
Abstract
Background
Trypanosoma conorhini and Trypanosoma rangeli, like Trypanosoma cruzi, are kinetoplastid protist parasites of mammals displaying divergent hosts, geographic ranges and lifestyles. Largely nonpathogenic T. rangeli and T. conorhini represent clades that are phylogenetically closely related to the T. cruzi and T. cruzi-like taxa and provide insights into the evolution of pathogenicity in those parasites. T. rangeli, like T. cruzi is endemic in many Latin American countries, whereas T. conorhini is tropicopolitan. T. rangeli and T. conorhini are exclusively extracellular, while T. cruzi has an intracellular stage in the mammalian host.
Results
Here we provide the first comprehensive sequence analysis of T. rangeli AM80 and T. conorhini 025E, and provide a comparison of their genomes to those of T. cruzi G and T. cruzi CL, respectively members of T. cruzi lineages TcI and TcVI. We report de novo assembled genome sequences of the low-virulent T. cruzi G, T. rangeli AM80, and T. conorhini 025E ranging from ~ 21–25 Mbp, with ~ 10,000 to 13,000 genes, and for the highly virulent and hybrid T. cruzi CL we present a ~ 65 Mbp in-house assembled haplotyped genome with ~ 12,500 genes per haplotype. Single copy orthologs of the two T. cruzi strains exhibited ~ 97% amino acid identity, and ~ 78% identity to proteins of T. rangeli or T. conorhini. Proteins of the latter two organisms exhibited ~ 84% identity. T. cruzi CL exhibited the highest heterozygosity. T. rangeli and T. conorhini displayed greater metabolic capabilities for utilization of complex carbohydrates, and contained fewer retrotransposons and multigene family copies, i.e. trans-sialidases, mucins, DGF-1, and MASP, compared to T. cruzi.
Conclusions
Our analyses of the T. rangeli and T. conorhini genomes closely reflected their phylogenetic proximity to the T. cruzi clade, and were largely consistent with their divergent life cycles. Our results provide a greater context for understanding the life cycles, host range expansion, immunity evasion, and pathogenesis of these trypanosomatids.
Similar content being viewed by others


Background
The class Kinetoplastea includes a broad spectrum of free-living and parasitic protists [1], all of which display unique features including trans-splicing, polycistronic transcription and RNA editing [2]. Trypanosoma cruzi is obligately parasitic, exhibits a broad mammalian host range, and is believed to have first infected and caused Chagas Disease in humans when the New World was populated ~ 15,000 years ago [3]. Usually spread by fecal contamination from an infected reduviid bug, the parasite replicates as intracellular amastigotes in a broad array of cell-types in its mammalian hosts [4]. It replicates as epimastigotes in the gut of its insect vectors, i.e. hemipterans of Triatominae such as species of the Rhodnius, Triatoma and Panstrongylus genera [5]. Clonal divergence [6, 7] and genetic exchange [8,9,10], have given rise to widely heterogeneous populations, termed Discrete Typing Units (DTU’s) TcI-TcVI and Tcbat (c.f. [11]). It is now generally believed that T. cruzi is a recent descendant of a phylogenetic lineage of closely related species of Trypanosoma tightly linked to bats [12, 13].
T. conorhini and T. rangeli are members of a phylogenetic group generally considered to be most closely related to the clade comprising T. cruzi and bat trypanosomes (T. cruzi-like) of the subgenus Schizotrypanum. Because of their phenotypes and the limited genetic information previously available, these two species of trypanosomes have been considered to occupy a phylogenetic position between the T. cruzi-like species and the African trypanosomes related to T. brucei. However, their close phylogenetic grouping is surprising given their strikingly different lifestyles. T. conorhini is spread to hosts in the feces of its vector the reduviid bug Triatoma rubrofasciata after replication in the insect gut [14, 15]. It is transmitted to a restricted host range in rats, where it causes a mild and transient infection, although it has also been reported to infect mice and non-human primates in the laboratory [15, 16]. The parasite and its vector are tropicopolitan, and there is a strong association of Tr. rubrofasciata with rats [14, 16]. In contrast to T. cruzi and like T. conorhini, T. rangeli exhibits an apparently exclusively extracellular lifestyle in its mammalian hosts. T. rangeli, like the African trypanosomes transmitted by tsetse flies, replicates as metacyclic trypomastigotes in the salivary glands of triatomine of the genus Rhodnius [17, 18], and is transmitted by a bite from an infected vector [19,20,21]. T. rangeli exhibits antigens in common with T. cruzi, and likewise is widely distributed in Central and South America with a broad mammalian host range that includes humans [18]. Five phylogenetic lineages of T. rangeli have been identified; TrA, C, D and E are phylogenetically close, but TrB (which includes the AM80 strain reported herein) is a more divergent lineage positioned basal to the clade formed by all lineages of T. rangeli [22,23,24,25,26,27]. T. rangeli isolates and local vectors have apparently co-evolved [18, 23, 24], with consequent parasite lineage association with Rhodnius complexes [18, 23,24,25, 28]. Infection of mammalian hosts by T. rangeli is non-pathogenic and induces low parasitaemia, but can persist for years [29]. Mammalian host-parasite interaction mechanisms remain largely unclear for both T. conorhini and T. rangeli.
Because of their taxonomic positions and diverse lifestyles, these parasites present an opportunity to identify the genetic bases of their differing abilities to invade cells, evade host immune responses, and cause disease, and their diverse host ranges and life cycles in mammals and vectors. Comparisons of dixenous trypanosomatids to free-living bodonids have suggested that most differences lie within genes encoding metabolic and surface proteins [30]. Genome analysis of Leishmania major Friedlin, Trypanosoma brucei TREU 927 and T. cruzi CL Brener [31,32,33,34], studies of lineage-specific features in T. cruzi Sylvio X10/1 (TcI) and T. cruzi CL Brener (TcVI) [35], and comparisons of T. cruzi and the bat-restricted T. cruzi marinkellei [36] suggest many differences are associated with differential multigene family expansion. More recently, a sequence draft of T. rangeli SC-58, a representative of the TrD lineage isolated from rodents and never found in humans [37], was presented [38]. The T. rangeli AM80 strain was isolated from a human source in the Amazon [39], where the TrB lineage, the basal and most divergent of all known T. rangeli lineages, is highly prevalent. Lineages TrA, prevalent from the northwestern region of South America (including Brazilian Amazonia) to Central America, and TrC spanning from the west of the Andes to Central America, have also been found in humans. Lineages TrD and TrE were rarely reported and so far only isolated from wild mammals and triatomines [22,23,24, 28, 40, 41]. Study of a TrB strain, e.g. AM80, would likely present one of the most topical and timely comparisons to other trypanosomatid groups in terms of relevance to human infection. Moreover, the ongoing rapid development of the Brazilian Amazon is likely to impact transmission of both T. rangeli and T. cruzi to humans, which are commonly co-infected with both parasites [23, 26, 40].
In this work, we sequence and compare the genomes of T. rangeli AM80, T. conorhini 025E, T. cruzi G (TcI) and T. cruzi CL (TcVI, a clone from the same parental strain as the published CL Brener strain [32]) (see Additional file 1: Table S1 for strain information). These two T. cruzi isolates present a disparate range of characteristics: T. cruzi G isolated from a marsupial, displays low parasitemia in vivo [42] and induces chronic infection of low virulence in mice [42], exhibits higher susceptibility to interferon-γ [43], and has a lower ratio of cruzipain to chagasin [44]. T. cruzi CL was derived from a triatomine bug captured in the residence of a chagasic person [45], is likely a hybrid of isolates from TcII/III lineages [9, 10, 46,47,48], and exhibits high parasitemias and virulence. T. cruzi G uses mucin-like glycoproteins to facilitate cell invasion, while the CL strain uses the stage-specific gp82 surface molecule and cruzipain [49, 50].
Our analyses of the sequences of these four parasites suggest disparate assembled genome sizes ranging from ~ 21–65 Mbp and extend previous observations that T. cruzi CL is a hybrid strain [11, 32, 48]. In contrast to many T. cruzi strains, we found no evidence of hybridization in the genomes of T. rangeli and T. conorhini. By comparing the genomes of these three species we aimed to infer how these genomes have evolved since their last common ancestor and to gain insight into the selective pressures acting upon them. Our data have led us to consider the hypothesis that higher levels of heterozygosity in protein-coding genes of T. cruzi CL impart an adaptive advantage. We further hypothesize that lower diversity in multigene families, and gene clusters defined by sequence similarity, may help explain the more restricted host range of T. conorhini 025E. Our results lay the groundwork for further studies to elucidate the genetic basis for the phenotypic differences among these closely related kinetoplastid taxa.
Results and discussion
Genome assemblies and molecular karyotypes
Molecular karyotypes
Pulsed Field Gel Electrophoresis (PFGE) under multiple conditions provided an estimate of the sizes and numbers of chromosomes in the genomes of T. conorhini 025E, T. rangeli AM80, T. cruzi G, T. cruzi CL, and an additional isolate, T. conorhini 30028, obtained from ATCC (Fig. 1, Table 1, and Additional file 1: Table S2). T. conorhini ATCC 30028 was isolated in Hawaii in 1947 from Triatoma rubrofasciata. Pixel intensity and area under the curve (“volume”) of each band were plotted against the distance migrated in the gel and compared to standard curves of presumed single-copy diploid chromosome band volumes to provide an estimate of the copy number of each chromosome. Bands with estimated areas that were half of that expected for a chromosome pair were found in all species and were assumed to be due to size differences in chromosome pairs, as has previously been observed in T. cruzi [51,52,53,54,55]. NGS estimates for T. cruzi genome size are given separately for its two haplotypes (Esmeraldo-like and Non-Esmeraldo-like).

Karyotypes from three PFGE runs. 1% Megabase agarose gels (Bio-Rad) were loaded with agarose plugs bearing lysates of ~ 1 × 107 epimastigotes of each trypanosomatid strain for electrophoresis at 13.5 °C using the CHEF DR III System (Bio-Rad). Run conditions used for karyotyping each species were based empirically on their individual distributions of chromosome sizes. For separation of smaller chromosome size ranges, we used the following program - Block 1: 5 V/cm, 20–200 s, 18 h, 120°. Block 2: 3 V/cm, 200–300 s, 32 h, 120°. Block 3: 1.5 V/cm 500–1100 s, 12 h, 120°. The program used for separation of the largest chromosome size ranges was as follows - Block 1: 2 V/cm, 1500 s, 12 h, 98°. Block 2: 2 V/cm, 1800 s, 12 h, 106°. Block 3: 3 V/cm, 500 s, 38 h, 106°. Block 4: 5 V/cm, 20–200 s, 23 h, 120°. Block 5: 3 V/cm, 200–400 s, 34 h, 120°. (a) T. rangeli AM80 vs. T. conorhini 025E using Saccharomyces cerevisiae chromosome size-markers (Bio-Rad). (b) T. conorhini 30028 vs. T. conorhini 025E. (c) T. cruzi G vs. T. cruzi CL. Schizosaccharomyces pombe, Hansenula wingei and Saccharomyces cerevisiae chromosomes (Bio-Rad) were used as markers for (b) and (c)
These analyses suggest that whereas T. cruzi CL and G each bear ~ 37 chromosomes, similar to that previously estimated for T. cruzi CL Brener [52]. T. rangeli AM80, T. conorhini 30028, and T. conorhini 025E have a slightly greater number; i.e. 40, 39.5 and 45 chromosomes, respectively. Although the chromosome numbers of the G and CL strains are quite conserved, the sizes of the individual chromosomes are not, following a trend previously predicted for T. cruzi strains [52]. Previous studies have revealed significant variation in PFGE patterns specific to distinct lineages of T. rangeli [26, 56]. Genome sizes determined as described (Table 1, Additional file 1: Table S2) are estimates, but closely match estimates from sequence analysis.
