
Abstract
α-Synuclein (AS)-positive inclusions are the pathological hallmark of Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), all belonging to the category of α-synucleinopathies. α-Synucleinopathies represent progressive neurodegenerative disorders characterised by increasing incidences in the population over the age of 65. The relevance of glial reactivity and dysfunction in α-synucleinopathies is highlighted by numerous experimental evidences. Glial AS inclusion pathology is prominent in oligodendroglia of MSA (glial cytoplasmic inclusions) and is a common finding in astroglial cells of PD and DLB, resulting in specific dysfunctional responses. Involvement of AS-dependent astroglial and microglial activation in neurodegenerative mechanisms, and therefore in disease initiation and progression, has been suggested. The aim of this review is to summarise and discuss the multifaceted responses of glial cells in α-synucleinopathies. The beneficial, as well as detrimental, effects of glial cells on neuronal viability are taken into consideration to draw an integrated picture of glial roles in α-synucleinopathies. Furthermore, an overview on therapeutic approaches outlines the difficulties of translating promising experimental studies into successful clinical trials targeting candidate glial pathomechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
General Background
α-Synuclein (AS) belongs to a distinct protein family including α-, β- and γ-synuclein. It is natively unfolded and consists of 140 amino acids. Its importance in synaptic structure and presynaptic terminal size was recently demonstrated in αβγ-knockout mice [1]. Furthermore, AS plays an important role in the mechanisms of folding and re-folding of synaptic proteins, acting in close connection with cysteine string protein-α and SNARE proteins [2].
The term α-synucleinopathies comprises progressive, neurodegenerative diseases including Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) with the major pathological hallmark of AS-positive inclusions in neuronal and glial cells. Neuronal inclusions, Lewy bodies (LBs) and Lewy neurites (LNs) are characteristic for PD and DLB, while AS-positive glial cytoplasmic inclusions (GCIs) are distinctive in MSA and occur predominantly in oligodendroglial cells [3,4]. Astroglial AS-positive inclusions may also occur in PD [5,6]. PD pathology has been partly related to point mutations [7,8] or duplications [9] and triplications [10,11] of the SNCA gene. Moreover, SNCA variants can increase the risk of developing PD and MSA [12,13]. AS inclusion formation may be related to posttranslational modifications of AS (nitration, ubiquitination and phosphorylation) which can lead to pathological accumulation of AS and enhance the progression of α-synucleinopathies [14–16]. Involvement of impaired AS clearance through autophagy pathways is also suggested to be involved in the generation of AS inclusions in PD and DLB [17,18]. A correlation between the aggregation of AS and neuronal cell loss and disease progression respectively was demonstrated in MSA [19] and also suggested in PD/DLB according to Braak staging [20]. Moreover, prion-like cell-to-cell propagation of AS has been proposed recently as a major contributor to disease progression in α-synucleinopathies [21–23].
Since the first description of glial cells (glia meaning glue) by Rudolf Virchow in 1864, the view of glial cells as mere substrate for neurons has changed by evidence, indicating the role of glial cells in the support of neuronal survival, synaptic function and local immunity [24,25]. Furthermore, the importance of glial cells is now extended towards a crucial role in the initiation and progression of different diseases of the CNS, including α-synucleinopathies [26–29]. Glial dysfunction in α-synucleinopathies not only comprises the above-mentioned AS-positive inclusion pathology in glia but also an over-activated state of microglial and astroglial cells, termed reactive microgliosis and astrogliosis. On different stimuli, e.g. injury or infection, microglial and astroglial cells get activated [30,31]. Activation is associated with morphological changes, release of trophic and inflammatory factors and, in regard to microglia, also clearance of dead or damaged cells [30–34]. These changes can be crucial for neuronal survival [32–34]. However, regarding chronic disease conditions of the CNS like neurodegenerative diseases, astroglia and microglia can get over activated. Reactive microgliosis and astrogliosis can lead to neurotoxicity and increased tissue damage after the release of (pro-)inflammatory cytokines, reactive oxygen species (ROS) and nitric oxide (NO) [35–40]. Alternatively, oligodendroglial cells show an increased vulnerability to oxidative stress and cytokines, resulting in demyelination, diminished trophic support, cellular dysfunction and cell death which affect neuronal survival [41,42].
Neuronal α-Synucleinopathies
α-Synucleinopathies show frequent incidences among the population over the age of 65. PD affects about 3 % of the general population over the age of 65 and, therefore, is the most common neurodegenerative movement disorder [43]. Furthermore, it is characterised by relentless disease progression [44]. DLB has a frequency of 20 % regarding all cases of dementia analysed by autopsy [45]. PD and DLB show various degrees of neurodegeneration of dopaminergic neurons in substantia nigra pars compacta (SNpc) and dopaminergic terminals in the striatum, as well as degeneration of extra-nigral structures including noradrenergic system, cholinergic system, serotonergic system, limbic structures and cerebral cortex [46–48]. The aggregation of AS in neuronal cells is the major pathological hallmark of PD and DLB, including LBs and LNs [4,49]. PD also features abnormal aggregations of AS in astroglial cells [5,6]. LB pathology is mostly present at the sites of neuronal loss [48]. However, evidence shows that the occurrence of LBs in the SNpc must not necessarily lead to neuronal death and a high number of neurons undergo apoptosis without the occurrence of AS aggregates [50,51]. This leads to the assumption that other factors may have a major influence on neuronal degeneration and, therefore, on the progression of these diseases. Hence, the role of microglial [28,52,53] and astroglial [5] activation in PD and DLB progression comes into consideration. However, the role of both cell types, microglia and astroglia, is still unclear in PD and DLB because of their controversial beneficial and toxic effects on neurons [54,55]. Furthermore, AS-positive inclusions were shown in oligodendroglial cells of PD brains [6]; however, in contrast to MSA, oligodendroglial cells seem to play an inferior role in the initiation of PD and DLB but may have a possible involvement in the late stages of disease progression [29].
Astroglial cells have been shown to get activated in PD and DLB. Different reports exist on astroglial activation, claiming no or mild astrogliosis [56,57] in contrast to massive astrogliosis [58] in post-mortem PD brains. Furthermore, astroglial cells show AS-positive inclusion pathology [6], which may lead to a different reactivity pattern in PD and DLB [5,19]. AS overexpression in murine astroglial cells leads to neuroinflammation and microglial activation, and in consequence to oxidative stress [59], providing a major link of AS astroglial pathology with neuroinflammation/microgliosis and oxidative stress that may also promote neurodegeneration. A possible explanation for the AS-positive inclusions in astroglial cells in PD brains was provided by Lee and colleagues [23]. They show that astroglial cells can endocytose AS released from neurons and form inclusions similar to LBs in a time-dependent manner. Importantly, the transfer of AS from neurons to astroglia leads to increased production of tumour necrosis factor α (TNF-α) and chemokine ligand 1 by astroglial cells, and results in enhanced neurodegeneration [23].
