
Abstract
Background
High grain yield is one of the most important traits requiring improvement in rice breeding programs. Consequently, the genetic basis of spikelets per panicle (SPP) and grain weight (TGW) have received much research focus because of their importance in rice yield.
Results
In this study, IL28, which is a near isogenic line (NIL) developed by introgressing chromosomal segments of the cultivar ‘Moroberekan’ into the cultivar ‘Ilpumbyeo’, showed a significant increase in the number of spikelets per panicle (SPP) and 1,000-grain weight (TGW) compared to the recurrent parent, Ilpumbyeo. Quantitative trait locus (QTL) analysis in 243 F2 plants derived from a cross between IL28 and Ilpumbyeo indicated that both qSPP6 and qTGW6 are located in the interval RM3430–RM20580. Following substitution mapping with 50 F3:4:5 lines, qSPP6 was mapped to a 429-kb interval between RM20521 and InDel-1, while qTGW6 was mapped to a 37.85-kb interval between InDel-1 and SNP--3 based on the japonica genome sequence. This result indicates that qSPP6 and qTGW6 are different genes. Yield trials with substitution lines indicated that lines harboring the homozygous Moroberekan segment at both the qSPP6 and qTGW6 region showed significantly higher grain yield than Ilpumbyeo.
Conclusion
Because the Moroberekan alleles for SPP and TGW have been shown to be beneficial in the genetic background of Ilpumbyeo, both the qSPP6 and qTGW6 alleles might prove valuable in improving rice yields. Closely linked SSR markers are expected to facilitate the cloning of genes that underlie these QTLs, as well as with marker-assisted selection for variation in SPP and TGW in rice breeding programs.
Similar content being viewed by others

Background
Rice (Oryza sativa L.) is the world’s most important cereal food crop. The anticipated rapid increase in the global human population, which is expected to reach 9.1 billion by 2050, might generate serious food shortage problems. Moreover, various factors, such as water scarcity, soil salinity, disease, climate change, and reduced arable land, will exacerbate food shortages in the next 50 years (Khush [1999] and Khush [2005]; Zhang [2007]). Therefore, researchers are focusing on increasing existing crop grain yield levels. Grain weight, spikelets per panicles, and the number of panicles per plant are the most important components of grain yield. However, the genetic analysis of these three yield components is difficult, because these traits are controlled by multiple genes, in addition to being influenced by the environment. Therefore, the advent of molecular maps for rice (Causse et al. [1994]) and quantitative trait locus (QTL) analysis approaches have facilitated the analysis of these quantitative traits.
Genes/QTLs for spikelets per panicle (SPP) have been reported in populations derived from inter-specific crosses (Xiong et al. [1999]; Thomson et al. [2003]; Suh et al. [2005]; Li et al. [2006] and Linh et al. [2008]), indica-indica crosses (Lin et al. [1996]; Zhuang et al. [1997]), and inter-subspecific crosses (Yamagishi et al. [2002]; Ando et al. [2008]). To date, a few QTLs associated with SPPs have been detected, including Gn1a, APO1, DEP1, and OsSPL14. Gn1a controls grain number in rice, and was found to encode cytokinin oxidase/dehydrogenase (OsCKX2), which is an enzyme that degrades the phytohormone cytokinin (Ashikari et al. [2005]). Higher expression of OsSPL14 during the reproductive stage of rice promotes panicle branching leading to higher grain yield (Miura et al. [2010]). DEP1, which alters panicle architecture, and hence increases grain yield, has also been identified (Huang et al. [2009]). Another approach to identify the genes related to panicle architecture is through the analysis of mutants that alter panicle structure. The characterization of the aberrant panicle organization 1 (apo1) mutant in rice revealed that APO1 positively controls spikelet number by suppressing the precocious conversion of inflorescence meristems to spikelet meristems (Ikeda et al. [2007]). Ookawa et al. ([2010]) also reported that a near isogenic line carrying APO1 enhanced culm strength and increased spikelet number per panicle.
Grain weight (GW) is another characteristic that is targeted to enhance rice yield. Many QTLs for grain size have been identified in rice populations based on crosses between divergent cultivars or accessions (Lin et al. [1996]; Cui et al. [2003]; Ishimaru [2003]; Zhang et al. [2009]). Several genes that regulate grain weight have been identified, including GS3, GS5, GW5, GW2, and TGW6. GW2 is a QTL for rice grain width and weight, which encodes a previously unknown RING-type E3 ubiquitin ligase that negatively regulates cell division by targeting its substrate(s) for degradation by the ubiquitin-proteasome pathway (Song et al. [2007]). Ishimaru et al. ([2013]) found that when the IAA-glucose hydrolase gene stops functioning, TGW6 enhances grain weight through pleiotropic effects on source organs, and leads to significant yield increases.
Numerous studies have reported that one genomic region harbors more than two independent genes that are closely linked, or a gene with pleiotropic effect on several traits (Xie et al. [2008]; Jin et al. [2009]; Lou et al. [2013]; Ji et al.,[2014]). Xie et al. ([2008]) reported that two QTLs, sn9.1 and gw9.1, for 1,000-grain weight and spikelets per panicle were identified in the 37.4-kb interval flanked by markers RM24718 and RM30005. Two QTLs acted in an additive manner in this region, resulting in the NIL with the O. rufipogon alleles exhibiting significantly higher SPP and TGW values compared to the recurrent parent, Hwaseongbyeo (Xie et al. [2008]). However, it remains unclear whether either of the two closely linked QTLs, or one pleiotropic QTL, are associated with the observed variation. A large-sized population combined with high-density mapping is required to determine whether target traits are controlled by closely linked QTLs, or one pleiotropic QTL. In previous studies (Ju et al. [2008]), IL28 was developed, which is an introgression line derived from a cross between the cultivars Ilpumbyeo and Moroberekan, showing higher SPP and TGW values compared to Ilpumbyeo. Kim et al. ([2013]) reported that a QTL for spikelets per panicle was located between RM20521 and RM20572 using F2:3 populations derived from a cross between Ilpumbyeo and IL28. The present study was carried out to detect a novel QTL for TGW, and to clarify its relationship to the qSPP6 QTL using F3:4:5 populations.