As expected, significant genome variability was observed among these karyotypes, although the two T. conorhini isolates show similar banding patterns (Fig. 1). Clearly, major chromosomal rearrangements, expansions, or deletions seem to have occurred during the evolution of these parasites. Interestingly, the two T. cruzi strains appear to have at least double the number of megabase-sized chromosomes as T. conorhini or T. rangeli, and although the latter parasites have more chromosomes than the T. cruzi strains, their overall genome sizes are reduced. Repeat expansions in individual chromosomes have previously been invoked to describe karyotype polymorphism across T. cruzi strains [53, 55, 57]. Despite these differences we found single copy ortholog genes to be highly syntenic in these genomes (data not shown).
Genome characteristics
Each of these genomes was sequenced as described in the Methods and analyzed for completion and integrity using an in-house genome completion assessment pipeline called GenoCIA, which demonstrated that all or nearly all of the genes from each of these organisms are represented full-length and intact in our assemblies (Additional file 2: Figure S1). The results are summarized in Table 2 and Additional file 1: Table S3.
The genome assembly sizes of these organisms range from ~ 21 Mbp for T. conorhini and T. rangeli to 25–65 Mbp for T. cruzi G and CL, respectively. Discrepancies between the assembly size in Table 2 and the estimated genome size from Table 1 were observed. As previously reported for the genome of T. cruzi CL Brener [32], this discrepancy has been ascribed to the collapse of near-identical repeats into fewer copies in the assembly. The genomes reported herein likewise contain such highly repetitive sequence. The genomes of T. cruzi G, T. conorhini, T. cruzi CL Esmeraldo-like and T. cruzi CL Non-Esmeraldo-like each exhibited collapsed repeats, with ~ 20–30% of all bases in their assemblies exhibiting > 1.5X, 5–15% >3X, and 0.5–0.8% > 10X coverage relative to the average coverage of their single copy orthologs. T. rangeli, which had the least discrepancy between assembly size and estimated genome size, had values of 12% > 1.5X, 4% >3X and 0.6% >10X the coverage of its single copy orthologs, and for T. cruzi CL Unassigned, which had the most discrepancy between the two sizes, these values were significantly higher, at 44%, 27% and 4%, respectively. Thus, we assume that these repetitive sequences largely explain the discrepancy between the genome assembly size and the estimated genome size from PFGE.
The GC content of T. conorhini was slightly higher at ~ 57% than for the other three genomes, which ranged from ~ 50–52%. The number of genes in T. conorhini and T. rangeli (~ 10,000) is less than the number in T. cruzi G or CL (13,000), and is largely consistent with that observed in other kinetoplastid protozoa [31,32,33] (see Additional file 1: Tables S4−7 for all genes and their annotation). T. cruzi, however, is recognized for having many large multigene families [32, 35] likely explaining the expanded repertoire in the G and CL strains. Additionally, the CL strain is considered a hybrid [48], with a larger genome size than typically found in TcI strains [57, 58], consistent with our observations of genome size and gene content.
The genomes of each of these organisms apparently contain genes required for meiosis, suggesting likely capacity for sexual reproduction (Additional file 1: Table S3). Counts of glycosylphosphatidylinositol (GPI) anchored proteins and proteins with transmembrane domains are very similar between T. conorhini 025E and T. rangeli AM80, and highest in the T. cruzi strains. 18S rRNA copy numbers show excellent agreement between bioinformatics estimates and measurements by quantitative PCR. We estimated that there are 4–7 copies in T. cruzi G and 2–3 copies in T. cruzi CL. The T. cruzi CL Brener genome assembly contains 12 fragments of 18S rRNA genes in TriTrypDB [59] v.28, all less than half the size of the predicted 18S genes from the CL and G strains (~ 2300 bp). Estimates based on sequence alignment to a 186 bp highly conserved region of the spliced leader gene transcript, suggested that the genomes of T. cruzi G, T. cruzi CL, T. rangeli AM80, and T. conorhini 025E have 66, 82, 44 and 13 copies of the spliced leader gene, respectively.
Sequence identity and phylogenetic analysis
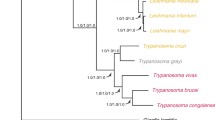
The relationships among these parasites and their phylogeny were explored using 139 single copy orthologs identified as outlined in the Methods from each of these parasites and six closely related trypanosomatids (Fig. 2, Additional file 1: Tables S8 and S9). Our analyses showed that T. conorhini 025E and T. rangeli exhibited ~ 84% identity to each other, and only 77% identity to T. cruzi isolates. Percent nucleotide identity between the TraB (AM80) and TrD (SC58) isolates of T. rangeli was 92%. As expected, the highest identities observed, i.e., 94–98%, were between the T. cruzi isolates, with two DTU I isolates, G and Sylvio, being the most similar, aside from T. cruzi strains CL and CL Brener, which are clones from the same strain and exhibit near 100% identity (not shown). Our observations support previous reports of T. brucei and T. cruzi marinkellei B7 percent identities to strains of T. cruzi [34, 36]. Interestingly, the ~ 92% percent identity between T. rangeli AM80 and T. rangeli SC-58 was similar to the results comparing T. c. marinkellei and the T. cruzi strains.

Maximum Likelihood tree from 139 aligned and concatenated amino acid sequences. Support values are calculated from 1000 bootstrap replicates, all bootstrap values were 100% and thus not displayed. Scale bar indicates mean number of substitutions per site. A break in the branch to T. brucei was used to aid visualization since the branch was too long to display
Phylogenetic analysis (see Methods, and Fig. 2) using amino acid sequences of these 139 orthologs confirmed previous phylogenetic analyses based on a few genes [12, 13, 26,27,28], showing a close relationship between T. rangeli and T. conorhini, and a greater evolutionary distance between them and T. brucei clades than the distance between the T. cruzi and T. brucei clades. As expected, the T. cruzi strains are the most closely related, with the closest relationship between the two DTU I isolates, G and Sylvio. The subspecies T. cruzi marinkellei is clearly divergent from the T. cruzi strains. Given the relatively higher number of genomes available within the T. cruzi clade, a greater taxon sampling of genomes closely related to T. conorhini, T. rangeli and species more closely related to the T. brucei clade, which are not yet available, would have provided a more accurate and complete phylogenetic reconstruction.
Gene cluster diversity
Our OrthoFinder analysis suggested that the majority of orthogroups are represented by single genes for each organism present (i.e. absence of paralogs), although the number of clusters containing two paralogs is higher in T. cruzi CL (Additional file 2: Figure S2). Interestingly, the percentage of clusters containing genes annotated as surface proteins was generally proportional to the number of predicted copies in the cluster (Additional file 2: Figure S2), consistent with previous observations that surface protein genes exposed to immune surveillance are often highly repetitive [32, 38, 60,61,62]. Examining the species represented in each gene cluster (Fig. 3) identified 7268 gene clusters common to all four of the genomes. In contrast, there were 11,418 clusters unique to either the G or CL strains of T. cruzi, and 12,781 gene clusters unique to T. cruzi. T. cruzi CL alone exhibited 10,487 unique clusters, probably due to its hybrid genome. T. rangeli AM80 shares a total of 7655 clusters with the two T. cruzi strains, and T. conorhini 025E shares 7810. These results are suggestive that the genomes of T. rangeli and T. conorhini have undergone less gene amplification and divergence than has been reported for T. cruzi strains [34].

Sequence diversity and functional enrichment across clustered genes. Called genes from each species were clustered using OrthoFinder v.0.7.1. This yielded 23,337 clusters, shown here using draw.quad.venn from the VennDiagram package of Rstudio v.3.0.2, together with percent hits to TriTrypDB v.24, KOG or PFAM databases in parentheses. Circles with solid lines indicate singleton genes (no paralogs or orthologs, i.e. classified as a ‘cluster of one’ for this analysis), ovals with dashed lines represent clusters of genes that have orthologs and paralogs
The most common BLASTp hits for the T. rangeli AM80 paralog-containing clusters include hypothetical proteins and the retrotransposon hotspot protein (RHS). For T. rangeli AM80 singletons, the most frequent possible homologs in NCBI’s non-redundant (nr) protein database are hypothetical proteins (208 clusters), trans-sialidase or T. rangeli sialidase (24 clusters), gp63 (20 clusters), and adenylate cyclase (10 clusters). The latter is important in the differentiation process of T. cruzi [63].
Around 20% of the 10,487 T. cruzi CL strain-specific gene clusters are from trans-sialidase, RHS, dispersed gene family 1 (DGF-1), mucin-associated surface protein (MASP), mucin and gp63 multigene families. It is interesting to note in T. rangeli AM80 the complete absence of BLAST hits <1e-5 for MASP and DGF-1 for singletons and clusters containing paralogs, and much fewer mucin and RHS hits than for T. cruzi CL. This result implies that in T. rangeli AM80 the MASP and DGF-1 genes have been lost or have diverged significantly. T. cruzi G, as for T. cruzi CL, contains many clusters with hits to trans-sialidase and RHS family members. The presence of many surface protein genes in the species-specific categories of T. rangeli AM80 and T. cruzi potentially contributes to their wide host range and ability to sustain infection in the mammalian host.
Multigene family copy number
Like previous reports about T. cruzi [35, 36, 64], both T. rangeli AM80 and T. conorhini have variable representations of genes in multigene families (Fig. 4 and Additional file 1: Table S10). We found trans-sialidase (TS) and GP63 genes are highly expanded in all genomes we examined herein. The TS family genes, which encode proteins that are linked to the cell membrane via GPI anchors, are very heterogeneous and form eight known groups [65,66,67]. The enzyme in T. cruzi transfers host sialic acids to parasite cell surface ligands, presenting a decoy to the host immune response and participating in the adhesion and internalization of the parasites into host cells [61, 68,69,70]. T. rangeli has a Group II sialidase that is a strict hydrolase lacking the ability to transfer sialic acid [38, 65, 71,72,73]. In both species, TS Group II enzymes likely participate in host cell adhesion and invasion, but for T. rangeli this activity is probably not required in the mammalian host, but may be relevant in the triatomine vector [71, 73]. The sequences of T. conorhini TS were the most divergent compared to those from T. cruzi, T. cruzi-like species and T. rangeli [65]. The findings from this and previous studies uncovering TS genes in all Trypanosoma species, many of which do not invade mammalian cells, suggest that, in addition to participation in host cell invasion and intracellular survival, TS may play other roles in parasite development, e.g. in their arthropod vectors [64]. GP63 proteins are zinc-dependent metalloproteases that are highly expressed in T. cruzi amastigotes, where they contribute to cell infection [62, 74]. This activity is consistent with our observation that pathogenic T. cruzi CL has the highest number of copies of this gene (~ 211), similar to the 174 copies predicted in T. cruzi CL Brener [32, 75]. However, the role(s) of GP63 in T. conorhini and T. rangeli are unknown.

