
Abstract
Background
Spinocerebellar ataxia type 31 (SCA31) is not usually associated with dementia, and autopsy in a patient with both conditions is very rare.
Case presentation
An 87-year-old male patient presented with ataxia and progressive dementia. Genetic testing led to a diagnosis of SCA31. Fifteen years after his initial symptoms of hearing loss and difficulty walking, he died of aspiration pneumonia. A pathological analysis showed cerebellar degeneration consistent with SCA31 and abundant argyrophilic grains in the hippocampal formation and amygdala that could explain his dementia.
Conclusions
This is the first autopsy report on comorbid argyrophilic grain disease with SCA31.
Similar content being viewed by others

Background
Spinocerebellar ataxia type 31 (SCA31) is an autosomal dominant pure cerebellar ataxia and is not usually associated with cognitive decline. Here we report autopsy findings from an 87-year-old Japanese male SCA31 patient with dementia. Argyrophilic grains were found to be responsible for the cognitive decline. Neuropathological examination showed findings compatible with SCA31 in the cerebellum accompanied by the finding of argyrophilic grains. This is an interesting condition of comorbid argyrophilic grain disease (AGD) with SCA31.
Case presentation
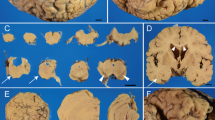
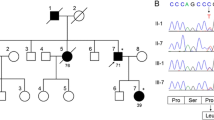
A male patient consulted with our hospital at the age of 77. He had had hypertension since age 67. Hearing loss appeared when he was 72 years old and difficulty in walking when he was 73. Dysarthria and difficulty with hand movements had gradually appeared. At presentation, limb and truncal ataxia and ataxic speech were seen, as was hyporeflexia in the distal extremities. Brain magnetic resonance imaging (MRI) showed atrophy of the upper vermis of the cerebellum (Fig. 1a,b), as well as moderate atrophy of the frontotemporal cortex, including the right-dominant medial temporal lobe (Fig. 1b,c). Two siblings and five nephews and nieces had similar dysarthria and gait disturbances (Fig. 2). Genetic analysis revealed a TGGAA repeat expansion in the SCA31 locus, and he was diagnosed with SCA31. An audiogram showed sensorineural deafness of the downward sloping type. His score on the Mini-Mental State Examination (MMSE) was 26/30. Neuropsychological assessment with the Miyake Memory Test and Rey-Osterrieth Complex Figure Test revealed short-term memory and performance impairment. As his activity and intelligence gradually declined, he needed home-visit medical treatment. His MMSE score was 16/30 at the age of 81 and he became irritated frequently. At the age of 84, he developed a resting tremor in the right hand. He died at home with aspiration pneumonia at the age of 87, after a disease duration of 15 years.

a: Sagittal view of a T1-weighted brain MR image showing mild atrophy of the upper vermis of the cerebellum. b,c: An axial MRI (fluid-attenuated inversion recovery image) showed mild atrophy in the upper vermis of the cerebellum and the right dominant anterior medial temporal lobe (b), and moderate atrophy of the frontotemporal cortex (c). d: Synaptophysin staining shows an eosinophilic halo-like structure (arrow) around a degenerated Purkinje cell in the right cerebellar hemisphere. e: Gallyas-positive, rod-like (arrowheads) or granular argyrophilic grains in the subiculum of the left hippocampus