Results
Characteristics of IL28
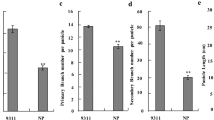
In our previous study (Kim et al. [2013]), the genetic map of IL28 was constructed using 134 markers that exhibited polymorphism between Ilpumbyeo and Moroberekan, with 2 Moroberekan introgression segments being detected on chromosomes 4 and 6. The phenotypic evaluation of agronomic traits for the introgression line, IL28, and the recurrent parent was conducted at two locations, Daejeon and Yeasan, in 2013. Comparison of 10 agronomic traits between IL28 and Ilpumbyeo obtained in the current study are shown in Table 1 and Figure 1. The results indicated that there were highly significant differences (P < 0.01) in panicle length (PL), secondary branch number (SBN), spikelets per panicle (SPP), first node width (FNW), second node width (SNW), and 1,000-grain weight between IL28 and Ilpumbyeo, while no significant difference was obtained for tiller number (TN) and culm length (CL). Because IL28 has a large endosperm and heavier grain, we investigated the grain milk-filling rate in IL28 and Ilpumbyeo. No difference was observed in either endosperm fresh weight or dry weight at 3 d and 6 d after fertilization (Figure 2). In comparison, both the fresh and dry weight of IL28 were significantly higher (P < 0.01) compared to those of Ilpumbyeo at 11 d after fertilization. These differences reached a maximum ~25 d after fertilization, at which point the fresh and dry weight of the endosperm of IL28 were 11.2% and 13.4% higher compared to Ilpumbyeo, respectively. Thus, an increase in grain weight in IL28 which is associated with higher grain milk-filling rate might be due to the Moroberekan segment on chromosome 6 considering that no QTL for grain width and weight was detected in the Moreberekan segment on chromosome 4 .

Phenotype difference between Ilpumbyeo and IL28 in (a) panicle, (b) grain size (scale bar: 3 mm), and (c) node width (scale bar: 3 mm). III, IV, and V indicate the third, fourth, and fifth nodes, respectively.

Characterization of grain filling in Ilpumbyeo and IL28. (a) Time-course of endosperm fresh weight increase. (b) Time-course of endosperm dry weight increase. Data are mean ± s.d. (n = 7 to 10 plants) in a and b.
Trait variation of the F2 population
In our previous study (Kim et al. [2013]), four traits (panicle length, numbers of primary branches and secondary branches, and spikelets per panicle) exhibited continuous and normal distributions in the 243 F2 plants. The 250-grain weight of Ilpumbyeo and IL28 was 5.51 and 6.14, respectively. In all traits measured, transgressive plants with higher or lower mean values compared to either parent were observed. The correlation between spikelets per panicle and grain weight was not significant (r = 0.102).
QTL analysis
Three significant QTLs for panicle length, secondary branch number, and spikelets per panicle were detected. These QTLs occurred in the same region, near the SSR marker RM3430 (Kim et al. [2013]). In addition, the QTL for grain weight was detected in the same region, on chromosome 6. The QTL for grain weight explained 8.3% of phenotypic variance. The Moroberekan alleles increased all trait values of panicle length, secondary branch number, spikelets per panicle and grain weight at this region.
Substitution mapping of qTGW6
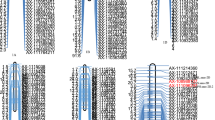
To confirm and narrow down the target region containing qTGW6, 1,120 F2 plants derived from a cross between Ilpumbyeo and IL28 were genotyped with 4 additional markers near the target region (RM20512, RM20521, RM20562, and RM20572) and one marker (RM551) located on chromosome 4. A total of 44 F2 homozygous plants with different cross-over breakpoints between RM7269 and RM20653 on chromosome 6 were selected and selfed to produce F3:4:5 lines. Genotype analysis indicated that these 44 F2 plants did not have Moroberekan segment on chromosome 4. According to the marker genotype, the 44 F3:4:5 lines were divided into 11 groups, and used for the substitution mapping of qTGW6 (Figure 3). The mean phenotypic values of each trait for each F3:4:5 line was compared to those of Ilpumbyeo and IL28 at the P < 0.05 level. In a previous study, the QTLs for SPP, PL, SBM, FNW, and SNW were consistently mapped between RM20521 and RM20572, which are 680-kb apart (Kim et al. [2013]). For the qTGW6 locus, groups D, E, F, G, H, I, and J showed significantly higher values than Ilpumbyeo for 1,000-grain weight, whereas no significance difference was detected between Ilpumbyeo and groups A, B, C, and K in 2011, 2012, and 2013 (Figure 3a). Thus, qTGW6 was located downstream of RM20521 and upstream of RM20580. Group D showed significantly higher values for SPP affected by qSPP6 and for TGW compared to Ilpumbyeo, while group J was significantly different from Ilpumbyeo for TGW but not SPP. This result indicates that qSPP6 and qTGW6 are different genes. Comparison of the genotypes of recombinants delimited the qTGW6 between markers RM20562 and RM20572, based on the finding that the TGW of both D and J groups were significantly different from that of Ilpumbyeo. The region between the two markers RM20562 and RM29572 for groups D and J shared common segment harboring of qTGW6. To further confirm the common segment shared by groups D and J, we genotyped the F3 lines using four markers (one InDel marker and three SNP markers) located between RM20562 and RM20572. The genotypes showed that qTGW6 was defined in a 37.85-kb region between InDel-1 and SNP3 (Figure 3b).