Multigene family copy numbers. Selected major multigene families shown are amastin, β-galactofuranosyl transferase (GALFT), surface protease GP63, retrotransposon hot spot (RHS) protein, mucin-associated surface protein (MASP), trans-sialidase (TS), and dispersed gene family protein 1 (DGF-1). Centers of plots represent 1 copy (0 in log10) and successive concentric circle values are shown by the log10 scale bar on the left
The most striking differences in copy number across the species are arguably in the mucin-associated surface protein (MASP) and dispersed gene family 1 (DGF-1) families, which are significantly less amplified in T. rangeli and T. conorhini than in the T. cruzi strains. In T. cruzi, MASP genes are often found in clusters with mucin and other surface protein genes [32], and the protein is localized to the surface of infective forms of the parasite [60]. Polymorphism of MASP amino acid sequence is high, which likely contributes to immune system evasion [60] and the ability to infect multiple cell types [64, 76, 77]. Thus, the smaller size of the MASP gene family in T. conorhini 025E and T. rangeli AM80 (and T. rangeli SC-58 [38]) may be related to their lack of host cell infectivity, and their inability to induce acute infections with high levels of parasitemia or long chronic infections. DGF-1 is less well represented in T. rangeli AM80 than in the other species, and shows lower diversity in gene cluster analysis in this study. Possession of only 16 copies of DGF-1 may contribute to the obligate extracellular nature of T. rangeli AM80 in the mammalian host, since this protein has been implicated in the ability of parasites to bind to extracellular matrix proteins of host cells [78]. T. rangeli SC-58 was previously estimated to have over 400 copies of this gene, despite less than 20 partial DGF-1 genes being annotated with genome coordinates [38]. If the latter copy number estimate is accurate, there is a striking inter-strain difference. T. conorhini, bearing only 23 copies, also shows significantly reduced numbers of DGF-1 compared to T. cruzi, which is likewise consistent with an extracellular lifestyle.
Cruzipain, a key player in cell invasion, and GALFT, which is involved in GPI anchor biosynthesis, are also highly differentially expanded. We find no evidence of cruzipain expansion in T. rangeli AM80 or T. conorhini 025E, although cruzipain homologs are present in these genomes. In T. rangeli, the homolog is known as rangelipain and is present in tandem repeats [26]. Amino acid identities for these genes are 76% between T. rangeli AM80 and T. conorhini 025E, 71% between T. conorhini 025E and the T. cruzi strains, and 69% between T. rangeli AM80 and the T. cruzi strains. We previously inferred network genealogies showing that cruzipain sequences of all DTUs of T. cruzi clustered tightly together and closer to T. c. marinkellei than to T. dionisii (T. cruzi-like species), but differed from homologs of T. rangeli and T. brucei, revealing DTU- and species-specific polymorphisms [79]. Cruzipain precursors are activated upon removal of the N-terminal prodomain, resulting in proteins linked to the invasion process that are thought to play a larger role in T. cruzi CL, where expression levels are higher during infection, than T. cruzi G [44]. However, we do not see a notable expansion of cruzipain precursors in the CL strain, which has ~ 50 copies, compared to ~ 38 copies in T. cruzi G.
We find a lower copy number of amastin in T. conorhini 025E (~ 3 copies) compared to T. rangeli AM80 (~ 18 copies), T. cruzi G and CL (~ 8–26 copies), and T. cruzi CL Brener (14 copies) [80]. Although the exact function of amastins remains unclear, they are thought to be abundantly expressed on the surface of intracellular T. cruzi amastigotes and apparently support intracellular survival [64, 81,82,83,84,85]. Since amastin is expressed in the intracellular mammalian amastigote stage of the parasite’s life cycles in T. cruzi and Leishmania, finding expansion of this immunogenic gene family in the extracellular T. rangeli AM80 (also previously reported in T. rangeli SC-58 [38]) was unexpected.
Motif analysis shows that the conserved amastin signature sequence of C-[IVLYF]-[TS]-[LFV]-[WF]-G-X-[KRQ]-X-[DENT]-C, which may be critical for amastin function [86], is present in all the species examined (Additional file 1: Table S11). Additionally, we found a motif, with consensus EAKKPAGSNEESPMSREALS, tandemly repeated 6 and 3 times respectively in two of the eight amastin genes analyzed from T. rangeli AM80. The function of this repeat is unknown, although we postulate that repeats may aid recombination and antigenic reshuffling associated with evasion of the host immune system [87]. Kinetoplastid Membrane Protein-11 (KMP-11) is encoded in Leishmania, T. brucei and T. cruzi. The observation that the KMP-11 genes are expanded in T. rangeli SC-58 [38] represented an unexpected result in this non-pathogenic strain. This finding is more unusual given that the gene is found in low numbers across other trypanosomatids [88, 89] and in our analysis we find just one copy in T. conorhini 025E, T. rangeli AM80 and T. cruzi CL, and none in T. cruzi G.
Finally, mucin, a family thought to confer immune system protection [61, 90], contains highly variable regions that make copy number estimation challenging. T. rangeli and T. conorhini appear to contain mostly mucin-like glycoproteins and little of the diversity of other mucin subgroups that is typical of T. cruzi, concurring with reports in other T. rangeli strains [91, 92]. The low gene copy numbers within this gene family in these two species are also consistent with previous genomic [38] and transcriptomic [72] data from T. rangeli, and likely contribute to their inability to invade mammalian cells [64]. Although likely underestimated here, we observe a larger gene family in T. cruzi CL compared to the other species, presumably contributing to the poorer immune system clearance of this strain.
Pseudogenes
Pseudogenes are defined herein as genes bearing in-frame stop codons or frameshifts, as well as the absence of features required for gene calling based on a non-supervised training model, such as upstream functional sites, start codons, nucleotide and amino acid composition, and length to the first in-frame stop codon. The number of putative pseudogenes predicted in the T. conorhini, T. rangeli, T. cruzi G, and T. cruzi CL genome assemblies were 113, 434, 942 and 2376, respectively (Additional file 1: Tables S12–15). The latter equates to 18% of total gene predictions (gene calls per haploid genome plus pseudogenes) in the CL strain. The T. cruzi CL Brener genome was previously estimated to have 3590 pseudogenes, or ~ 16% of all its genes. Over 2000 of these were attributed to large multigene families [32]. We analyzed the NCBI nr annotated functions of our panels of predicted pseudogenes and found over 300 copies of putative pseudogenes from multigene families in T. cruzi CL. In both T. cruzi G and CL, the most frequent putative pseudogenes were of the trans-sialidase, RHS and MASP gene families, and hypothetical proteins.
The pseudogenes in these genomes may provide a repertoire of genetic information for producing variation, especially in multigene-families. T. cruzi was the first species in which a tandem array of pseudogenes, consisting of six mucin genes each with an in-frame stop codon, was discovered [93]. These were postulated to be selectively maintained in the genome, possibly to generate mucin gene diversity. A diversifying role has been suggested for the numerous pseudogenes of variable surface glycoproteins (VSGs) in Trypanosoma equiperdum and African trypanosomes that undergo rapid antigenic variation through gene recombination [94,95,96,97]. Additionally, TS gene and pseudogene organization, flanked by RHS genes at subtelomeric regions, in strain CL Brener is reminiscent of regions next to T. brucei VSG genes [98]. Pseudogenes could also play a role in post-transcriptional control of gene expression. Some pseudogenes transcribed in T. brucei have been proposed to participate in RNAi-based natural antisense suppression [99]. The genes responsible for RNAi machinery are absent in all strains of T. cruzi examined to date and may only be present as pseudogenes in T. rangeli SC-58, but are present and intact in both T. rangeli AM80 and T. conorhini 025E [100]. Analysis of the transcriptional activities and structural organization of these pseudogenes is beyond the scope of this study, but may clarify their roles in generation of protein diversity or post-transcriptional regulation.
Heterozygosity
The four organisms described herein are thought to be primarily diploid, although some T. cruzi strains, e.g., CL and CL Brener, are hybrid strains in which ploidy is less well defined [58, 101, 102]. Thus, we examined levels of apparent heterozygosity in these strains using a set of 6394 conserved single copy orthologs, covering ~ 9 million sites in each genome. The number of heterozygous genes with at least one SNP varied from ~ 42% in T. cruzi G to ~ 88% in T. cruzi CL, whereas T. conorhini and T. rangeli have an intermediate 55–60% of apparently heterozygous genes. The average percent of heterozygous bases varied from ~ 0.1–0.3% in T. rangeli, T. conorhini and T. cruzi G to ~ 1.6% in T. cruzi CL (Fig. 5a and b). The low percentage of heterozygous positions for T. conorhini, T. rangeli and T. cruzi G are close to estimates of heterozygosity in T. c. cruzi Sylvio X10 (~ 0.22%) and T. c. marinkellei B7 (0.19%) [36], although those analyses were not restricted to single-copy orthologs, which may have positively biased their estimates. The high level of heterozygosity in T. cruzi CL is very likely mostly due to the fact that it is a hybrid in which a significant fraction of its genes are derived from two distantly related progenitors. The distributions of heterozygous genes falling into discrete mean levels of percent heterozygous positions were unimodal for all species (Fig. 5b), suggesting that the genes examined were not of biased origin. We found < 0.1% tri-alleles, and 0% tetra-alleles at the heterozygous sites of each organism, consistent with the genomes being largely diploid.

Heterozygosity of single copy orthologs. (a) Summary values from 6394 shared single copy ortholog genes. Percent heterozygous genes indicate percentage of genes with at least one heterozygous position, mean percent heterozygous positions were calculated by dividing the number of heterozygous sites by the total number of positions. (b) Histogram showing the distribution of heterozygosity values among heterozygous genes. Red vertical dashed lines represent the mean values
There was no significant enrichment of any KOG categories or enzyme E.C. numbers in genes with high or low heterozygosity values (data not shown). The overlap of highly heterozygous genes from the 6394 orthologs in each species was limited, and we found no evidence of synteny in heterozygosity patterns across contigs in any pairwise species comparison (data not shown). Together, these observations suggest that generation of heterozygosity in these organisms is largely a stochastic process.
T. cruzi displays strong linkage disequilibrium and features of a mainly clonal species [103]. However, the presence of natural T. cruzi hybrids such as those of TcV and TcVI and conservation of meiosis-related orthologs in the genomes suggest the capacity for sexual reproduction. Notably, the genome of T. cruzi G in particular exhibits overall low levels of heterozygosity. These levels do not seem to fit with a strictly clonal model of evolution, where diversity is expected to accumulate independently between alleles in an individual over time (the Meselson effect). Moreover, long-term clonality without mechanisms to attenuate the impact of high mutational load (Müller’s ratchet) would seem to be detrimental to these species.
A comprehensive analysis of heterozygosity in sequences spanning the genomes compared to expected heterozygosity is beyond the scope of the present study. However, our analysis of single copy orthologs identified an apparent mosaic pattern of heterozygosity across these genomes, especially in T. cruzi G and T. rangeli AM80, where continuous regions of homozygosity often exceeding 50 Kbp interspersed with heterozygous clusters were identified (Additional file 1: Table S16). Mosaic heterozygosity has been seen before in Naegleria gruberi [104], and clustering of heterozygosity has also been described in T. c. marinkellei [36]. Precise mechanisms that control heterozygosity in trypanosomes have yet to be elucidated. Many regions of low heterozygosity in all species have average coverage for single copy orthologs (Additional file 1: Table S16). Therefore, loss of heterozygosity via chromosome loss seems unlikely in these species, although this process cannot be ruled out. Mutational hotspots, mitotic recombination, mitotic gene conversion, and segmental duplication are also possible sources of differentially heterozygous regions of T. cruzi G and T. rangeli AM80 chromosomes.
Members of DTU TcVI, including hybrid strain T. cruzi CL, are reported to have a high degree of fixed heterozygosity but low intralineage diversity [105]. The higher heterozygosity originates at least in part from the distances between the Esmeraldo-like (TcII) and non-Esmeraldo-like (TcIII) alleles, provided by the ‘parental strains’ of DTU TcVI. As previously suggested [9], these hybrid genotypes were likely stabilized through long term asexual reproduction. Increased heterozygosity has been linked to hybrid vigor, which has been reported in Leishmania [106], and is consistent with an enhanced host range and the ability to invade cells, replicate, and cause pathogenicity.
