
Abstract
The renin angiotensin system (RAS) consists of the systemic hormone system, critically involved in regulation and homeostasis of normal physiological functions [i.e. blood pressure (BP), blood volume regulation], and an independent brain RAS, which is involved in the regulation of many functions such as memory, central control of BP and metabolic functions. In general terms, the RAS consists of two opposing axes; the ‘classical axis’ mediated primarily by Angiotensin II (Ang II), and the ‘alternative axis’ mediated mainly by Angiotensin-(1–7) (Ang-(1–7)). An imbalance of these two opposing axes is thought to exist between genders and is thought to contribute to the pathology of cardiovascular conditions such as hypertension, a stroke co-morbidity. Ischaemic stroke pathophysiology has been shown to be influenced by components of the RAS with specific RAS receptor antagonists and agonists improving outcome in experimental models of stroke. Manipulation of the two opposing axes following acute ischaemic stroke may provide an opportunity for protection of the neurovascular unit, particularly in the presence of pre-existing co-morbidities where the balance may be shifted. In the present review we will give an overview of the experimental stroke studies that have investigated pharmacological interventions of the RAS.
Similar content being viewed by others

Background
In the UK, stroke is the fourth leading cause of death and one of the largest contributors towards long-term disability, affecting approximately 152,000 people every year (https://www.stroke.org.uk; State of the Nation 2015) [1]. In the past 40 years the number of stroke fatalities has been decreasing, however, it is estimated that over two-thirds of stroke survivors require daily medical care and over half are left disabled [2], resulting in an annual cost of nearly £4 billion and accounting for approximately 4–6 % of total NHS expenditure [3].
Recombinant tissue plasminogen activator (rt-PA; Alteplase), is the only thrombolytic treatment currently available for acute ischaemic stroke (AIS). It acts by breaking down the clot or thrombus obstructing the cerebral vessel, thus, re-establishing blood flow. However, it has a narrow therapeutic time window of 4.5 h from stroke onset, resulting in only 2–5 % of ischaemic strokes being treated globally, and can have detrimental side effects, including haemorrhage [4]. Recent results from a number of randomised clinical trials of mechanical thrombectomy have demonstrated efficacy for this intervention up to 6 h after stroke onset [5]. The positive results from these trials have reinvigorated the stroke community and open up new possibilities for adjunctive protective strategies.
Failure to translate effective therapeutic strategies from the ‘bench to bedside’ may partly be attributed to the use of animal models that do not incorporate non-modifiable risk factors such as gender and many of the stroke co-morbidities observed in the clinical stroke population, such as hypertension, diabetes, obesity, etc. For instance, hypertension is the single most important modifiable risk factor for stroke, acting as a contributing factor in over 75 % of first time stroke patients [6] with hypertension during acute stroke is associated with poorer clinical outcome [7].
The renin angiotensin system (RAS), a peptide hormone system intrinsically involved in blood pressure regulation and blood volume homeostasis in the circulation, has been shown to be present as a local paracrine system in the brain [8]. The RAS is reported to be involved in the pathology of AIS and its risk factors [8, 9], therefore, emerging as a potential therapeutic target. This review discusses the therapeutic potential of the RAS following AIS, emphasising the importance of cerebral RAS receptor targeting and its relevance in the presence of known stroke risk factors.
Brain RAS: classical and alternative axis
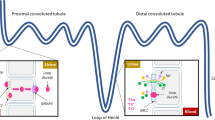
In the circulation, a drop in blood pressure (systemic hypotension) and/or blood volume results in juxtaglomerular cells within the kidneys to release renin (protease) whereas increased blood pressure (hypertension) inhibits renin release under normal circumstances. Circulating angiotensinogen is hydrolysed by renin to produce Angiotensin I (Ang I), which is then further converted by angiotensin converting enzyme (ACE) to generate biological active octapeptide, Ang II (Fig. 1). Ang II, a potent vasoconstrictor, acts by stimulating Ang II type 1 and type 2 receptors (AT1R and AT2R) [10]. Ang II exhibits a higher affinity to the widely expressed AT1R whereby it exerts its main physiological effects by constricting blood vessels, increasing BP, and stimulating aldosterone release from adrenal glands, promoting water and salt reabsorption in the kidneys, thus, raising blood volume levels [10]. In the last decade, an ‘alternative axis’ has been identified involving the monocarboxypeptidase, ACE2, the biologically active peptide Ang-(1–7) and its G-protein coupled receptor, Mas (MasR). Ang-(1–7) is formed by the direct actions of ACE2 on Ang II or via ACE2 induced cleavage of Ang I, generating the nine amino acid peptide Ang-(1–9), which is further converted to Ang (1–7) by ACE or peptidases such as neprilysin (NEP) [8].

Simplified overview of the renin angiotensin system (RAS). Angiotensinogen is cleaved by renin generating Angiotensin I (Ang I) which is then formed into Angiotensin II (Ang II) via the actions of angiotensin converting enzyme (ACE). Ang II preferentially binds to Angiotensin II type I receptor (AT1R) (‘classical axis’) inducing vasoconstriction, inflammation, oxidative stress, apoptosis and cell proliferation. Ang II can also activate the Angiotensin II type II (AT2R) and is metabolised by angiotensin converting enzyme 2 (ACE2) to generate Angiotensin-(1–7). Ang-(1–7) activates the Mas receptor (MasR). Ang-(1–7) can be formed by the actions of ACE or neprilysin (NEP) on Angiotensin-(1–9) or Angiotensin I. AT2R and ACE2/Ang-(1–7)/MasR form the ‘alternative axis’ and its activation is thought to counteract the detrimental effects induced by AT1R by leading to vasodilation, angiogenesis and preventing inflammation, oxidative stress and apoptosis
All the components of the ‘classical axis’ (angiotensinogen, renin, ACE, Ang II) have been identified within the brain parenchyma (see reviews [8, 11]). In addition, there is evidence that the ‘alternative axis’ is locally produced within the brain as ACE2 has been localised in neurons [12], astrocytes [13] and within the cerebrovasculature [14]. Similarly, the receptor subtypes responsible for mediating the functional effects of RAS peptides are expressed in neuronal and glial cells throughout the brain. For example, AT1 and AT2 receptors have been shown to be present in dopaminergic neurons, astrocytes and microglia from both human and primate brain tissue [15] and MasR shown to be expressed in neurons, astrocytes, microglia and cerebral endothelial cells in rodents [16, 17].