Development of a high density map of qTGW6 and structures of candidate genes for qTGW6. (a) Graphical representation of F3:4:5 lines and a fine scale map of the target region on chromosome 6. Apo1 located between RM3430 and RM20562 might be allelic to qSPP6 detected in this study. White and black portions of the graph are homozygous Ilpumbyeo, homozygous Moroberekan, respectively and dotted regions are where crossing-over occurred. Genotypes of the 44 lines were double checked at F3 generation. The table to the right of the graphical genotypes indicates mean values of traits. Numbers followed by the same letter in each column are not significantly different at P = 0.05 based on Duncan’s multiple range test. & No. in ( ) indicate the number of F3 lines. P1: Ilpumbyeo, P2: IL28. (b) High density map of qTGW6 using two groups (D and J) and four markers. Four genes were predicted in the target 37.85-kb region. The arrowheads indicate the position of a single non-synonymous nucleotide substitution. (c) Structures of two genes, Loc_Os06g45550 and Loc_Os06g45560. Loc_Os06g45550 consists of five exons (black boxes) and four introns and Loc_Os06g45560 consists of 8 exons and 7 introns. Numbers on the structure of two genes show the position of non-synonymous nucleotide substitutions from the start codon.
Evaluation of the grain shape trait
To determine which grain shape traits are associated with increase in grain weight, three grain shape traits (specifically, grain length [GL], width [GW], and thickness [GT]) were evaluated for the two parents and F5 lines (Figure 3). The mean phenotypic values of each F5 line were compared to those of Ilpumbyeo and IL28 at the P < 0.05 level. The GL of Ilpumbyeo and IL28 were 4.88 mm and 5.13 mm, respectively. The GW values for Ilpumbyeo and IL28 were 2.93 mm and 3.15 mm, respectively. The GL and GW values of IL28 were significantly higher compared to Ilpumbyeo (P < 0.05), whereas no significant difference was detected for GT. The GL and GW of the groups with qTGW6 (D, E, F, G, H, I, and J) were significantly different from those of the groups without qTGW6 (A, B, C, and K). This finding indicates that qTGW6 variation is caused by variation in GL and GW at qTGW6. Grain weight was significantly correlated with GL and GW (P < 0.01 for both characteristics), but not with GT (r = −0.06).
Cross-section of the central part of the spikelet hull
Given that the hull of the IL28 spikelet was wider than that of Ilpumbyeo before fertilization, the cross-section of the central part of the spikelet hull for groups D and J was analyzed in comparison to Ilpumbyeo to investigate the origins of the observed size differences (Figure 4). The outer glume cell layer of groups D and J contained substantially more cells (16.5% and 15.3%, respectively) compared to that of Ilpumbyeo, with only a 1.3% and 1.4% increase in cell length, respectively. These data indicate that the increased width of group D and J spikelet hulls mainly results from an increase in cell number, rather than cell size, indicating that qTGW6 may be involved in the regulation of cell division.

Histological analyses of spikelet hull 3 days before heading in Ilpumbyeo, group D, and group J. (a) Cross-section of spikelet hull. Upper: spikelets (scale bar, 3 mm). Low: cross-section of spikelet hull (scale bar, 500um). Dotted line indicates position of cross-section (b) Magnified view of spikelet hull cross-section boxed in a. (c) Comparison of total cell number and mean cell length in the outer glume cell layers of spikelet hull of Ilpumbyeo, group D, and group J. (n = 5 to 8 spikelets). Numbers followed by the same letter on each box are not significantly different at P = 0.05 based on Duncan’s multiple range test.
Impact of the QTL cluster on yield per plant
Eleven groups containing 44 F5 lines were used for the yield trials, together with Ilpumbyeo and IL28 in 2013 (Figure 3). IL28 had significantly higher grain yield compared to Ilpumbyeo. IL28 produced 8% higher grain yield on average compared to Ilpumbyeo. All groups with the Moroberekan segment at both qSPP6 and qTGW6 (i.e., groups D, E, F, G, H, and I) had significantly higher grain yield values compared with the groups without the Moroberekan segment at both qSPP6 and qTGW6 (i.e., groups A, B, C, and K) and Ilpumbyeo (Figure 3). Also, the yield per plant of group J with qTGW6 and without qSPP6 was higher than that of group C and Ilpumbyeo at P < 0.1 (P = 0.068, P = 0.067, respectively), and this result implies the contribution of qTGW6 to the yield increase. However, we could not determine the effect of qSPP6 on yield due to the lack of line(s) only with the Moroberekan segment at qSPP6.