Ratios of non-synonymous to synonymous SNPs within the set of single copy orthologs were vastly different across the organisms. Despite having a similar overall number of polymorphic sites, T. rangeli AM80 and T. conorhini 025E had non-synonymous to synonymous SNP ratios of 0.68:1 and 0.9:1 respectively. The lower rate of non-synonymous SNPs in T. rangeli AM80 is perhaps suggestive of greater purifying selection or functional constraint within proteins. The two T. cruzi strains also displayed differences, with a ratio of 0.95:1 for T. cruzi G and 0.8:1 for T. cruzi CL. This suggests that analysis of dN/dS ratios, especially for sites of selected gene groups within each species, would likely be an interesting next step to determine the extent and specificity of selective pressures.
Repetitive elements
The genomes were analyzed for known trypanosome repeats, i.e., non-Long Terminal Repeat (non-LTR) elements, LTR elements, and satellites, using Repbase [107]. Repeat profiles (Additional file 1: Table S3 and S17–20) are similar for T. conorhini 025E and T. rangeli AM80. The most common satellite sequence in all species is SZ23_TC. Retroelements are markedly increased in the T. cruzi strains. Analysis of T. conorhini 025E and T. rangeli AM80 LTR- and non-LTR elements identified only 22 and 35 known trypanosomal elements, respectively. We hypothesize that these organisms may lack “copy and paste” type retrotransposons. To show that fewer repetitive element copies in the genomes of T. rangeli and T. conorhini was not just due to collapse of reads in highly similar repeats, we calculated the average coverage of the de novo repeat finder predictions in each repeat class. T. cruzi CL has coverage estimates close to the genomic average for every repeat class. T. cruzi G and T. conorhini 025E have around two-fold higher average coverage for the non-LTR and satellite sequences than the genomic average, and T. rangeli AM80 has two-fold higher coverage for just non-LTR sequences than the genomic average. The collapse of these highly similar repeats in the assembly may suggest mainly recent repeat expansion, although the numbers are still much lower than in T. cruzi CL. Retroelements may have wide-ranging implications on generation of genomic diversity, and their greater number in T. cruzi may have potentiated antigenic variation in this complex parasite [108]. It is interesting to speculate that the presence or absence of RNAi in these organisms may be connected to retroelement counts, as RNAi has been proposed to have a defensive role against transposable elements [109]. T. conorhini and T. rangeli have RNAi and possess fewer retroelements, whereas the T. cruzi strains lack RNAi and have a large repertoire of retroelements.
Metabolic pathways analysis
We examined the metabolic capacity predicted by the genomes of these organisms (Additional file 1: Table S21). Below, we outline some of the more significant observations from these analyses.
Fatty acid metabolism
Trypanosomatids possess a unique set of elongase enzymes for de novo fatty acid synthesis [110]. T. cruzi amastigotes utilize lipid-dependent energy metabolism [75], but the functional importance of fatty acid oxidation in trypanosomatids is not fully understood. The organisms analyzed herein appear to be capable of synthesizing and oxidizing fatty acids (Additional file 1: Table S21). Glycerol dehydrogenase (EC 1.1.1.6), which is involved in converting glycerol to dihydroxyacetone, is present in T. rangeli and T. conorhini, but absent in the T. cruzi strains. This enzyme was reportedly acquired by lateral gene transfer in Leishmania, Crithidia and Leptomonas spp. [111], enabling the parasites to use glycerol as a carbon source.
Amino acid metabolism
In T. cruzi, amino acids are relevant in energy metabolism [112,113,114,115], host-cell invasion [116], stress resistance [117, 118], and differentiation [115, 119]. Proline, in particular, plays a fundamental role in these processes, including energy support during the parasite’s intracellular stages [115].
T. cruzi, unlike T. brucei, can metabolize D-proline using a putative proline racemase (PRAC). Although the T. conorhini genome bears a PRAC gene, as we have previously described [27] T. rangeli AM80, and strains of all other known T. rangeli lineages [27] contain only a pseudogene for this enzyme. Interestingly, genes for 5-oxoprolinase, which is involved in L-proline metabolism, are absent in T. conorhini 025E and T. rangeli AM80. Analysis of the available genomes of TriTrypDB [59] showed only intracellular-replicating species seem to possess this enzyme, which makes its apparent loss in these two species expected. L-proline metabolism via 5-oxoprolinase produces L-glutamate in the glutathione-mediated stress response pathway. T. rangeli AM80 also lacks enzymes that use oxygen as an acceptor (EC 1.4.3.-), which further limits the pathways available for glutamate synthesis. Absence of these enzymes may shed new light on the recent finding that T. rangeli SC-58 is particularly susceptible to oxidative stress [38]. However, there appear to be alternative pathways to produce glutamate and glutathione in both T. rangeli AM80 and T. conorhini 025E, e.g. glutamate dehydrogenase, which converts α-ketoglutarate to glutamate.
Consistent with previous reports in T. cruzi [120, 121] all of these species lack ornithine and arginine decarboxylase genes, indicating that they are unable to generate putrescine or other polyamines and must salvage them from their hosts. Primary-amine oxidase, which is significant for amino acid metabolism and alkaloid biosynthesis, is absent in T. rangeli AM80. The T. rangeli AM80 and T. conorhini 025E genomes encode branched-chain amino acid aminotransferase, which is required for synthesis and degradation of valine, leucine and isoleucine. That T. cruzi strains lack this gene [31] is interesting since leucine is reported to act as a negative regulator of proline-dependent metacylogenesis [122]. Additionally, the ability of this parasite to use the intact leucine skeleton, presumably obtained from the host [123], for isoprenoid and sterol formation would confer advantages in energy economy [124]. T. cruzi and T. rangeli can interconvert serine and glycine, a capacity not found in T. brucei [31]. T. conorhini 025E, like T. brucei, lacks the glycine hydroxymethyltransferase gene for conversion of glycine to L-serine and tetrahydrofolate or vice versa, although alternative routes exist in this organism for synthesis of these compounds.
Carbohydrate metabolism
Kinetoplastids compartmentalize a variety of enzymes involved in carbohydrate metabolism within organelles known as glycosomes [125]. Glucose is the predominant carbohydrate utilized by T. cruzi [126] and T. brucei [127], although Leishmania spp. and Phytomonas spp. have developed adaptations to metabolize plant-derived carbon sources [31, 128]. Genes for NADP-alcohol dehydrogenase (EC 1.1.1.2), which participates along with other enzymes in acetaldehyde to ethanol interconversion in glycolysis [129], are absent in T. conorhini and T. rangeli, but present in both T. cruzi strains. Several bacterial-type sugar kinases (glucokinase, galactokinase and L-ribulokinase), which contain targeting signals for import into glycosomes, are encoded in all four of the genomes of this study. However, genes for many other sugar metabolism enzymes, i.e. beta-glucosidase, fructuronate reductase, xylulokinase, and mannitol 2-dehydrogenase are only present in T. conorhini and T. rangeli. Proteins encoded by beta-glucosidase genes, for example, convert glucoside to α–D-glucose, and cellulose derivatives cellobiose and 1,4-β-D-Glucan to β –D-Glucose [129, 130]. These observations are consistent with the hypothesis that the latter two parasites are better adapted to environments more enriched in exogenous sugars and complex carbohydrates, an adaptation inconsistent with replication in the glucose-rich bloodstream. A nutritional role of plants in triatomines appears possible given demonstration of Rhodnius phytophagy [131]. Adaptation to vector diet may therefore have played a more important role in the evolution of these species than in T. cruzi, which is interesting given that the latter commonly co-infects the same triatomine vector as T. rangeli and also shares the same vector species as T. conorhini.
Overall metabolic potential
The metabolic potentials of T. rangeli and T. conorhini are more similar to each other than either is to T. cruzi. Each has around 20 differences in enzyme presence/absence compared to T. cruzi. All have complete pathways for glycolysis/gluconeogenesis, mannose metabolism, and pyruvate metabolism, although D-lactate dehydrogenase genes are absent in the T. cruzi strains. Glyoxylate and dicarboxylate metabolism appears deficient in all species, since isocitrate lyase and malate synthase, the two enzymes characteristic of the glyoxylate cycle, are absent. Interestingly, CAAX prenyl protease 1 (STE24 endopeptidase), presumably a membrane-associated protein [132] involved in terpenoid backbone synthesis, is present in the T. cruzi strains, but absent in T. rangeli AM80 (and also the T. rangeli SC-58 assembly of TriTrypDB v.24) and T. conorhini 025E. This gene is widely conserved in eukaryotes and highly diverged from CAAX prenyl protease 2, suggesting lack of redundancy. Terpenoids are precursors of steroids and sterols, possibly suggesting a role in host-parasite interaction [76]. Several genes common to T. rangeli, T. conorhini and the T. cruzi strains i.e. genes encoding galactokinase, glutamate dehydrogenase (NADP), serine acetyltransferase and l-ribulokinase and 2-aminoethylphosphonate-pyruvate aminotransaminase (AEP transaminase), have purportedly been passed to trypanosomes via horizontal gene transfer, and are absent in T. brucei [31]. Genes for aminoethylphosphonate (AEP) offer an alternative to ethanolamine phosphate for linkage of mucins to their GPI anchors [133]. Enzymes for the synthesis of AEP from phosphoenol pyruvate are conserved in all four genomes.
Conclusions
Herein, we showed that genomes of T. rangeli AM80, T. conorhini 025E and T. cruzi strains G and CL, range from ~ 30–70 Mbp and contain between 10,000 and 13,000 genes. We characterized multigene families, the heterozygosity, and pseudogene content of these genomes, and used multi-gene strategies to explore their phylogenetic relationships. Our results show that T. conorhini and T. rangeli have less complex genomes, fewer genes, a decreased representation of multigene families, and fewer pseudogenes, than the T. cruzi strains. These observations generally are consistent with the simpler, non-intracellular lifestyles of these parasites. Genes and gene families, including amastin, MASP, and DGF-1, and others, are represented in these parasites in ways that support their association with pathogenicity, intracellular life cycle and host range. The metabolic potentials of these organisms provide clues as to the basis of these biological capabilities, with T. rangeli and T. conorhini bearing a greater number of enzymes for utilizing complex carbohydrates and glycerol as carbon sources, and displaying highly divergent amino acid metabolism to T. cruzi. Heterozygosity levels suggest less allelic diversity in T. rangeli AM80, T. conorhini 025E and T. cruzi G, than in T. cruzi CL. Phylogenetic distance in substitutions per site between the T. rangeli strains SC-58 and AM80 is about the same as T. cruzi strains to T. c. marinkellei, and the distance of T. rangeli AM80 to the T. cruzi strains is just over twice the distance between T. rangeli AM80 and T. conorhini 025E.
Methods
Parasites and culture
Parasites were obtained from the Trypanosomatid Culture Collection (TCC) at the University of Sao Paulo, the American Type Culture Collection (ATCC), and Nobuko Yoshida (Universidade Federal de São Paulo) as shown (see Additional file 1: Table S1). Parasites were cultured, and DNA was isolated and sequenced essentially as previously described [134]. Briefly, epimastigote form parasites were cultured at 28 °C in liver-infusion tryptose (LIT) medium, supplemented with 20% fetal bovine serum (FBS) with 20 μg/ml hemin for T. rangeli AM80, and 10% fetal bovine serum (FBS) with 10 μg/ml hemin for all other species, and harvested in log phase at ~ 1 X 107/ ml. Total DNA was isolated, and depleted of kinetoplast DNA (kDNA), by gel electrophoresis as previously described [134].