An upregulation of interferon-γ (IFN-γ) receptor on astroglia in PD post-mortem brains suggested a neurotoxic reaction after INF-γ activation [54,60]. Furthermore, astroglial cells in the ventral midbrain of PD brains show an enhanced expression of myeloperoxidase (MPO), a key enzyme related to oxidative stress during inflammation [61]. However, astroglial cells also seem to function in a contrary direction by the production of anti-oxidative and anti-inflammatory agents. A beneficial function of astroglial cells in PD and DLB seems to be the release of neurotrophic factors, e.g. brain-derived neurotrophic factor [55]. Moreover, the activity of glutathione peroxidase (GPx), a crucial protective enzyme against oxidative damage, has been associated with astrocytic proliferation and showed an enhancement of 30 % in the substantia nigra (SN) of PD brains [62]. Enhanced levels of glial fibrillary acidic protein were associated with increased GPx activity in PD brains [63], suggesting a fundamental role of astroglia in neuronal protection against oxidative stress.
Experimental models of PD reveal further the involvement of astrocytes not only in neurotoxicity but also in neuronal protection. Astroglial cells in Parkinsonian monkeys, intoxicated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), show an upregulated expression of the IFN-γ receptor similar to human post-mortem brains. Moreover, TNF-α immunoreactivity was observed almost exclusively in astroglial cells, associated with an increased number of astrocytes even years after the MPTP intoxication, suggesting a role in neurodegeneration [64]. Selective astroglial expression of mutant A53T AS in an inducible mouse model led to rapidly progressive paralysis most likely caused by widespread astrogliosis, degeneration of spinal cord motor and dopaminergic neurons [59]. An alternative pathogenic pathway of astroglia-mediated neurotoxicity could be related to morphological and functional alterations in astroglial mitochondria and a disturbed secretion of factors crucial for neuronal differentiation as demonstrated in a genetic mouse model overexpressing mutant AS [65]. In a recent study, cerebrospinal fluid of PD patients was added to an astroglial cell culture, and a decrease in proliferation rate as well as increased contents of AS on day 7 was observed [66]. Upregulated expression of interleukine-6 (IL-6) by astroglial cells upon AS treatment has been shown in vitro, supporting the evidence of astroglia-triggered neuroinflammatory response [67]. However, astroglial release of glial cell line-derived neurotrophic factor (GDNF) may favour neuronal protection in SNpc [68]. Glutathione, another agent reported to have an antioxidant character with beneficial functions in PD, was demonstrated to be released by astroglial cells activated by 6-OHDA-injured dopaminergic neurons [69]. Hydrogen sulphide, a potential anti-inflammatory and neuroprotective agent produced by astroglial cells, was found downregulated upon inflammatory activation of astroglia or microglia, suggesting a possible mechanism relevant to PD pathogenesis [70].
In summary, it is speculated that astroglia may play a dominant role at least in the initiation of PD related to astroglial AS inclusion pathology [29]. Later on, astroglia may mediate the progression, releasing inflammatory agents and recruiting microglial cells (see Fig. 1). On the other hand, astroglia-mediated secretion of trophic and antioxidant factors should also be taken into account, even though there are insufficient data regarding AS-dependent astroglial neuroprotection. Considering the neuroprotective features of astroglial cells in oxidative stress situations, possible therapeutic options to regulate the astroglial response to AS and chronic disease conditions may be of major interest.

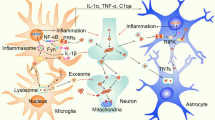
Characteristic cellular changes in the CNS during disease initiation and progression in neuronal (PD/DLB) and oligodendroglial (MSA) α-synucleinopathies. In the healthy brain, microglial cells (red) are present in a quiescent or resting state. Their main task is to scan the environment for injury or infection. Astroglial cells (green) are involved in synaptic transmission support, nutrient support and control of extracellular homeostasis, thereby crucial for neuronal viability. Neurotrophic support is also provided by microglial, astroglial and oligodendroglial (myelinating and non-myelinating) cells. Myelinating oligodendroglia (blue) are essentially involved in maintaining the myelin sheet and trophic support of myelinated neurites. Neuronal α-synucleinopathies: Early in disease, AS aggregations (yellow) in neurons (grey) and astroglial cells occur, leading to a decreased neuronal viability. Moreover, astroglial cells get activated, resulting in an enhanced release of neurotoxic pro-inflammatory factors. The recruitment of microglial cells starts even before neuronal cell loss occurs, and their arrival at the site of AS accumulation facilitates the production of pro-inflammatory cytokines, as well as oxidative stress. However, beneficial phagocytic microglial activity may be involved in the early clearance of extracellular AS. Later in disease progression, full-blown neuronal inclusion pathology develops, including the formation of LBs and LNs. The build-up of AS in astroglia leads to dysfunctionality and increased neurotoxic activity. The phagocytic microglia appears inefficient to clear extracellular AS and to stop disease progression. Accumulation of AS may even occur in non-myelinating oligodendroglial cells late in disease. All these dramatic changes in the CNS lead to chronic overactivation of glial cells and an enhanced neuronal cell loss. Oligodendroglial α-synucleinopathies: At the beginning of MSA, oligodendroglial cells start to accumulate AS in the cytoplasm (origin is still unresolved). Demyelination, oligodendroglial and neuronal degeneration are initiated. Again, activated microglia and astroglia are attracted to the sites of GCI accumulation, and through the release of pro-inflammatory cytokines and oxidative stress, promote the disease. However, microglial phagocytic activity may provide an effort to reduce extracellular AS levels in the CNS. Final stage MSA is presented by AS-positive GCIs, massive oligodendroglial dysfunction (demyelination and disturbed trophic support) and prominent reactive gliosis. Moreover, accumulation of AS in the cytoplasm and nucleus of neurons is frequent. In consequence of all these cellular changes, secondary axonal degeneration and neuronal cell death occur
Microglial cells have been shown to be activated in all α-synucleinopathies. In PD, different studies report an accumulation of reactive microglia around AS-positive LBs using in vivo imaging techniques [28] or post-mortem brain analysis [53,71]. In DLB as well, a correlation between microglial activation and LBs in different brain regions has been demonstrated [52]. Microglial activation is shown in different brain regions of PD end-stage cases, including SN, putamen, hippocampus, transentorhinal, cingulated and temporal cortex [72], in all areas of the limbic system, in particular, the dentate gyrus and the CA2/3 region of the hippocampus [73]. To clarify if there is an involvement of microgliosis in PD initiation, Ouchi and colleagues [74] used [11C](R)-PK11195 PET scans to image microglial activation in early-stage drug-naïve PD patients [74]. They demonstrated that enhanced microglial activation in midbrain correlates with the loss of dopaminergic terminals in PD. The re-scan of some patients in a follow-up study presented a more prevalent distribution of microgliosis, also affecting extra-striatal regions of the brain [74].