Involvement in ischaemic stroke
Over-activation of the ACE/Ang II/AT1R axis is thought to contribute to the pathogenesis of AIS through its vasoconstrictor effects on cerebral vessels as well as its pro-inflammatory, pro-fibrotic and increased oxidative stress effects in the parenchyma [8]. For instance, in the brain, AT1R knockout (KO) mice subjected to permanent middle cerebral artery occlusion (pMCAO) exhibit a larger metabolic penumbra volume (cerebral protein synthesis and ATP mismatch) and higher CBF within the core and penumbra when compared to wild type (WT) mice whereas mice over-expressing human renin and angiotensinogen genes have larger infarcts [18, 19]. Furthermore, Ang II has been shown to enhance the contractile response in isolated middle cerebral arteries (MCA) following MCAO via the AT1R [20], therefore, worsening cerebral perfusion following ischaemia.
Interestingly, emerging evidence identifies the ‘alternative axis’ as an endogenous protective system, which acts to counteract the effects of the ‘classical axis’ by mediating vasodilation and anti-inflammatory, anti-oxidant and anti-apoptotic effects via MasR and AT2R activation [21, 22]. Following focal cerebral ischaemia in the rat, AT2Rs have been shown to be upregulated in the peri-infarct region of the cortex, and also within the selectively vulnerable regions of the cortex and hippocampus following global cerebral ischaemia [23, 24]. AT2R KO mice exhibit less ischaemic damage compared to WT control animals following MCAO [25] suggesting a protective role for this receptor in the setting of cerebral ischaemia. In support of a role for this pathway following acute ischaemic stroke the ACE2/Ang-(1–7)/Mas axis has been shown to be upregulated. Brain expression (mRNA and protein levels) of both ACE2 and MasR are upregulated from as early as 6 h following permanent MCAO in the rat, peaking at 24 h post MCAO. This was associated with an concomitant increase in both circulating serum and cerebral Ang-(1–7) levels [16]. This increase occurs during the first critical hours after stroke when ischaemic damage and loss of potentially salvageable penumbra is taking place, suggesting a potential role for this pathway in the pathogenesis. Increasing exogenous Ang-(1–7) levels in the brain by central infusion prior to stroke has been shown to decrease infarct volume in rats [26], providing evidence of a protective role for this pathway post-stroke.
The current evidence suggests that there is an imbalance in the RAS following stroke with an enhanced activation of the ACE/Ang II/AT1R pathway and that targeting the counter-regulatory ACE2/Ang-(1–7)/Mas axis may provide protection. This imbalance in the RAS may be exacerbated in the presence of known stroke risk factors such as gender and hypertension and therefore provide a potential therapeutic target in a subset of patients.
RAS and stroke co-morbidities
Men have a higher incidence of stroke, however, after the menopause the incidence of stroke in women rapidly increases and this is co-incident with the decrease in female sex hormones [27]. In addition, to sex differences in stroke incidence, it is also well established in experimental stroke studies that female animals have less ischaemic injury than their male counterparts and that this protection is lost after ovariectomy [28, 29]. Furthermore, cells exhibit sex specific differences in the specific mechanisms of cell death following cerebral ischaemia [30] therefore it is not unlikely that sex differences will exist in the RAS in the brain and elsewhere. It is widely accepted that the RAS is influenced by sex hormones with males and females exhibiting differential responses to stimulation and inhibition of the RAS [31]. Differences in vascular reactivity to Ang II and receptor expression have been shown to be influenced by sex hormones, with testosterone stimulating the ‘classical axis’ and oestrogen lowering the AT1R:AT2R ratio, thereby enhancing vasodilatation [32–34]. Interestingly, in experimental animal models, female rats exhibit enhanced activity of the ACE2/Ang-(1–7)/Mas axis compared to males [31], showing increased renal Ang-(1–7) levels [35]. These findings may translate to humans, where normotensive healthy adult females have been shown to have higher plasma levels of Ang-(1–7) than their male counterparts [36]. These combined findings suggest that oestrogen may confer some degree of protection against stroke in premenopausal women by promoting increased activation of the ACE2/Ang-(1–7)/Mas axis and therefore, the loss of oestrogen associated with menopause may contribute to increased stroke risk through a loss of this enhanced protective pathway. In terms of brain expression, AT1R expression has been shown to be lowered in rats treated with oestrogen plus ovariectomy compared to ovariectomy alone. In contrast, oestrogen treatment in ovariectomised rats resulted in an increased expression of the AT2R compared to ovariectomised rats [37]. Similarly, mRNA expression of the AT1R and ACE are increased in the brains of hypertensive ovariectomised rats compared to intact females [38].