Genome sequencing and assembly
The purified DNA was used to prepare shotgun and 3 Kbp mate pair libraries (8 Kbp mate pair libraries in the case of T. cruzi CL) for sequencing on the Roche 454 GS FLX+ platform as indicated by the manufacturer. Reads aligning with a minimum of 50% identity and over 50% length to kDNA from TriTrypDB were removed, and only those reads with at least 70% bases with a PHRED quality score greater than 25 and a minimum read length of 40 bp were kept using NGS QC toolkit [135] version 2.3. This yielded ~ 3–5 million reads for each organism, with average read lengths of 330–360 bp (Additional file 1: Table S3). Assembly was performed using the Newbler version 2.9 assembler (Roche, Inc.), which limits the size of scaffolds to a minimum of 2 Kbp. Hence, all contigs larger than 500 bp that were not part of any scaffold were appended to the scaffolded assemblies for completeness. The highly repetitive and heterozygous T. cruzi CL genome was assembled using the T. cruzi CL Brener assembly from TriTrypDB v.24 as a reference. Reads were mapped to the entire T. cruzi CL Brener genome with BWA v.0.7.12 [136], and reads aligning to each haplotype were extracted for use in individual haplotype assembly runs. Contigs less than 500 bp in length were removed from the final assemblies. Reads were realigned to the final assemblies using BWA [136] and the average genome-wide coverage was calculated to range between 14-50X, depending on the strain (Additional file 1: Table S3). The in-house tool Genome assembly Completion and Integrity Analyzer (GenoCIA) was used to estimate assembly completion and gene calling integrity. This tool performs two tasks: (i) randomly selects 2, 4, 6, 8, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 99% of the reads and performs assemblies using Newbler with these read subsets; and (ii) uses tBLASTn to determine the presence of a curated set of 2217 kinetoplastid orthologous single copy genes at 25, 50, 75, 90 and 99% alignment lengths (merging reference gene alignment lengths over multiple contigs or genes where necessary). BLASTn was used to further characterize whether these genes were complete or fragmented on contigs or gene calls. The general characteristics of these genomes were determined using an in house Genome Annotation Pipeline (GAP). Briefly, genes were called using GeneMarkS v.4.7b [137]; tRNAscan-SE [138] v.1.23 was used to detect tRNAs; and 5S/18S/28S sequences were detected using RNAmmer [139] v.1.2. SignalP [140] v.4.1 (default settings) identified signal peptides and anchors in called genes. TMHMM [141] v2.0 (default settings) determined genes with at least one transmembrane domain. KOHGPI v.1.5 of GPI-SOM [142] was employed with the default training set and settings to predict genes with GPI anchors. BLAST [143] searches against Pfam [144], KOG [145], TriTrypDB and NCBI’s nr databases were performed to determine validity and integrity of the gene calls, and ascertain probable gene functions and inferred annotations. Collapse of repetitive sequences in the assemblies was assessed from the Newbler assembler coverage histogram, within the Newbler output file 454NewblerMetrics.txt.
Molecular karyotypes
Genomic DNA isolation and pulsed field gel electrophoresis (PFGE) were performed essentially as previously described [55]. See Fig. 1 for run conditions. Band sizing based on standard curves of marker chromosome migration and densitometry for each gel was performed using GelAnalyzer v. 2010a [146], with rolling ball background subtraction. Briefly, we obtained the “volume” of each presumed single diploid chromosomal band by multiplying pixel area by the sum of the pixel intensities within the boundary assigned to the band, and then adjusted for background pixel intensity to get “adjusted volume.” A standard curve of adjusted volume vs. size using marker chromosomes was used for reading off the expected volume of each observed band of a specific size. This was then compared to the actual volume to get diploid chromosome copy number, assuming a linear correlation between copy number and volume at a specific band size.
18S rRNA copy number via qPCR
qPCR and the relative threshold algorithm on ViiA 7 (Thermo Fisher Scientific) were employed using MGB primers and probes specific for conserved regions of the18S gene in each species where the highest number of reads mapped (Additional file 1: Table S22), and DHFR as a single copy reference gene for normalization. Estimations were taken at two different dilutions of gDNA sample, each in triplicate, with three biological replicates performed in separate runs. Wells with no DNA served as no template controls, and standard curves indicated equivalency of primer/probe set efficiencies.
Sequence identity and phylogenetic analysis
Annotated proteins from T. brucei TREU 927, T. brucei gambiense DAL 972, T. grayi ANR4, T. evansi STIB805, T. rangeli SC-58, T. cruzi marinkellei B7, T. cruzi Sylvio-X10, T. cruzi Dm28c, Leishmania mexicana MHOM/GT/2001/U1103, Leishmania major Friedlin, T. congolense IL3000 and T. vivax Y486 were downloaded from TriTrypDB v.24. OrthoFinder v.0.7.1 [147] processing (default parameters) using the data from TriTrypDB and the gene calls from our sequenced genomes, identified 224 annotated single copy orthologs that are present in all species. Clustalo v.1.2 [148] alignments with Gblocks v0.91b [149] editing (parameters: b4 = 5, b5 = h) of these genes in 10 selected species were checked to ensure no alignment had > 50% of positions filtered out or had a length of < 100 amino acids. EMBOSS infoalign [150] and a custom Perl script were used to remove any edited alignments that contained a sequence > 25% shorter than the median alignment length to avoid including partial or broken genes. Thirty-seven orthologs were removed in this analysis. Visual inspection of the remaining 187 edited alignments identified 48 that contained at least one poorly aligning sequence and were therefore removed, leaving a final set of 139 orthologous genes present in all 10 organisms. These gene alignments were concatenated using FASconCAT v1.0 [151], and the resulting supermatrices were used for phylogenetic reconstruction. ProtTest v.3.4 [152] Bayesian Information Criterion (BIC) determined that 88% of these proteins best fit the JTT substitution model, and 85% of proteins had gamma as the best model for rate heterogeneity. RAxML v.8.1.17 [153] PROTGAMMAJTT, which applies a gamma distribution with four discrete rate categories allowing for different rates of evolution at different sites, was used for building 200 maximum likelihood (ML) trees on distinct randomized stepwise addition parsimony starting trees to obtain the tree with the best likelihood. Support values for the tree were then obtained by rapid bootstrap analysis with 1000 replicates. Bootstrap values were then used to draw bipartitions on the best ML tree. TreeGraph 2.4.0 [154] and Inkscape 0.91 [155] were used for tree visualization and editing, with mid-point rooting on T. brucei. The 139 amino acid alignments without Gblocks editing were used by PAL2NAL v.14 [156] to obtain corresponding codon alignments. These were both concatenated with FASconCAT v1.0 and used for average pairwise percent identity calculation with a custom Python script incorporating the AlignIO utility of Biopython [157] .
Multigene family and 18S in silico analysis
Multigene family copy number: We selected 13 multigene families for analysis based on gene cluster diversity analyses of this study and literature searches. Called genes by GeneMarkS v.4.7b [137] were grouped into multigene families based on choosing the best non-hypothetical protein annotation out of the top 10 E-value hits to NCBI’s non-redundant protein database via BLASTp (E-value threshold 1e-5). Gene coordinates were then converted to GFF format for reads mapping with BWA v.0.7.12 [136] (default parameters). Copy number for each multigene family was calculated based on read depth using SAMtools v1.2 [158]. Average per base coverage was calculated then divided by average coverage of a set of 6394 single copy orthologs (same gene set as for heterozygosity analysis). A representative complete gene length for each multigene family was selected based on the consensus longest trypanosome gene length for each multigene family from UniProtKB full-length genes (as described in Additional file 1: Table S10), and used to correct copy number estimates for fragmented genes. Fragmentation of genes is a common problem in copy number estimation of complex and incomplete genomes [159], and given that portions of genes in the genome may not assemble our estimates are likely conservative. As a validation for our read-based approach we obtained values of 1 for dihydrofolate reductase, poly (A) polymerase and DNA topoisomerase type IB, which are widely considered to be single copy genes in trypanosomatids, using the same methodology.
Amastin motif analysis: motif prediction was performed on translated gene sequences using MEME [160] v.4.10.0 with the anr option and a maximum width of 20.
18S copy number: estimated as described above for multigene family analysis, except that gene coordinates were predicted by RNAmmer v1.2 [139] and aligned bases were only calculated at positions of q-score over 25 with a minimum two-fold coverage.
Pseudogenes
Longest nr database hits for each genomic coordinate were predicted by gapped BLAST with the program lastal [161] v.744 (parameters -F15, −l5 –K20, −X 150 –P0), followed by selection of hits containing frameshifts or premature stop codons. Coordinates were converted to GFF format, removing any overlapping genes called by GeneMarkS v.4.7b [137] using BEDTools v.2.19.1 intersect. As described above, since over 98% of 2217 single copy orthologs shared between T. brucei, T. vivax, T. congolense, T. dionisii, T. cruzi and Leishmania species were present, likelihood of finding false positives due to uncalled genes was low.
Heterozygosity
High quality sequence reads, i.e., reads with 70% of the bases with quality score ≥ 25, were aligned to each genome assembly. Polymorphic positions were then quantified in each of 6394 single copy OrthoFinder v.0.7.1-generated orthologs present in each of the four genomes examined, using SAMtools v1.2 [158] to generate and index bam files, FreeBayes v1.0.1 [162] to detect variants (parameters --ploidy 2 --vcf) and VCFtools v.0.1.9 [163] to summarize SNP results (parameters --remove-indels --recode-INFO-all). Synteny of the distribution of heterozygosity values at local areas of the genomes was assessed by Spearman’s Rank followed by adjusting the p-values using the Bonferroni correction using Rstudio. Windows of distance (bp) and number of genes had to be similar for pairwise species comparisons. To assess the percentage of bi- tri- and tetra-allelic sites the FreeBayes VCF output was subjected to alternative allele counts (e.g. --min-alleles 3 --max-alleles 3 for tri-alleles). SnpEff v4.3T [164] was used to assess synonymous to non-synonymous changes at SNP sites (default parameters).
Repetitive elements
Repeat counts in intergenic regions of the assemblies were identified by performing a Cross Match v. 0990329 search and categorization with RepeatMasker [165] v. 4.0.6. RepeatMasker library sequences of Trypanosoma species derived from Repbase (20150807 download) were used as a database for the search. De novo repeats were predicted using RepeatMasker with a library built using RepeatModeler [166] v1.0.8. The latter identified and modeled de novo repeat families from the four genomes using RECON v.1.08, RepeatScout v.1.0.5 and Tandem Repeat Finder v.4.0.4 [167,168,169], with an RMBLASTn [166] v.1.2 search of Repbase.
Metabolic pathways analysis
Database reference genes from UniRef100 [170] and the Kyoto Encyclopedia of Genes and Genomes KEGG [129] were located on assembly contigs and mapped to metabolic pathways using ASGARD [171]. Enzymes found to be differentially present among the four species were subjected to an additional tBLASTn analysis of the sequencing reads, requiring > 60% of the reference gene sequence to be covered by at least four reads with an E-value <1e-5 to indicate presence.
Abbreviations
- DGF:
-
Dispersed gene family
- DHFR:
-
Dihydrofolate reductase
- DTU:
-
Discrete typing unit
- GPI:
-
Glycophosphatidylinositol
- KMP:
-
Kinetoplast membrane protein
- KOG:
-
Clusters of orthologous groups for eukaryotes
- LINE:
-
Non-long terminal repeat retrotransposon
- MASP:
-
Mucin-associated surface protein
- PFGE:
-
Pulsed field gel electrophoresis
- RHS:
-
Retrotransposon hot spot
- RNAi:
-
RNA interference
- SNP:
-
Single nucleotide polymorphism
- STXBP:
-
Syntaxin-binding protein
- TS:
-
Trans-sialidase
- VSG:
-
Variant surface glycoprotein
References
Lukeš J, Skalický T, Týč J, Votýpka J, Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol. 2014;195:115–22.