The observations in PD/DLB patients support the recently suggested hypothesis that microglial activation, stimulated by extracellular AS or astroglia, occurs before neurodegeneration in SNpc and is therefore a major participant in the initiation of PD and DLB [29,75]. Furthermore, microglia are suggested to be crucial in the ongoing progression of PD and DLB including, e.g. the secretion of different pro-inflammatory agents [75–78]. Therefore, different experimental models characterise microglial activation by AS and modified forms of AS. The overexpression of wild-type AS in mice presented early microglial activation [78]. The neuronal overexpression of mutant AS forms (A53T and A30P homozygous double mutants) may even enhance microgliosis [75]. In a rat PD model with rAAV-based overexpression of AS in the midbrain, the cell number of microglia increased with the level of AS expression [79]. In addition, dopaminergic cell death influenced the AS-induced microglial activation: Occurrence of dopaminergic cell death lead to a delayed and long-lasting microglial activation, whereas the absence of dopaminergic cell death induced an early and transient activation. Furthermore, the activation profile upon AS overexpression was related to four different types of microglial activation associated with different stages of progression of the neurodegenerative process [79]. In a PD-like mouse model with rAAV-based overexpression of human AS, microglia and the adaptive immune system were activated due to AS alone [80]. AS led to NF-κB/p65 expression, release of pro-inflammatory cytokines and neurodegeneration triggered by microglial cells. The microglial activation was attenuated by the lack of the Fc gamma receptor, suggesting an important role of the adaptive immune system in AS-mediated microglial activation and neurodegeneration [81].
Cell culture models demonstrated that microglial-conditioned release of pro-inflammatory cytokines upon AS treatment was dose-dependent [76,78]. Moreover, microglia treated with mutant (A30P, E46K and A53T) AS resulted in an enhanced microglial activation with increased release of cytokines (IL-6 and IL-10) and chemokines (RANTES and MCP-1) compared to wild-type AS treatment [77]. Wild-type and mutant AS released by neurons led to an enhanced pro-inflammatory response of the mouse microglia cell line BV2 [82], and mutant AS-overexpressing BV2 cells showed an increased release of inflammatory cytokines (e.g. TNF-α, IL-6) [83]. Oxidative stress is another neurotoxic event that occurs after AS-induced microglial activation and may play a crucial role in PD and DLB disease progression. The activation of NADPH oxidase and the production of ROS by AS stimulated microglia led to dopaminergic neuronal loss [84]. Furthermore, mutant AS-overexpressing BV2 cells produced an increased amount of NO [83]. Aggregated and nitrated forms of AS not only led to inflammatory events and oxidative stress but also to enhanced neuronal cell death, as shown in mesencephalic neuron–microglia co-cultures [84,85].
However, activated microglial cells are not only involved in the neuroinflammation and neurodegeneration processes in PD and DLB, they may also play a fundamental role in the clearance of damaged or dead cells and AS [32,84,86], thereby supporting neuronal survival. Microglial cells show an enhanced phagocytic activity when treated with monomeric AS; on the contrary, aggregated AS inhibits microglial phagocytosis in vitro [86]. Microglial cells are capable of fast AS degradation with an intracellular AS half-life of about 4 h, compared to astroglial and neuronal degradation comprising less than half of the degradation time [87]. Regarding microglial phagocytosis of AS, Toll-like receptors (TLRs) seem to play an important part in recognition and internalisation of AS. The pattern-recognition receptors TLR2 and TLR4 were identified as important players in microglial activation [88,89]. Recently, it was demonstrated that TLR4 ablation leads to a disturbed clearance of AS by microglia [89].
To summarise, microglia plays a fundamental but dual role in neuronal α-synucleinopathies. On the one hand, microglial cells recognise extracellular AS released by damaged neurons; moreover, they are crucial in the clearance of this protein [86,87]. On the other hand, AS may trigger microglial overactivation and, further, this can lead to the production of pro-inflammatory agents and to oxidative stress and as a consequence to enhanced neurodegeneration and neuronal cell death [84].
The role of oligodendroglial pathology in PD and DLB does not seem to be a leading one. Occasional oligodendroglial AS-positive inclusions have been reported in clinically overt PD cases [6,90]. Moreover, complement-activated oligodendroglial cells were found in some brain regions of PD and DLB cases [91,92]. Oligodendrocytes show a higher susceptibility in PD and DLB regarding the lack or decrease of myelinated axons of AS-affected neurons [93,94]. These data indicate that oligodendroglial cells could be involved in the late disease progression of PD and DLB. However, an involvement of oligodendroglial pathology in disease initiation and/or progression in neuronal α-synucleinopathies lacks sufficient evidence at present [29].
Oligodendroglial α-Synucleinopathy
MSA is a progressive neurodegenerative disease of unknown aetiology with a prevalence of about 4.4 new cases per 100,000 year−1 [95], and the mean age at onset of first symptoms is 52–57 years [96,97]. The term MSA was first used by Graham and Oppenheimer in 1969 to merge the variable diagnoses of striatonigral degeneration, olivopontocerebellar ataxia, Shy–Drager syndrome and orthostatic hypotension [98,99]. Now, two major clinical subtypes are defined: (1) the MSA-C subtype presents with predominant cerebellar ataxia and (2) the MSA-P subtype shows prevalent Parkinsonism [100]. Progressive autonomic failure is present in both motor subtypes correlating with neuronal degeneration in autonomic brainstem centres, intermediolateral cell columns and Onuf’s nucleus in the spinal cord [19,101]. The main pathological and diagnostic hallmark of MSA are AS-positive GCIs present in oligodendroglial cells and involving different areas of the brain, including pons, medulla, putamen, SN, cerebellum and preganglionic autonomic structures [3,102–105]. Moreover, astrogliosis and microgliosis were found to be involved in this presumably primary oligodendrogliopathy [19,27,106].
