotics: meta-analysis and implications. Mol Psychiatry. 2015;20:1108–19.
Barron M, Gartlon J, Dawson LA, Atkinson PJ, Pardon MC. A state of delirium: deciphering the effect of inflammation on tau pathology in Alzheimer’s disease. Exp Gerontol. 2017;94:103–7.
O’Donovan A, Ahmadian AJ, Neylan TC, Pacult MA, Edmondson D, Cohen BE. Current posttraumatic stress disorder and exaggerated threat sensitivity associated with elevated inflammation in the Mind Your Heart Study. Brain Behav Immun. 2017;60:198–205.
Kirkpatrick B, Miller BJ. Inflammation and schizophrenia. Schizophr Bull. 2013;39:1174–9.
Fernandes BS, Steiner J, Bernstein HG, Dodd S, Pasco JA, Dean OM, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry. 2016;21:554–64.
Madore C, Leyrolle Q, Lacabanne C, Benmamar-Badel A, Joffre C, Nadjar A, et al. Neuroinflammation in autism: plausible role of maternal inflammation, dietary omega 3, and microbiota. Neural Plast. 2016;2016:3597209.
De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, et al. Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun Rev. 2016;15:1005–11.
Hong S, Banks WA. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun. 2015;45:1–12.
McKee CA, Lukens JR. Emerging roles for the immune system in traumatic brain injury. Front Immunol. 2016;7:556.
Selmi C, Barin JG, Rose NR. Current trends in autoimmunity and the nervous system. J Autoimmun. 2016;75:20–9.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–83.
Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200.
Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36:764–85.
Fernandes BS, Steiner J, Molendijk ML, Dodd S, Nardin P, Gonçalves CA, et al. C-reactive protein concentrations across the mood spectrum in bipolar disorder: a systematic review and meta-analysis. Lancet Psychiatry. 2016;3:1147–56.
Slyepchenko A, Maes M, Köhler CA, Anderson G, Quevedo J, Alves GS, et al. T helper 17 cells may drive neuroprogression in major depressive disorder: proposal of an integrative model. Neurosci Biobehav Rev. 2016;64:83–100.
Andreazza AC, Kauer-Sant’anna M, Frey BN, Bond DJ, Kapczinski F, Young LT, et al. Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord. 2008;111:135–44.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Köhler CA, Freitas TH, Maes M, de Andrade NQ, Liu CS, Fernandes BS, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand. 2017;135:373–87.
Hannestad J, DellaGioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology. 2011;36:2452–9.
Eller T, Vasar V, Shlik J, Maron E. Pro-inflammatory cytokines and treatment response to escitaloprsam in major depressive disorder. Prog Neuropsychopharmacology Biol Psychiatry. 2008;32:445–50.
Pasco JA, Pasco JA, Jacka FN, Williams LJ, Henry MJ, Nicholson GC, Kotowicz MA, et al. Clinical implications of the cytokine hypothesis of depression: the association between use of statins and aspirin and the risk of major depression. Psychother Psychosom. 2010;79:323–5.
Valkanova V, Ebmeier KP, Allan CL. CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord. 2013;150:736–44.
Wium-Andersen MK, Oørsted DD, Nordestgaard BG. Elevated C-reactive protein, depression, somatic diseases, and all-cause mortality: a mendelian randomization study. Biol Psychiatry. 2014;76:249–57.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression. JAMA Psychiat. 2013;70:31–41.
Makino M, Kitano Y, Hirohashi M, Takasuna K. Enhancement of immobility in mouse forced swimming test by treatment with human interferon. Eur J Pharmacol. 1998;356:1–7.
Makino M, Kitano Y, Komiyama C, Takasuna K. Human interferon-alpha increases immobility in the forced swimming test in rats. Psychopharmacology (Berl). 2000;148:106–10.
Ping F, Shang J, Zhou J, Zhang H, Zhang L. 5-HT(1A) receptor and apoptosis contribute to interferon-α-induced ‘depressive-like’ behavior in mice. Neurosci Lett. 2012;514:173–8.
Fischer CW, Eskelund A, Budac DP, Tillmann S, Liebenberg N, Elfving B, et al. Interferon-alpha treatment induces depression-like behaviour accompanied by elevated hippocampal quinolinic acid levels in rats. Behav Brain Res. 2015;293:166–72.
Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11:680–4.
Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects. JAMA Psychiat. 2014;71:1381–91.
Uher R, Carver S, Power RA, Mors O, Maier W, Rietschel M, et al. Non-steroidal anti-inflammatory drugs and efficacy of antidepressants in major depressive disorder. Psychol Med. 2012;42:2027–35.
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35.
Menter A, Augustin M, Signorovitch J, Yu AP, Wu EQ, Gupta SR, et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J Am Acad Dermatol. 2010;62:812–8.
Ertenli I, Ozer S, Kiraz S, Apras SB, Akdogan A, Karadag O, et al. Infliximab, a TNF-alpha antagonist treatment in patients with ankylosing spondylitis: the impact on depression, anxiety and quality of life level. Rheumatol Int. 2012;32:323–30.
Langley RG, Feldman SR, Han C, Schenkel B, Szapary P, Hsu MC, et al. Ustekinumab significantly improves symptoms of anxiety, depression, and skin-related quality of life in patients with moderate-to-severe psoriasis: results from a randomized, double-blind, placebo-controlled phase III trial. J Am Acad Dermatol. 2010;63:457–65.
Karson A, Demirtaş T, Bayramgürler D, Balci F, Utkan T. Chronic administration of infliximab (TNF-α inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin Pharmacol Toxicol. 2013;112:335–40.
Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21:227–31.
Fond G, Hamdani N, Kapczinski F, Boukouaci W, Drancourt N, Dargel A, et al. Effectiveness and tolerance of anti-inflammatory drugs’ add-on therapy in major mental disorders: a systematic qualitative review. Acta Psychiatr Scand. 2014;129:163–79.
Berk M, Dean O, Drexhage H, McNeil JJ, Moylan S, O’Neil A, et al. Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Med. 2013;11:74.
Almeida OP, Flicker L, Yeap BB, Alfonso H, McCaul K, Hankey GJ. Aspirin decreases the risk of depression in older men with high plasma homocysteine. Transl Psychiatry. 2012;2, e151.
Berk M, Dean O, Cotton SM, Gama CS, Kapczinski F, Fernandes BS, et al. The efficacy of N-acetylcysteine as an adjunctive treatment in bipolar depression: an open label trial. J Affect Disord. 2011;135:389–94.
Berk M, Dean OM, Cotton SM, Jeavons S, Tanious M, Kohlmann K, et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2014;75:628–36.
Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M. N-acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. 2016;77:e457–66.
Wood WG, Mΰller WE, Eckert GP. Statins and neuroprotection: basic pharmacology needed. Mol Neurobiol. 2014;50:214–20.
Li Q, Zhuang QK, Yang JN, Zhang YY. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: the potential molecular mechanisms. Eur Rev Med Pharmacol Sci. 2014;18:1113–26.
Malfitano AM, Marasco G, Proto MC, Laezza C, Gazzerro P, Bifulco M. Statins in neurological disorders: an overview and update. Pharmacol Res. 2014;88:74–83.
O’Neil A, Sanna L, Redlich C, Sanderson K, Jacka F, Williams LJ, et al. The impact of statins on psychological wellbeing: a systematic review and meta-analysis. BMC Med. 2012;10:154.
Young-Xu Y, Chan KA, Liao JK, Ravid S, Blatt CM. Long-term statin use and psychological well-being. J Am Coll Cardiol. 2003;42:690–7.
Salagre E, Fernandes BS, Dodd S, Brownstein DJ, Berk M. Statins for the treatment of depression: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Affect Disord. 2016;200:235–42.
Appleton KM, Rogers PJ, Ness AR. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91:757–70.
Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–6.
Wu Y, Wang X, Shen X, Tan Z, Yuan Y. The I/D polymorphism of angiotensin-converting enzyme gene in major depressive disorder and therapeutic outcome: a case-control study and meta-analysis. J Affect Disord. 2012;136:971–8.
López-León S, Janssens AC, González-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13:772–85.
Baghai TC, Binder EB, Schule C, Salyakina D, Eser D, Lucae S, et al. Polymorphisms in the angiotensin-converting enzyme gene are associated with unipolar depression, ACE activity and hypercortisolism. Mol Psychiatry. 2006;11:1003–15.