Clayton CE. Life without transcriptional control? From fly to man and back again. EMBO J. 2002;21:1881–8.
Mulligan CJ, Szathmáry EJE. The peopling of the Americas and the origin of the Beringian occupation model. Am J Phys Anthropol. 2017;162:403–8.
Fernandes MC, Andrews NW. Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol Rev. 2012;36:734–47.
Garcia ES, Ratcliffe NA, Whitten MM, Gonzalez MS, Azambuja P. Exploring the role of insect host factors in the dynamics of Trypanosoma cruzi-Rhodnius prolixus interactions. J Insect Physiol. 2007;53:11–21.
Tibayrenc M, Ward P, Moya A, Ayala FJ. Natural populations of Trypanosoma cruzi, the agent of Chagas disease, have a complex multiclonal structure. Proc Natl Acad Sci U S A. 1986;83:115–9.
Tibayrenc M, Ayala FJ. The clonal theory of parasitic protozoa: 12 years on. Trends Parasitol. 2002;18:405–10.
Bogliolo AR, Lauria-Pires L, Gibson WC. Polymorphisms in Trypanosoma cruzi: evidence of genetic recombination. Acta Trop. 1996;61:31–40.
Machado CA, Ayala FJ. Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc Natl Acad Sci U S A. 2001;98:7396–401.
Brisse S, Henriksson J, Barnabe C, Douzery EJP, Berkvens D, Serrano M, et al. Evidence for genetic exchange and hybridization in Trypanosoma cruzi based on nucleotide sequences and molecular karyotype. Infect Genet Evol. 2003;2:173–83.
Zingales B, Miles MA, Campbell DA, Tibayrenc M, Macedo AM, Teixeira MMG, et al. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect Genet Evol. 2012;12:240–53.
Hamilton PB, Teixeira MMG, Stevens JR. The evolution of Trypanosoma cruzi: the “bat seeding” hypothesis. Trends Parasitol. 2012;28:136–41.
Lima L, Espinosa-Alvarez O, Hamilton PB, Neves L, Takata CSA, Campaner M, et al. Trypanosoma livingstonei: a new species from African bats supports the bat seeding hypothesis for the Trypanosoma cruzi clade. Parasit Vectors. 2013;6:221.
Deane MP, Deane LM. Studies on the life cycle of Trypanosoma conorrhini. “In vitro” development and multiplication of the bloodstream trypanosomes. Rev Inst Med Trop São Paulo. 1961;3:149–60.
Hoare CA. The trypanosomes of mammals. Oxford and Edinburgh: Blackwell Scientific Publications; 1972.
Deane LM, Deane MP, Lourenco-de-Oliveira R. Are Asian monkeys the original mammalian hosts of Trypanosoma conorhini? Mem Inst Oswaldo Cruz. 1986;81:127–9.
Azambuja P, Garcia ES. Trypanosoma rangeli interactions within the vector Rhodnius prolixus: a mini review. Mem Inst Oswaldo Cruz. 2005;100:567–72.
Vallejo GA, Guhl F, Schaub GA. Triatominae-Trypanosoma cruzi/T. rangeli: vector-parasite interactions. Acta Trop. 2009;110:137–47.
Gomes SAO, Fonseca-de-Souza AL, Silva BA, Kiffer-Moreira T, Santos-Mallet JR, Santos ALS, et al. Trypanosoma rangeli: differential expression of cell surface polypeptides and ecto-phosphatase activity in short and long epimastigote forms. Exp Parasitol. 2006;112:253–62.
Fonseca-de-Souza AL, Dick CF, Dos-Santos ALA, Fonseca FV, Meyer-Fernandes JR. Trypanosoma rangeli: a possible role for ecto-phosphatase activity on cell proliferation. Exp Parasitol. 2009;122:242–6.
Dick CF, Dos-Santos ALA, Fonseca-de-Souza AL, Rocha-Ferreira J, Meyer-Fernandes JR. Trypanosoma rangeli: differential expression of ecto-phosphatase activities in response to inorganic phosphate starvation. Exp Parasitol. 2010;124:386–93.
Grisard EC, Campbell DA, Romanha AJ. Mini-exon gene sequence polymorphism among Trypanosoma rangeli strains isolated from distinct geographical regions. Parasitology. 1999;118(Pt 4):375–82.
Maia da Silva F, Rodrigues AC, Campaner M, Takata CSA, Brigido MC, Junqueira ACV, et al. Randomly amplified polymorphic DNA analysis of Trypanosoma rangeli and allied species from human, monkeys and other sylvatic mammals of the Brazilian Amazon disclosed a new group and a species-specific marker. Parasitology. 2004;128:283–94.
Maia da Silva F, Junqueira ACV, Campaner M, Rodrigues AC, Crisante G, Ramirez LE, et al. Comparative phylogeography of Trypanosoma rangeli and Rhodnius (Hemiptera: Reduviidae) supports a long coexistence of parasite lineages and their sympatric vectors. Mol Ecol. 2007;16:3361–73.
Maia da Silva F, Marcili A, Lima L, Cavazzana MJ, Ortiz PA, Campaner M, et al. Trypanosoma rangeli isolates of bats from Central Brazil: genotyping and phylogenetic analysis enable description of a new lineage using spliced-leader gene sequences. Acta Trop. 2009;109:199–207.
Ortiz PA, Maia da Silva F, Cortez AP, Lima L, Campaner M, EMF P, et al. Genes of cathepsin L-like proteases in Trypanosoma rangeli isolates: markers for diagnosis, genotyping and phylogenetic relationships. Acta Trop. 2009;112:249–59.
Caballero ZC, Costa-Martins AG, Ferreira RC, P Alves JM, Serrano MG, Camargo EP, et al. Phylogenetic and syntenic data support a single horizontal transference to a Trypanosoma ancestor of a prokaryotic proline racemase implicated in parasite evasion from host defences. Parasit Vectors. 2015;8:115–8.
Maia da Silva F, Noyes H, Campaner M, Junqueira ACV, Coura JR, Añez N, et al. Phylogeny, taxonomy and grouping of Trypanosoma rangeli isolates from man, triatomines and sylvatic mammals from widespread geographical origin based on SSU and ITS ribosomal sequences. Parasitology. 2004;129:549–61.
Eger-Mangrich I, de Oliveira MA, Grisard EC, de Souza W, Steindel M. Interaction of Trypanosoma rangeli Tejera, 1920 with different cell lines in vitro. Parasitol Res. 2001;87:505–9.
Jackson AP, Quail MA, Berriman M. Insights into the genome sequence of a free-living Kinetoplastid: Bodo saltans (Kinetoplastida: Euglenozoa). BMC Genomics. 2008;9:594.
Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–22.
El-Sayed NM. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–15.
Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–42.
El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–9.
Franzén O, Ochaya S, Sherwood E, Lewis MD, Llewellyn MS, Miles MA, et al. Shotgun sequencing analysis of Trypanosoma cruzi I Sylvio X10/1 and comparison with T. cruzi VI CL Brener. PLoS Negl Trop Dis. 2011;5:e984–9.
Franzén O, Talavera-Lopez C, Ochaya S, Butler CE, Messenger LA, Lewis MD, et al. Comparative genomic analysis of human infective Trypanosoma cruzi lineages with the bat-restricted subspecies T. cruzi marinkellei. BMC Genomics. 2012;13:1–1.
Steindel M, Pinto JC, Toma HK, Mangia RH, Ribeiro-Rodrigues R, Romanha AJ. Trypanosoma rangeli (Tejera, 1920) isolated from a sylvatic rodent (Echimys dasythrix) in Santa Catarina Island, Santa Catarina state: first report of this trypanosome in southern Brazil. Mem Inst Oswaldo Cruz. 1991;86:73–9.
Stoco PH, Wagner G, Talavera-López C, Gerber A, Zaha A, Thompson CE, et al. Genome of the avirulent human-infective trypanosome—Trypanosoma rangeli. PLoS Negl Trop Dis. 2014;8:e3176–17.
D 'Alessandro A, Saravia NG. Trypanosoma rangeli. In: Kreier J, Baker JR, editors. editors Parasitic Protozoa. 2nd ed. New York: Academic Press; 1992. p. 1–54.
Maia da Silva F, Naiff RD, Marcili A, Gordo M, D'Affonseca Neto JA, Naiff MF, et al. Infection rates and genotypes of Trypanosoma rangeli and T. cruzi infecting free-ranging Saguinus bicolor (Callitrichidae), a critically endangered primate of the Amazon rainforest. Acta Trop. 2008;107:168–73.
Hamilton PB, Lewis MD, Cruickshank C, Gaunt MW, Yeo M, Llewellyn MS, et al. Identification and lineage genotyping of south American trypanosomes using fluorescent fragment length barcoding. Infect Genet Evol. 2011;11:44–51.
Yoshida N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An Acad Bras Cienc. 2006;78:87–111.
Rodrigues AA, Saosa JSS, da Silva GK, Martins FA, da Silva AA, Souza Neto CPDS, et al. IFN-γ plays a unique role in protection against low virulent Trypanosoma cruzi strain. PLoS Negl Trop Dis. 2012;6:e1598–9.
Santos CC, Sant'anna C, Terres A, Cunha-e-Silva NL, Scharfstein J, de A Lima APC. Chagasin, the endogenous cysteine-protease inhibitor of Trypanosoma cruzi, modulates parasite differentiation and invasion of mammalian cells. J Cell Sci. 2005;118:901–15.
Vargas N, Pedroso A, Zingales B. Chromosomal polymorphism, gene synteny and genome size in T. cruzi I and T. cruzi II groups. Mol Biochem Parasitol. 2004;138:131–41.
de Freitas JM, Augusto-Pinto L, Pimenta JR, Bastos-Rodrigues L, Gonçalves VF, Teixeira SMR, et al. Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog. 2006;e24:2.
Westenberger SJ, Barnabé C, Campbell DA, Sturm NR. Two hybridization events define the population structure of Trypanosoma cruzi. Genetics. 2005;171:527–43.
Lewis MD, Llewellyn MS, Yeo M, Acosta N, Gaunt MW, Miles MA. Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Negl Trop Dis. 2011;5:e1363.
Ruiz RC, Favoreto SJ, Dorta ML, Oshiro ME, Ferreira AT, Manque PM, et al. Infectivity of Trypanosoma cruzi strains is associated with differential expression of surface glycoproteins with differential Ca2+ signalling activity. Biochem J. 1998;330(Pt 1):505–11.
Maeda FY, Cortez C, Izidoro MA, Juliano L, Yoshida N. Fibronectin-degrading activity of Trypanosoma cruzi cysteine proteinase plays a role in host cell invasion. Infect Immun. 2014;82:5166–74.
Cano MI, Gruber A, Vazquez M, Cortéz A, Levin MJ, González A, et al. Molecular karyotype of clone CL Brener chosen for the Trypanosoma cruzi genome project. Mol Biochem Parasitol. 1995;71:273–8.
Ghedin E, Bringaud F, Peterson J, Myler P, Berriman M, Ivens A, et al. Gene synteny and evolution of genome architecture in trypanosomatids. Mol Biochem Parasitol. 2004;134:183–91.
Henriksson J, Aslund L, Pettersson U. Karyotype variability in Trypanosoma cruzi. Parasitol Today. 1996;12:1–7.
