
Abstract
The intracellular deposition and intercellular transmission of α-synuclein (α-syn) are shared pathological characteristics among neurodegenerative disorders collectively known as α-synucleinopathies, including Parkinson's disease (PD). Although the precise triggers of α-synucleinopathies remain unclear, recent findings indicate that disruption of microglial homeostasis contributes to the pathogenesis of PD. Microglia play a crucial role in maintaining optimal neuronal function by ensuring a homeostatic environment, but this function is disrupted during the progression of α-syn pathology. The involvement of microglia in the accumulation, uptake, and clearance of aggregated proteins is critical for managing disease spread and progression caused by α-syn pathology. This review summarizes current knowledge on the interrelationships between microglia and α-synucleinopathies, focusing on the remarkable ability of microglia to recognize and internalize extracellular α-syn through diverse pathways. Microglia process α-syn intracellularly and intercellularly to facilitate the α-syn neuronal aggregation and cell-to-cell propagation. The conformational state of α-synuclein distinctly influences microglial inflammation, which can affect peripheral immune cells such as macrophages and lymphocytes and may regulate the pathogenesis of α-synucleinopathies. We also discuss ongoing research efforts to identify potential therapeutic approaches targeting both α-syn accumulation and inflammation in PD.
Video Abstract
Similar content being viewed by others

Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by selective vulnerability of dopaminergic neurons in the midbrain substantia nigra pars compacta (SNpc) [1]. Degeneration of dopaminergic neurons in the SNpc that project to the striatum reduces dopamine input to the basal ganglia, resulting in motor symptoms such as bradykinesia and rigidity [2]. Another neuropathological feature of PD is the presence of Lewy bodies (LBs) and Lewy neurites (LNs), which are formed by abnormal deposition of α-synuclein (α-syn), dysmorphic organelles, vesicular structures, and lipids in the cytoplasm of neurons in several different brain regions [3, 4]. Although PD was initially described as a movement disorder closely related to dopaminergic neuron degeneration, other cell types throughout the central and peripheral autonomic nervous system are also involved. This may lead to various symptoms, including nonmotor hyposmia, sleep disorders, constipation, and cognitive and psychiatric symptoms such as dementia and depression [5].
Although most PD cases are sporadic, a substantial proportion of PD cases are attributed to variants or alterations in specific monogenic genes. To date, mutations in at least 20 genes have been identified to be responsible for PD [6]. Additionally, variants of these genes collectively play a role in the development of sporadic PD [7]. Intraneuronal protein aggregates consisting primarily of α-syn are found in most patients with PD. According to the findings of a large-scale genome-wide association study (GWAS), polymorphisms in noncoding regions of the α-syn gene, SNCA, are significant risk factors for idiopathic PD [8]. α-Syn can deviate from its native state, forming amyloid fibrils, ultimately resulting in the formation of cytoplasmic inclusions, which are associated with several neurodegenerative diseases, including PD [9, 10], dementia with Lewy bodies (DLB) [11, 12], and multiple system atrophy (MSA) [13, 14]. Various pathological α-syn aggregates can form from the same precursor protein, for instance, LBs in neurons [15] and GCIs (glial cytoplasmic inclusions) in glial cells [16], leading to the diverse clinical and pathological characteristics of α-synucleinopathies [17].
Microglia are the primary type of innate immune cells in the central nervous system (CNS), accounting for approximately 5–10% of all glial cells in the healthy human brain [18]. Mammalian microglia originate from myeloid precursor cells in yolk sac tissue before migrating to and colonizing the brain. In the CNS, microglia are considered to reach a stable state and are responsible for maintaining immune homeostasis and protecting the brain against diseases and pathogens [19]. α-Syn aggregation has been demonstrated to not only evoke the innate immune response but also recruit and activate the adaptive arms of the immune system in PD to promote neuroinflammation [20, 21], and microglial activation is the initial step in this process [22]. Additionally, microglial activation induced by inflammagen such as lipopolysaccharide (LPS), stimulates the aggregation of insoluble α-syn and exacerbates neuroinflammation [23, 24]. This implies that microglial activation and inflammation engage in a self-perpetuating cycle. Similar to macrophages, microglia play a crucial role in eliminating invading pathogenic bacteria, cell debris, and abnormal proteins from the CNS [25, 26]. Investigations involving positron emission tomography (PET) have revealed that microglia are activated in the brains of individuals with PD [27]. Recently, emerging evidence has suggested that microglia perform a variety of distinct roles, exhibiting a spectrum of phenotypes in the pathogenesis of PD. For example, under inflammatory conditions, microglia can secrete cytotoxic proinflammatory cytokines and directly promote dopaminergic neuron death; alternatively, activated microglia can scavenge debris and toxic metabolites from damaged neurons and other cells. Activated microglia can clear pathological α-syn through phagocytosis, and microglia activated by IL-6 may attenuate the number of α-syn inclusions in animal models [28]. Microglial phagocytosis can be selectively activated by the monomeric form of α-syn but is suppressed by aggregated α-syn and promotes α-syn-induced dopaminergic neurotoxicity, suggesting that microglia contribute to the pathological process of PD [29, 30]. In addition, several PD-related genes, such as leucine-rich repeat kinase 2 (LRRK2) and DJ-1, are expressed in microglia and regulate microglial clearance [31,32,33], suggesting that microglial phagocytosis contributes to the development and progression of PD pathology. These findings demonstrate that phagocytosis of α-syn cooperates with intracellular events involved in α-syn processing by microglia, which is involved in neuronal deposition, spreading, and disease progression. Therefore, elucidating the association between microglial activation and α-syn aggregation and propagation may provide insight into the pathological progression of PD, which is critical for developing future therapies.