Angunsri R, Sritharathikhun T, Suttirat S, Tencomnao T. Association of angiotensin-converting enzyme gene promoter single nucleotide polymorphisms and haplotype with major depression in a northeastern Thai population. J Renin Angiotensin Aldosterone Syst. 2009;10:179–84.
Firouzabadi N, Shafiei M, Bahramali E, Ebrahimi SA, Bakhshandeh H, Tajik N. Association of angiotensin-converting enzyme (ACE) gene polymorphism with elevated serum ACE activity and major depression in an Iranian population. Psychiatry Res. 2012;200:336–42.
Ancelin ML, Carrière I, Scali J, Ritchie K, Chaudieu I, Ryan J. Angiotensin-converting enzyme gene variants are associated with both cortisol secretion and late-life depression. Transl Psychiatry. 2013;3, e322.
Bondy B, Baghai TC, Zill P, Schule C, Eser D, Deiml T, et al. Genetic variants in the angiotensin I-converting-enzyme (ACE) and angiotensin II receptor (AT1) gene and clinical outcome in depression. Prog Neuropsychopharmacology Biol Psychiatry. 2005;29:1094–9.
Kondo DG, Speer MC, Krishnan KR, McQuoid DR, Slifer SH, Pieper CF, et al. Association of AGTR1 with 18-month treatment outcome in late-life depression. Am J Geriatr Psychiatry. 2007;15:564–72.
Zill P, Baghai TC, Schüle C, Born C, Früstück C, Büttner A, et al. DNA methylation analysis of the angiotensin converting enzyme (ACE) gene in major depression. PLoS One. 2012;7, e40479.
Bahramali E, Firouzabadi N, Yavarian I, Shayesteh MR, Erfani N, Shoushtari AA, et al. Influence of ACE gene on differential response to sertraline versus fluoxetine in patients with major depression: a randomized controlled trial. Eur J Clin Pharmacol. 2016;72:1059–64.
Baghai TC, Schule C, Zill P, Deiml T, Eser D, Zwanzger P, et al. The angiotensin I converting enzyme insertion/deletion polymorphism influences therapeutic outcome in major depressed women, but not in men. Neurosci Lett. 2004;363:38–42.
Hou Z, Yuan Y, Zhang Z, Hou G, You J, Bai F, et al. The D-allele of ACE insertion/deletion polymorphism is associated with regional white matter volume changes and cognitive impairment in remitted geriatric depression. Neurosci Lett. 2010;479:262–6.
Wang Z, Yuan Y, Bai F, You J, Li L, Zhang Z. Abnormal default-mode network in angiotensin converting enzyme D allele carriers with remitted geriatric depression. Behav Brain Res. 2012;230:325–32.
Fudalej S, Fudalej M, Kostrzewa G, Kuźniar P, Franaszczyk M, Wojnar M, et al. Angiotensin-converting enzyme polymorphism and completed suicide: an association in caucasians and evidence for a link with a method of self-injury. Neuropsychobiology. 2009;59:151–8.
Sparks DL, Hunsaker 3rd JC, Amouyel P, Malafosse A, Bellivier F, Leboyer M, et al. Angiotensin I-converting enzyme I/D polymorphism and suicidal behaviors. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:290–4.
Sonino N, Tomba E, Genesia ML, Bertello C, Mulatero P, Veglio F, et al. Psychological assessment of primary aldosteronism: a controlled study. J Clin Endocrinol Metab. 2011;96:E878–83.
Künzel HE. Psychopathological symptoms in patients with primary hyperaldosteronism - Possible pathways. Horm Metab Res. 2012;44:202–7.
Hlavacova N, Wes PD, Ondrejcakova M, Flynn ME, Poundstone PK, Babic S, et al. Subchronic treatment with aldosterone induces depression-like behaviours and gene expression changes relevant to major depressive disorder. Int J Neuropsychopharmacol. 2012;15:247–65.
Hlavacova N, Jezova D. Chronic treatment with the mineralocorticoid hormone aldosterone results in increased anxiety-like behavior. Horm Behav. 2008;54:90–7.
Morris MJ, Na ES, Grippo AJ, Johnson AK. The effects of deoxycorticosterone-induced sodium appetite on hedonic behaviors in the rat. Behav Neurosci. 2006;120:571–9.
Hlavacova N, Jezova D. Effect of single treatment with the antihypertensive drug eplerenone on hormone levels and anxiety-like behaviour in rats. Endocr Regul. 2008;42:147–53.