Candidate genes for qTGW6
qTGW6 was defined in a 37.85-kb region between InDel-1 and SNP3. On the basis of available sequence annotation databases (http://www.gramene.org), there are four predicted genes in the target region. The functional annotations of the four genes are as follows: Loc_Os06g45550 (five exons and four introns) and Loc_Os06g45560 (eight exons and seven introns) are retrotransposon protein putative expressed genes; Loc_Os06g45570 (one exon) is a VQ domain-containing protein putative expressed gene; Loc_Os06g45580 (one exon) is a RING-H2 zinc finger putative protein expressed gene (Figure 3). We compared the genomic sequences of the open reading frame (ORF) and promoter region of the four genes between Ilpumbyeo and Moroberekan. Comparison of the genomic sequence of two genes (Loc_Os06g45550 and Loc_Os06g45560) among Nipponbare, Ilpumbyeo, and Moroberekan revealed the existence of a single non-synonymous SNP in the third and fourth exon of Loc_Os06g45550 and Loc_Os06g45560, respectively. The non-synonymous SNP at the third exon of Loc_Os06g45550 is characterized by the nucleotide Cytosine in Ilpumbyeo and the nucleotide Adenine in Moroberekan. The change in the nucleotide base from Cytosine to Adenine resulted in missense mutation from cysteine to tyrosine. The non-synonymous SNP at the fourth exon of Loc_Os06g45560, is characterized by the nucleotide Adenine in Ilpumbyeo and the nucleotide Guanine in Moroberekan. The nucleotide substitution A (adenine) to G (guanine) in Loc_Os06g45560 resulted in a missense mutation from Threonine to Alanine. No sequence variation in open reading frames and the promoter region of two genes was detected for Loc_Os06g45570 and Loc_Os06g45580 among the three varieties, Nipponbare, Ilpumbyeo, and Moroberekan.
Discussion
There has been much debate as to whether similar genomic locations of QTLs that affect different traits are caused by the pleiotropy of a single gene or the tight linkage of several genes that individually influence specific traits. The question of pleiotropy versus tight linkage might be resolved by using a large-size population combined with high-density mapping, because two linked QTLs segregate independently in NIL-F2:3 populations. Recently, several QTLs controlling the same or multiple traits have been confirmed to be closely linked in a small genomic region. For example, the rice photoperiod sensitivity gene, Hd3 was originally detected as a heading date-related quantitative trait locus that is localized on chromosome 6 in the F2 population (Yano et al. [1997]). For the fine-scale mapping of Hd3, high-resolution linkage analysis using a large segregating population derived from advanced backcross progeny between the japonica rice variety, Nipponbare and the indica variety, Kasalath, was carried out. As a result of the high-resolution linkage mapping of Hd3, two tightly linked loci, Hd3a and Hd3b, which promote flowering under short-day conditions and inhibit heading under long-day conditions, were identified in the Hd3 region (Monna et al. [2002]). Lou et al. ([2013]) mapped two QTLs (qSPP5 and qTGW5) for spikelets per panicle and 1,000-grain weight to the same region on chromosome 5. Substitution mapping with the BC5F3 and BC5F4 populations derived from a cross between QTL-NIL and the recurrent parent, demonstrated that two tightly linked QTLs (qSPP5 and qTGW5) were different. Several QTLs with large pleiotropic effects on multiple traits have also been found and cloned (APO1, Ghd7, and Ghd8 or DTH8). For instance, Hua et al. ([2002]) reported that a QTL cluster influences multiple traits (namely, grain per panicle, plant height, and heading date) on chromosome 7 in a primary population. High-resolution mapping with a large-sized segregating population and complementation tests revealed that these three traits are controlled by a single locus, designated as Ghd7 (Xue et al. [2008]).
The original target of this study was qSPP6, which was mapped on the long arm of chromosome 6 (Ju et al. [2008]). In the process of the fine-scale mapping, the QTL for additional yield components (including panicle length, node width, and 1,000-grain weight) were consistently detected in the same region. A total of 243 F2 plants derived from a cross between NIL (IL28) and Ilpumbyeo for genetic analysis was not large enough to clarify the relationship between SPP and TGW. To confirm the precise location of the two QTLs, substitution mapping was carried out. Following substitution mapping with 44 F3:4:5 lines, qSPP6 was mapped to a 431-kb interval between RM20521 and SNP1, while qTGW6 was mapped to a 37.85-kb interval between InDel-1 and SNP3, based on the japonica genome sequence. Therefore, the development of NIL and substitution mapping has enabled us to map SPP and TGW at the fine-scale.
No TGW-associated QTL was detected near the qTGW6 QTL in this study, indicating that qTGW6 is a novel QTL for TGW. Among the four predicted genes in the target region between InDel-1 and SNP-3 (37.85-kb), Loc_Os06g45570 encodes a VQ domain-containing protein. VQ domains are believed to be critical for the defense response to pathogens by interacting with WRKY transcription factors (Tao et al. [2011]). Loc_Os06g45580 encodes the RING-H2 zinc finger protein. Recent studies reported a critical role of the ubiquitin pathway in grain development in rice (Song et al. [2007]; Weng et al. [2008]). GW2, a major QTL for rice grain width and weight encodes a new RING-type E3 ubiquitin ligase. GW5, another major QTL underlying rice width and weight, is likely to act in the ubiquitine-proteasome pathway to regulate cell division during cell division (Weng et al. [2008]). qTGW6 exhibited common function for grain width with GW2 and GW5 in that it was associated with increased cell numbers, a wider spikelet hull and an increased grain milk-filling rate. This raises a possibility that Loc_Os06g45580 is a candidate for qTGW6. However, no sequence variation in open reading frames and the promoter regions of two genes, Loc_Os06g45570 and Loc_Os06g45580 was observed between Ilpumbyeo