Similar to PD and DLB, the role of astroglial and microglial activation in MSA is not entirely resolved, taking neuroprotective and neurotoxic functions into account. Extensive astrogliosis in MSA brains was reported by Ozawa and colleagues [19] and by Jellinger et al. [107]. Astroglia in MSA brains undergo pathological changes regarding their morphology featuring enlarged cell bodies and distorted processes [108]. However, in contrast to PD and DLB, AS accumulation in astroglia does not seem to occur in MSA. Experimental MSA models provide further evidence on the involvement of astrogliosis in MSA-like neurodegeneration. Overexpression of human AS in oligodendroglia under the control of the murine myelin basic protein (MBP) promoter triggered neurodegeneration and prominent astrogliosis detected at 6 months of age [109]. Exposure to 3-nitropropionic acid in a transgenic mouse model overexpressing human AS in oligodendroglial cells under the control of the proteolipid protein (PLP) promoter not only lead to striatonigral degeneration and olivopontocerebellar atrophy but also to widespread astrogliosis accompanying the neurodegeneration [110]. Most of the in vitro data on astroglia and AS referenced for PD/DLB, as described above, may be also relevant for MSA. In MSA, astrocytes show high reactivity, and astrogliosis may lead to oxidative stress and neurotoxicity, similar to PD/DLB. However, insufficient data on astroglial activation and its mechanisms in MSA allow only speculations on the role of astroglial cells. Further studies would be very valuable for a complete understanding of the astroglial role in MSA.
Microglial activation in MSA is a common finding. In MSA patients, Gerhard and colleagues reported microglial activation in the dorsolateral prefrontal cortex, putamen, pallidum, pons and SN using [11C](R)-PK11195 PET imaging [27]. Moreover, an enhanced microgliosis was found in motor-related brain structures associated with GCI pathology, including cerebellar input, extrapyramidal motor and pyramidal motor structures [111]. Microglial activation was reproduced in the transgenic MSA mouse model overexpressing AS under a PLP promoter in oligodendroglial cells [110]. The progressive upregulation of microglial activation in this MSA transgenic mouse model resulted in neuroinflammation and oxidative stress correlating with dopaminergic neuronal loss in SNpc [112]. Moreover, the microglial activation was associated with an upregulation of TLR4 in these mice, as also detected in human MSA [112]. In double transgenic mice with oligodendroglial overexpression of AS and lack of functional TLR4, the efficiency of microglial AS clearance was diminished and resulted in enhanced nigral dopaminergic neurodegeneration [89]. These data suggest that TLR4 is a crucial mediator of microglial AS clearance, and the enhanced expression of this receptor in post-mortem brains may represent an augmented effort of AS clearance by microglial cells in MSA. In vitro data confirm that AS activates microglial cells, triggers the release of pro-inflammatory agents [76,82], increases oxidative stress through the release of ROS [84] and may be of equal relevance to the disease progression in PD and DLB as well as MSA. In summary, similar to PD and DLB, microglial cells in MSA display positive (phagocytosis) and negative (oxidative stress and inflammation) features, and further studies are warranted to elucidate the complete spectrum of microglial activation in disease initiation and progression.
It is considered that oligodendroglial cells play a leading role in MSA, due to the AS inclusions present in these cells. Oligodendrocytes seem to be initiators of the disease as regarded to the distribution of GCIs [106,113], which may even represent the primary injury in MSA [106,114]. GCIs are distributed throughout large proportions of the CNS [3,102–104]. However, the source of AS accumulation, the main component of GCIs, in oligodendroglial cells is not resolved yet. The prevalent assumption is that oligodendroglial cells actively incorporate and accumulate AS released by neighbouring neurons [115]. This hypothesis becomes highly relevant regarding the data on cell-to-cell propagation of AS in different studies [21–23]. Furthermore, the release of AS by neuronal cells into the extracellular space was confirmed recently [22,116]. Primary oligodendroglial dysfunction related to abnormal endocytic activity as suggested by the ectopic expression of Rab5 and Rabaptin-5 in GCIs [117] may represent an early event in MSA pathogenesis preceding pathological uptake and accumulation of AS in oligodendroglia. However, there are currently no studies demonstrating AS propagation to oligodendroglia [22,118]. Another possibility of AS aggregation in oligodendroglia could be an enhanced expression of AS, and further, a defective degradation mechanism could lead to accumulation of AS in the cell [119,120]. Yet, no mRNA expression of AS could be found in human oligodendroglial cells of MSA brains [113,121]. However, AS is a major trigger of oligodendroglial protein inclusion formation, and the absence of AS prevents accumulation of tau and αB-crystallin, further components of GCIs [122]. The oligodendroglial phosphoprotein p25α (tubulin polymerization promoting protein) induces AS aggregation in vitro [123]; furthermore, in MSA, p25α may relocate to oligodendroglial soma, suggesting an involvement of early oligodendroglial dysfunction in MSA initiation and GCI formation [124]. In support of these data, co-expression of human AS and p25α in rat oligodendroglia led to disorganisation of the microtubular cytoskeleton and apoptosis [125]. Inhibition of AS-Ser129 phosphorylation abolished these effects, suggesting an important role for Ser129 phosphorylation in the formation of AS oligomers and oligodendroglial apoptosis [125]. Recently, the cytoplasmic enzyme histone deacetylase 6 (HDAC6) was found in over 98 % of all GCIs of MSA post-mortem brains [126]. The exact role of HDAC6 in the pathogenic cascade of MSA is currently unclear. HDAC6 is identified to regulate the transport of ubiquitinated misfolded proteins, the formation of aggresomes [127] and aggresome degradation [128] as well as the control of autophagy pathways [129], and its accumulation in MSA may represent another sign of oligodendroglial injury.
Different studies demonstrate that GCIs affect oligodendroglial function and viability, suggesting an important role in MSA progression. Cell culture experiments, using glial cells overexpressing AS, revealed increased susceptibility to oxidative stress and TNF-α which may represent further events in the pathogenesis of MSA [130,131]. Overexpression of AS in oligodendroglial cells reduced the adhesion to fibronectin, leading to impaired cell–extracellular matrix interactions [132]. Oligodendroglial overexpression of AS in transgenic mice resulted in neurodegeneration in different brain areas including SNpc, locus coeruleus, nucleus ambiguous, pedunculopontine tegmental nucleus, laterodorsal tegmental nucleus and Onuf’s nucleus [110,133]. Moreover, MSA transgenic mouse models demonstrated that oligodendroglial AS inclusions may cause myelin disruption and mitochondrial dysfunction [109,112,134]. The expression of neurotrophic factors, especially GDNF, was decreased in the MBP-AS mouse model, but not in transgenic mice with neuronal overexpression of AS, suggesting MSA-specific oligodendroglial dysfunction, related to reduced trophic support of neurons [135].