Hypertension is the single most important modifiable risk factor for the development of stroke, clinical outcome is poorer in patients with hypertension during acute stroke and in experimental animal models of stroke, ischaemic damage is significantly increased in hypertensive animals [39, 40]. Dysregulation of the RAS has been implicated in the development of hypertension, where hyperactivity of Ang II and other RAS components lead to enhanced oxidative stress and inflammation. Preclinical models of genetic hypertension have demonstrated increased AT1R expression in the vasculature of spontaneously hypertensive rats (SHR) compared to age-matched normotensive controls [41]. In addition to hyperactivity of the ‘classical axis’, dampening of the protective counter-regulatory axis is also evident, where hypertensive rat strains exhibit decreased ACE2 mRNA and protein expression compared to normotensive controls [42]. Chronic treatment of diabetic SHR rats with either an AT1R blocker (olmesartan) or ACE inhibitor (enalapril) reverses the microcirculatory changes that occur in pial vessels (functional and structural rarefaction) of the brain resulting in an improved cerebral perfusion and reduced cerebral oxidative stress [43]. Candesartan treatment in salt loaded SHRSP rats was shown to increase endothelial cell progenitor (EPC) colony number and reduce oxidative stress levels in mononuclear cells. This study suggests that ARB treatment may also act to improve endothelial cell function and angiogenesis in the presence of hypertension [44]. These results demonstrate the influence of the ACE/Ang II/AT1R pathway on remodelling of the cerebral microvasculature and suggest that overactivation of this pathway may contribute to the pathology.
Therefore, manipulation of the RAS towards the protective ACE2/Ang-(1–7)/MasR pathway in the presence of co-morbidities may shift the balance to prevent the exacerbation of ischaemic damage following AIS.
Therapeutic targeting of the RAS following stroke
AT1R blockers
Angiotensin type 1 receptor blockers (ARBs or “sartans”) have been widely used as a successful and established therapy for the treatment of clinical hypertension [45]. As a result, this class of drug has been assessed for possible neuroprotective effects in a number of experimental stroke studies (Table 1) [46–56]. Central administration of irbesartan (ARB) prior to transient middle cerebral artery occlusion (tMCAO) has been shown to improve neurological outcome with no effect on blood pressure, however one limitation was the lack of any measure of infarct volume [43]. Follow up studies demonstrated that administration of ARB’s prior to MCAO could in fact reduce infarct volume and this was associated with a reduction in the number of activated microglia and macrophages (ED-1 staining) as well as a reduction in markers of apoptosis (TUNEL, PARP P85 staining and caspase 3) [47, 56].
The cerebral vasodilatory potential of ARB’s have been extensively investigated and studies have examined its effects on isolated cerebral vessels as well as the cerebral blood flow (CBF) response in vivo. Candesartan in particular, has shown the potential to increase cerebral perfusion following MCAO in both normotensive as well as hypertensive rats (SHR). In normotensive rats, administration of Candesartan as an i.v bolus (2 h prior to MCAO) was reported to increase CBF in the ipsilateral hemisphere both at baseline and during MCAO [57]. In SHRs, chronic candesartan infusion for 28 days prior to MCAO reduced infarct volume and this was associated with an improved CBF compared to vehicle treated rats, particularly in the cortical areas at the periphery of the infarct. In addition, isolated vessels taken at the end of the chronic treatment protocol demonstrated an increased MCA diameter and reduced media thickness suggesting chronic changes to cerebral vessels resulting in reduced hypertension induced remodelling and enhanced collateral flow [55]. Similarly, Nishimura and colleagues demonstrated that chronic pre-treatment with Candesartan improved cerebrovascular autoregulation and decreased infarct size, an outcome associated with reduced AT1R binding in the MCA with Ang II autoradiography [54].
Other reported effects of AT1R blockade include pro-angiogenic/neurogenic effects. For example, Candesartan pre-treatment (at a dose with no BP effect) reduced infarct volume and increased mRNA expression of brain derived neurotrophic factor (BDNF) and its associated receptor (tropomyosin receptor kinase B; TrkB) 48 h after tMCAO [46]. Similarly, in SHR rats candesartan treatment following MCAO was shown to increase BDNF protein levels [58], suggesting, a potential involvement in neuronal cell regeneration. Despite indications of neuroprotection with blockade of the AT1R, not all experimental studies attenuated infarct evolution independently of BP lowering effects (Table 1).
AT2R agonism
It has been proposed that the neuroprotective mechanisms induced by ARB’s may partly involve increased Ang II binding to the AT2 receptor [59]. Consequently, selective AT2R agonists have been developed and investigated in models of experimental stroke (Table 2) [60–67]. In normotensive rats, central and systemic administration of compound 21 (C21), a selective non-peptide and orally active AT2R agonist, prior to and post ET-1 induced MCAO, reduces infarct size and improves neurological deficit. This protective effect was attributed to a decrease in inflammatory markers, inducible nitric oxide synthase (iNOS) and C–C motif chemokine receptor type 2 (CCR2) mRNA expression in the cerebral cortex following tMCAO, an effect blocked by the AT2R selective antagonist PD123319 [62].
In conscious SHR rats, the AT2R agonist, CGP42112, attenuates lesion progression and improves motor function following tMCAO. These effects were independent of BP alterations and possibly due to enhanced AT2R receptor level expression, increased microglial activation and reduced superoxide production within the peri-infarct region [63, 64]. These findings are supported by in vitro data in primary cortical neurons where CGP42112 administration was shown to attenuate cell death following oxygen glucose depravation (OGD) [66]. Similarly, C21, was shown to dose dependently reduce infarct volume in SHR when administered centrally for 5 days prior to endothelin 1 (ET-1) induced MCAO. This was associated with an increase in microglia activation within the infarct core and peri-infarct, however, when administered 6 h post-stroke the protective effect was still observed but there was no enhancement of microglial activation [65].