and Moroberekan and this result seems to suggest that they are not candidate genes for qTGW6. The other two genes in the target region are retrotransposon proteins, Loc_Os06g45550 and Loc_Os06g45560. Loc_Os06g45560 was transcribed in seedling and embryo/endosperm 25 days after pollination but Loc_Os06g45550 was not transcribed based on the available gene expression database (RGAP, http://rice.plantbiology.msu.edu/expression.shtml). More than 40% of rice genome is composed of repetitive sequences and transposable elements (Goff et al. [2002]). A recent study showed that the expression of Tos17, a copia-like retrotransposon was affected by overexpression or down-regulation of several rice SUVH (Su(var)3–9 homologs) genes and RNAi plants with down-regulated SDG728, a rice SUVH gene showed a reduced seed size phenotype (Qin et al. [2010]). This raises a possibility that a relationship between altered seed morphology and overexpression of Tos17 might exist. Interestingly, our alignment analysis of GW5 a.a sequence using BLASTP program showed that GW5 protein has high homology with two retrotransposon proteins encoded by LOC_Os08g26160.1 and LOC_Os05g47720.1 (data not shown), although it has been reported that the protein encoded by the GW5 ORF had no sequence homology with any proteins of known molecular function (Weng et al., [2008]). GW5 protein showed 56% homology to the retrotransposon protein with 194 a.a encoded by LOC_Os08g26160.1 and 47-50% homologies to the transposon protein with 411 a.a. by LOC_Os05g47720.1. Numerous Indels and single-nucleotide polymorphisms (SNPs) were detected between Nipponbare (japonica) and 9311 (indica) and Indels are mainly caused by transposons (Feng et al. [2002]). Indels and SNPs in some key domestication genes including qSH1 and GW2 are responsible for morphological changes occurred during domestication (Konishi et al. [2006]; Song et al. [2007]). Also, the difference in grain width between japonica and indica cultivars is associated with a 1212-bp deletion harboring the GW5 ORF which was possibly caused by transposons (Weng et al. [2008]). These observations support our hypothesis that the protein encoded by the GW5 ORF might be a retrotransposon and Loc_Os06g45560 is a candidate for qTGW6. However, the presence of SNPs in the upstream regions of the genes between two parents suggests the possibility that a long-distance regulation is involved in controlling the qTGW6 (data not shown). Konishi et al. ([2006]) reported that the casual mutation in qSH1 underlying the seed shattering was found in the 12-kb upstream region from the ORF of the gene and caused the absence of the abscission layer in Nipponbare. Additional fine mapping experiments are underway to determine the functional relationship between qTGW6 and GW5 in controlling seed development. Also, loss-of-function mutant lines of Loc_Os06g45560 will be evaluated for grain morphology traits and the gene expression. These informations on the molecular mechanism of qTGW6 would be necessary to get more insights into the mechanism of grain development.
Although the candidate gene of qTGW6 and its genetic mechanism was not elucidated here, we found that the Moroberekan chromosomal segment at this region (a tropical japonica) harbors QTLs for yield component traits leading to an yield increase in japonica cultivar. Yield trials confirmed that introgression lines containing the Moroberekan segment at the qSPP6 and qTGW6 loci produced significantly higher yields than Ilpumbyeo (P < 0.05). The grain yield per plant in IL28 was 8% higher than that of Ilpumbyeo. Moroberekan alleles in the target region on chromosome 6 had a favorable effect on yield, by increasing the number of spikelets per panicle and 1,000-grain weight, in addition to lodging tolerance by increasing the node width, whereas it had no negative effect on heading date and plant height. Thus, the qSPP6 and qTGW6 alleles are potentially valuable for improving rice yield. It is proposed that NILs containing the target segment associated with positive QTLs from Moreberekan should be developed from this population, and evaluated in a wide range of environments, to assess the interaction between QTLs and the environment. Tightly linked SSR markers are expected to facilitate the cloning of genes underlying these QTLs, in addition to marker-assisted selection for variation in the SPP and TGW in rice breeding programs.
Conclusion
In this study, we demonstrated that two QTLs, qSPP6 for spikelets per panicle (SPP) and qTGW6 for 1,000-grain weight (TGW) are tightly linked on chromosome 6. Because Moroberekan alleles for the SPP and TGW have been shown to be beneficial in the genetic background of Ilpumbyeo, the qSPP6 and qTGW6 alleles may prove valuable for improving the yield potential of japonica rice cultivars. Tightly linked SSR markers are expected to facilitate the cloning of genes underlying these QTLs, in addition to marker-assisted selection for variation in SPP and TGW in rice breeding programs.
Methods
Plant materials
In a previous study (Kim et al. [2013]), IL28 was shown to exhibit a higher number of spikelets per panicle and heavier grain compared to the recurrent parent, and contained the Moroberekan chromosomal segment at the qSPP6 region on chromosome 6; hence, it was selected for the fine-scale mapping of qSPP6 in the current study. IL28 was crossed with the cultivar Ilpumbyeo, with the resulting F1 plants being self-pollinated to obtain an F2 population (234 plants in 2009 and 1,150 plants in 2010). To confirm qSPP6, 44 F2 homozygous plants with different cross-over breakpoints between RM7269 and RM20653 were selected and selfed to produce F3 lines for the phenotyping and substitution mapping of qSPP6. In the process of the fine-scale mapping of qSPP6, a QTL for 1,000-grain weight (TGW) was consistently detected in the same region. To confirm the precise location of qSPP6 and qTGW6, these 44 F3 lines were advanced to F4 and F5 lines, which were used to evaluate spikelets per panicle, grain weight, grain morphology traits, and grain yield.
