
Abstract
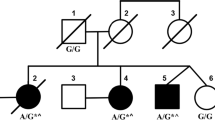
Spinocerebellar ataxia type 21 (SCA21) is a rare subtype of autosomal dominant cerebellar ataxias, which was first identified in a French family and has been reported almost exclusively in French ancestry so far. We here report the first Japanese family with SCA21, in which all affected members examined carried a heterozygous c.509C > T:p.Pro170Leu variant in TMEM240. Their clinical features were summarized as a slowly progressive ataxia of young-adult onset (5–48 years) associated with various degree of psychomotor retardation or cognitive impairment. The MR images revealed atrophy in the cerebellum, but not in the cerebrum or brainstem. These clinical findings were consistent with those in the original French families with SCA21. Neuropathological findings in one autopsied patient showed a prominent decrease of cerebellar Purkinje cells, but no specific abnormalities outside the cerebellum.



Similar content being viewed by others

References
Delplanque J, Devos D, Huin V, Genet A, Sand O, Moreau C, et al. TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain. 2014;137:2657–63. https://doi.org/10.1093/brain/awu202.
Zeng S, Zeng J, He M, Zeng X, Zhou Y, Liu Z, et al. Spinocerebellar ataxia type 21 exists in the Chinese Han population. Sci Rep. 2016;6:19897. https://doi.org/10.1038/srep19897.
Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. 2017;62:525–9. https://doi.org/10.1038/jhg.2016.163.
Devos D, Schraen-Maschke S, Vuillaume I, Dujardin K, Nazé P, Willoteaux C, et al. Clinical features and genetic analysis of a new form of spinocerebellar ataxia. Neurology. 2001;56:234–8.
Delplanque J, Devos D, Vuillaume I, De Becdelievre A, Vangelder E, Maurage CA, et al. Slowly progressive spinocerebellar ataxia with extrapyramidal signs and mild cognitive impairment (SCA21). Cerebellum. 2008;7(2):179–83. https://doi.org/10.1007/s12311-008-0014-3.
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–9.
Satake T, Yamashita K, Hayashi K, Miyatake S, Tamura-Nakano M, Doi H, et al. MTCL1 plays an essential role in maintaining Purkinje neuron axon initial segment. EMBO J. 2017;36:1227–42. https://doi.org/10.15252/embj.201695630.
Acknowledgements
The authors thank Ms. Misuzu Netsu for her technical assistance on neuropathological examinations.
Funding
This work was supported from Research Committee of the Ataxia, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health and Labour Sciences Research Grants from the Ministry of Health, Labour, and Welfare (MHLW), Japan (Yoshida K), the grants for Research on Measures for Intractable Diseases grant (Matsumoto N); the Comprehensive Research on Disability Health and Welfare grant (Matsumoto N); the Strategic Research Program for Brain Science (Matsumoto N); the Practical Research Project for Rare/Intractable Diseases (Matsumoto N), from the Japan Agency for Medical Research and Development (AMED); a grant-in-aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT); grants-in-aid for Scientific Research (A, and C) (Matsumoto N, Doi H, Miyatake S); the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency (JST) (Matsumoto N); and grants from the Ministry of Health, Labor and Welfare (MHLW), Japan (Matsumoto N), grant provided by The Ichiro Kanehara Foundation (Miyatake S), and the Takeda Science Foundation (Matsumoto N).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Written informed consent was obtained from all the individuals and molecular analyses were approved by the Committee for Ethical Issues at both Shinshu University School of Medicine and Yokohama City University School of Medicine.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Yahikozawa, H., Miyatake, S., Sakai, T. et al. A Japanese Family of Spinocerebellar Ataxia Type 21: Clinical and Neuropathological Studies. Cerebellum 17, 525–530 (2018). https://doi.org/10.1007/s12311-018-0941-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-018-0941-6