
Abstract
Spinocerebellar ataxia type 34 (SCA34) is an autosomal dominant inherited ataxia due to mutations in ELOVL4, which encodes one of the very long-chain fatty acid elongases. SCA38, another spinocerebellar ataxia, is caused by mutations in ELOVL5, a gene encoding another elongase. However, there have been no previous studies describing the neuropathology of either SCA34 or 38. This report describes the neuropathological findings of an 83-year-old man with SCA34 carrying a pathological ELOVL4 mutation (NM_022726, c.736T>G, p.W246G). Macroscopic findings include atrophies in the pontine base, cerebellum, and cerebral cortices. Microscopically, marked neuronal and pontocerebellar fiber loss was observed in the pontine base. In addition, in the pontine base, accumulation of CD68-positive macrophages laden with periodic acid-Schiff (PAS)-positive material was observed. Many vacuolar lesions were found in the white matter of the cerebral hemispheres and, to a lesser extent, in the brainstem and spinal cord white matter. Immunohistological examination and ultrastructural observations with an electron microscope suggest that these vacuolar lesions are remnants of degenerated oligodendrocytes. Electron microscopy also revealed myelin sheath destruction. Unexpectedly, aggregation of the four-repeat tau was observed in a spatial pattern reminiscent of progressive supranuclear palsy. The tau lesions included glial fibrillary tangles resembling tuft-shaped astrocytes and neurofibrillary tangles and pretangles. This is the first report to illustrate that a heterozygous missense mutation in ELOVL4 leads to neuronal loss accompanied by macrophages laden with PAS-positive material in the pontine base and oligodendroglial degeneration leading to widespread vacuoles in the white matter in SCA34.
Similar content being viewed by others


Introduction
Spinocerebellar ataxias (SCAs) are a group of autosomal dominant disorders that typically show progressive ataxic symptoms, including truncal and limb ataxias and dysarthria. Patients with SCAs exhibit a variety of neurological symptoms and signs, including oculomotor disturbances, pyramidal and extrapyramidal tract signs, and autonomic failures. Currently, SCA1–48 have been described and over 30 causative genes have been identified [18]. Recent reports show that mutations in the ELOVL fatty acid elongases 4 and 5 genes (ELOVL4 and ELOVL5) cause SCA34 (OMIM #133,190) and SCA38 (#615,957), respectively [12, 15, 19, 35]. The prevalence of SCA34 is rare [36] but SCA34 patients from the USA, Brazil, Canada, and Japan, have been identified [8, 9, 12, 19, 35, 36, 47]. To date, five heterozygous mutations (c.504G>C, p.L168F; c.736T>G, p.W246G; c.512T>C, p.I171T; c.539A>C, p.Q180P; c.698C>T, p.T233M in ELOVL4) causing SCA34 have been reported. Prototypal manifestations of SCA34 include slowly progressive gait ataxia with erythrokeratodermia skin lesions, which are observed in patients with the mutations c.504G>C, p.L168F; c.539A>C, p.Q180P; c.698C>T, p.T233M. Patients with SCA34 show a broad spectrum of signs and symptoms, including pyramidal and extrapyramidal tract signs, autonomic dysfunction, peripheral neuropathy, and possible cognitive decline and psychomotor symptoms [4, 19, 35, 36]. A recent report on SCA34 pedigree with c.512T>C, p.I171T mutation indicated retinal degeneration [47]. As described in our previous study of Japanese pedigrees with c.736T>G, p.W246G mutation, patients with the original mutation c.504G>C, p.L168F in Canada also displayed pontine hot cross bun sign hyperintensity on brain magnetic resonance imaging (MRI) [4].
ELOVL4 mutations are known to cause allelic diseases. For example, mutations leading to carboxy-terminal truncations of ELOVL4 cause autosomal dominant Stargardt macular dystrophy type 3 [49]. Ichthyosis, spastic quadriplegia, and mental retardation (ISQMR) is a recessive disease caused by nonsense and frameshift mutations in ELOVL4 [3].
ELOVL4 encodes the 314 amino acid multipass transmembrane protein, ELOVL4. ELOVL4 is expressed in the retina, skin, brain, testis, and other organs [1] and is mainly localized in the endoplasmic reticulum. ELOVL4 is a key enzymatic component in the elongation of saturated and unsaturated very long-chain (C26 to C28 or longer) fatty acids. Of the seven elongases (ELOVL1 to ELOVL7) in humans, ELOVL4 plays an important role in the synthesis of fatty acids with the longest carbon chains, which can be over C30. The elongated fatty acids are utilized for the synthesis of important lipids, such as sphingomyelin, ceramide, phosphatidylcholine, and cholesterol esters [1, 25]. Repertoires of fatty acids synthesized by the ELOVL4 enzyme and the complex lipids which contain the synthesized fatty acids, differ in a tissue-specific manner. And, different mutations in ELOVL4 may preferentially affect the synthesis of various lipids. For example, an experimental study using knock-in rats with p.W246G mutation indicated preferential disturbance in synthesizing very long-chain saturated fatty acids [2]. In the lipid-rich human brain, lipid substances, including phospholipids, glycolipids, and cholesterol, account for 70% of the dry weight of the myelin sheath surrounding axons [26]. Thus, myelin may be affected by ELOVL4 mutations in SCA34. In animal models, very long-chain saturated fatty acids synthesized by ELOVL4 are enriched in neuronal synaptic vesicles and are involved in the modulation of presynaptic release [14, 21]. However, no autopsy studies and, thus, no information on the neuropathological consequences of ELOVL4 mutations in humans with SCA34 have been reported. The mechanisms leading to neurodegeneration and subsequent neurological signs and symptoms in humans are not known.
For the first time, we describe the autopsy of a patient with SCA34, with a known c.736T>G, p.W246G (NM_022726.4) mutation in ELOVL4. Neuropathological findings include marked neuronal loss and pontocerebellar fiber degeneration in the pontine base, CD68-positive macrophages laden with periodic acid-Schiff (PAS) and Gallyas-positive material, and oligodendrocyte and myelin degeneration with frequent cerebral white matter vacuoles. Furthermore, four-repeat tauopathy with neurofibrillary tangles and glial fibrillary tangles was unexpectedly observed in a spatial pattern compatible with progressive supranuclear palsy (PSP). The glial fibrillary tangles include those which resembled tuft-shaped astrocytes, which is a distinctive feature of PSP. These findings demonstrate that a heterozygous ELOVL4 missense mutation can cause various distinct neuropathological features of neuronal and oligodendroglial degeneration. The four-repeat tauopathy compatible with PSP may be an incidental co-occurrence or one of the neuropathological consequences of the ELOVL4 mutation in SCA34.
Case presentation
We studied an 83-year-old man who presented with slowly progressive gait ataxia at the age of 46 years old. He was an index case from our previous report of a Japanese pedigree with SCA34 [patient II-1 in family A] caused by a heterozygous c.736T>G, p.W246G mutation in ELOVL4 [35]. As previously described, the patient had ophthalmoplegia more obvious in the vertical direction than the horizontal direction, nystagmus, dysarthria, pyramidal tract signs, truncal and limb ataxia, trivial akinesia, and autonomic symptoms, including bladder disturbance and constipation [35]. The patient had been wheelchair bound since his sixties and had a gastrostomy due to dysphagia. The brain MRI showed marked atrophy in the pontine base and cerebellum, hot cross bun sign, mild atrophy in the cerebral cortices, and incidental small infarcts in the basal ganglia, as described in our previous report [35]. The patient had recurrent urinary tract infections and cellulitis. He had gastric carcinoma, urinary tract carcinoma, and subsequent urinary tract obstructions. The patient succumbed to death due to postrenal kidney failure at the age of 83. He was autopsied at a postmortem interval of 8 h 34 min, after obtaining written consent from his family.