Field trial and trait evaluation
A total of 243 F2 plants and 1150 F2 plants (2009 and 2010), and 41 F3:4:5 lines (2011, 2012, and 2013) and the parental lines were grown at the experimental field of Chungnam National University, Daejeon, Korea. In 2009, the 243 plants and the parents were planted at distances of 30 × 15 cm, and evaluated for grain (brown rice) weight (150 out of 243 plants were used to evaluate grain weight). In 2010, for the substitution mapping of qTGW6, 1,150 F2 plants were genotyped with four additional markers being located between RM7269 and RM20653 (RM20512, RM20521, RM20652, and RM20572) and one marker (RM551) located on chromosome 4. The experiment using 44 F3:4:5 lines derived from 44 F2 plants followed a completely randomized block design with two replications, one row per plot, and 25 plants per row in 2011, 2012, and 2013. The middle 10 plants from each line were chosen for the evaluation, and the average of the measurements was used for the phenotype of each line for the spikelets per panicle (SPP) and 1,000-grain weight (TGW). SPP were measured by averaging the three major panicles per plants. Grains with hulls were allowed to dry naturally after harvesting, and partial or unfilled seeds were removed by soaking grains in water. Fully filled seeds were re-dried in an oven at 30°C for 24 h. The TGW was evaluated by measuring the weight of 250 randomly selected, de-hulled grains per plant (10 plants per line). In 2013, grain length (GL), grain width (GW), grain thickness (GT), and yield per plants (YD) were measured. The grain length (GL), grain width (GW), and grain thickness (GT) were measured for 50 grains (brown rice) per plant (10 plants per line) using a 150-mm Vernier caliper (Mitutoyo Corp., Japan). Yield per plant (YD) was measured by averaging the grain yield (g) of 10 plants that were randomly selected from the center of each plot per block. The 1,000-grain weight and the yield per plant were corrected for the 12% grain moisture content.
DNA extraction and SSR analysis
DNA was extracted from the leaf tissue of the F2 population using a chloroform-based DNA extraction protocol (Causse et al., [1994]). A 20-μL reaction mixture was used, containing 5.0 μL (5 ng/μL) of template DNA, 0.1 μl (5 Unit/μL) Taq polymerase, 0.8 μl dNTP (2.5 mM each), Forward + Reverse primer 1 μl (10 pmol each), 2.0 μl 10× PCR buffer (10 mM Tris–HCl PH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.1% gelatin), and 11.1 μl triple-distilled water. Amplification was achieved using a Thermo Cycler (Bio-Rad) based on the step-cycle program set for denaturation at 94°C for 5 min, subsequent denaturation was performed at 94°C for 1 min, annealing at 55°C for 1 min, extension at 72°C for 1 min; steps 2 to 4 were repeated for a total of 35 cycles, with a final extension step at 72°C for 5 min. PCR products were run on 4% polyacrylamide denaturing gels for 1–2 h at 1800–2000 V, and marker bands were revealed by silver staining (Panaud et al. [1996]). The orientation of the SSR markers was based on the SSR maps (McCouch et al. [2002]). Due to the lack of markers showing polymorphism in the region between RM20562 and RM20572, additional genotyping of the F3 lines was conducted with four targeted SNPs and one Insertion/Delition marker. To detect the SNPs and InDels of Ilpumbyeo and Moroberekan, resequencing with NGS (Next-Generation Sequencing) was conducted using the two parents according to Jeong et al. ([2013]). Through the comparison of genomic sequences corresponding to the region between RM20562 and RM20572, a total of 28 polymorphisms (14 SNPs, and 14 Insertion/Deletions) were detected between Ilpumbyeo and Moroberekan. The polymorphisms were assayed by the direct sequencing of the four regions. Information on the primers used is presented in Table 2. The primers were designed according to the Nipponbare sequence (http://rgp.dna.affrc.go.jp/E/IRGSP/Build5/build5.html). The first InDel, hereafter referred to as InDel-1, occurs at the 27,565,774th position based on the Nipponbare sequence (www.gramene.org), and is characterized by nucleotide - (1 bp deletion) in Ilpumbyeo and nucleotide A in Moroberekan. The first SNP, SNP-1, that occurs at the 27,572,767th position, is characterized by nucleotide G in Ilpumbyeo and nucleotide A in Moroberekan. The second SNP, SNP-2, that occurs at the 27,586,202th position, is characterized by nucleotide A in Ilpumbyeo and nucleotide G in Moroberekan. The third SNP, SNP-3, that occurs at the 27,629,080th position, is characterized by nucleotide A in Ilpumbyeo and nucleotide C in Moroberekan.
Statistical analysis
Statistical analysis was performed using Qgene software version 2.30 for Macintosh (Nelson [1997]) and SAS (SAS Institute). Single point analysis (SPA) was performed to determine the effect of each marker on each trait. In SPA, a QTL was confirmed if the phenotype was associated with a marker locus at P < 0.005, or with two adjacent marker loci at P < 0.01. The proportion of total phenotypic variation explained by each QTL was calculated as an R2 value, from the regression of each marker/phenotype combination. QTLs were mapped at a fine-scale by comparing the phenotypic means of two genotypic classes of recombinants (Ilpumbyeo homozygote and Moroberekan homozygote) within the target region by using the SAS statistical software package.
Histological analysis
Paraffin-embedded sections of spikelet samples were prepared according to Li et al. ([2009]), with minor modifications. Plant materials were fixed in FAA (50% ethanol, 5% glacial acetic acid and 5% formaldehyde) and stored at 4°C for 24 h. The fixed spikelets were dehydrated by soaking them for 6 h in a gradient ethanol series (60%, 70%, 80%, 90%), and were then incubated in 100% ethanol overnight. Dehydrated spikelets were embedded in Paraplast (Sigma). Tissue sections (8 μm thick) were cut using a rotary microtome, transferred onto gelatin-coated glass slides, and dried at 42°C for 1 day. The sections were de-paraffinized with 100% xylene for 5 min, followed by soaking for 2 min in 50% ethanol/50% xylen, 100% ethanol, and sterile water. The samples were stained with toluidine blue for 30 s, and washed twice with sterile water. The samples were then soaked for 2 min in 70% ethanol, 80% ethanol, 90% ethanol, and 100% ethanol. Finally, the samples were cleaned by soaking them twice in 100% xylen. The sections were photographed under an Olympus BH2-RFCA using a U-PMTVC camera.