In this review, we summarize recent findings related to microglial activation and microglia-associated neuroinflammation that are relevant to α-syn aggregation and propagation. We focus our discussions on research developments related to cellular processes associated with inflammation, describing the current understanding of the connection between α-syn and the disruption of microglial homeostasis in the pathogenesis of PD. In addition, we also discuss current therapeutic approaches targeting microglia-mediated inflammation for preventing disease progression from the perspective of these new findings.
Structure and conformation of α-syn
The α-syn protein, encoded by the SNCA gene, is abundantly expressed in neuronal presynaptic terminals [34, 35]. Although its precise biological function is unclear, accumulating evidence indicates that α-syn plays a crucial role in regulating synaptic plasticity [36], synaptic vesicle release [37], molecular chaperoning [38], apoptosis [39], and oxidative stress [40], and it contributes to the pathogenesis of PD. α-Syn is a small 140-amino acid protein consisting of three distinct domains: the N-terminal domain (amino acids 1–65), the nonamyloid component of plaques (NAC) domain (amino acids 66–95), and the C-terminal domain (amino acids 96–140) [41,42,43] (Fig. 1a).

A Schematic of the domain structures, genetic mutations, and posttranscriptional modifications of α-syn related to Parkinson’s disease (PD). The α-synuclein (α-syn) protein comprises three domains: the N-terminal amphipathic region, the nonamyloidogenic component (NAC) domain, and the C-terminal acidic domain. All identified disease-linked mutations are in the N-terminal domain. Mutation sites related to diseases are shown in red; these sites can be posttranslationally modified. B Transition of the monomeric form of α-syn to a pathological aggregate. Native forms of α-syn exist physiologically and can be folded into oligomers. Monomers form fibrils through a nucleated polymerization mechanism, and an equilibrium between fibrils and monomeric α-syn is maintained. The aggregation of amyloid fibrils results in the formation of intracellular deposits referred to as Lewy bodies (LBs)
The N-terminus is a highly conserved domain with four copies of an imperfect 11-amino acid repeat that displays a KTKEGV consensus sequence; another three copies of this repeat are present in the NAC domain [44, 45]. This domain is amphiphilic, is unstructured in solution, and allows binding to phospholipid vesicles and the plasma membrane [46]. In addition, previous reports have indicated that several point mutations of in the N-terminus of α-syn are associated with autosomal dominant forms of familial PD [47]. The A53T and E46K mutations lead to a predisposition to α-syn fibril formation [48, 49], and the A30P mutation leads to a predisposition to the accumulation of α-syn oligomers [50, 51]. The NAC domain of α-syn, a central hydrophobic sequence, is essential for promoting α-syn aggregation [52], especially the NPC61-95 and NPC71-82 domains [53, 54]. A further study reported that the NAC domain's sixth and seventh 11-mer repeats can promote α-syn aggregation [55] and may serve as critical structures for acting on the β-amyloid (Aβ) protein to initiate the formation of amyloid plaques, a pathological feature of Alzheimer’s disease (AD) [56, 57]. The C-terminus of α-syn contains a high concentration of acidic amino acids [58] and 16 amino acid repeats essential for Ca2+ binding [59]. Compared to those exposed to full-length α-syn, neurons exposed to preformed fibrils (PFFs) containing residues 1–99 of α-syn exhibited a decrease in endogenous α-syn accumulation, indicating that the C-terminus of α-syn may facilitate aggregation [60]. C-terminal deletion mutants of α-syn without acidic repeats (residues 125–140) are vital for chaperone protein interactions and promote α-syn aggregation by eliminating chaperone activity [61, 62]. The phosphorylation and nitration of the second acidic repeat in the C-terminus can also disrupt the chaperone activity of α-syn [63, 64].