Apart from an anti-inflammatory role, C21 has also been shown to promote angiogenesis. In primary cortical neurones, C21 treatment 24 h post OGD challenge, enhances vascular endothelial growth factor (VEGF) via mechanistic targeting of rapamycin (mTOR) pathway activation [68]. Similarly, in in vivo models, 28 day C21 treatment in mice subjected to either transient or permanent MCAO resulted in increased angiogenesis via VEGF upregulation and effect which the authors hypothesise is through an AT2R mediated activation of the P13K-Akt-mTOR pathway [61]. Additionally, in ex vivo studies, vasodilation and increased perfusion seems to be dependent on the animal model used. Using wire myography, C21 treatment in isolated basilar arteries causes cerebral vessel relaxation, promoting vasodilation [59], an outcome supported by in vivo studies where mice subjected to pMCAO with C21 pre-treatment had an improved CBF in the ischaemic hemisphere at days 1 and 3 following pMCAO. This was associated with a decreased infarct size, and attenuated blood brain barrier (BBB) breakdown as measured by Evans blue extravasation [67]. On the contrary, in mice subjected to tMCAO, C21 did not induce any acute changes in CBF when administered following reperfusion [68].
In contrast to the protective effects of C21 discussed above, a recent study showed that C21 did not affect infarct volume in mice 4 days following tMCAO; however, it did improve neurological score and mortality rates. Interestingly, the neurological improvement observed was associated with an increase in anti-apoptotic and regenerative molecules BDNF, TrkB and growth associated protein 43 (GAP-43) in peri-infarct regions when compared to vehicle [68].
MasR agonism
Evidence suggests that AT2R interacts with other RAS mediators and its effects may be partly mediated by an interaction with the Mas receptor, which is activated by Ang-(1–7) [69]. Recently, the interaction between ACE2/Ang-(1–7)/MasR in AIS has been studied and shown to be protective in several animal models (Table 3) [17, 26, 70–73]. Mecca and colleagues were one of the first groups to identify the potential neuroprotective effects of this peptide and its receptor. They demonstrated that central administration of Ang-(1–7) for 7 days prior to ET-1 induced MCAO, reduced infarct volume via MasR activation and decreased cortical iNOS mRNA expression [26]. Further evidence now proposes that Ang-(1–7) might have direct anti-inflammatory properties and specifically target microglia. In primary microglial cell cultures under basal conditions, MasR activation modulates inflammatory marker expression by attenuating pro-inflammatory genes [74]. Following tMCAO injury, Ang-(1–7) treatment not only reduces iNOS mRNA and protein levels in ipsilateral cerebral cortex but attenuates chemokine C-X-C motif ligand 12 (CXCL12) levels at 6 h post tMCAO and interleukin (IL)-1β, IL-6 and cluster differentiation 11b (CD11b) at 24 h [17]. These findings are further supported by pMCAO studies, where Ang-(1–7) decreased nuclear factor kappa B (NFκB) phosphorylation and cyclooxygenase-2 (COX-2) protein levels in peri-infarct regions when compared to vehicle [71].
The vasodilatory properties of Ang-(1–7) in cerebral vessels are conflicting. In canine MCA and piglet pial arterioles, Ang-(1–7) induces vasodilation in intact vessels only at very high concentrations [75, 76]. However, Durand and colleagues demonstrated that in normotensive isolated rat MCA's, Ang-(1–7) dose dependently induced a vasodilator response which was blocked by Mas and AT2 receptor antagonists [77]. Ang-(1–7) mediated vasodilatation may vary depending on animal species. Nevertheless, in rodents, it is possible that Ang-(1–7) might induce vasodilation in cerebral vessels due to increases in NO and/or bradykinin (BK) release. Central administration of Ang-(1–7) in a rat transient MCAO model has been shown to enhance nitric oxide (NO) release between 3 and 72 h after transient MCAO when compared to vehicle treated rats and this was associated with an increased mRNA and protein expression of endothelial nitric oxide synthase (eNOS) in ischaemic brain tissue at these acute time points following MCAO. In addition, concentration levels of BK and its receptors were shown to be upregulated in ischaemic cortex following Ang-(1–7) treatment between 6 and 48 h post tMCAO [70, 78].
Similarly, 4-week chronic Ang-(1–7) infusion prior to MCAO was shown to increase protein levels of eNOS and NO concentration in the ischaemic hemisphere whereas iNOS and neuronal (nNOS) expression were unchanged. Interestingly, brain VEGF protein levels were also elevated, an outcome associated with an increase in angiogenic markers (CD31 and EdU). Following pMCAO, Ang-(1–7) reduced infarct volume and improved CBF an effect specific to the MasR, suggesting that Ang-(1–7) activates Mas/eNOS signalling pathways, improving angiogenesis and cerebral perfusion [72]. Still, the impact of Ang-(1–7) on CBF is under debate as Mecca et al. [26] reported that central infusion of Ang-(1–7) for 7 days prior to ET-1 induced MCAO did not affect CBF when measured during MCAO.
Targeting the MasR with Ang-(1–7) is showing promising results in experimental stroke models however, Ang-(1–7) has a 20 s half life in the bloodstream and is unlikely to cross the BBB therefore necessitating specific receptor agonists to be developed with improved pharmacokinetic profiles [79]. As a result, in order to see an effect following AIS, it has had to be administered centrally, a route of administration which is not clinically feasible. At present, Mas agonist, AVE0991 has been developed, however, following tMCAO in mice, i.p AVE0991 post-treatment failed to induce similar neuroprotective as observed in Ang-(1–7) treated studies [73].