References
Ando T, Yamamoto T, Shimizu T, Ma XF, Shomura A, Takeuchi Y, Lin SY, Yano M: Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosomal segment substitution lines in rice. Theor Appl Genet 2008, 116: 881–890. 10.1007/s00122-008-0722-6
Ashikari M, Sakakibara H, Lin SY, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M: Cytokinin oxidase regulates rice grain production. Science 2005, 309: 741–745. 10.1126/science.1113373
Causse MA, Fulton TM, Cho YG, Ahn SN, Chunwonse J, Wu K, Xiao J, Yu Z, Ronald PC, Harrington SE, Second G, McCouch SR, Tanksley SD: Saturated molecular map of the rice genome based on an interspecific backcross population. Genetics 1994, 138: 1251–1274.
Cui KH, Peng SB, Xing YZ, Yu SB, Xu CG, Zhang Q: Molecular dissection of the genetic relationships of source-sink and transport tissue with yield traits in rice. Theor Appl Genet 2003, 106: 649–658.
Feng Q, Zhang Y, Hao P, Wang S, Fu G, Huang Y, Li Y, Zhu J, Liu Y, Hi X, Jia P, Zhang Y, Zhao Q, Ying K, Yu S, Tang Y, Weng Q, Zhang L, Lu Y, Mu J, Lu Y, Zhang LS, Yu Z, Fan D, Liu X, Lu T, Li C, Wu Y, Sun T, Lei H, et al.: Sequence and analysis of rice chromosome 4. Nature 2002, 420: 316–320. 10.1038/nature01183
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, et al.: A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296: 92–100. 10.1126/science.1068275
Hua JP, Xing YZ, Xu CG, Sun XL, Yu SB, Zhang Q: Genetic dissection of an elite rice hybrid reveal that heterozygotes are not always advantageous for performance. Genetics 2002, 162: 1885–1895.
Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X: Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 2009, 41: 494–497. 10.1038/ng.352
Ikeda K, Ito M, Nagasawa N, Kyozuka J, Nagato Y: Rice ABERRANT PANICLE ORGANIZATION 1, encoding an F-box protein, regulates meristem fate. Plant J 2007, 51: 1030–1040. 10.1111/j.1365-313X.2007.03200.x
Ishimaru K: Identification of a locus increasing rice yield and physiological analysis of its function. Plant Physiol 2003, 133: 1083–1090. 10.1104/pp.103.027607
Ishimaru K, Hirotsu N, Madoka Y, Murakami N, Hara N, Onodera H, Kashiwagi T, Kazuhiro U, Shimizu B, Onishi A, Miyagawa H, Katoh E: Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat Genet 2013, 45: 707–711. 10.1038/ng.2612
Jeong IS, Yoon UH, Lee GS, Ji HS, Lee HJ, Han CD, Hahn JH, An GH, Kim TH: SNP-based analysis of genetic diversity in anther-derived rice by whole genome sequencing. Rice 2013, 6: 6. 10.1186/1939-8433-6-6
Ji SD, Luo XL, Ahn SN: Mapping QTL for ratooning ability in advanced backcross lines from an Oryza sativa x O. rufipogon cross. CNU J Agricultural Sci 2014, 41(1):1–7. 10.7744/cnujas.2014.41.1.001
Jin F, Kim DM, Ju HG, Ahn SN: Mapping quantitative trait loci for awnness and yield component tratis in isogenic lines derived from an Oryza sativa / O. rufipogon cross. J Crop Sci Biotech 2009, 12(1):9–16. 10.1007/s12892-009-0061-4
Ju HG, Kim DM, Kang JW, Kim MK, Kim YG, Ahn SN: Mapping QTLs for agronomic traits using an introgression line population from a cross between Ilpumbyeo and Moroberekan in rice. Korean J Breed 2008, 40(4):414–421.
Kim DM, Kim JH, Kang JW, Lee HS, Ahn SN: Fine mapping of a quantitative trait loci controlling the number of spikelets per panicle in rice. Korean J Breed 2013, 44(4):462–469.