Several reports have shown that the conformational convergence of the α-helix with its β-sheet is essential for α-syn seeding and toxicity. Although oligomers with different β-sheet contents and degrees of hydrophobicity exist, they all possess a hollow cylindrical architecture with similarities with certain types of fibrils, suggesting that the β-sheet geometry is acquired in the early stages of oligomeric α-syn self-assembly [65], which generally requires approximately 30 monomers. However, the minimum number of α-syn monomers for stable fibril generation is approximately 70, which yields fibrils approximately 40 nm in size [66]. The existence of a bidirectional equilibrium between oligomers and higher molecular weight fibrillar assemblies suggests that cellular toxicity and seeding competency could be caused by different-sized conformers or assemblies [67] (Fig. 1b). Another important consideration is that conformational polymorphs of α-syn in distinct synucleinopathies and even specific cells spreading in different regions may support the spatiotemporal spread of α-syn within the brain [68].
Heterogenous phenotypes of microglia responding to α-syn
Microglia are thought to maintain immune homeostasis in the CNS in response to various stimuli. Generally, activated microglia have been classically divided into two distinct phenotypes, the proinflammatory phenotype (M1 microglia) and the anti-inflammatory phenotype (M2 microglia) [69]. Rapidly emerging evidence suggests that microglia cannot be simply divided into two distinct categories; instead, they exist on a spectrum, with their phenotype changing along a continuum depending on the environment they inhabit [70,71,72]. Microglial phenotypic shift is related to neuroinflammation, as activated microglia are mainly responsible for the release of inflammatory cytokines, including IL-1β, IL-6, and TNFα [73, 74]. Lentivirus-mediated selective α-syn accumulation in microglia was used to investigate the transcriptome of model mice, and the results showed that the microglia in which α-syn accumulated exhibited heightened reactivity, characterized by phagocytic exhaustion and the presence of proinflammatory molecules [75]. Although α-syn is a potent activator of microglia, it is challenging to accurately assess microglial states throughout the various stages of PD due to the influence of the assessment method and timing of collection [70]. In addition, the complex and variable nature of microglial activation makes it challenging to determine activation status based solely on the stage and severity of neurodegeneration [76]. For example, monomeric α-syn-treated microglia exhibit significantly decreased iNOS expression and increased ARG-1 and CD206 expression levels, indicating the protective role of microglia under specific conditions. Microglia gradually transform into inflammatory microglia with prolonged α-syn incubation and increased iNOS and CD16/32 expression [77]. Recently, methodological advances in single-cell omics assays and integrative gene and protein expression analyses have enabled a deeper understanding of the molecular processes that underlie particular microglial activation states in a given context [78, 79]. Integrating published GWAS data with cell-type enrichment data, Reynolds et al. discovered that PD heritability through disease-relevant genes operates in a pathway-specific manner, with the lysosome-related gene set highly expressed in a microglial subpopulation [80]. To understand the involvement of glial cell types in PD and their impact on the severity of the disease, it is essential to utilize single-cell omics combined with advanced techniques and analysis tools in the future [81].
Highly polarized microglia are closely related to neuronal survival, and active microglia are related to disease type, disease stage, and even brain region [82,83,84]. The distribution of microglia is uneven across different brain areas, with the rat substantia nigra having abundant microglia [85]. Furthermore, evidence has suggested that microglia are proliferative and activated in the substantia nigra in PD patients [86] and animal models [87]. In addition to changes in synaptic function in the early stages, PD mouse models also exhibit differences in the number and location of microglia [88]. These findings suggest that microglia are responsible for the pathological process underlying dopamine neuron vulnerability and stage-dependent differences in PD progression.
Involvement of α-syn in microglial pathology
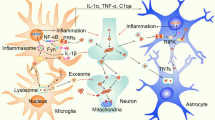
Experimental and clinical studies have provided evidence that α-syn propagation contributes to the anatomical spread of Lewy pathology. Although α-syn is typically considered an intracellular protein, several studies have identified its presence in the cerebrospinal fluid (CSF) and plasma of individuals with PD [89, 90]. It has been observed that primary cortical neurons from rats and neuron cell lines can secrete extracellular vesicles containing α-syn [91]. Furthermore, when intracellular protein transport through lysosomes is blocked, there is an increase in the release of extracellular vesicles containing α-syn, suggesting that modulating intracellular α-syn processing may significantly contribute to its release [92,93,94]. Accumulating research has highlighted the crucial role of microglia in transmitting harmful protein aggregates between cells, which is thought to contribute to the spreading of α-syn neuropathology and underlie the pathogenesis of PD [95]. Microglia have a strong capacity to take up surrounding α-syn containing exosomes, leading to their activation and subsequent intracellular processing [96, 97]. Blocking exosome activation in microglia has been found to decrease the spread of α-syn to neurons [98]. In pharmacological ablation experiments targeting host microglia in the striatum, it was observed that α-syn accumulation was increased in α-syn-expressing dopaminergic neurons, and α-syn was transferred to neurons via a process modulated by microglial inflammatory activation [99]. These studies provide insight into the noncell-autonomous effect associated with the intracellular accumulation of α-syn and its interneuronal transmission, indicating that microglia are functionally involved in the disease pathogenesis (Fig. 2).