Methods for neuropathological analyses
This study was conducted with permission from the institutional review boards of Tokyo Medical and Dental University, Japan (G2000-198). Written consent was obtained from his family for this study. The whole brain and spinal cord were fixed in formalin. Samples from the left side of the supratentorial regions and bilateral infratentorial regions of the brain were analyzed, including the superior and middle frontal gyrus, pre- and post-central gyrus, mammillary body and basal nucleus of the Meynert, superior temporal gyrus, temporal pole, hippocampus, amygdala, caudate nucleus, putamen, globus pallidus, thalamus, visual cortex, cerebellar cortices, and deep cerebellum. In addition, the whole brainstem, cervical and lumbar spinal cord, and pituitary were analyzed. Sections (5 μm) of paraffin-embedded samples were subjected to Hematoxylin–Eosin (HE), Klüver-Barrera (KB), and PAS staining and immunohistochemistry. Antibodies against Neurofilament H phosphorylated (1:5000, mouse monoclonal, SMI-31, BioLegend, USA, RRID:AB_10124140), glial fibrillary acidic protein (1:1000, rabbit polyclonal, DAKO, USA, RRID:AB_10013382), myelin basic protein (MBP) (1:1000, rabbit polyclonal, IBL, Japan, RRID not available/IBL Cat # 16,141), Olig2 (1:1000, rabbit polyclonal, IBL, Japan, RRID not available/IBL Cat # 18,953), CD68 (1:1000, clone KP1, mouse monoclonal, DAKO, USA, RRID:AB_2314148), Iba1 (1:500, rabbit polyclonal, WAKO, Japan, AB_839504), ubiquitin (1:1000, rabbit polyclonal, DAKO, USA, RRID:AB_2315524), RD3 (3-repeat tau) (1:3000, mouse monoclonal, MILLIPORE, USA, RRID:AB_310013), RD4 (4-repeat tau) (1:1000, mouse monoclonal, MILLIPORE, USA, RRID:AB_310014), alpha-synuclein (1:10,000, mouse monoclonal clone 5G4, Roboscreen, Germany, RRID not available/ Order #847–0,102,004,001), and tau (tau2) (1:1000, clone TAU-2, mouse monoclonal, Sigma, USA, RRID:AB_477581) were used. Free-floating sections from formalin-fixed samples were subsequently stained with oil-red, Sudan black, and OsO4 for light microscopy, and anti-Neurofilament H phosphorylated antibody (SMI-31), 4′,6-diamidino-2-phenylindole (DAPI), and fluoroMyelin (1:300, Invitrogen, USA) for immunofluorescence microscopy. Light microscopes (NanoZoomer S210, Hamamatsu Photonics, Japan and BX-7/DP-5, Olympus, Japan) and a confocal immunofluorescence microscope (A1R, Nikon Corp, Japan) were used for observation and digital recording of the prepared samples.
For electron microscopy, a small part of the formalin-fixed corpus callosum at the coronal section at the mammillary body level was excised, fixed in 2% glutaraldehyde buffer, and post-fixed in 1% OsO4. The section was dehydrated and embedded in Epon. Ultrathin sections were made and stained with uranyl acetate and lead citrate. Sections were examined with a transmission electron microscope (H-7100/XR41, Hitachi, Japan). We examined the variant table from exome-sequencing in this patient [35], and checked for any mutations in genes MAPT, PRKN, TARDBP, and LRRK2.
General pathology findings
General autopsy findings included urothelial carcinoma of the urinary bladder, which invaded the prostate gland with no metastases to other organs, early-stage carcinoma of the stomach (tubular adenocarcinoma), pyelonephritis, bilateral postrenal renal failure, and atherosclerosis of the coronary artery and abdominal aorta. No evidence of metastases was found.
Neuropathological findings
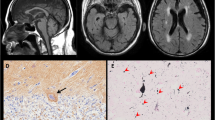
Total brain weight was 974 g (990.7 g after formalin fixation, the brainstem and cerebellum weighed 11.2 and 71.8 g, respectively). Macroscopic observation revealed marked atrophy in the brainstem, especially in the pontine base. The ventral parts of the midbrain and medulla were more atrophied than the dorsal parts. The bilateral crus cerebri were remarkably atrophic and the pyramis in the medulla was barely detected (Fig. 1c). In addition, cerebral hemispheres predominantly in the bilateral medial temporal lobes and cerebellum were atrophied (Fig. 1a, b, e, and f).

Macroscopic views of the brain of an autopsy case with SCA34. (a and b) Superior and inferior views, respectively, of brain surfaces. P denotes posterior pole to show direction. Atrophy of cerebral cortices, especially in the medial temporal lobes, was noted (see also d). (c) Transverse sections of the brainstem, from the medulla oblongata to the midbrain. White arrows indicate severe atrophy in the pontine base. In addition, the ventral parts of the midbrain and medulla were more atrophied than the dorsal parts. Depigmentation of the substantia nigra was also noted (arrowheads). (d) Coronal section of the cerebrum showed largely preserved caudate nucleus, putamen, and globus pallidus. Arrowheads indicates moderately atrophied corpus callosum. Mild atrophy of the cerebral white matter was observed (arrow). (e) Horizontal sections of right cerebellar hemisphere and sagittal sections of left hemisphere. Arrowheads show atrophied white matter. Arrow indicates obscured hilum of dentate nucleus. (f) Inferior view of the brain shows marked atrophy of the basis pontis (arrowheads). Scale bar = 1 cm (a to f)
Moderate atrophy was observed in the corpus callosum, while mild atrophy was found in the cerebral white matter (Fig. 1d). The caudate nucleus, globus pallidus, and amygdale were macroscopically normal. A small infarction in the left putamen was detected (not shown). The basal ganglia were otherwise unremarkable.
The midbrain was atrophic, especially in the cerebral peduncles, with mild enlargement of the aqueduct. Depigmentation was found in the substantia nigra (Fig. 1c). Superior cerebellar peduncles and the red nucleus were unremarkable. Remarkable atrophy was found in the pons, especially in the pontine base, and in the middle cerebellar peduncles (Fig. 1c). The inferior olivary nuclei were preserved. Despite the marked atrophy of the cerebellar white matter, the cerebellar folia and the dentate nuclei were relatively preserved (Fig. 1e). The spinal cord and roots were not atrophic.
Microscopic observations
The histological findings can be summarized in the following three changes.
-
1.