In conclusion, oligodendroglial cells may play a major role in the initiation and progression of MSA (see Fig. 1). The accumulation of AS in these cells leads to altered oligodendroglial function including reduced trophic support and demyelination and in consequence to neurodegeneration. However, the source of AS in GCIs and the mechanisms of GCI formation in oligodendroglia remain unclear and need further elucidation to gain a deeper insight into MSA disease mechanisms.
Therapeutic Approaches Targeting Glial Dysfunction in α-Synucleinopathies: Where Are We Now?
Increasing body of evidence confirms the relevance of glial dysfunction in the pathogenesis of α-synucleinopathies. However, the broad spectrum of activation profiles of microglial and astroglial cells makes it still difficult to obtain a clear-cut overall picture of all glial features and their mode of action in these diseases. The wide variety of glial functions offers diverse therapeutic targets. Yet, due to the insufficient knowledge on the exact chronology and relevance of the beneficial and detrimental roles of glia in α-synucleinopathies, researchers are currently confronted with discrepancies between findings on neuroprotection in experimental setups and clinical settings.
Neuroinflammatory responses and oxidative stress mediated by microglial or astroglial cells are prior targets in therapeutic approaches regarding neuroprotection in α-synucleinopathies. Non-steroidal anti-inflammatory drugs (NSAIDs) showed neuroprotective effects in toxin-induced PD-like neurodegeneration in rodents [136,137] and, furthermore, resulted in a decreased AS aggregation in vitro [138]. Recently, eicosanyl-5-hydroxytryptamide treatment lead to a repressed astro- and microglial activation and inducible nitric oxide synthase (iNOS) expression in AS-overexpressing mice [139]. Moreover, different strategies on the inhibition of micro- and astroglial activation were followed in various experimental approaches leading to neuroprotection, including iNOS and NADPH oxidase inhibition [140,141], suppression of the peroxisome proliferator-activated receptor γ pathway via pioglitazone [142–144] or inhibition of the enzyme MPO, which is involved in ROS production and upregulated in PD and MSA [61,145]. Minocycline, a tetracycline derivative known for its antimicrobial activity and the inhibition of protein synthesis, revealed additional anti-neuroinflammatory and anti-apoptotic efficacy [146]. In different experimental studies of PD and MSA, minocycline had various neuroprotective effects probably dependent on the timing of therapy onset within the course of neurodegeneration [112,147–149]. The modulation of TLR4-dependent microglial activation through a TLR4 antagonist naloxone has been suggested to prevent microgliosis-associated dopaminergic neurodegeneration [150,151]. However, in light of the recent finding that TLR4 is also an important modulator of AS clearance by microglia [89], caution and further studies are needed to justify such a therapeutic approach in α-synucleinopathies. Anti-neuroinflammatory strategies with the goal of modifying glial responses towards neuroprotection currently fail to translate into successful clinical trials. The application of NSAIDs in PD resulted in contradictory outcomes. In an epidemiological study, NSAID treatment was associated with decreased risk of PD [152]; however, in a recent study using a UK cohort of PD cases and controls [153] and in observational studies using meta-analysis [154,155], these results were not confirmed. A similar conflicting outcome was obtained after minocycline therapy in PD and MSA patients [156,157]. Minocycline treatment of MSA patients in a prospective, randomised, double-blind clinical trial lead to a significant downregulation of microglial activation after 24 weeks of therapy; however, no effect on disease progression was demonstrated, suggesting that an early therapy onset may be preferable [156].
Alternative therapeutic strategies, like immunomodulation, AS-reducing strategies and neurotrophic factor delivery and modulation, targeting glial dysfunction have been approached. In an experimental immunisation study, the adoptive transfer of copolymer-1 immune cells resulted in decreased microglial activation and enhanced local expression of astroglia-associated GDNF amongst other effects [158,159]. However, transfer of T cells from nitrated AS-immunised mice lead to neuroinflammation in correlation with neuronal loss [160]. In contrast, AS vaccination in a PD mouse model yielded a decrease in microglial and astroglial activation and enhanced neuroprotection, as well as reduced AS inclusion pathology [161]. The strategy of using AS-reducing agents for the therapy of α-synucleinopathies was further expanded by the application of rifampicin in a transgenic mouse model of MSA. The results indicated that the successful lowering of AS levels in the brain of MBP-AS mice resulted in neuroprotection associated with suppressed astroglial activation [162]. Clinical proof of concept is currently awaited for the efficacy of these strategies. Finally, the delivery of neurotrophic factors is a relevant approach related to glial dysfunction in α-synucleinopathies. Genetically modified macrophages were used for the delivery of GDNF inducing neuroprotection in the MPTP model of PD [163]. However, AAV gene delivery of a GDNF analogue in the putamen of PD patients failed to exert beneficial effects [164].
In conclusion, the divergence between the clinical and the experimental outcomes on therapies targeting glial dysfunction in α-synucleinopathies may be resolved only by further in-depth studies on the role of glial cells in disease initiation and progression. The role of glia should be further analysed in association with the basic changes that occur in CNS related to normal ageing, which may play a crucial predisposing/promoting role in AS-related neurodegeneration.