Family tree of the patient. Arrow shows the proband. Black boxes indicate family members who had cerebellar ataxia
The autopsy confirmed bilateral pneumonia as the direct cause of death. The brain weighed 1060 g. Macroscopically, the cerebellum was severely atrophic and cerebral atrophy was mild. Although the volume of the hippocampus and amygdala were relatively well preserved, white matter atrophy was accentuated in the parahippocampal gyrus. Histologically, marked neuronal loss of Purkinje cells was seen in most of the cerebellar cortex. In the remaining Purkinje cells, degeneration accompanied by cytoplasmic atrophy and nuclear aggregation was noticeable. An eosinophilic halo-like structure was present around the degenerated Purkinje cells and showed positive staining for synaptophysin (Fig. 1d). These findings are consistent with those previously reported for SCA31 [1]. Numerous Gallyas-positive, rod-like or granular argyrophilic grains were found in the ambient gyrus, amygdala, hippocampus CA1, parahippocampal gyrus, and subiculum of the hippocampus (Fig. 1e). Apparent spongiform degeneration and neuron loss with gliosis were observed in the ambient gyrus and amygdala, compatible with stage II AGD [2].
Neurofibrillary tangles were limited (Braak neurofibrillary stage II) and senile plaques were seen in the neocortex, parahippocampal gyrus, and precentral gyrus (Braak neurofibrillary stage B-C).
Very few alpha-synuclein–positive deposits were present in the dorsal nucleus of the vagus nerve, locus coeruleus, and substantia nigra.
Discussion and conclusions
SCA31 is characteristically a pure cerebellar ataxia, but this patient presented clinically with dementia as well as progressive cerebellar ataxia. In previous reports, dementia was noted in 0% [3], 5% [4], 11.9% [5], and 58% [6] of patients with SCA31.
The only reported autopsy of a patient with SCA31 and dementia [7] showed moderate tau and amyloid pathology in the neocortex that might have led to dementia at the terminal stage. However, in our case, neither the amyloid deposits nor the neurofibrillary pathology was sufficient to explain the patient’s dementia. Instead, abundant argyrophilic grains may better explain the cognitive decline in this case. The asymmetric atrophy of the anterior medial temporal lobes seen with MRI is a diagnostic feature of AGD and is consistent with the diagnosis in this case. SCA31 usually has an older age at onset than other autosomal dominant hereditary ataxias, which provides more chance to develop age-related cognitive decline such as Alzheimer disease. However, our case shows that it is necessary to consider argyrophilic grains as a pathological substrate for cognitive decline in patients with SCA31 even though the coexistence of SCA31 and AGD is extremely rare.
This is the first autopsy report on comorbid AGD with SCA31, providing a clue for neuroradiological diagnosis of grain disease.
Availability of data and materials
Not applicable.
Abbreviations
- SCA31:
-
Spinocerebellar ataxia type 31
- MMSE:
-
Mini-Mental State Examination
- AGD:
-
Argyrophilic grain disease
- MRI:
-
Magnetic resonance imaging
References
Owada K, Ishikawa K, Toru S, et al. A clinical, genetic, and neuropathologic study in a family with 16q-linked ADCA type III. Neurology. 2005;65:629–32.
Saito Y, Ruberu NN, Sawabe M, et al. Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol. 2004;63:911–8.
Basri R, Yabe I, Soma H, et al. Spectrum and prevalence of autosomal dominant spinocerebellar ataxia in Hokkaido, the northern island of Japan: a study of 113 Japanese families. J Hum Genet. 2007;52:848–55.
Nozaki H, Ikeuchi T, Kawakami A, et al. Clinical and genetic characterizations of 16q-linked autosomal dominant spinocerebellar ataxia (AD-SCA) and frequency analysis of AD-SCA in the Japanese population. Mov Disord. 2007;22:857–62.
Nakamura K, Yoshida K, Matsushima A, et al. Natural history of spinocerebellar ataxia type 31: a 4-year prospective study. Cerebellum. 2017;16:518–24.
Hayashi M, Adachi Y, Mori M, et al. Clinical and genetic epidemiological study of 16q22.1-linked autosomal dominant cerebellar ataxia in western Japan. Acta Neurol Scand. 2007;116:123–7.
Adachi T, Kitayama M, Nakano T, et al. Autopsy case of spinocerebellar ataxia type 31 with severe dementia at the terminal stage. Neuropathology. 2015;35:273–9.
Acknowledgements
None.
Funding
This work was supported for the fee of writing the manuscript and English proofreading by a grant from the Japanese Ministry of Education, Sports and Culture (K.I.), the Japan Society for Promotion of Science (JSPS) (K.I.), from the Health and Labour Sciences Research Grants on Ataxic Diseases (K.I.) of the Japanese Ministry of Health, Labour and Welfare, Japan and from Japan Agency for Medical Research and Development (AMED),
Author information
Authors and Affiliations
Contributions
ST was an attending doctor and a major contributor in writing the manuscript. SI, TU, KH, and MK performed the pathological examination. KI performed the genetic analysis. SI, TU, KH, MK and KI interpreted the data. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was provided by Nitobe Memorial Nakano General Hospital ethics committee and written and signed consent to participate was obtained from the patient’s daughter.
Consent for publication
Written consent for publication including other family information was obtained from the patient’s daughter on behalf of the family.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Toru, S., Ishida, S., Uchihara, T. et al. Comorbid argyrophilic grain disease in an 87-year-old male with spinocerebellar ataxia type 31 with dementia: a case report. BMC Neurol 20, 136 (2020). https://doi.org/10.1186/s12883-020-01723-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-020-01723-2