Büttner M, Jezova D, Greene B, Konrad C, Kircher T, Murck H, et al. Target-based biomarker selection - mineralocorticoid receptor-related biomarkers and treatment outcome in major depression. J Psychiatr Res. 2015;66–7:24–37.
Segeda V, Izakova L, Hlavacova N, Bednarova A, Jezova D. Aldosterone concentrations in saliva reflect the duration and severity of depressive episode in a sex dependent manner. J Psychiatr Res. 2017;91:164–8.
Hallberg L, Westrin A, Isaksson A, Janelidze S, Träskman-Bendz L, Brundin L. Decreased aldosterone in the plasma of suicide attempters with major depressive disorder. Psychiatry Res. 2011;187:135–9.
Franklin M, Hlavacova N, Babic S, Pokusa M, Bermudez I, Jezova D. Aldosterone signals the onset of depressive behaviour in a female rat model of depression along with SSRI treatment resistance. Neuroendocrinology. 2015;102:274–87.
Berk M, Nierenberg AA. Three paths to drug discovery in psychiatry. Am J Psychiatry. 2015;172:412–4.
Thöne-Reineke C, Steckelings UM, Unger T. Angiotensin receptor blockers and cerebral protection in stroke. J Hypertens. 2006;24:S115–21.
Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–57.
Kume K, Hanyu H, Sakurai H, Takada Y, Onuma T, Iwamoto T. Effects of telmisartan on cognition and regional cerebral blood flow in hypertensive patients with Alzheimer’s disease. Geriatr Gerontol Int. 2012;12:207–14.
Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465.
Okuyama S, Sakagawa T, Sugiyama F, Fukamizu A, Murakami K. Reduction of depressive-like behavior in mice lacking angiotensinogen. Neurosci Lett. 1999;261:167–70.
Giardina WJ, Ebert DM. Positive effects of captopril in the behavioral despair swim test. Biol Psychiatry. 1989;25:697–702.
Martin P, Massol J, Puech AJ. Captopril as an antidepressant? Effects on the learned helplessness paradigm in rats. Biol Psychiatry. 1990;27:968–74.
Ayyub M, Najmi AK, Akhtar M. Protective effect of irbesartan an angiotensin (AT1) receptor antagonist in unpredictable chronic mild stress induced depression in mice. Drug Res (Stuttg). 2017;67:59–64.
Ping G, Qian W, Song G, Zhaochun S. Valsartan reverses depressive/anxiety-like behavior and induces hippocampal neurogenesis and expression of BDNF protein in unpredictable chronic mild stress mice. Pharmacol Biochem Behav. 2014;124:5–12.
Aswar U, Chepurwar S, Shintre S, Aswar M. Telmisartan attenuates diabetes induced depression in rats. Pharmacol Reports. 2017;69:358–64.
Saavedra JM, Armando I, Bregonzio C, Juorio A, Macova M, Pavel J, et al. A centrally acting, anxiolytic angiotensin II AT1 receptor antagonist prevents the isolation stress-induced decrease in cortical CRF1 receptor and benzodiazepine binding. Neuropsychopharmacology. 2006;31:1123–34.
Llano López LH, Caif F, García S, Fraile M, Landa AI, Baiardi G, et al. Anxiolytic-like effect of losartan injected into amygdala of the acutely stressed rats. Pharmacol Reports. 2012;64:54–63.
Srinivasan J, Suresh B, Ramanathan M. Differential anxiolytic effect of enalapril and losartan in normotensive and renal hypertensive rats. Physiol Behav. 2003;78:585–91.
Costall B, Domeney AM, Gerrard PA, Horovitz ZP, Kelly ME, Naylor RJ, et al. Effects of captopril and SQ29,852 on anxiety-related behaviours in rodent and marmoset. Pharmacol Biochem Behav. 1990;36:13–20.
Golding BJ, Overall ADJ, Gard PR. Strain differences and the role of AT1 receptor expression in anxiety. Int J Mol Epidemiol Genet. 2011;2:51–5.
Kangussu LM, Almeida-Santos AF, Moreira FA, Fontes MAP, Santos RAS, Aguiar DC, et al. Reduced anxiety-like behavior in transgenic rats with chronically overproduction of angiotensin-(1-7): role of the Mas receptor. Behav Brain Res. 2017;331:193–8.