References
Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS (2010) Alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A 107(45):19573–19578
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123(3):383–396
Beyer K, Ariza A (2007) Protein aggregation mechanisms in synucleinopathies: commonalities and differences. J Neuropathol Exp Neurol 66(11):965–974. doi:10.1097/nen.0b013e3181587d64
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251(3):205–208
Braak H, Sastre M, Del Tredici K (2007) Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114(3):231–241. doi:10.1007/s00401-007-0244-3
Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H (2000) NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol 99(1):14–20
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55(2):164–173. doi:10.1002/ana.10795
Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N (2006) Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann Neurol 59(2):298–309. doi:10.1002/ana.20753
Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2:440. doi:10.1038/ncomms1453
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. doi:10.1126/science.1090278
Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74(2):97–109
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65(5):610–614. doi:10.1002/ana.21685
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281(40):29739–29752
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290(5493):985–989
Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG (2003) Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 278(45):44405–44411. doi:10.1074/jbc.M308041200
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC (2010) alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190(6):1023–1037
Xilouri M, Stefanis L (2011) Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 13:e8
Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, Healy DG, Wood NW, Lees AJ, Holton JL, Revesz T (2004) The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 127(Pt 12):2657–2671
Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 249(Suppl 3):III/1–5
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106(31):13010–13015
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121(2):715–725. doi:10.1172/JCI43366
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285(12):9262–9272
Hauser DN, Cookson MR (2011) Astrocytes in Parkinson’s disease and DJ-1. J Neurochem 117(3):357–358. doi:10.1111/j.1471-4159.2011.07217.x
Webster H, Astrom KE (2009) Gliogenesis: historical perspectives, 1839–1985. Adv Anat Embryol Cell Biol 202:1–109
Fellner L, Jellinger KA, Wenning GK, Stefanova N (2011) Glial dysfunction in the pathogenesis of alpha-synucleinopathies: emerging concepts. Acta Neuropathol 121(6):675–693. doi:10.1007/s00401-011-0833-z
Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN, Turkheimer F, Good CD, Mathias CJ, Quinn N, Schwarz J, Brooks DJ (2003) [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61(5):686–689
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21(2):404–412
Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26(1):6–17. doi:10.1002/mds.23455
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318
Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M (2006) Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A 103(46):17513–17518
del Rio-Hortega P (1932) Microglia. In: Penfield W (ed) Cytology and cellular pathology of the nervous system. Hoeber, PB, New York, pp 483–534
Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW (2004) Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem 89(5):1092–1100. doi:10.1111/j.1471-4159.2004.02420.x
van Rossum D, Hanisch UK (2004) Microglia. Metab Brain Dis 19(3–4):393–411
Dean JM, Wang X, Kaindl AM, Gressens P, Fleiss B, Hagberg H, Mallard C (2010) Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain Behav Immun 24(5):776–783
Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, Ghorpade A (2005) Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res 7(3):183–192
Mizuno T, Kuno R, Nitta A, Nabeshima T, Zhang G, Kawanokuchi J, Wang J, Jin S, Takeuchi H, Suzumura A (2005) Protective effects of nicergoline against neuronal cell death induced by activated microglia and astrocytes. Brain Res 1066(1–2):78–85
Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA (2002) Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci 22(3):854–862
Qian L, Flood PM (2008) Microglial cells and Parkinson’s disease. Immunol Res 41(3):155–164. doi:10.1007/s12026-008-8018-0
Qian L, Flood PM, Hong JS (2010) Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J Neural Transm 117(8):971–979. doi:10.1007/s00702-010-0428-1
Jurewicz A, Matysiak M, Tybor K, Kilianek L, Raine CS, Selmaj K (2005) Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain 128(Pt 11):2675–2688
Thorburne SK, Juurlink BH (1996) Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 67(3):1014–1022
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68(5):384–386
Fahn S (2003) Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 991:1–14
McKeith IG, Burn DJ, Ballard CG, Collerton D, Jaros E, Morris CM, McLaren A, Perry EK, Perry R, Piggott MA, O’Brien JT (2003) Dementia with Lewy bodies. Semin Clin Neuropsychiatry 8(1):46–57. doi:10.1053/scnp.2003.50006
Jellinger KA (2003) Neuropathological spectrum of synucleinopathies. Mov Disord 18(Suppl 6):S2–12
Jellinger KA (2007) Lewy body disorders. In: Youdim MBH, Riederer P, Mandel SA, Battistin L, Lajtha A (eds) Degenerative diseases of the nervous system. Springer Science, New York, pp 267–343
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120(2):131–143. doi:10.1007/s00401-010-0711-0
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152(4):879–884
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55(3):259–272
Tompkins MM, Hill WD (1997) Contribution of somal Lewy bodies to neuronal death. Brain Res 775(1–2):24–29
Mackenzie IR (2000) Activated microglia in dementia with Lewy bodies. Neurology 55(1):132–134
Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM (2005) A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128(Pt 11):2665–2674
Hashioka S, Klegeris A, Schwab C, McGeer PL (2009) Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol Aging 30(12):1924–1935
Knott C, Stern G, Kingsbury A, Welcher AA, Wilkin GP (2002) Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Parkinsonism Relat Disord 8(5):329–341
Mirza B, Hadberg H, Thomsen P, Moos T (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95(2):425–432
Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S (2001) The role of glial cells in Parkinson’s disease. Curr Opin Neurol 14(4):483–489
Hirsch EC, Hunot S, Hartmann A (2005) Neuroinflammatory processes in Parkinson’s disease. Parkinsonism Relat Disord 11(Suppl 1):S9–S15
Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H (2010) Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 3:12
Hashioka S, Klegeris A, Schwab C, Yu S, McGeer PL (2010) Differential expression of interferon-gamma receptor on human glial cells in vivo and in vitro. J Neuroimmunol 225(1–2):91–99
Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, Jackson-Lewis V, Vila M, Vonsattel JP, Heinecke JW, Przedborski S (2005) Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J Neurosci 25(28):6594–6600
Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52(1):1–6
Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK (2011) Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem Res 36(8):1452–1463. doi:10.1007/s11064-011-0471-9
Barcia C, Ros CM, Annese V, Gomez A, Ros-Bernal F, Aguado-Yera D, Martinez-Pagan ME, de Pablos V, Fernandez-Villalba E, Herrero MT (2011) IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis 2:e142
Schmidt S, Linnartz B, Mendritzki S, Sczepan T, Lubbert M, Stichel CC, Lubbert H (2011) Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum Mol Genet 20(6):1197–1211
Schiess MC, Barnes JL, Ellmore TM, Poindexter BJ, Dinh K, Bick RJ (2010) CSF from Parkinson disease patients differentially affects cultured microglia and astrocytes. BMC Neurosci 11:151