Neuronal loss, pontocerebellar fiber loss, and CD68-positive macrophages laden with periodic acid-Schiff positive material in the pontine base
Microscopic findings underlying the pontine base atrophy included marked neuronal loss in the pontine nucleus, destruction of transverse fibers, and gliosis (Fig. 2a to c). Cells with swollen cytoplasm (staining violet with KB) in the pontine base were observed. These cells were found in the pontine nucleus and transverse fibers and occasionally around vessels (Fig. 2d, e). These cells were CD68 positive and, thus, considered macrophages (Fig. 2g, h). The macrophages contained pale gray substances on HE-stained sections (Fig. 2f), which was positive for PAS (Fig. 2i). Double labeling for PAS and CD68 also supports this interpretation (Fig. 2j). Double staining for PAS and either Olig2 (see Additional file 1 Fig. 1A) or Iba1 (Additional file 1 Fig. 1B) indicated that these macrophages (CD68 positive cells) were not microglia or oligodendrocytes. These cells were glial fibrillary acidic protein–negative and, thus, were not astrocytes (Additional file 1 Fig. 1C). Accumulated PAS-positive substances suggested the presence of polysaccharides, such as glycogen, glycoproteins, or, most importantly, glycolipids.
The macrophages in the pontine base unexpectedly exhibited argyrophilia on Gallyas silver impregnation method (Fig. 2k and Additional file 1 Fig. 1G and H). A previous report showed that loss of function for the yeast homolog of ELOVL4 promoted alpha-synuclein aggregation in a yeast model of Parkinson’s disease [27]. We examined whether the mutation in ELOVL4 led to alpha-synuclein aggregation in the current case with SCA34. Immunohistochemistry illustrated an absence of phosphorylated alpha-synuclein aggregation in the pontine macrophages, other pontine structures, basal ganglia, and motor cortex (Additional file 1 Fig. 1D, E, and F). Campbell–Switzer silver impregnation method, which can stain Alzheimer’s disease neurofibrillary tangles, senile plaques, and Lewy bodies [45], did not stain the pontine macrophages (Additional file 1 Fig. 1I). The macrophages did not exhibit positive staining for anti-three-repeat, four-repeat, or phosphorylated tau (Additional file 1 Fig. 1J, K, and L). Moderate but uneven tau2 antibody staining of the macrophages was observed (Additional file 1 Fig. 1M). The tau2 antibody presumably stains an atypical conformation of tau protein, for instances in a previous study, in microglia/macrophages of cerebral infarction or inflammation [44]. The CD68 positive macrophages laden with PAS-positive material were not observed in any brain structures other than the pontine base.
In accordance with the severe degeneration of the pontine nucleus and pontocerebellar fibers, the cerebellar white matter was atrophied, especially in the terminal lobular areas compared to the deep cerebellar white matter (Fig. 2l). Purkinje cells were mildly to moderately reduced and atrophied (Additional file 1 Fig. 1N). Mild and uneven demyelination in the cerebellar white matter was also noted (arrow in Fig. 2l). The cerebellar cortices were relatively preserved compared to the white matter. The molecular layer was thin, while the granular layer was preserved.
-
2.
Widespread vacuolar lesions and myelin degeneration consistent with oligodendrocyte degeneration
Many vacuoles (Fig. 3a to m) were found in the cerebral white matter in the frontal, temporal, parietal, and posterior lobes, optic tract, corpus callosum, and internal capsule in HE stained sections. Vacuoles were also found along the intramedullary fibers of the ophthalmic nerve roots in the midbrain and the pontine tegmental area, including the medial lemniscus, superior cerebellar peduncle, trigeminal nerve root, and, to a lesser extent, the pyramidal tract (Fig. 3a, b, and Additional file 2 Fig. 2A to D). The vacuoles were notably absent in the atrophic pontocerebellar fibers of the pontine base, including the middle cerebellar peduncles. The vacuoles were rare in the anterior, lateral, and posterior columns of the spinal cord, but often observed along intramedullary fibers of anterior and posterior roots (Additional file 2 Fig. 2E to G).
The vacuoles were mostly 15 to 50 μm in diameter in the paraffin-embedded sections and were very rare in gray matter throughout the brain. Solitary vacuoles and multiple adhered vacuoles were observed (Fig. 3c to g). The vacuoles did not appear to contain solid substances and were often associated with cells in a line harboring swollen clear cytoplasm and round nuclei (arrows in Fig. 3c and e). The nuclei remaining in these vacuoles or associated cells were often positive for Olig2, one of the oligodendroglial nuclear markers (Fig. 3j to l). Therefore, the vacuoles were likely derived from oligodendrocytes.
The intravacuolar substances were negative for lipophilic staining, including oil red, Sudan-black, or OsO4 (Fig. 3h and Additional file 2 Fig. 2H and I). The thin lining of rims and septum of vacuoles were positive for MBP, suggesting components of myelin surround vacuoles (arrowhead in Fig. 3i). Confocal immunofluorescence microscopy using anti-Neurofilament H antibody, which stains the neuronal axons, in conjunction with fluoroMyelin, which stains the lipid contents of myelin, indicated that vacuoles displaced neural fibers, including myelin (arrows in Fig. 3m). Because MBP is one of the cytoplasmic components of myelin, these findings are compatible with the view that the vacuoles originated in oligodendroglia. Sections prepared using freezing methods from formalin-fixed tissues showed smaller vacuoles.
To evaluate the ultrastructural features of vacuoles, oligodendrocytes, and myelin, we analyzed the corpus callosum, a part of the cerebral white matter, with electron microscopy. We found swollen oligodendrocytes with disrupted myelin (Fig. 4a to d); however, the large vacuolar lesions, which were observed using light microscopy, were rarely found in the electron microscopy. Loose membrane structures in swollen oligodendrocytes, which likely reflect disrupted myelin, were observed (arrowheads in Fig. 4e and f). These loose membranes were absent in paraffin-embedded sections, probably reflecting the fragility of the structures that were lost during processing. In summary, electron microscopy showed destructed myelin structures associated with swollen oligodendrocytes and degraded fine myelin inside the cells, which were not seen under the light microscope.
-
3.
Tau accumulation in the brain of the SCA34 patient
Because we found that PAS-positive macrophages of the pontine base were also positive for Gallyas silver impregnation but negative for alpha-synuclein accumulation, we next sought to evaluate the accumulation of tau protein. Tau is another major protein that can form pathologic aggregations exhibiting argyrophilia on Gallyas silver impregnation. The macrophages in the pontine base were negative for three- or four-repeat tau (3R tau and 4R tau) or phosphorylated tau (p-tau) staining, as described above; however, we unexpectedly found widespread neuronal and glial fibrillary tangles positive for phosphorylated tau in the brain, including the frontal cortex, subthalamic nucleus, putamen, globus pallidus, substantia nigra, and midbrain tegmentum, where four-repeat tau was dominant (see Table 1 and Fig. 5a to j).
Morphological characteristics of the glial fibrillary tangles positive for four-repeat tau resembled tuft-shaped astrocytes, coiled bodies, and threads (Fig. 5b to g), which characterize progressive supranuclear palsy, while some tau-positive lesions were difficult to classify. In addition, we observed comparable three- and four-repeat tau positive neurofibrillary tangles in the parahippocampus, hippocampus, locus ceruleus, and, to a lesser extent, in the temporal cortex and amygdala (Braak neurofibrillary tangles (NFT) stage II) [10]. Trace amounts or no staining of senile plaque, which is suggestive of early-stage Alzheimer’s disease or possibly consistent with primary age-related tauopathy (PART) [13], were observed in these areas using anti-beta amyloid immunostaining (Fig. 5l). Part of the intraaxial fibers of the left oculomotor nerve was positive for anti-four-repeat tau staining (Fig. 5k).