Wang L, de Kloet AD, Pati D, Hiller H, Smith JA, Pioquinto DJ, et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology. 2016;105:114–23.
Bild W, Ciobica A. Angiotensin-(1-7) central administration induces anxiolytic-like effects in elevated plus maze and decreased oxidative stress in the amygdala. J Affect Disord. 2013;145:165–71.
Almeida-Santos AF, Kangussu LM, Moreira FA, Santos RA, Aguiar DC, Campagnole-Santos MJ. Anxiolytic- and antidepressant-like effects of angiotensin-(1-7) in hypertensive transgenic (mRen2)27 rats. Clin Sci (Lond). 2016;130:1247–55.
Torika N, Asraf K, Roasso E, Danon A, Fleisher-Berkovich S. Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J Neuroimmune Pharmacol. 2016;11:774–85.
Torika N, Asraf K, Danon A, Apte RN, Fleisher-Berkovich S. Telmisartan modulates glial activation: in vitro and in vivo studies. PLoS One. 2016;11, e0155823.
Torika N, Asraf K, Cohen H, Fleisher-Berkovich S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav Immun. 2017;64:80–90.
Sanchez-Lemus E, Murakami Y, Larrayoz-Roldan IM, Moughamian AJ, Pavel J, Nishioku T, et al. Angiotensin II AT1 receptor blockade decreases lipopolysaccharide-induced inflammation in the rat adrenal gland. Endocrinology. 2008;149:5177–88.
Sanchez-Lemus E, Benicky J, Pavel J, Larrayoz IM, Zhou J, Baliova M, et al. Angiotensin II AT1 blockade reduces the lipopolysaccharide-induced innate immune response in rat spleen. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1376–84.
Ambrosino SV. Depressive reactions associated with reserpine. NY State J Med. 1974;74:860–4.
Bevacqua BK, Fattouh M, Backonja M. Depression, night terrors, and insomnia associated with long-term intrathecal clonidine therapy. Pain Pract. 2007;7:36–8.
Ghanizadeh A. Insomnia, night terror, and depression related to clonidine in attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2008;28:725–6.
Goldstein BI, Carnethon MR, Matthews KA, McIntyre RS, Miller GE, Raghuveer G, et al. Major depressive disorder and bipolar disorder predispose youth to accelerated atherosclerosis and early cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132:965–86.
Scalco AZ, Scalco MZ, Azul JBS, Lotufo NF. Hypertension and depression. Clinics (Sao Paulo). 2005;60:241–50.
Johansen A, Holmen J, Stewart R, Bjerkeset O. Anxiety and depression symptoms in arterial hypertension: the influence of antihypertensive treatment. The HUNT study, Norway. Eur J Epidemiol. 2012;27:63–72.
Huffman JC, Stern TA. Neuropsychiatric consequences of cardiovascular medications. Dialogues Clin Neurosci. 2007;9:29–45.
Long J, Duan G, Tian W, Wang L, Su P, Zhang W, et al. Hypertension and risk of depression in the elderly: a meta-analysis of prospective cohort studies. J Hum Hypertens. 2015;29:478–82.
Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry. 1998;55:580–92.
Meng L, Chen D, Yang Y, Zheng Y, Hui R. Depression increases the risk of hypertension incidence. J Hypertens. 2012;30:842–51.
Zubenko GS, Nixon RA. Mood-elevating effect of captopril in depressed patients. Am J Psychiatry. 1984;141:110–1.
Deicken RF. Captopril treatment of depression. Biol Psychiatry. 1986;21:1425–8.
Germain L, Chouinard G. Treatment of recurrent unipolar major depression with captopril. Biol Psychiatry. 1988;23:637–41.
Hertzman M, Adler LW, Arling B, Kern M. Lisinopril may augment antidepressant response. J Clin Psychopharmacol. 2005;25:618–20.
Rathmann W, Haastert B, Roseman JM, Giani G. Cardiovascular drug prescriptions and risk of depression in diabetic patients. J Clin Epidemiol. 1999;52:1103–9.
Williams LJ, Pasco JA, Kessing LV, Quirk SE, Fernandes BS, Berk M. Angiotensin converting enzyme inhibitors and risk of mood disorders. Psychother Psychosom. 2016;85:250–2.