Henriksson J, Dujardin JC, Barnabé C, Brisse S, Timperman G, Venegas J, et al. Chromosomal size variation in Trypanosoma cruzi is mainly progressive and is evolutionarily informative. Parasitology. 2002;124:277–86.
Souza RT, Lima FM, Barros RM, Cortez DR, Santos MF, Cordero EM, et al. Genome size, karyotype polymorphism and chromosomal evolution in Trypanosoma cruzi. PLoS One. 2011;6:e23042–14.
Henriksson J, Solari A, Rydaker M, Sousa OE, Pettersson U. Karyotype variability in Trypanosoma rangeli. Parasitology. 1996;112(Pt 4):385–91.
Reis-Cunha JL, Rodrigues-Luiz GF, Valdivia HO, Baptista RP, Mendes TAO, de Morais GL, et al. Chromosomal copy number variation reveals differential levels of genomic plasticity in distinct Trypanosoma cruzi strains. BMC Genomics. 2015;16:499.
Lewis MD, Llewellyn MS, Gaunt MW, Yeo M, Carrasco HJ, Miles MA. Flow cytometric analysis and microsatellite genotyping reveal extensive DNA content variation in Trypanosoma cruzi populations and expose contrasts between natural and experimental hybrids. Int J Parasitol. 2009;39:1305–17.
Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38:D457–62.
Bartholomeu DC, Cerqueira GC, Leão ACA, DaRocha WD, Pais FS, Macedo C, et al. Genomic organization and expression profile of the mucin-associated surface protein (masp) family of the human pathogen Trypanosoma cruzi. Nucleic Acids Res. 2009;37:3407–17.
Buscaglia CA, Campo VA, Frasch ACC, Di Noia JM. Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nat Rev Microbiol. 2006;4:229–36.
Cuevas IC, Cazzulo JJ, Sánchez DO. gp63 homologues in Trypanosoma cruzi: surface antigens with metalloprotease activity and a possible role in host cell infection. Infect Immun. 2003;71:5739–49.
Gonzales-Perdomo M, Romero P, Goldenberg S. Cyclic AMP and adenylate cyclase activators stimulate Trypanosoma cruzi differentiation. Exp Parasitol. 1988;66:205–12.
Bartholomeu DC, de Paiva RMC, Mendes TAO, DaRocha WD, Teixeira SMR. Unveiling the intracellular survival gene kit of trypanosomatid parasites. PLoS Pathog. 2014;10:e1004399.
Chiurillo MA, Cortez DR, Lima FM, Cortez C, Ramírez JL, Martins AG, et al. The diversity and expansion of the trans-sialidase gene family is a common feature in Trypanosoma cruzi clade members. Infect Genet Evol. 2016;37:266–74.
Burgos JM, Risso MG, Breniere SF, Barnabe C, Campetella O, Leguizamon MS. Differential distribution of genes encoding the virulence factor trans-sialidase along Trypanosoma cruzi discrete typing units. PLoS One. 2013;8:e58967.
Freitas LM, Santos dos SL, Rodrigues-Luiz GF, Mendes TAO, Rodrigues TS, Gazzinelli RT, et al. Genomic analyses, gene expression and antigenic profile of the trans-sialidase superfamily of Trypanosoma cruzi reveal an undetected level of complexity. PLoS One. 2011;6:e25914.
Scudder P, Doom JP, Chuenkova M, Manger ID, Pereira ME. Enzymatic characterization of beta-D-galactoside alpha 2,3-trans-sialidase from Trypanosoma cruzi. J Biol Chem. 1993;268:9886–91.
Ferrero-Garcia MA, Trombetta SE, Sanchez DO, Reglero A, Frasch AC, Parodi AJ. The action of Trypanosoma cruzi trans-sialidase on glycolipids and glycoproteins. Eur J Biochem. 1993;213:765–71.
Buschiazzo A, Amaya MF, Cremona ML, Frasch AC, Alzari PM. The crystal structure and mode of action of trans-sialidase, a key enzyme in Trypanosoma cruzi pathogenesis. Mol Cell. 2002;10:757–68.
Añez-Rojas N, Peralta A, Crisante G, Rojas A, Añez N, Ramírez JL, et al. Trypanosoma rangeli expresses a gene of the group II trans-sialidase superfamily. Mol Biochem Parasitol. 2005;142:133–6.
Grisard EC, Stoco PH, Wagner G, Sincero TCM, Rotava G, Rodrigues JB, et al. Transcriptomic analyses of the avirulent protozoan parasite Trypanosoma rangeli. Mol Biochem Parasitol. 2010;174:18–25.
Peña CP, Lander N, Rodriguez E, Crisante G, Añez N, Ramirez JL, et al. Molecular analysis of surface glycoprotein multigene family TrGP expressed on the plasma membrane of Trypanosoma rangeli epimastigotes forms. Acta Trop. 2009;111:255–62.
Kulkarni MM, Olson CL, Engman DM, McGwire BS. Trypanosoma cruzi gp63 proteins undergo stage-specific differential posttranslational modification and are important for host cell infection. Infect Immun. 2009;77:2193–200.
Atwood JA, Weatherly DB, Minning TA, Bundy B, Cavola C, Opperdoes FR, et al. The Trypanosoma cruzi proteome. Science. 2005;309:473–6.
Romano MC, Jiménez P, Miranda-Brito C, Valdez RA. Parasites and steroid hormones: corticosteroid and sex steroid synthesis, their role in the parasite physiology and development. Front Neurosci. 2015;9:224.
Burleigh BA, Woolsey AM. Cell signalling and Trypanosoma cruzi invasion. Cell Microbiol. 2002;4:701–11.
Kawashita SY, da Silva CV, Mortara RA, Burleigh BA, Briones MRS. Homology, paralogy and function of DGF-1, a highly dispersed Trypanosoma cruzi specific gene family and its implications for information entropy of its encoded proteins. Mol Biochem Parasitol. 2009;165:19–31.
Lima L, Ortiz PA, da Silva FM, Alves JMP, Serrano MG, Cortez AP, et al. Repertoire, genealogy and genomic organization of cruzipain and homologous genes in Trypanosoma cruzi, T. cruzi-like and other trypanosome species. PLoS One. 2012;7:e38385–15.
Kangussu-Marcolino MM, Cardoso de Paiva RM, Araújo PR, de Mendonça-Neto RP, Lemos L, Bartholomeu DC, et al. Distinct genomic organization, mRNA expression and cellular localization of members of two amastin sub-families present in Trypanosoma cruzi. BMC Microbiol. 2013;13:1–11.
Cruz MC, Souza-Melo N, da Silva CV, DaRocha WD, Bahia D, Araújo PR, et al. Trypanosoma cruzi: role of δ-Amastin on extracellular amastigote cell invasion and differentiation. PLoS One. 2012;7:e51804–11.
Jackson AP. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol Biol Evol. 2010;27:33–45.
Rafati S, Hassani N, Taslimi Y, Movassagh H, Rochette A, Papadopoulou B. Amastin peptide-binding antibodies as biomarkers of active human visceral leishmaniasis. Clin Vaccine Immunol. 2006;13:1104–10.
Salotra P, Duncan RC, Singh R, Subba Raju BV, Sreenivas G, Nakhasi HL. Upregulation of surface proteins in Leishmania donovani isolated from patients of post kala-azar dermal leishmaniasis. Microbes Infect. 2006;8:637–44.
Stober CB, Lange UG, Roberts MTM, Gilmartin B, Francis R, Almeida R, et al. From genome to vaccines for leishmaniasis: screening 100 novel vaccine candidates against murine Leishmania major infection. Vaccine. 2006;24:2602–16.
Rochette A, McNicoll F, Girard J, Breton M, Leblanc E, Bergeron MG, et al. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol Biochem Parasitol. 2005;140:205–20.
Mendes TAO, Lobo FP, Rodrigues TS, Rodrigues-Luiz GF, daRocha WD, Fujiwara RT, et al. Repeat-enriched proteins are related to host cell invasion and immune evasion in parasitic protozoa. Mol Biol Evol. 2013;30:951–63.
Diez H, Thomas MC, Uruena CP, Santander SP, Cuervo CL, Lopez MC, et al. Molecular characterization of the kinetoplastid membrane protein-11 genes from the parasite Trypanosoma rangeli. Parasitology. 2005;130:643–51.
Ramirez JR, Berberich C, Jaramillo A, Alonso C, Velez IV. Molecular and antigenic characterization of the Leishmania (Viannia) panamensis kinetoplastid membrane protein-11. Mem Inst Oswaldo Cruz. 1998;93:247–54.
Schenkman S, Ferguson MA, Heise N, de Almeida ML, Mortara RA, Yoshida N. Mucin-like glycoproteins linked to the membrane by glycosylphosphatidylinositol anchor are the major acceptors of sialic acid in a reaction catalyzed by trans-sialidase in metacyclic forms of Trypanosoma cruzi. Mol Biochem Parasitol. 1993;59:293–303.
Abate T, Rincon M, Diaz-Bello Z, Spencer L, Rodriguez-Acosta A. A mucin like gene different from the previously reported members of the mucin like gene families is transcribed in Trypanosoma cruzi but not in Trypanosoma rangeli. Mem Inst Oswaldo Cruz. 2005;100:391–5.
Wagner G, Yamanaka LE, Moura H, Lückemeyer DD, Schlindwein AD, Stoco PH, et al. The Trypanosoma rangeli trypomastigote surfaceome reveals novel proteins and targets for specific diagnosis. J Proteome. 2013;82:52–63.
Allen CL, Kelly JM. Trypanosoma cruzi: mucin pseudogenes organized in a tandem array. Exp Parasitol. 2001;97:173–7.
Thon G, Baltz T, Eisen H. Antigenic diversity by the recombination of pseudogenes. Genes Dev. 1989;3:1247–54.
Barnes RL, McCulloch R. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 2007;35:3478–93.
Borst P, Bitter W, Blundell PA, Chaves I, Cross M, Gerrits H, et al. Control of VSG gene expression sites in Trypanosoma brucei. Mol Biochem Parasitol. 1998;91:67–76.
Boothroyd CE, Dreesen O, Leonova T, Ly KI, Figueiredo LM, Cross GAM, et al. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature. 2009;459:278–81.
Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One. 2008;e3527:3.
Wen Y-Z, Zheng L-L, Qu L-H, Ayala FJ, Lun Z-R. Pseudogenes are not pseudo any more. RNA Biol. 2012;9:27–32.
Matveyev AV, Alves JMP, Serrano MG, Lee V, Lara AM, Barton WA, et al. The evolutionary loss of RNAi key determinants in kinetoplastids as a multiple sporadic phenomenon. J Mol Evol. 2017;84:104–15.
Branche C, Ochaya S, Aslund L, Andersson B. Comparative karyotyping as a tool for genome structure analysis of Trypanosoma cruzi. Mol Biochem Parasitol. 2006;147:30–8.
Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM. Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16-kb GC-rich transcriptional “strand-switch” domain as a major feature. Genome Res. 2005;15:36–43.
Tibayrenc M, Ayala FJ. Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc Natl Acad Sci U S A. 2012;109:E3305–13.
Fritz-Laylin LK, Prochnik SE, Ginger ML, Dacks JB, Carpenter ML, Field MC, et al. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell. 2010;140:631–42.
Yeo M, Mauricio IL, Messenger LA, Lewis MD, Llewellyn MS, Acosta N, et al. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl Trop Dis. 2011;5:e1049.
Volf P, Benkova I, Myskova J, Sadlova J, Campino L, Ravel C. Increased transmission potential of Leishmania major/Leishmania infantum hybrids. Int J Parasitol. 2007;37:589–93.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Wickstead B, Ersfeld K, Gull K. Repetitive elements in genomes of parasitic protozoa. Microbiol Mol Biol Rev. 2003;67:360–75.
Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond Ser B Biol Sci. 2009;364:99–115.
Lee SH, Stephens JL, Englund PT. A fatty-acid synthesis mechanism specialized for parasitism. Nat Rev Microbiol. 2007;5:287–97.
Kraeva N, Butenko A, Hlavacova J, Kostygov A, Myskova J, Grybchuk D, et al. Leptomonas seymouri: adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with Leishmania donovani. PLoS Pathog. 2015;11:e1005127.
Cazzulo JJ. Intermediate metabolism in Trypanosoma cruzi. J Bioenerg Biomembr. 1994;26:157–65.
Lisvane Silva P, Mantilla BS, Barison MJ, Wrenger C, Silber AM. The uniqueness of the Trypanosoma cruzi mitochondrion: opportunities to identify new drug target for the treatment of Chagas disease. Curr Pharm Des. 2011;17:2074–99.
Silber AM, Colli W, Ulrich H, Alves MJM, Pereira CA. Amino acid metabolic routes in Trypanosoma cruzi: possible therapeutic targets against Chagas' disease. Curr Drug Targets Infect Disord. 2005;5:53–64.
Tonelli RR, Silber AM, Almeida-de-Faria M, Hirata IY, Colli W, Alves MJM. L-proline is essential for the intracellular differentiation of Trypanosoma cruzi. Cell Microbiol. 2004;6:733–41.
Martins RM, Covarrubias C, Rojas RG, Silber AM, Yoshida N. Use of L-proline and ATP production by Trypanosoma cruzi metacyclic forms as requirements for host cell invasion. Infect Immun. 2009;77:3023–32.
Pereira CA, Alonso GD, Torres HN, Flawia MM. Arginine kinase: a common feature for management of energy reserves in African and American flagellated trypanosomatids. J Eukaryot Microbiol. 2002;49:82–5.
Pereira CA, Alonso GD, Ivaldi S, Silber AM, Alves MJM, Torres HN, et al. Arginine kinase overexpression improves Trypanosoma cruzi survival capability. FEBS Lett. 2003;554:201–5.
Contreras VT, Salles JM, Thomas N, Morel CM, Goldenberg S. In vitro differentiation of Trypanosoma cruzi under chemically defined conditions. Mol Biochem Parasitol. 1985;16:315–27.
Carrillo C, Cejas S, Huber A, Gonzalez NS, Algranati ID. Lack of arginine decarboxylase in Trypanosoma cruzi epimastigotes. J Eukaryot Microbiol. 2003;50:312–6.
Ariyanayagam MR, Oza SL, Mehlert A, Fairlamb AH. Bis (glutathionyl) spermine and other novel trypanothione analogues in Trypanosoma cruzi. J Biol Chem. 2003;278:27612–9.
Homsy JJ, Granger B, Krassner SM. Some factors inducing formation of metacyclic stages of Trypanosoma cruzi. J Protozool. 1989;36:150–3.
Manchola NC, Rapado LN, Barison MJ, Silber AM. Biochemical characterization of branched chain amino acids uptake in Trypanosoma cruzi. J Eukaryot Microbiol. 2016;63:299–308.
Ginger ML, Prescott MC, Reynolds DG, Chance ML, Goad LJ. Utilization of leucine and acetate as carbon sources for sterol and fatty acid biosynthesis by Old and New World Leishmania species, Endotrypanum monterogeii and Trypanosoma cruzi. Eur J Biochem. 2000;267:2555–66.
Opperdoes FR, Borst P. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome. FEBS Lett. 1977;80:360–4.
Cannata JJ, Cazzulo JJ. The aerobic fermentation of glucose by Trypanosoma cruzi. Comp Biochem Physiol B. 1984;79:297–308.
Fairlamb AH, Opperdoes FR. Carbohydrate metabolism in African trypanosomes, with special reference to the glycosome. In: Morgan MJ, editor. Carbohydrate metabolism in cultured cells. Boston, MA: Springer US; 1986. p. 183–224.
Chaumont F, Schanck AN, Blum JJ, Opperdoes FR. Aerobic and anaerobic glucose metabolism of Phytomonas sp. isolated from Euphorbia characias. Mol Biochem Parasitol. 1994;67:321–31.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34.
Shewale JG. Beta-glucosidase: its role in cellulase synthesis and hydrolysis of cellulose. Int J BioChemiPhysics. 1982;14:435–43.
Diaz-Albiter HM, Ferreira TN, Costa SG, Rivas GB, Gumiel M, Cavalcante DR, et al. Everybody loves sugar: first report of plant feeding in triatomines. Parasit Vectors. 2016;9:114.
Pryor EEJ, Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, et al. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339:1600–4.
Previato JO, Jones C, Xavier MT, Wait R, Travassos LR, Parodi AJ, et al. Structural characterization of the major glycosylphosphatidylinositol membrane-anchored glycoprotein from epimastigote forms of Trypanosoma cruzi Y-strain. J Biol Chem. 1995;270:7241–50.
Alves JMP, Serrano MG, Maia da Silva F, Voegtly LJ, Matveyev AV, Teixeira MMG, et al. Genome evolution and phylogenomic analysis of Candidatus Kinetoplastibacterium, the betaproteobacterial endosymbionts of Strigomonas and Angomonas. Genome Biol Evol. 2013;5:338–50.
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–18.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Lagesen K, Hallin P, Rodland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Petersen TN, Brunak S, Heijne von G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Krogh A, Larsson B, Heijne von G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Fankhauser N, Maser P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics. 2005;21:1846–52.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinf. 2003;4:41.
Skosyrev VS, Vasil'eva GV, Lomaeva MG, Malakhova LV, Antipova VN, Bezlepkin VG. Specialized software product for comparative analysis of multicomponent DNA fingerprints. Genetika. 2013;49:531–7.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157.
Sievers F, Higgins DG. Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 2014;1079:105–16.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–7.
Kuck P, Meusemann K. FASconCAT: convenient handling of data matrices. Mol Phylogenet Evol. 2010;56:1115–8.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Stover BC, Muller KF. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 2010;11:7.
Inkscape. http://inkscape.org/. Accessed 12 Dec 2015.
Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–12.
Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–3.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Denton JF, Lugo-Martinez J, Tucker AE, Schrider DR, Warren WC, Hahn MW. Extensive error in the number of genes inferred from draft genome assemblies. PLoS Comput Biol. 2014;10:e1003998–9.
Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Mach Learn. 1995;21:51–80.
Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–93.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing https://arxiv.org/abs/1207.3907. Accessed 30 Sept 2017.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0 2013–2015. http://www.repeatmasker.org. Accessed 21 May 2017.
Smit AFA, Hubley R. RepeatModeler Open-1.0. 2008-2015. http://www.repeatmasker.org.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–76.
Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–8.
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–8.
Alves JMP, Buck GA. Automated system for gene annotation and metabolic pathway reconstruction using general sequence databases. Chem Biodivers. 2007;4:2593–602.
Acknowledgements
The authors greatly thank Dr. José R. Coura and Angela C. V. Junqueira for providing T. rangeli AM80, Dr. Yoshida Nobuko for providing T. cruzi strains, Marta Campaner for culture of T. rangeli AM80 and T. conorhini, Flavia Maia da Silva for DNA preparation, Logan Voegtly for library preparation and sequencing, Ana Lara for parasite culture, and Ruth Carvalho for assisting with qPCR. Sequencing was performed in the Nucleic Acids Research Facility at VCU. We also appreciate Dr. Paul Fawcett and Dr. Andrew Eckert for their constructive comments.
Funding
This work has been funded by a grant DEB-#080056 from the National Science Foundation’s Assembling the Tree of Life program to GAB. and grants from the Brazilian agencies CNPq and CAPES to MMGT. JMPA is supported by grant #2013/14622–3, São Paulo Research Foundation (FAPESP). OSA was a PhD student supported by the Brazilian agency CNPq, and AGCM and PAO have a Post-Doctoral fellowship from CAPES.
Availability of data and materials
Genome sequences and annotations are available from GenBank. The data can be accessed through GenBank BioProject PRJNA315397: Assembling the Tree of Life: Phylum Euglenozoa, under the following BioSample and GenBank accession numbers:
SAMN04566061, MKGL00000000 Trypanosoma rangeli AM80; SAMN04565988, MKKU00000000 Trypanosoma conorhini 025E and SAMN04566062, MKKV00000000 Trypanosoma cruzi G. Trypanosoma cruzi CL has the BioSample accession SAMN04566063 and was originally deposited under accession MKQG00000000, the version described in this paper is version MKQG01000000.
Scripts from custom analyses are available through GitHub:
https://github.com/kbradwell/comparative_trypanosoma_paper/tree/v1.0.0.
Zenodo DOI http://doi.org/10.5281/zenodo.1442351.
Author information
Authors and Affiliations
Contributions
GAB, KRB, VNK, MGS, and MMGT designed the study; VL led library preparation and genome sequencing; VNK performed genome assembly, genome completion analysis, and provided some of the bioinformatics tools and pipelines for general genome statistics (GAP) and heterozygosity measurements; KRB participated in genome assembly development and assessment, provided tools and pipelines, and performed bioinformatics analysis; KRB and AVM conducted the molecular experiments; KRB analyzed the molecular experiment data; BH and HP contributed comments on analyses; KRB wrote the manuscript; GAB, VNK, JMPA and MMGT edited the manuscript; KRB, and MGS coordinated and conducted the data deposition. OSA, PAO and AGCM collaborated with biological and molecular characterization, and previous phylogenetic studies used to select T. rangeli AM80 and T. conorhini for this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no completing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Strain information. Table S2. Densitometry of PFGE. Table S3. Sequencing and genome statistics summary. Tables S4-S7. Annotation of genes. Table S8. Orthologous genes used for phylogenetic reconstruction. Table S9. Percent identity matrix. Table S10. Multigene family copy number analysis and full gene lengths from UniProtKB. Table S11. Amastin motif analysis. Tables S12-S15. Putative pseudogenes for each species. Table S16. Orthologous gene heterozygosity values. Tables S17–20. Coordinates of repetitive elements. Table S21. Metabolic analysis. Table S22. SSU rRNA qPCR primers and probes. (XLSX 18721 kb)
Additional file 2:
Figure S1. Genome assembly Completion and Integrity Analysis (GenoCIA). (A) Genome assemblies are comprehensive. Sequential assemblies were performed from 2, 4, 6, 8, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100% of the sequence reads generated for each of the species, and the percent of 2217 single copy orthologs shared between T. brucei, T. vivax, T. congolense, T. dionisii, T. cruzi and Leishmania species found in the assemblies was determined. (i) shows the percent of the orthologs that have a hit with 50% alignment length, (ii) shows the percent that have a hit with 90% alignment length. (B) Integrity of the gene calls. The genes called with GeneMark for each of the genomes analyzed herein were queried with the set of 2217 single copy orthologs, and the percent of orthologs that align at any length (at least), 25%, 50%, 90% or 99% length of the query gene/protein is shown. Figure S2. Distribution of the number of genes per OrthoFinder cluster. Percentage of clusters containing discrete gene counts, grouped by organism. The colour gradient and percentages over bars indicate the percent of clusters in each size bin that contain at least one gene with a TMHMM, KOHGPI or SignalP designation as surface-located or secreted. (PPTX 99 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bradwell, K.R., Koparde, V.N., Matveyev, A.V. et al. Genomic comparison of Trypanosoma conorhini and Trypanosoma rangeli to Trypanosoma cruzi strains of high and low virulence. BMC Genomics 19, 770 (2018). https://doi.org/10.1186/s12864-018-5112-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5112-0