-
4.
Other microscopic observations of the SCA34 patient brain and spinal cord

Microscopic observation of the pons and cerebellum from an autopsy case with SCA34. a Semi-macroscopic view of the pons after Klüver-Barrera (KB) staining. Arrowheads indicate severe atrophy of the pontine base. An arrow indicates demyelination of the transverse fibers. b Transverse fibers between the medial lemniscus and pyramidal tract showed severe degeneration (magnified from the rectangle area in a). c Further magnified view of the transverse fibers between the medial lemniscus and pyramidal tract (magnified from the rectangle area in B). Very scarce and degenerated fibers were observed. d, e, and f After KB (d and e) and Hematoxylin–Eosin (HE) (f) staining, the most ventral part of the pontine base also showed severe degeneration of pontocerebellar fibers and neuronal loss. Cells with irregular shaped dense nuclei and violet cytoplasm after KB staining as shown in e (magnified from the rectangle area in d) or pale gray after HE staining (arrows in f and magnified in its inset) were observed. g and h immunostaining for CD68. Strongly CD68-positive cells were widespread (white and black arrowheads in g) in the pontine base, excluding the pyramidal tracts. These cells occasionally accumulated around the vasculature (white arrowheads in g and h). h is a magnified view from the rectangle in g. The inset in H illustrates a magnified view from the rectangular area in the center. i The cytoplasm of the CD68-positive cells was positively stained with periodic acid-Schiff (PAS) (arrowheads). V denotes vessels. j Double staining with PAS (red) and anti-CD68 (brown) illustrates co-staining for PAS and CD68 in the same cells (arrowheads). The double-staining is shown as a dark-brown color, except for wall of the vasculature, which only stained for PAS. k These macrophages were positive for Gallyas silver impregnation method. L Semi-macroscopic view of the cerebellum showing white matter atrophy and mild and uneven demyelination (arrow). Scale bar = 5 mm (a and l), 500 μm (b, d, and g), 100 μm (c, f, h, and k), 50 μm (e and l), and 20 μm (j)

Light microscopic observation of white matter vacuoles in an autopsy case with SCA34. a Hematoxylin–Eosin (HE) staining of the frontal cortex showing vacuoles in the white matter (arrowheads) but few in the gray matter. WM and GM denote white and gray matter, respectively. b Vacuoles in the right superior cerebellar peduncle are shown. c A magnified view showing single vacuoles and adhered multiple vacuoles (arrowhead) (magnified from the rectangular area in a). Cells with swollen cytoplasm and round and bright nuclei, often accompanied by one or more similar adjacent cells (arrows in c and e), suggest that these vacuoles were derived from oligodendrocytes. d, e, f, and g Various forms of vacuoles in the cerebral white matter were observed. A thin septum was often seen between adjacent vacuoles (arrowheads in d and f). In some vacuoles, flattened nuclei were observed in the rim (arrows in g). h Oil red staining did not reveal lipid substance in vacuoles (arrowhead). i, Anti-MBP staining of thin rims and septum, suggesting that components of myelin surround the vacuoles (arrowhead). j, k, l Nuclei inside the rims of vacuoles were often Olig2-positive. m Confocal immunofluorescence microscopy using anti-Neurofilament H antibody (axonal neuronal protein, in red), fluoroMyelin (staining lipid contents of myelin, in green), and 4′,6-diamidino-2-phenylindole (DAPI) (nuclear staining, in blue). Arrows indicate the neuronal fibers, including myelin circumscribing a vacuole and an oligodendrocyte. Scale bar = 250 μm (a and b), 50 μm (c to g), 20 μm (h to m)

Electron microscopic observation of white matter abnormalities in an autopsy case with SCA34. a, b, c, d, e, and f, transmission electron microscopy of the corpus callosum. a Vacuoles (asterisks) associated with discontinuous and degenerated myelin, which ran nearby (large and small arrows, respectively), are shown. b A ballooned cytoplasmic space (asterisk) and nucleus are at the outermost space of the oligodendroglial cytoplasm. Arrows indicate deformed myelin. Myelin digestion is shown (arrowhead). c An enlarged view from the same site as B showing disrupted myelin fibers (arrows). d Deformed and disrupted myelin (arrows) accompanying cytoplasmic digestion of myelin (arrowheads) is shown. e and f Loose membrane structures, which likely reflect disrupted myelin, in swollen oligodendrocytes, can be observed (arrowheads). Scale bar = 10 μm (e and f), 4 μm (a and b), 2 μm (c and d)

Tauopathy in SCA34. a and b The subthalamic nucleus showing frequent glial fibrillary tangles (arrows) stained with anti-four-repeat tau antibody. Magnified view (b) from the rectangular area in a showing some of the tangles that resemble a tuft-shaped astrocyte (arrow). C A tuft-shaped astrocyte exhibiting argyrophilia on Gallyas silver impregnation in the subthalamic nucleus. d Coiled bodies (arrows) in the subthalamic nucleus stained with the anti-phosphorylated tau antibody, AT8. e A tuft-shaped astrocyte (arrow) in the prefrontal cortex exhibiting argyrophilia on Gallyas silver impregnation. f A tuft-shaped astrocyte in the putamen stained with anti-four-repeat tau antibody. g Glial fibrillary tangles akin to a tuft-shaped astrocyte (arrow) and thread (arrowhead) in the external segment of the globus pallidus, stained with anti-four-repeat tau antibody. h Glial fibrillary tangles in the frontal cortex stained with anti-phosphorylated tau antibody AT8. i Pretangle (arrow) and neurofibrillary tangle (arrowhead) in the substantia nigra pars compacta stained with anti-phosphorylated tau antibody AT8. j The superior colliculus in the midbrain showing frequent glial fibrillary tangles stained with anti-four-repeat tau. k The intraaxial oculomotor nerve partly stained with anti-four-repeat tau antibody in the midbrain (arrowheads). L Few amyloid plaques stained with anti-amyloid antibody in the insular cortex (magnified view of a plaque in inset). Scale Bar = 250 μm (a and k), 100 μm (h and l), 50 μm (b, c, e, f, g, and i), 25 μm (D and inset in L)
The volume of the substantia nigra pars compacta was decreased, while the substantia nigra pars reticulata was relatively preserved. Mild gliosis was observed in the red nuclei. The locus ceruleus nuclei were preserved but neurofibrillary tangles were observed, as described above. In the medulla oblongata, the inferior olivary nucleus, nuclei ambiguus, and spinal tract nucleus of the trigeminal nerve were preserved. In the spinal cord, motor neurons were spared.
Discussion and conclusions
We present the first autopsy report of an SCA34 case caused by an ELOVL4 mutation. The neuropathological findings included remarkable neuronal loss in the pontine nucleus, pontocerebellar fiber loss, pontine macrophages laden with PAS-positive material, widespread vacuoles in the white matter with oligodendroglial degeneration, and, unexpectedly, four-repeat tauopathy reminiscent of progressive supranuclear palsy. This peculiar combination of characteristic lesions among neurons, oligodendrocytes, and macrophages, and tau accumulation suggests that the molecular pathology of SCA34 is distinct and pleiotropic. Furthermore, the findings might provide important clues to understand how mutations in a series of elongase genes lead to neurodegeneration.