The effect of ARBs in clinical trials
In clinical trials, the effectiveness of ARB treatment in preventing vascular events and mortality following AIS is under debate. The LIFE trial compared losartan (AT1R antagonist) and atenolol (selective β1 receptor blocker) treatment for preventing cerebral ischaemic events in patients with a clinical history of hypertension and left ventricular hypertrophy. After a follow up time of 4.8 years, the authors demonstrated that the use of losartan was associated with a decrease in the frequency of stroke [80]. Moreover the ACCESS trial was designed to assess the efficacy of a modest blood pressure reduction acutely after stroke (day 1 post stroke for 7 days) with patients receiving either candesartan cilexetil (AT1R antagonist) or placebo treatment. Although the trial was stopped early it did demonstrate a reduced number of vascular events and decreased mortality in the candesartan treatment group [81]. In following years, the MOSES study investigated the effects of eprosartan or nitrendipine (calcium channel blocker) in hypertensive patients who had a cerebrovascular event within the last 24 months prior to recruitment. After 2.5 years follow up, it was identified that eprosartan group showed significantly less cases of cardiovascular and cerebrovascular events [82]. Furthermore, telmisartan, as an alternative therapy for cardiovascular disease patients who are intolerant to ACE inhibitors, was shown to induce a modest reduction in stroke incidence [83].
Despite the benefits of AT1R antagonism observed, other studies showed contradictory effects. For instance, a study assessing whether blood pressure lowering with telmisartan treatment in ischaemic stroke patients affected the risk of recurrent cerebral events concluded after a 2.5 year follow up that telmisartan did not affect the risk of recurrent stroke nor other cardiovascular events [84]. More recently, the SCAST clinical trial investigated whether blood pressure lowering with candesartan in acute stroke patients is beneficial. After 6 months follow up, no beneficial effect of blood pressure lowering with candesartan was observed with no difference in the occurrence of vascular events. Plus, for functional outcome there was a less favourable modified Rankin score in the candesartan group albeit this was not statistically significant [85]. The results from clinical trials to date have primarily investigated the influence of modulation of the RAS in terms of stroke incidence or blood pressure lowering strategies following stroke however, the effect of RAS modulation acutely after ischaemic stroke in terms of outcome (i.e. penumbral salvage, lesion volume) has yet to be fully investigated.
Conclusions
The RAS is currently a therapeutic target for the treatment of AIS. Pathological activation of the ‘classical axis’ contributes towards ischaemic injury development and the ‘alternative axis’ is thought to counteract these effects leading to protection. Interestingly, the pathology of stroke co-morbidities such as hypertension and gender are all influenced by RAS dysfunction, where there is an increased/overactive ACE/Ang II/AT1R pathway. Consequently, targeting the RAS could act as a preventive line of therapy for the development of stroke and attenuate injury following stroke by diminishing brain RAS imbalances or providing direct neuroprotection.
Several preclinical studies have identified the potential neuroprotective effect of AT1R blockers, AT2R and MasR agonists. ARBs have been extensively studied in the context of experimental stroke due to its current clinical use in hypertension management. Although there were indications of a level of neuroprotection induced by ARBs, its clinical potential has been set aside due to clinical trials that showed that ARBs may induce harmful effects in ischaemic stroke patients. On the other hand, AT2R and MasR agonism are showing promising effects in preclinical stroke models; however, further studies have to be conducted to identify the actual mechanisms induced following stroke, addressing several limitations associated to preclinical stroke.
Further understanding of the role of the RAS following ischaemic stroke, in particular the role of the ACE2/Ang-(1–7)/Mas pathway and development of improved pharmacological drugs targeting the central RAS components are needed before any successful translation would be possible.
Abbreviations
- ACE:
-
angiotensin converting enzyme
- ACE2:
-
angiotensin converting enzyme 2
- AIS:
-
acute ischaemic stroke
- ARB:
-
angiotensin type 1 receptor blockers
- AT1R:
-
angiotensin type 1 receptor
- AT2R:
-
angiotensin type 2 receptor
- BBB:
-
blood brain barrier
- BP:
-
blood pressure
- CBF:
-
cerebral blood flow
- C21:
-
compound 21, AT2 receptor agonist
- EPC:
-
endothelial progenitor cell
- KO:
-
knockout
- MasR:
-
Mas receptor
- pMCAO:
-
permanent middle cerebral artery occlusion
- tMCAO:
-
transient middle cerebral artery occlusion
- NEP:
-
neprilysin
- NO:
-
nitric oxide
- OGD:
-
oxygen glucose deprivation
- RAS:
-
renin angiotensin system
- SHR:
-
spontaneously hypertensive rat
- WT:
-
wild type
- VEGF:
-
vascular endothelial growth factor
References
State of the Nation: stroke statistics 2015. https://www.stroke.org.uk/sites/default/files/stroke_statistics_2015.pdf.
Adamson J, Beswick A, Ebrahim S. Is stroke the most common cause of disability? J Stroke Cerebrovasc Dis. 2004;13:171–7.
Saka Ö, Mcguire A, Wolfe C. Cost of stroke in the United Kingdom. Age Ageing. 2009;38:27–32.
Broussalis E, Killer M, McCoy M, Harrer A, Trinka E, Kraus J. Foundation review: current therapies in ischemic stroke. Part A. Recent developments in acute stroke treatment and in stroke prevention. Drug Discov Today. 2012;17:296–309.
Goyal M, Demchuk AM, Menon BK, Eesa M, Rempel JL, Thornton J, Roy D, Jovin TG, Willinsky RA, Sapkota BL, Dowlatshahi D, Frei DF, Kamal NR, Montanera WJ, Poppe AY, Ryckborst KJ, Silver FL, Shuaib A, Tampieri D, Williams D, Bang OY, Baxter BW, Burns PA, Choe H, Heo JH, Holmstedt CA, Jankowitz B, Kelly M, Linares G, Mandzia JL, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. 2015;372:1019–30.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres J-P, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. American heart association statistics committee and stroke statistics subcommitte: heart disease and stroke statistics—2015 update: a report from the American heart association. Circulation. 2015;131:e29–322.