Boal AH, Smith DJ, McCallum L, Muir S, Touyz RM, Dominiczak AF, et al. Monotherapy with major antihypertensive drug classes and risk of hospital admissions for mood disorders. Hypertension. 2016;68:1132–8.
Habra ME, Baker B, Frasure-Smith N, Swenson JR, Koszycki D, Butler G, et al. First episode of major depressive disorder and vascular factors in coronary artery disease patients: baseline characteristics and response to antidepressant treatment in the CREATE trial. J Psychosom Res. 2010;69:133–41.
Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry. 2006;163:1161–72.
Hunter AM, Cook IA, Leuchter AF. Does prior antidepressant treatment of major depression impact brain function during current treatment? Eur Neuropsychopharmacol. 2012;22:711–20.
Cohen LM, Anderson G, Firnhaber WR. Enalapril and hypertension. Am J Psychiatry. 1984;141:1012–3.
Croog SH, Levine S, Testa MA, Brown B, Bulpitt CJ, Jenkins CD, et al. The effects of antihypertensive therapy on the quality of life. N Engl J Med. 1986;314:1657–64.
Testa MA, Anderson RB, Nackley JF, Hollenberg NK. Quality of life and antihypertensive therapy in men. A comparison of captopril with enalapril. The Quality-of-Life Hypertension Study Group. N Engl J Med. 1993;328:907–13.
Pavlatou MG, Mastorakos G, Lekakis I, Liatis S, Vamvakou G, Zoumakis E, et al. Chronic administration of an angiotensin II receptor antagonist resets the hypothalamic–pituitary–adrenal (HPA) axis and improves the affect of patients with diabetes mellitus type 2: preliminary results. Stress. 2008;11:62–72.
Callender JS, Hodsman GP, Hutcheson MJ, Lever AF, Robertson JI. Mood changes during captopril therapy for hypertension. A double-blind pilot study. Hypertension. 1983;5:III90–3.
Deary I, Capewell S, Hajducka C, Muir A. The effects of captopril vs atenolol on memory, information processing and mood: a double-blind crossover study. Br J Clin Pharmacol. 1991;32:347–53.
Omvik P, Thaulow E, Herland OB, Eide I, Midha R, Turner RR. Double-blind, parallel, comparative study on quality of life during treatment with amlodipine or enalapril in mild or moderate hypertensive patients: a multicentre study. J Hypertens. 1993;11:103–13.
Fletcher AE, Bulpitt CJ, Chase DM, Collins WC, Furberg CD, Goggin TK, et al. Quality of life with three antihypertensive treatments. Cilazapril, atenolol, nifedipine. Hypertension. 1992;19:499–507.
Weir MR, Elkins M, Liss C, Vrecenak AJ, Barr E, Edelman JM. Efficacy, tolerability, and quality of life of losartan, alone or with hydrochlorothiazide, versus nifedipine GITS in patients with essential hypertension. Clin Ther. 1996;18:411–28.
Acknowledgements
Not applicable.
Funding
BSF is supported by a postdoctoral fellowship from Deakin University, Australia. CAK is supported by a postdoctoral scholarship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; Brazil). AFC is supported by a research fellowship award from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Brazil). MB is supported by a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship (grant number 1059660). The Department of Psychiatry and Behavioral Sciences of McGovern Medical School (USA) funds the Translational Psychiatry Program. The University of Texas Health Science Center at Houston (UTHealth). Laboratory of Neurosciences (Brazil) is a National Institute for Molecular Medicine centre, and a member of the Center of Excellence in Applied Neurosciences of Santa Catarina. Its research is supported by grants from CNPq (JQ), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (JQ); Instituto Cérebro e Mente (JQ) and Universidade do Extremo Sul Catarinense (JQ). JQ is a 1A CNPq Research Fellow. MB is supported by an NHMRC Senior Principal Research Fellowship (GNT1059660).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors participated in the design of this review. JV, CP, and VC performed the systematic review. JV, CP, VC, and BSF wrote the first draft of the manuscript. BSF and MB critically reviewed the first draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Vian, J., Pereira, C., Chavarria, V. et al. The renin–angiotensin system: a possible new target for depression. BMC Med 15, 144 (2017). https://doi.org/10.1186/s12916-017-0916-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-017-0916-3