The most prominent neurological deficit in SCA34 was ataxia [12, 35]; one of the remarkable radiological findings in the brain MRI of SCA34 patients, including this case, is atrophy in the pontine base and cerebellum and the hot cross bun sign in the pons. In this patient, neuronal loss and destruction of transverse fibers (pontocerebellar tract) in the pontine base may explain the symptoms and signs of ataxia and the radiological features. These neuropathological findings were notably similar to other cerebellar ataxias that have pontine base atrophy and the hot cross bun sign, such as cerebellar type of multiple system atrophy (MSA-c) and spinocerebellar ataxia type 2 [20]. However, the CD68 positive macrophages laden with PAS-positive material in the pontine base pathology of SCA34 was not described in MSA-c or spinocerebellar ataxia type 2. The cardinal macroscopic neuropathological findings of MSA-c include atrophy of the pontine base, middle cerebellar peduncles, inferior olivary nucleus, and pontocerebellar fibers representing as cruciform demyelination in the pons and white matter atrophy in the cerebellum. Additionally, depigmentation in the substantia nigra and variable discoloration and atrophy in the putamen can also be found macroscopically [29]. In this study, these findings except for atrophy in the putamen and inferior olivary nucleus were observed in SCA34. And we did not notice glial cytoplasmic inclusions, a hallmark of MSA, or any other abnormal inclusions by anti-alpha-synuclein staining. Nevertheless, MSA may originate from abnormal lipid metabolism [6]. Because distinct involvement of the pontine base neurons and pontocerebellar fibers are common to both SCA34 and MSA-c, these diseases might share a common pathway involving aberrant lipid metabolism. SCA38, a spinocerebellar ataxia caused by mutations in ELOVL5, exhibits cerebellar atrophy but no abnormality in the brainstem on MRI [7]. Because the ELOVL5 protein is also a fatty acid elongating enzyme but has non-overlapping substrate specificity and functions as well as catalyzes elongation of shorter carbon chain (between 18 and 22) fatty acids [14], further studies are needed to explain how mutations in ELOVL4 and ELOVL5 lead to distinct spatial patterns of neurodegeneration, by analyzing elongases and lipids in the brain. A recent report indicated that a rat model of SCA34 with c.736T>G, p.W246G mutation had a motor impairment and synaptic dysfunction but without overt neurodegeneration in the cerebellar cortex [30]. This rodent model harbors significant similarities to our case and would be useful for further investigations.
When we review aged cases of other SCAs to compare neuropathological characteristics with this SCA34 case, in a rare SCA2 case of 85 years old [23], for example, neuropathological features consistent with SCA2, such as neuronal loss and gliosis distinct in the pontine nucleus, inferior olivary nucleus, cerebellar cortex, posterior column nuclei, substantia nigra, dentate nucleus of the cerebellum, were reportedly observed combined with diffuse neurofibrillary tangles and senile plaques, which are compatible with Alzheimer’s disease pathology (Braak NFT stage IV). Additionally, in an SCA7 case of a 77 year old patient harboring very short 39 CAG repeats in the mutated allele of the SCA7 gene [38], neuropathological changes in cerebral, cerebellar, and brainstem regions were more widespread than in previously reported SCA7 cases with longer repeats and of younger-onset. However, this case only indicated NFT stage I of Alzheimer’s disease pathology. And in an 87 year old autopsy case of SCA31 [43], which is one of the purely cerebellar forms of SCAs and generally prolonged clinical course, comorbid with argyrophilic grain disease, which is also one of the frequent dementing diseases [29], exhibited cerebellar degeneration consistent with SCA31 combined with argyrophilic grains. This case had NFT stage II of neurofibrillary tangles and senile plaques in the neocortex, parahippocampal gyrus, and precentral gyrus and very few alpha-synuclein-positive deposits. Thus, neuropathological findings significantly differ between SCA types and individual cases, and comorbid neuropathological changes related to aging can exist, rather independently of the pathological severity of SCAs. And typical PSP has a much faster clinical course (median survival time 5.6 years in one study) compared to our case [28], although there is a report of an extremely rare autopsy case of PSP with an unusually slow and prolonged disease course (24 years) [24], where a 79 year old woman exhibited widespread neurodegeneration and tauopathy in typically involved regions, and Braak stage III NFTs, senile plaques, but without glial cytoplasmic inclusion. Pathological changes related to PSP were much milder in our SCA34 case compared with the case.
Macrophages, which were CD68 positive and laden with PAS-positive material in the pontine base, were reminiscent of X-linked adrenoleukodystrophy (X-ALD), although we were unable to examine the cleft-like trilamellar cytoplasmic inclusions, which are found in perivascular macrophages in X-ALD, because of our specimen limitation [32]. These macrophages may contain myelin remnants, as suggested in X-ALD [46], considering the marked destruction of myelin in the pontocerebellar fibers of the pontine base. In the current case, we can raise several possible explanations of how ELOVL4 gene mutations resulted in the generation of such phagocytosed materials in macrophages. First, due to the missense mutation in ELOVL4, myelin content might be abnormal and difficult to digest within macrophages. Second, the ELOVL4 mutation might disrupt organelle functions within macrophages, thus affecting the metabolism of myelin. Third, ongoing massive myelin degeneration might simply surpass the myelin digestion capacity of macrophages. This might be similar, though milder, to the demyelination plaques of multiple sclerosis, where infiltration of massive macrophages laden with myelin is typically seen [37]. Further studies are warranted to clarify the altered function of the markedly abnormal pontine macrophages in SCA34.
Another remarkable finding in this neuropathological study is the frequent vacuoles in the white matter, predominantly in the supratentorial regions. These vacuoles did not abnormally store lipids but were compatible with remnants of degenerated oligodendrocytes, based on anti-Olig2 immunohistochemical and ultrastructural observations. In addition, myelin remnants revealed under the electron microscope also suggests abnormalities in myelin integrity and composition. Although a detailed analysis of lipid metabolism abnormalities is lacking, one plausible explanation for the degenerated myelin in SCA34 is an abnormal or reduced synthesis of complex lipid substances, such as phospholipids and glycolipids, resulting in myelin destruction. Knockout mice genetically lacking ceramide synthase 2 (CerS2) developed vacuoles in the white matter due to myelin ballooning [5, 22]. Curiously, CerS2 interacts with ELOVL4 and other elongase proteins in cultured cells [33]. Mitochondrial encephalopathies, such as Kearns-Sayre syndrome (KSS) [34], also exhibit vacuoles in the white and gray matter. One neuropathological study of KSS suggested that intracytoplasmic oligodendroglial vacuoles of the white matter could be the structural origin of the vacuoles in KSS [41]. The vacuoles in KSS morphologically resemble the vacuoles in our study in that the oligodendroglial nuclei reside within or in contact with single or multiloculated vacuoles and the rims surrounding the vacuoles are positive for MBP. Therefore, a common cascade may cause vacuole formation from oligodendroglial degeneration in both the mitochondrial disease and SCA34.