Ishitsuka K, Kamouchi M, Hata J, Fukuda K, Matsuo R, Kuroda J, Ago T, Kuwashiro T, Sugimori H, Nakane H, Kitazono T. High blood pressure after acute ischemic stroke is associated with poor clinical outcomes: Fukuoka stroke registry. Hypertension. 2014;63:54–60.
Wright JW, Harding JW. The brain renin–angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch. 2013;465:133–51.
Volpe M. The renin–angiotensin system as a risk factor and therapeutic target for cardiovascular and renal disease. J Am Soc Nephrol. 2002;13(Suppl. 3):S173–8.
Schalekamp MA, Danser AH. How does the angiotensin II type 1 receptor “trump” the type 2 receptor in blood pressure control? J Hypertens. 2013;31:705–12.
McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FAO, Chai SY. The brain renin–angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35:901–18.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin–angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292:R373–81.
Gallagher PE, Chappell MC, Errario CM, Tallant EA, Patricia E. Distinct roles for ANG II and ANG-(1–7) in the regulation of angiotensin-converting enzyme 2 in rat astrocytes. Am J Physiol Cell Physiol. 2006;1032:420–6.
Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Garrido-Gil P, Valenzuela R, Villar-Cheda B, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: an intracellular renin–angiotensin system in the nigra. Brain Struct Funct. 2013;218:373–88.
Lu J, Jiang T, Wu L, Gao L, Wang Y, Zhou F, Zhang S, Zhang Y. The expression of angiotensin-converting enzyme 2-angiotensin-(1–7)-Mas receptor axis are upregulated after acute cerebral ischemic stroke in rats. Neuropeptides. 2013;47:289–95.
Regenhardt RW, Desland F, Mecca AP, Pioquinto DJ, Afzal A, Mocco J, Sumners C. Anti-inflammatory effects of angiotensin-(1–7) in ischemic stroke. Neuropharmacology. 2013;71:154–63.
Walther T, Olah L, Harms C, Maul B, Bader M, Hörtnagl H, Schultheiss H-P, Mies G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2002;16:169–76.
Inaba S, Iwai M, Tomono Y, Senba I, Furuno M, Kanno H, Okayama H, Mogi M, Higaki J, Horiuchi M. Exaggeration of focal cerebral ischemia in transgenic mice carrying human renin and human angiotensinogen genes. Stroke. 2009;40:597–603.
Stenman E, Edvinsson L. Cerebral ischemia enhances vascular angiotensin AT1 receptor-mediated contraction in rats. Stroke. 2004;35:970–4.
Regenhardt RW, Bennion DM, Sumners C. Cerebroprotective action of angiotensin peptides in stroke. Clin Sci. 2014;126:195–205.
Gaspari TA, Vinh A, Jones ES, Widdop RE. Ganging up on angiotensin II type 1 receptors in vascular remodeling. Hypertension. 2012;60:17–9.
Zhu YZ, Chimon GN, Zhu YC, Lu Q, Li B, Hu HZ, Yap EH, Lee HS, Wong PT. Expression of angiotensin II AT2 receptor in the acute phase of stroke in rats. Neuroreport. 2000;11:1191–4.
Makino I, Shibata K, Ohgami Y, Fujiwara M, Furukawa T. Transient upregulation of the AT2 receptor mRNA level after global ischemia in the rat brain. Neuropeptides. 1996;30:596–601.
Iwai M, Liu H-W, Chen R, Ide A, Okamoto S, Hata R, Sakanaka M, Shiuchi T, Horiuchi M. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation. 2004;110:843–8.
Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C. Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96:1084–96.
Appelros P, Stegmayr B, Terént A. Sex differences in stroke epidemiology: a systematic review. Stroke. 2009;40:1082–90.
Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–52.
Baskerville TA, Macrae IM, Holmes WM, McCabe C. The influence of gender on “tissue at risk” in acute stroke: a diffusion-weighted magnetic resonance imaging study in a rat model of focal cerebral ischaemia. J Cereb Blood Flow Metab. 2016;36:381–6.
Herson PS, Palmateer J, Hurn PD. Biological sex and mechanisms of ischemic brain injury. Transl Stroke Res. 2013;4:413–9.
Sullivan JC. Sex and the renin–angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1220–6.
Silva-Antonialli MM, Tostes RCA, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MHC, Fortes ZB, Nigro D. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res. 2004;62:587–93.
Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension. 2008;52:666–71.
Sampson AK, Hilliard LM, Moritz KM, Thomas MC, Tikellis C, Widdop RE, Denton KM. The arterial depressor response to chronic low-dose angiotensin II infusion in female rats is estrogen dependent. AJP Regul Integr Comp Physiol. 2012;302:R159–65.
Zimmerman MA, Baban B, Tipton AJ, O’Connor PM, Sullivan JC. Chronic ANG II infusion induces sex-specific increases in renal T cells in Sprague–Dawley rats. Am J Physiol Ren Physiol. 2015;308:F706–12.
Sullivan JC, Rodriguez-Miguelez P, Zimmerman MA, Harris RA. Differences in angiotensin (1–7) between men and women. Am J Physiol Heart Circ Physiol. 2015;308:H1171–6.
Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Lanciego JL, Labandeira-Garcia JL. Estrogen and angiotensin interaction in the substantia nigra. Relevance to postmenopausal Parkinson’s disease. Exp Neurol. 2010;224:517–26.
Dai S-Y, Zhang Y-P, Peng W, Shen Y, He J-J. Central infusion of angiotensin II type 2 receptor agonist compound 21 attenuates DOCA/NaCl-induced hypertension in female rats. Oxid Med Cell Longev. 2016;2016:3981790. doi:10.1155/2016/3981790.