Khush GS: Green revolution: preparing for the 21st century. Genome 1999, 42: 646–655. 10.1139/g99-044
Khush GS: What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol 2005, 59: 1–6. 10.1007/s11103-005-2159-5
Konishi S, Izawa T, Lin S, Ebana K, Fukuta Y, Sasaki T, Yano M: An SNP caused loss of seed shattering during rice domestication. Science 2006, 312: 1392–1396. 10.1126/science.1126410
Li C, Zhou A, Sang T: Gentetic analysis of rice domestication syndrome with the wild annual species. Oryza Nivara New Phytol 2006, 170: 185–194. 10.1111/j.1469-8137.2005.01647.x
Li H, Wue D, Gao Z, Yan M, Xu W, Xing Z, Huang D, Qian Q, Wue Y: A putative lipase gene EXTRA GLUME1 regulates both empty-glume fate and spikelet development in rice. Plant J 2009, 57: 593–605. 10.1111/j.1365-313X.2008.03710.x
Lin HX, Qian HR, Zhuang JY, Lu J, Min SK, Xiong ZM, Huang N, Zheng KL: RFLP mapping of QTLs for yield and related characters in rice ( Oryza sativa L.). Theor Appl Genet 1996, 92: 920–927. 10.1007/BF00224031
Linh HL, Hang NT, Jin FX, Kang KH, Lee YT, Kwon KH, Ahn SN: Introgression of a quantitative trait locus for spikelets per panicle from Oryza minuta to the O. sativa cultivar Hwaseongbyeo. Plant Breed 2008, 127: 262–267. 10.1111/j.1439-0523.2007.01462.x
Lou X, Ji SD, Yuan PR, Lee HS, Kim DM, Balkunde S, Kang JW, Ahn SN: QTL mapping reveals a tight linkage between QTLs for grain weight and panicle spikelet number in rice. Rice 2013, 6: 33. 10.1186/1939-8433-6-33
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein D: Development and mapping of 2240 new SSR markers for rice ( Oryza sativa L.). DNA Res 2002, 9: 199–207. 10.1093/dnares/9.6.199
Miura K, Ikeda M, Matsubara A, Song XJ, Ito M, Asano K, Matsuoka M, Kitano H, Ashikari M: OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat Genet 2010, 42: 545–549. 10.1038/ng.592
Monna L, Lin X, Kojima S, Sasaki T, Yano M: Genetic dissection of a genomic region for a quantitative trait locus, Hd3, into two loci, Hd3a and Hd3b, controlling heading date in rice. Theor Appl Genet 2002, 104: 772–778. 10.1007/s00122-001-0813-0
Nelson JC: QGENE: software for marker-based genomic analysis and breeding. Mol Breed 1997, 3: 239–245. 10.1023/A:1009604312050
Ookawa T, Hobo T, Yano M, Murata K, Ando T, Miura H, Asano K, Ochiai Y, Ikeda M, Nishitani R, Ebitani T, Ozaki H, Angeles ER, Hirasawa T, Matsuoka M: New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat Comms 2010, ᅟ: 1–132. doi:10. 1038/ncomms1132 doi:10. 1038/ncomms1132 10.1038/ncomms1090
Panaud O, Chen X, McCouch SR: Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice ( Oryza sativa L.). Mol Gen Genet 1996, 252: 597–607.
Qin FJ, Sun QW, Huang LM, Chen XS, Zhou DX: Rice SUVH histone methyltransferase genes display specific functions in chromatin modification and retrotransposon repression. Mol Plant 2010, 3(4):773–782. 10.1093/mp/ssq030
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX: A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 2007, 39: 623–630. 10.1038/ng2014
Suh JP, Ahn SN, Cho YC, Suh HS, Hwang HG: Mapping of QTLs for yield traits using an advanced backcross population from a cross between Oryza sativa and O. glaberrima . Korean J Breed 2005, 37(4):214–220.
Tao Z, Kou Y, Liu H, Li X, Xiao J, Wang S: OsWRKY45 alleles play different roles in abscisic acid signaling and salt stress tolerance but similar roles in drought and cold tolerance in rice. J Exp Bot 2011, 62: 4863–4874. 10.1093/jxb/err144
Thomson MJ, Tai T, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR: Mapping quantitative trait loci for yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 2003, 107: 479–493. 10.1007/s00122-003-1270-8
Weng J, Gu S, Wan X, Gao H, Guo T, Su N, Lei C, Zhang X, Cheng Z, Guo X, Wang J, Zhai H, Wan J: Isolation and initial characterization of GW5 , a major QTL associated with rice grain width and weight. Cell Res 2008, 18: 1199–1209. 10.1038/cr.2008.307
Xie X, Jin F, Song MH, Suh JP, Hwan HG, Kim YG, McCouch SR, Anh SN: Fine mapping of a yield-enhancing QTL cluster associated with transgressive variation in an Oryza sativa X O. rufipogon cross. Theor Appl Genet 2008, 116: 613–622. 10.1007/s00122-007-0695-x
Xiong LZ, Liu KD, Dai XK, Xu CG, Zhang Q: Identification of genetic factors controlling domestication related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon . Theor Appl Genet 1999, 98: 243–251. 10.1007/s001220051064
Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q: Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 2008, 40: 761–767. 10.1038/ng.143
Yamagishi M, Takeuchi Y, Kono I, Yano M: QTL analysis for panicle characteristics in temperate japonica rice. Euphytica 2002, 128: 219–224. 10.1023/A:1020893731249
Yano M, Harushima Y, Nagamura Y, Kurata N, Minobe Y, Sasaki T: Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor Appl Genet 1997, 95: 1025–1032. 10.1007/s001220050658
Zhang Q: Strategies for developing green super rice. Proc Natl Acad Sci U S A 2007, 104: 16402–16409. 10.1073/pnas.0708013104
Zhang Y, Luo L, Liu T, Xu C, Xing Y: Four rice QTLs controlling number of spikelets per panicle expressed the characteristics of single Mendelian gene in near isogenic backgrounds. Theor Appl Genet 2009, 118: 1035–1044. 10.1007/s00122-008-0960-7
Zhuang JY, Lin HX, Lu J, Qian HR, Hittalmani S, Huang N, Zheng KL: Analysis of QTL X environment interaction for yield components and plant height in rice. Theor Appl Genet 1997, 95: 799–808. 10.1007/s001220050628
Acknowledgements
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund)(KRF-2010-0024118) and the Next-Generation Biogreen 21 Program (Plant Molecular Breeding Center No. PJ008136), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DK participated in phenotyping, genotyping, drafting the manuscript. HL, MF and JK participated in phenotyping. SK participated in sequencing, primer design and genotyping. SA conceived of the study, drafted proposal and corrected manuscript. All authors have read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kim, DM., Lee, HS., Kwon, SJ. et al. High-density mapping of quantitative trait loci for grain-weight and spikelet number in rice. Rice 7, 14 (2014). https://doi.org/10.1186/s12284-014-0014-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-014-0014-5