A previous study in mice showed that ELOVL4 proteins are primarily expressed in neurons throughout the brain and in white matter oligodendrocytes [40]. Thus, it is surprising that only pontine base neurons were severely reduced, whereas other neurons were largely spared in this case. For example, the cerebellar granular layer, which was highly positive by anti-ELOVL4 antibodies in rodents in previous reports [30, 40], was surprisingly preserved in this study. Of note, pontocerebellar fibers, which were most severely affected, showed almost no vacuoles. In contrast, the vacuoles were widespread in the supratentorial white matter of the brain. Thus, cell type- and location-specific metabolic components may be important in the subsequent morphological consequences. In fact, lipid compositions differ from site to site in the brain [31]. Therefore, a more detailed study on lipid metabolism is necessary to fully understand the metabolic alterations in the SCA34 brain. Such future studies may include biochemical analyses for very long-chain fatty acids and perhaps glycolipids in the brain.
Surprisingly, PSP-like pathology was observed in the SCA34 case. When we retrospectively looked back at neurological signs of this case, vertically dominant gaze palsy and trivial akinesia could be clinical features potentially suggestive of PSP. Four repeat tauopathy exhibiting tuft-shaped astrocytes, coiled bodies, and neurofibrillary tangles and pretangles, which are distinctive neuropathological characteristics of PSP, were observed in the regions typically affected in PSP, such as the subthalamic nucleus, substantia nigra, putamen, globus pallidus, and the frontal cortex, although the extent of neuronal loss was none to very mild in those regions. Other typically involved areas, such as midbrain tegmentum, locus ceruleus, red nucleus, nuclei of III, IV, X, XII cranial nerves, and dentate nucleus of the cerebellum were generally preserved. On the one hand, PSP in the current case may be an incidental co-occurrence. A previous study reported that neuropathological features compatible with PSP were found in 45 cases with and 5 cases without clinically diagnosed PSP out of 706 autopsied elderly patients analyzed [16]. Also, in serial forensic autopsies of 998 Japanese cases [48], 4.6% of cases over 60 years old were neuropathologically diagnosed with PSP. Thus, PSP is not an extremely rare entity in the elderly. On the other hand, the four-repeat tauopathy compatible with PSP in the current case may have been caused by the ELOVL4 mutation. In the literature, at least 19 genetic disorders other than MAPT-associated ones displayed tauopathies [42]. At least three cases show some, if not all, of the important neuropathological features of PSP. A case with hereditary Parkinson’s disease/Park8 caused by the LRRK2 mutation (p.R1441C) showed PSP-like tuft-shaped astrocytic lesions but without the density and distribution diagnostic of PSP [50]. Another case with semantic dementia harboring the TARDBP mutation (p.Ile383Val) showed tau-positive astrocytic lesions in the amygdala but not in the subthalamus or basal ganglia, which are usually involved in PSP [17]. One case with the heterozygous PRKN mutation (p.C212Y) showed hyperphosphorylated tau lesions consisting of globose-type neurofibrillary tangles and tuft-shaped astrocytes, with distribution consistent with PSP, whereas other family members harboring this mutation (p.C212Y) and another PRKN mutation in the compound heterozygous state suffered juvenile-onset Parkinson’s disease [39]. Thus, hereditary disorders that display PSP-like neuropathological features have been reported, although they are very rare. Although PSP is caused by primary alteration of the tau protein, PSP-like deposition of tau may be programmed in the brain as a template that is triggered by aging or other genetic abnormalities like those listed above. We reviewed the exome-sequencing data of this patient in our previous genetic study [35] and discovered no mutations in MAPT, PRKN, TARDBP, and LRRK2. Furthermore, aberrant interactions of alpha-synuclein and phosphorylated tau with lipids in a specific subset of neurotransmitter-containing vesicles were described in aging mouse brain [11]; thus, ELOVL4 mutations may promote tauopathy through abnormal lipid content. Further investigation is needed to determine whether ELOVL4 mutations can cause PSP-like pathology.
One of the most critical limitations in this study is that this report only describes a single case. The observed neuropathological features should be considered with caution before more autopsied cases of SCA34 are accumulated. When considering the background medical conditions of this patient, the age of 83 years old is an inevitably essential factor that can affect brain neuropathology. For example, white matter volume in the cerebrum decreases with age [29]. This may explain the white matter volume decrease in the current case, at least in part. Among specific neurodegenerative processes that arise with age, Alzheimer’s disease and related disorders are frequent and essential from the viewpoint of tauopathy. In this study, neurofibrillary tangles positively stained with three and four repeat tau antibodies were observed in the limbic system. However, only trace amounts of senile plaques existed, and neuronal loss was not evident in the areas. Thus, these neuropathological changes can be regarded as the early phase of Alzheimer’s disease or a recently proposed entity, primary age-related tauopathy (PART), which exhibits marked neurofibrillary tangles compatible with Alzheimer’s disease in the hippocampus and medial temporal lobes in the absence of amyloid plaques, especially in the elderly [13]. And for tauopathy, subpial astrocytic lesions called thorn-shaped astrocytes along lateral ventricles are often observed in the elderly, and were remarkably observed in this study. However, these age-related changes or diseases cannot explain the currently described four repeat tauopathy or pontine atrophy in the current case. And for alpha-synucleinopathies, such as Parkinson’s disease, Lewy body disease, and multiple system atrophy, we found no evidence of alpha-synuclein accumulation by immunohistochemistry. Potential neuropathological changes caused by atherosclerosis that progresses with age include dilatation of perivascular spaces, microinfarcts, and changes of vessel walls, and white matter changes around vessels such as myelin loss and astrocytosis [29]. Oligodendroglial degeneration and resulting vacuoles are very distinctive lesions that cannot be explained only by atherosclerosis and aging. Myelin degeneration could be influenced by such factors, and requires further investigations in accumulated cases with SCA34 in the future. Finally, kidney failure with a uremic state can accompany neuropathological changes with the presence of Alzheimer type II astrocytes and possibly perivascular neuronal degeneration and demyelination [29]. These changes were not observed in this case, and this medical condition is unlinked to the widespread oligodendroglial degeneration. However, we have to be careful until more autopsied cases with SCA34 are accumulated, and effects by the above-mentioned conditions are considered in those cases in the future.
A recent report described psychomotor deficits in SCA34 patients with another mutation (c.504G>C, p.L168F) [4]. The widespread white matter vacuoles and macroscopic atrophy, especially in frontal, temporal, and parietal lobes, might be associated with the psychiatric symptoms.
We describe the first autopsy case of SCA34 caused by an ELOVL4 mutation, showing remarkable neuronal loss and pontocerebellar fiber loss in the pontine base. These findings are compatible with the clinical and radiological features in this patient. We also unexpectedly found an accumulation of macrophages laden with PAS-positive material in the pontine base and oligodendroglial degeneration with widespread vacuoles in the white matter. Four-repeat tauopathy that displayed neuronal and glial fibrillary tangles reminiscent of PSP was observed, although whether the tauopathy is coincidental or caused by the ELOVL4 mutation is unknown. Taken together, these findings suggest that the ELOVL4 mutation led to neuronal and glial degeneration through pleiotropic pathways in a cell type- and location-specific manner in SCA34.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request and with permission of the patient’s family and the institutional review boards, if applicable.