McCabe C, Gallagher L, Gsell W, Graham D, Dominiczak AF, MacRae IM. Differences in the evolution of the ischemic penumbra in stroke-prone spontaneously hypertensive and Wistar–Kyoto rats. Stroke. 2009;40:3864–8.
Willmot M, Leonardi-Bee J, Bath PMW. High blood pressure in acute stroke and subsequent outcome: a systematic review. Hypertension. 2004;43:18–24.
Pei F, Wang X, Yue R, Chen C, Huang J, Huang J, Li X, Zeng C. Differential expression and DNA methylation of angiotensin type 1A receptors in vascular tissues during genetic hypertension development. Mol Cell Biochem. 2015;402:1–8.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8.
Estato V, Obadia N, Carvalho-Tavares J, Freitas FS, Reis P, Neto HCF, Lessa MA, Tibiriçá E. Blockade of the renin–angiotensin system improves cerebral microcirculatory perfusion in diabetic hypertensive rats. Microvasc Res. 2013;87:41–9.
Yu Y, Fukuda N, Yao E-H, Matsumoto T, Kobayashi N, Suzuki R, Tahira Y, Ueno T, Matsumoto K. Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens. 2008;21:72–7.
Catanzaro DF, Frishman WH. Angiotensin receptor blockers for management of hypertension. South Med J. 2010;103:669–73.
Dai W-J, Funk A, Herdegen T, Unger T, Culman J, Armstead WM. Blockade of central angiotensin AT1 receptors improves neurological outcome and reduces expression of AP-1 transcription factors after focal brain ischemia in rats editorial comment. Stroke. 1999;30:2391–9.
Lou M, Blume A, Zhao Y, Gohlke P, Deuschl G, Herdegen T, Culman J. Sustained blockade of brain AT1 receptors before and after focal cerebral ischemia alleviates neurologic deficits and reduces neuronal injury, apoptosis, and inflammatory responses in the rat. J Cereb Blood Flow Metab. 2004;24:536–47.
Fagan SC, Kozak A, Hill WD, Pollock DM, Xu L, Johnson MH, Ergul A, Hess DC. Hypertension after experimental cerebral ischemia: candesartan provides neurovascular protection. J Hypertens. 2006;24:535–9.
Liu H, Kitazato KT, Uno M, Yagi K, Kanematsu Y, Tamura T, Tada Y, Kinouchi T, Nagahiro S. Protective mechanisms of the angiotensin II type 1 receptor blocker candesartan against cerebral ischemia: in vivo and in vitro studies. J Hypertens. 2008;26:1435–45.
Krikov M, Thone-Reineke C, Müller S, Villringer A, Unger T. Candesartan but not ramipril pretreatment improves outcome after stroke and stimulates neurotrophin BNDF/TrkB system in rats. J Hypertens. 2008;26:544–52.
Hosomi N, Nishiyama A, Ban CR, Naya T, Takahashi T, Kohno M, Koziol JA. Angiotensin type 1 receptor blockage improves ischemic injury following transient focal cerebral ischemia. Neuroscience. 2005;134:225–31.
Mecca AP, Connor TEO, Katovich MJ, Sumners C. Candesartan pretreatment in cerebroprotective in a rat model of endothelin-1-induced middle cerebral artery occlusion. Exp Physiol. 2009;94:937–46.
Kim CK, Yang X-L, Kim Y-J, Choi I-Y, Jeong H-G, Park H-K, Kim D, Kim TJ, Jang H, Ko S-B, Yoon B-W. Effect of long-term treatment with fimasartan on transient focal ischemia in rat brain. Biomed Res Int. 2015;2015:1–9.
Nishimura Y, Ito T, Saavedra JM. Angiotensin II AT(1) blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke. 2000;31:2478–86.
Ito T, Yamakawa H, Bregonzio C, Terrón JA, Falcón-Neri A, Saavedra JM. Protection against ischemia and improvement of cerebral blood flow in genetically hypertensive rats by chronic pretreatment with an angiotensin II AT1 antagonist. Stroke. 2002;33:2297–303.
Li JM, Mogi M, Iwanami J, Min LJ, Tsukuda K, Sakata A, Fujita T, Iwai M, Horiuchi M. Temporary pretreatment with the angiotensin II type 1 receptor blocker, valsartan, prevents ischemic brain damage through an increase in capillary density. Stroke. 2008;39:2029–36.
Engelhorn T, Goerike S, Doerfler A, Okorn C, Forsting M, Heusch G, Schulz R. The angiotensin II type 1—receptor blocker candesartan increases cerebral blood flow, reduces infarct size, and improves neurologic outcome after transient cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:467–74.
Alhusban A, Kozak A, Ergul A, Fagan SC. AT1 receptor antagonism is proangiogenic in the brain: BDNF a novel mediator. J Pharmacol Exp Ther. 2013;344:348–59.
Faure S, Bureau A, Oudart N, Javellaud J, Fournier A, Achard J-M. Protective effect of candesartan in experimental ischemic stroke in the rat mediated by AT2 and AT4 receptors. J Hypertens. 2008;26:2008–15.
Alhusban A, Fouda AY, Pillai B, Ishrat T, Soliman S, Fagan SC. Compound 21 is pro-angiogenic in the brain and results in sustained recovery after ischemic stroke. J Hypertens. 2015;33:170–80.
Mateos L, Perez-Alvarez MJ, Wandosell F. Angiotensin II type-2 receptor stimulation induces neuronal VEGF synthesis after cerebral ischemia. Biochim Biophys Acta Mol Basis Dis. 2016;1862:1297–308.