Klegeris A, Giasson BI, Zhang H, Maguire J, Pelech S, McGeer PL (2006) Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J 20(12):2000–2008
Saavedra A, Baltazar G, Santos P, Carvalho CM, Duarte EP (2006) Selective injury to dopaminergic neurons up-regulates GDNF in substantia nigra postnatal cell cultures: role of neuron-glia crosstalk. Neurobiol Dis 23(3):533–542
Sandhu JK, Gardaneh M, Iwasiow R, Lanthier P, Gangaraju S, Ribecco-Lutkiewicz M, Tremblay R, Kiuchi K, Sikorska M (2009) Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol Dis 33(3):405–414
Lee M, Schwab C, Yu S, McGeer E, McGeer PL (2009) Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol Aging 30(10):1523–1534
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1291
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 106(6):518–526. doi:10.1007/s00401-003-0766-2
Imamura K, Hishikawa N, Ono K, Suzuki H, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2005) Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol 109(2):141–150. doi:10.1007/s00401-004-0919-y
Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T (2005) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57(2):168–175. doi:10.1002/ana.20338
Su X, Federoff HJ, Maguire-Zeiss KA (2009) Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox Res 16(3):238–254. doi:10.1007/s12640-009-9053-x
Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL (2008) Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging 29(5):739–752
Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Fernandez-Montesinos R, Caro M, Lachaud CC, Waudby CA, Delgado M, Dobson CM, Pozo D (2010) Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson’s disease-linked mutants. PLoS One 5(10):e13481. doi:10.1371/journal.pone.0013481
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29(11):1690–1701
Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M (2010) Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One 5(1):e8784. doi:10.1371/journal.pone.0008784
Theodore S, Cao S, McLean PJ, Standaert DG (2008) Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol 67(12):1149–1158. doi:10.1097/NEN.0b013e31818e5e99
Cao S, Theodore S, Standaert DG (2010) Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol Neurodegener 5:42
Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ (2011) Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69(4):337–342
Rojanathammanee L, Murphy EJ, Combs CK (2011) Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J Neuroinflammation 8:44
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, Zhang J (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19(6):533–542
Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE (2008) Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem 104(6):1504–1525
Park JY, Paik SR, Jou I, Park SM (2008) Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia 56(11):1215–1223. doi:10.1002/glia.20691
Lee HJ, Suk JE, Bae EJ, Lee SJ (2008) Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun 372(3):423–428
Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ (2000) Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J Immunol 165(2):618–622
Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK (2011) Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol 179(2):954–963
Arai T, Ueda K, Ikeda K, Akiyama H, Haga C, Kondo H, Kuroki N, Niizato K, Iritani S, Tsuchiya K (1999) Argyrophilic glial inclusions in the midbrain of patients with Parkinson’s disease and diffuse Lewy body disease are immunopositive for NACP/alpha-synuclein. Neurosci Lett 259(2):83–86
Yamada T, McGeer PL, McGeer EG (1992) Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol 84(1):100–104
Yamada T, McGeer PL, McGeer EG (1992) Some immunohistochemical features of argyrophilic grain dementia with normal cortical choline acetyltransferase levels but extensive subcortical pathology and markedly reduced dopamine. J Geriatr Psychiatry Neurol 5(1):3–13
Braak H, Del Tredici K (2004) Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging 25(1):19–23
Braak H, Del Tredici K (2009) Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv Anat Embryol Cell Biol 201:1–119
Schrag A, Ben-Shlomo Y, Quinn NP (1999) Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 354(9192):1771–1775
O’Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, Revesz T, Lees AJ (2008) Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain 131(Pt 5):1362–1372
Schrag A, Wenning GK, Quinn N, Ben-Shlomo Y (2008) Survival in multiple system atrophy. Mov Disord 23(2):294–296. doi:10.1002/mds.21839
Graham JG, Oppenheimer DR (1969) Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 32(1):28–34
Wenning GK, Colosimo C, Geser F, Poewe W (2004) Multiple system atrophy. Lancet Neurol 3(2):93–103
Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, Kaufmann H, Klockgether T, Lang AE, Lantos PL, Litvan I, Mathias CJ, Oliver E, Robertson D, Schatz I, Wenning GK (1999) Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 163(1):94–98
Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP (1997) Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 12(2):133–147. doi:10.1002/mds.870120203
Jellinger KA, Lantos PL (2010) Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol 119(6):657–667. doi:10.1007/s00401-010-0672-3
Nishie M, Mori F, Fujiwara H, Hasegawa M, Yoshimoto M, Iwatsubo T, Takahashi H, Wakabayashi K (2004) Accumulation of phosphorylated alpha-synuclein in the brain and peripheral ganglia of patients with multiple system atrophy. Acta Neuropathol 107(4):292–298. doi:10.1007/s00401-003-0811-1
Papp MI, Lantos PL (1994) The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 117(Pt 2):235–243
Ubhi K, Low P, Masliah E (2011) Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci
Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG (2008) Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol 64(3):239–246. doi:10.1002/ana.21465
Jellinger KA, Seppi K, Wenning GK (2005) Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord 20(Suppl 12):S29–36. doi:10.1002/mds.20537
Song YJ, Halliday GM, Holton JL, Lashley T, O’Sullivan SS, McCann H, Lees AJ, Ozawa T, Williams DR, Lockhart PJ, Revesz TR (2009) Degeneration in different Parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68(10):1073–1083. doi:10.1097/NEN.0b013e3181b66f1b
Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, Masliah E (2005) Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci 25(46):10689–10699
Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle PJ, Wenning GK (2005) Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol 166(3):869–876
Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T (2004) Microglial activation parallels system degeneration in multiple system atrophy. J Neuropathol Exp Neurol 63(1):43–52
Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK (2007) Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov Disord 22(15):2196–2203. doi:10.1002/mds.21671
Ozawa T, Okuizumi K, Ikeuchi T, Wakabayashi K, Takahashi H, Tsuji S (2001) Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol 102(2):188–190
Wenning GK, Quinn N, Magalhaes M, Mathias C, Daniel SE (1994) “Minimal change” multiple system atrophy. Mov Disord 9(2):161–166. doi:10.1002/mds.870090206
Wakabayashi K, Takahashi H (2006) Cellular pathology in multiple system atrophy. Neuropathology 26(4):338–345
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30(20):6838–6851
Nakamura S, Kawamoto Y, Nakano S, Akiguchi I (2000) Expression of the endocytosis regulatory proteins Rab5 and Rabaptin-5 in glial cytoplasmic inclusions from brains with multiple system atrophy. Clin Neuropathol 19(2):51–56
Stefanova N, Hainzer M, Stemberger S, Couillard-Despres S, Aigner L, Poewe W, Wenning GK (2009) Striatal transplantation for multiple system atrophy—are grafts affected by alpha-synucleinopathy? Exp Neurol 219(1):368–371
Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K (2002) Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 176(1):98–104
Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM (2000) alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res 62(1):9–14. doi:10.1002/1097-4547(20001001)62:1<9::AID-JNR2>3.0.CO;2-U
Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB (2005) Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm 112(12):1613–1624. doi:10.1007/s00702-005-0378-1
Riedel M, Goldbaum O, Richter-Landsberg C (2009) alpha-Synuclein promotes the recruitment of tau to protein inclusions in oligodendroglial cells: effects of oxidative and proteolytic stress. J Mol Neurosci 39(1-2):226–234. doi:10.1007/s12031-009-9190-y
Lindersson E, Lundvig D, Petersen C, Madsen P, Nyengaard JR, Hojrup P, Moos T, Otzen D, Gai WP, Blumbergs PC, Jensen PH (2005) p25alpha stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J Biol Chem 280(7):5703–5715
Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Hojrup P, Otzen D, Halliday GM, Jensen PH (2007) p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol 171(4):1291–1303
Kragh CL, Lund LB, Febbraro F, Hansen HD, Gai WP, El-Agnaf O, Richter-Landsberg C, Jensen PH (2009) {alpha}-Synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J Biol Chem 284(15):10211–10222
Miki Y, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K (2011) Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology. doi:10.1111/j.1440-1789.2011.01200.x
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115(6):727–738
Iwata A, Riley BE, Johnston JA, Kopito RR (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280(48):40282–40292
Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131(Pt 8):1969–1978
Stefanova N, Klimaschewski L, Poewe W, Wenning GK, Reindl M (2001) Glial cell death induced by overexpression of alpha-synuclein. J Neurosci Res 65(5):432–438. doi:10.1002/jnr.1171
Stefanova N, Schanda K, Klimaschewski L, Poewe W, Wenning GK, Reindl M (2003) Tumor necrosis factor-alpha-induced cell death in U373 cells overexpressing alpha-synuclein. J Neurosci Res 73(3):334–340. doi:10.1002/jnr.10662
Tsuboi K, Grzesiak JJ, Bouvet M, Hashimoto M, Masliah E, Shults CW (2005) Alpha-synuclein overexpression in oligodendrocytic cells results in impaired adhesion to fibronectin and cell death. Mol Cell Neurosci 29(2):259–268
Stemberger S, Poewe W, Wenning GK, Stefanova N (2010) Targeted overexpression of human alpha-synuclein in oligodendroglia induces lesions linked to MSA-like progressive autonomic failure. Exp Neurol 224(2):459–464
Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM (2005) Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45(6):847–859
Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E (2010) Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci 30(18):6236–6246
Aubin N, Curet O, Deffois A, Carter C (1998) Aspirin and salicylate protect against MPTP-induced dopamine depletion in mice. J Neurochem 71(4):1635–1642
Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G (2007) Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp Neurol 205(2):295–312
Hirohata M, Ono K, Morinaga A, Yamada M (2008) Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. Neuropharmacology 54(3):620–627
Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, Fernandez JR, Chao Y, Masliah E, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM (2011) Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J Neurosci 31(19):6963–6971
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM (2008) Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci 28(30):7687–7698
Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS (2011) HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci 31(3):1081–1092
Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002) Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J Neurochem 82(3):615–624
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88(2):494–501
Randy LH, Guoying B (2007) Agonism of peroxisome proliferator receptor-gamma may have therapeutic potential for neuroinflammation and Parkinson’s disease. Curr Neuropharmacol 5(1):35–46
Stefanova N, Eriksson H, Georgievska B, Poewe W, Wenning GK (2010) Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Mov Disord 25(suppl 3):625
Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM (2003) Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A 100(18):10483–10487. doi:10.1073/pnas.1832501100
Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA (2006) Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem 97(4):934–946
Stefanova N, Mitschnigg M, Ghorayeb I, Diguet E, Geser F, Tison F, Poewe W, Wenning GK (2004) Failure of neuronal protection by inhibition of glial activation in a rat model of striatonigral degeneration. J Neurosci Res 78(1):87–91. doi:10.1002/jnr.20233
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22(5):1763–1771
Kong LY, McMillian MK, Hudson PM, Jin L, Hong JS (1997) Inhibition of lipopolysaccharide-induced nitric oxide and cytokine production by ultralow concentrations of dynorphins in mixed glia cultures. J Pharmacol Exp Ther 280(1):61–66
Lu X, Bing G, Hagg T (2000) Naloxone prevents microglia-induced degeneration of dopaminergic substantia nigra neurons in adult rats. Neuroscience 97(2):285–291
Gagne JJ, Power MC (2010) Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology 74(12):995–1002
Becker C, Jick SS, Meier CR (2011) NSAID use and risk of Parkinson disease: a population-based case–control study. Eur J Neurol. doi:10.1111/j.1468-1331.2011.03399.x
Polazzi E, Monti B (2010) Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92(3):293–315
Samii A, Etminan M, Wiens MO, Jafari S (2009) NSAID use and the risk of Parkinson’s disease: systematic review and meta-analysis of observational studies. Drugs Aging 26(9):769–779. doi:10.2165/11316780-000000000-00000
Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, Trenkwalder C, Sixel-Doring F, Herting B, Kamm C, Gasser T, Sawires M, Geser F, Kollensperger M, Seppi K, Kloss M, Krause M, Daniels C, Deuschl G, Bottger S, Naumann M, Lipp A, Gruber D, Kupsch A, Du Y, Turkheimer F, Brooks DJ, Klockgether T, Poewe W, Wenning G, Schade-Brittinger C, Oertel WH, Eggert K (2010) Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord 25(1):97–107. doi:10.1002/mds.22732
Racette B (2008) A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin Neuropharmacol 31(3):141–150. doi:10.1097/WNF.0b013e3181342f32
Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE (2004) Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 101(25):9435–9440. doi:10.1073/pnas.0400569101
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL (2007) Neuroprotective activities of CD4 + CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol 82(5):1083–1094
Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE (2008) Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One 3(1):e1376. doi:10.1371/journal.pone.0001376
Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46(6):857–868
Ubhi K, Rockenstein E, Mante M, Patrick C, Adame A, Thukral M, Shults C, Masliah E (2008) Rifampicin reduces alpha-synuclein in a transgenic mouse model of multiple system atrophy. Neuroreport 19(13):1271–1276. doi:10.1097/WNR.0b013e32830b3661
Biju K, Zhou Q, Li G, Imam SZ, Roberts JL, Morgan WW, Clark RA, Li S (2010) Macrophage-mediated GDNF delivery protects against dopaminergic neurodegeneration: a therapeutic strategy for Parkinson’s disease. Mol Ther 18(8):1536–1544
Marks WJ Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, Vitek J, Stacy M, Turner D, Verhagen L, Bakay R, Watts R, Guthrie B, Jankovic J, Simpson R, Tagliati M, Alterman R, Stern M, Baltuch G, Starr PA, Larson PS, Ostrem JL, Nutt J, Kieburtz K, Kordower JH, Olanow CW (2010) Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 9(12):1164–1172
Acknowledgement
This work was supported by grants of the Austrian Science Fund (FWF) P19989-B05 and F4404-B19.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fellner, L., Stefanova, N. The Role of Glia in Alpha-Synucleinopathies. Mol Neurobiol 47, 575–586 (2013). https://doi.org/10.1007/s12035-012-8340-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8340-3