Abbreviations
- HE:
-
Hematoxylin-Eosin
- KSS:
-
Kearns-Sayre syndrome
- KB:
-
Klüver-Barrera
- MRI:
-
Magnetic resonance imaging
- MSA-c:
-
Cerebellar type of multiple system atrophy
- MBP:
-
Myelin basic protein
- PAS:
-
Periodic acid-Schiff
- PSP:
-
Progressive supranuclear palsy
- SCA:
-
Spinocerebellar ataxia
- X-ALD:
-
X-linked adrenoleukodystrophy
References
Agbaga MP, Mandal NA, Anderson RE (2010) Retinal very long-chain PUFAs: new insights from studies on ELOVL4 protein. J Lipid Res 51:1624–1642. https://doi.org/10.1194/jlr.R005025
Agbaga MP, Stiles MA, Brush RS, Sullivan MT, Machalinski A, Jones KL et al (2020) The Elovl4 spinocerebellar ataxia-34 mutation 736T>G (p. W246G) impairs retinal function in the absence of photoreceptor degeneration. Mol Neurobiol 57:4735–4753. https://doi.org/10.1007/s12035-020-02052-8
Aldahmesh MA, Mohamed JY, Alkuraya HS, Verma IC, Puri RD, Alaiya AA et al (2011) Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 89:745–750. https://doi.org/10.1016/j.ajhg.2011.10.011
Beaudin M, Sellami L, Martel C, Touzel-Deschenes L, Houle G, Martineau L et al (2020) Characterization of the phenotype with cognitive impairment and protein mislocalization in SCA34. Neurol Genet 6:e403. https://doi.org/10.1212/nxg.0000000000000403
Ben-David O, Pewzner-Jung Y, Brenner O, Laviad EL, Kogot-Levin A, Weissberg I et al (2011) Encephalopathy caused by ablation of very long acyl chain ceramide synthesis may be largely due to reduced galactosylceramide levels. J Biol Chem 286:30022–30033. https://doi.org/10.1074/jbc.M111.261206
Bleasel JM, Wong JH, Halliday GM, Kim WS (2014) Lipid dysfunction and pathogenesis of multiple system atrophy. Acta Neuropathol Commun 2:15. https://doi.org/10.1186/2051-5960-2-15
Borroni B, Di Gregorio E, Orsi L, Vaula G, Costanzi C, Tempia F et al (2016) Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38). Parkinsonism Relat Disord 28:80–86. https://doi.org/10.1016/j.parkreldis.2016.04.030
Bourassa CV, Raskin S, Serafini S, Teive HA, Dion PA, Rouleau GA (2015) A New ELOVL4 Mutation in a Case of Spinocerebellar Ataxia With Erythrokeratodermia. JAMA Neurol 72:942–943. https://doi.org/10.1001/jamaneurol.2015.0888
Bourque PR, Warman-Chardon J, Lelli DA, Laberge L, Kirshen C, Bradshaw SH et al (2018) Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet 4:e263. https://doi.org/10.1212/nxg.0000000000000263
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. https://doi.org/10.1007/s00401-006-0127-z
Brekk OR, Moskites A, Isacson O, Hallett PJ (2018) Lipid-dependent deposition of alpha-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci Rep 8:15207. https://doi.org/10.1038/s41598-018-33474-z
Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, Martin C, Meloche C, Gravel M et al (2014) Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 71:470–475. https://doi.org/10.1001/jamaneurol.2013.6337
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. https://doi.org/10.1007/s00401-014-1349-0
Deák F, Anderson RE, Fessler JL, Sherry DM (2019) Novel cellular functions of very long chain-fatty acids: insight from ELOVL4 mutations. Front Cell Neurosci 13:428. https://doi.org/10.3389/fncel.2019.00428
Di Gregorio E, Borroni B, Giorgio E, Lacerenza D, Ferrero M, Lo Buono N et al (2014) ELOVL5 mutations cause spinocerebellar ataxia 38. Am J Hum Genet 95:209–217. https://doi.org/10.1016/j.ajhg.2014.07.001
Evidente VG, Adler CH, Sabbagh MN, Connor DJ, Hentz JG, Caviness JN et al (2011) Neuropathological findings of PSP in the elderly without clinical PSP: possible incidental PSP? Parkinsonism Relat Disord 17:365–371. https://doi.org/10.1016/j.parkreldis.2011.02.017
Gelpi E, Van Der Zee J, Turon Estrada A, Van Broeckhoven C, Sanchez-Valle R (2014) TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol 40:225–230. https://doi.org/10.1111/nan.12063
Genis D, Ortega-Cubero S, San Nicolas H, Corral J, Gardenyes J, De Jorge L et al (2018) Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91:e1988–e1998. https://doi.org/10.1212/wnl.0000000000006550
Giroux J, Barbeau A (1972) Erythrokeratodermia with ataxia. Arch Dermatol 106:183–188. https://doi.org/10.1001/archderm.1972.01620110019005
Higashi M, Ozaki K, Hattori T, Ishii T, Soga K, Sato N et al (2018) A diagnostic decision tree for adult cerebellar ataxia based on pontine magnetic resonance imaging. J Neurol Sci 387:187–195. https://doi.org/10.1016/j.jns.2018.02.022
Hopiavuori BR, Deak F, Wilkerson JL, Brush RS, Rocha-Hopiavuori NA, Hopiavuori AR et al (2018) Homozygous expression of mutant ELOVL4 leads to seizures and death in a novel animal model of very long-chain fatty acid deficiency. Mol Neurobiol 55:1795–1813. https://doi.org/10.1007/s12035-017-0824-8
Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J et al (2009) Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem 284:33549–33560. https://doi.org/10.1074/jbc.M109.031971
Ishida C, Komai K, Yonezawa K, Sakajiri K, Nitta E, Kawashima A et al (2011) An autopsy case of an aged patient with spinocerebellar ataxia type 2. Neuropathology 31:510–518. https://doi.org/10.1111/j.1440-1789.2010.01176.x
Iwasaki Y, Mori K, Ito M, Mimuro M, Yoshida M (2012) An autopsied case of progressive supranuclear palsy presenting with slow progression and unusually prolonged disease duration. Rinsho Shinkeigaku 52: 156–60 Japanese.. https://doi.org/10.5692/clinicalneurol.52.156
Kihara A (2012) Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem 152:387–395. https://doi.org/10.1093/jb/mvs105
Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD (eds) (1999) Basic neurochemistry: molecular, cellular, and medical aspects, 6th edn. Lippincott-Raven, Philadelphia
Lee YJ, Wang S, Slone SR, Yacoubian TA, Witt SN (2011) Defects in very long chain fatty acid synthesis enhance alpha-synuclein toxicity in a yeast model of Parkinson’s disease. PLoS ONE 6:e15946. https://doi.org/10.1371/journal.pone.0015946
Litvan I, Mangone CA, Mckee A, Verny M, Parsa A, Jellinger K et al (1996) Natural history of progressive supranuclear palsy (Steel-Rechardson-Olszewski syndrome) and clinical predictors of survival: A clinicopathological study. J Neurol Neurosurg Psychiatry 61:615–620. https://doi.org/10.1136/jnnp.60.6.615
Love S, Perry A, Ironside JW, Budka H (eds) (2015) Greenfield’s Neuropathology, 9th edn. CRC Press, Florida
Nagaraja RY, Sherry DM, Fessler JL, Stiles MA, Li F, Multani K et al (2021) W246G Mutant ELOVL4 Impairs Synaptic Plasticity in Parallel and Climbing Fibers and Causes Motor Defects in a Rat Model of SCA34. Mol Neurobiol published online. https://doi.org/10.1007/s12035-021-02439-1
Nampei M, Horikawa M, Ishizu K, Yamazaki F, Yamada H, Kahyo T et al (2019) Unsupervised machine learning using an imaging mass spectrometry dataset automatically reassembles grey and white matter. Sci Rep 9:13213. https://doi.org/10.1038/s41598-019-49819-1