Joseph JP, Mecca AP, Regenhardt RW, Bennion DM, Rodríguez V, Desland F, Patel NA, Pioquinto DJ, Unger T, Katovich MJ, Steckelings UM, Sumners C. The angiotensin type 2 receptor agonist compound 21 elicits cerebroprotection in endothelin-1 induced ischemic stroke. Neuropharmacology. 2014;81:134–41.
McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;40:1482–9.
McCarthy CA, Vinh A, Broughton BRS, Sobey CG, Callaway JK, Widdop RE. Angiotensin II type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension. 2012;60:1531–7.
McCarthy CA, Vinh A, Miller AA, Hallberg A, Alterman M, Callaway JK, Widdop RE. Direct angiotensin AT2 receptor stimulation using a novel AT2 receptor agonist, compound 21, evokes neuroprotection in conscious hypertensive rats. PLoS One. 2014;9:e95762.
Lee S, Brait VH, Arumugam TV, Evans MA, Kim HA, Widdop RE, Drummond GR, Sobey CG, Jones ES. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp Transl Stroke Med. 2012;4:16.
Min LJ, Mogi M, Tsukuda K, Jing F, Ohshima K, Nakaoka H, Kan-No H, Wang XL, Chisaka T, Bai HY, Iwanami J, Horiuchi M. Direct stimulation of angiotensin II type 2 receptor initiated after stroke ameliorates ischemic brain damage. Am J Hypertens. 2014;27:1036–44.
Schwengel K, Namsolleck P, Lucht K, Clausen BH, Lambertsen KL, Valero-esquitino V, Thöne-reineke C, Müller S, Widdop RE, Denton KM, Horiuchi M. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J Mol Med. 2016;94:957–66.
Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, Unger T, Bader M, Santos RA, Sumners C, Steckelings UM. Angiotensin type 2 receptor (AT 2 R) and receptor Mas: a complex liaison. Clin Sci. 2015;128:227–34.
Zhang Y, Lu J, Shi J, Lin X, Dong J, Zhang S, Liu Y, Tong Q. Central administration of angiotensin-(1–7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats. Neuropeptides. 2008;42:593–600.
Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y. Suppressing inflammation by inhibiting the NF-IB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br J Pharmacol. 2012;167:1520–32.
Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L, Wang HF, Lu J, Gao Q, Zhang YD, Tan L. Angiotensin-(1–7) induces cerebral ischaemic tolerance by promoting brain angiogenesis in a Mas/eNOS-dependent pathway. Br J Pharmacol. 2014;171:4222–32.
Lee S, Evans MA, Chu HX, Kim HA, Widdop RE, Drummond GR, Sobey CG. Effect of a selective Mas receptor agonist in cerebral ischemia in vitro and in vivo. PLoS One. 2015;10:e0142087.
Liu M, Shi P, Sumners C. Direct anti-inflammatory effects of angiotensin-(1–7) on microglia. J Neurochem. 2016;136:163–71.
Feterik K, Smith L, Katusic ZS. Angiotensin-(1–7) causes endothelium-dependent relaxation in canine middle cerebral artery. Brain Res. 2000;873:75–82.
Meng W, Busija DW. Comparative effects of angiotensin-(1–7) and angiotensin II on piglet pial arterioles. Stroke. 1993;24:2041–4 (discussion 2045).
Durand MJ, Raffai G, Weinberg BD, Lombard JH. Angiotensin-(1–7) and low-dose angiotensin II infusion reverse salt-induced endothelial dysfunction via different mechanisms in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol. 2010;299:H1024–33.
Lu J, Zhang Y, Shi J. Effects of intracerebroventricular infusion of angiotensin-(1–7) on bradykinin formation and the kinin receptor expression after focal cerebral ischemia-reperfusion in rats. Brain Res. 2008;1219:127–35.
Iusuf D, Henning RH, van Gilst WH, Roks AJM. Angiotensin-(1–7): pharmacological properties and pharmacotherapeutic perspectives. Eur J Pharmacol. 2008;585:303–12.
Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003.
Schrader J, Lüders S, Kulschewski A, Berger J, Zidek W, Treib J, Einhäupl K, Diener HC, Dominiak P. The ACCESS study: evaluation of acute candesartan cilexetil therapy in stroke survivors. Stroke. 2003;34:1699–703.
Schrader J, Lüders S, Kulschewski A, Hammersen F, Plate K, Berger J, Zidek W, Dominiak P, Diener HC. Morbidity and mortality after stroke, eprosartan compared with nitrendipine for secondary prevention: principal results of a prospective randomized controlled study (MOSES). Stroke. 2005;36:1218–24.
Telmisartan Randomised AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND) Investigators, Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–83.
Yusuf S, Diener H-C, Sacco RL, Cotton D, Ôunpuu S, Lawton WA, Palesch Y, Martin RH, Albers GW, Bath P, Bornstein N, Chan BPL, Chen S-T, Cunha L, Dahlöf B, De Keyser J, Donnan GA, Estol C, Gorelick P, Gu V, Hermansson K, Hilbrich L, Kaste M, Lu C, Machnig T, Pais P, Roberts R, Skvortsova V, Teal P, Toni D, et al. Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med. 2008;359:1225–37.
Sandset EC, Bath PMW, Boysen G, Jatuzis D, Kõrv J, Lüders S, Murray GD, Richter PS, Roine RO, Terént A, Thijs V, Berge E. The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo-controlled, double-blind trial. Lancet. 2011;377:741–50.
Authors’ contributions
MMCA, ER and CM all contributed to the review of the literature, critical analysis and drafting of the manuscript. MMCA prepared the figure and tables. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and material
Data sharing not applicable to this article as no datasets were generated or analysed during the current study. All the references have been cited in the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Arroja, M.M.C., Reid, E. & McCabe, C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp & Trans Stroke Med 8, 8 (2016). https://doi.org/10.1186/s13231-016-0022-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13231-016-0022-1