Ogaki K, Koga S, Aoki N, Lin W, Suzuki K, Ross OA et al (2016) Adult-onset cerebello-brainstem dominant form of X-linked adrenoleukodystrophy presenting as multiple system atrophy: case report and literature review. Neuropathology 36:64–76. https://doi.org/10.1111/neup.12230
Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, al, (2010) ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci USA 107:18439–18444. https://doi.org/10.1073/pnas.1005572107
Oldfors A, Fyhr IM, Holme E, Larsson NG, Tulinius M (1990) Neuropathology in Kearns-Sayre syndrome. Acta Neuropathol 80:541–546. https://doi.org/10.1007/bf00294616
Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, et al (2015) A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 72:797–805. https://doi.org/10.1001/jamaneurol.2015.0610
Ozaki K, Ansai A, Nobuhara K, Araki T, Kubodera T, Ishii T et al (2019) Prevalence and clinicoradiological features of spinocerebellar ataxia type 34 in a Japanese ataxia cohort. Parkinsonism Relat Disord 65:238–242. https://doi.org/10.1016/j.parkreldis.2019.05.019
Popescu BF, Lucchinetti CF (2012) Pathology of demyelinating diseases. Annu Rev Pathol 7:185–217. https://doi.org/10.1146/annurev-pathol-011811-132443
Rüb U, Brunt ER, Gierga K, Seidel K, Schultz C, Schöls L et al (2005) Spinocerebellar ataxia type 7 (SCA7): first report of a systematic neuropathological study of the brain of a patient with a very short expanded CAG-repeat. Brain Pathol 15:287–295. https://doi.org/10.1111/j.1750-3639.2005.tb00113.x
Sánchez MP, Gonzalo I, Avila J, De Yébenes JG (2002) Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J Alzheimers Dis 4:399–404. https://doi.org/10.3233/jad-2002-4506
Sherry DM, Hopiavuori BR, Stiles MA, Rahman NS, Ozan KG, Deak F et al (2017) Distribution of ELOVL4 in the developing and adult mouse brain. Front Neuroanat 11:38. https://doi.org/10.3389/fnana.2017.00038
Szalardy L, Molnar M, Torok R, Zadori D, Vecsei L, Klivenyi P et al (2016) Histopathological comparison of Kearns-Sayre syndrome and PGC-1alpha-deficient mice suggests a novel concept for vacuole formation in mitochondrial encephalopathy. Folia Neuropathol 54:9–22. https://doi.org/10.5114/fn.2016.58911
Tacik P, Sanchez-Contreras M, Rademakers R, Dickson DW, Wszolek ZK (2016) Genetic disorders with tau pathology: a review of the literature and report of two patients with tauopathy and positive family histories. Neurodegener Dis 16:12–21. https://doi.org/10.1159/000440840
Toru S, Ishida S, Uchihara T, Hirokawa K, Kitagawa M, Ishikawa K (2020) Comorbid argyrophilic grain disease in an 87-year-old male with spinocerebellar ataxia type 31 with dementia: a case report. BMC Neurol 20:136. https://doi.org/10.1186/s12883-020-01723-2
Uchihara T, Duyckaerts C, Seilhean D, Nakamura A, Lazarini F, Hauw JJ (2005) Exclusive induction of tau2 epitope in microglia/macrophages in inflammatory lesions-tautwopathy distinct from degenerative tauopathies. Acta Neuropathol 109:159–164. https://doi.org/10.1007/s00401-004-0922-3
Uchihara T (2007) Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol 113:483–499. https://doi.org/10.1007/s00401-007-0200-2
Weinhofer I, Zierfuss B, Hametner S, Wagner M, Popitsch N, Machacek C et al (2018) Impaired plasticity of macrophages in X-linked adrenoleukodystrophy. Brain 141:2329–2342. https://doi.org/10.1093/brain/awy127
Xiao C, Binkley EM, Rexach J, Knight-Johnson A, Khemani P, Fogel BL et al (2019) A family with spinocerebellar ataxia and retinitis pigmentosa attributed to an ELOVL4 mutation. Neurol Genet 5:e357. https://doi.org/10.1212/nxg.0000000000000357
Yoshida K, Hata Y, Kinoshita K, Takashima S, Tanaka K, Nishida N (2017) Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol 133:809–823. https://doi.org/10.1007/s00401-016-1665-7
Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z et al (2001) A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 27:89–93
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. https://doi.org/10.1016/j.neuron.2004.11.005
Acknowledgements
The authors would like to thank M. Higashi and H. Aoki for technical assistance.
Funding
This work was supported by Grants-in-Aid for Scientific Research, Early-Career Scientists from the Japan Society for the Promotion of Science, Japan (JSPS KAKENHI Grant Number JP18K15441) (KO) and 2018 Novartis Research Grants from Novartis Pharma KK (KO). Neither JSPS or Novartis Pharma KK had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
KO conceptualized and initiated the study, performed clinical and neuropathological examinations, acquired funding, curated data, supervised the project, and participated in writing the manuscript. TI managed the patient, did project administration, conceptualized the study, and conducted clinical assessment and data curation. TU conceptualized the study, performed data curation and supervision, and substantially contributed to detailed neuropathological evaluation and analysis. AY conducted neuropathological examinations, contributed to the methodology, and curated the data. AN conducted all special neuropathological staining and data curation. TM managed the patient and did clinical assessments. SI conducted clinical assessments and data curation. HS did neuropathological examinations and data curation. MY conducted general pathological assessments. YT conducted general pathological assessment and management of pathological specimens. YO conducted data curation, assisted funding acquisition, and supervised the study. KI has initiated the study and conducted conceptualization, clinical assessment, neuropathological examination, data curation, supervision. KI also contributed substantially to writing the manuscript. TY conducted data curation and supervision of the whole study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted under permission from institutional review boards in Tokyo Medical and Dental University, Japan (G2000-198). A written consent was obtained from the patient’s family for this study.
Consent for publication
Consent for publication of this study was provided by the family of the deceased patient.
Competing interests
KO received funding (2018 Novartis Research Grants) from Novartis Pharma KK. Other authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Additional characterization of the pontine pathology in SCA34. Description of data: Contains additional images and legends, describing additional staining of the pontine pathology in SCA34.
Additional file 2
: Description of data: Contains additional images and legends, describing abnormal vacuoles in the brain and spinal cord of the SCA34 patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ozaki, K., Irioka, T., Uchihara, T. et al. Neuropathology of SCA34 showing widespread oligodendroglial pathology with vacuolar white matter degeneration: a case study. acta neuropathol commun 9, 172 (2021). https://doi.org/10.1186/s40478-021-01272-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01272-w