Schematic overview of the involvement of α-synucleinopathies and microglia in the pathogenesis of PD. α-Syn has the propensity to form polymeric and fibrillar structures, and the accumulation of α-syn in neurons can be toxic, resulting in neuronal degeneration and cell death. α-Syn in various conformations is released from neurons and recognized and internalized by microglia, leading to microglial homeostasis disruption. Microglia spatiotemporally release cytokines and cell survival factors, such as IL-4, IL-10, and BDNF, to restore the function of damaged neurons and the cytotoxic factors IL-1β, IL-6, and IFN-γ and substances such as reactive oxygen species (ROS), NO, and COX2 to promote α-syn aggregation and neuronal damage
Pathways by which microglia internalize α-syn
The internalization of α-syn by resting and activated microglia is a critical step that drives neuroinflammatory responses, and multiple processes underlying this function have been proposed. Endocytosis is thought to be the primary process by which pathological proteins are internalized. Endocytosis can be divided into two categories: clathrin-independent and clathrin-dependent endocytosis. Particles that are larger than 0.5 μm can be internalized by clathrin-independent endocytic mechanisms, such as micropinocytosis and phagocytosis [100], and particles that are smaller than 200 nm can be internalized through dynamin-dependent or dynamin-independent pathways [101].
Microglial phagocytosis is a process by which microglia can recognize and take up extracellular targets, including abnormally aggregated proteins, via specific receptors [102]. Compared with that of wild-type microglia, the phagocytic capability of microglia from SNCA−/− mice was found to be reduced, suggesting that endogenous α-syn might exert a positive effect on the phagocytic ability of microglia [103]. Monomeric α-syn, but not β- or γ-syn, was observed to boost microglial phagocytosis in a dose- and time-dependent manner. Several receptors are critical for activating microglial phagocytosis and the ability of microglia to remove α-syn [29, 104, 105]. Microglial TLR4 deficiency was found to diminish α-syn phagocytic activity. TLR-4 mediates microglial phagocytosis of full-length soluble α-syn, fibrillary α-syn, and C-terminally truncated α-syn, and the latter has been identified as have more potent effects than the other forms [106]. Nevertheless, the uptake of α-syn by astrocytes with TLR4 deficiency did not abolish phagocytosis, indicating a potential cell type-specific difference in the uptake of α-syn. The expression of CD22, a negative regulator of microglial homeostasis, is significantly elevated in microglia-conditioned α-syn-expressing CX3CR1-SNCA mice, and CD22 significantly abolishes microglial phagocytosis [75, 107]. According to one study, monomeric α-syn can enhance microglial phagocytosis, while aggregated α-syn can suppress this effect [29], suggesting that microglia show different responses when they phagocytose distinct forms of α-syn. A further investigation discovered that the aggregated form of α-syn directly binds to and facilitates FcγRIIB expression, resulting in the activation of SHP-1, a suppressor of microglial phagocytosis [108], supporting the observation that highly complex form of α-syn may attenuate microglial phagocytosis. An in vitro study demonstrated that immortalized BV2 microglia-like cells take up α-syn via ganglioside GM1 by linking to lipid rafts at the plasma membrane [109] or directly binding to macrophage antigen-1 (Mac-1) receptor [110] (Fig. 3).

Involvement of α-syn in microglia-mediated cellular processes in PD. Microglia recognize and internalize monomeric, oligomeric, and fibrillary α-syn through cell surface receptors, including the Toll-like receptors (TLRs) 1/2, 4, and 5, and intracellular proinflammatory pathways are subsequently activated via the transcription factor NF-κB/P65. Additionally, α-syn upregulates NLRP3 (NOD, LRR-and pyrin domain-containing 3), which results in the assembly of apoptosis-associated speck-like protein containing a CARD (ASC) into specks and activation of caspase 1 to form the inflammasome. Alternatively, the binding of different forms of α-syn to TLR4 or CD11b induces mitochondrial dysfunction and facilitates the release of mtROS, thus inducing the assembly and activation of the NLRP3 inflammasome. α-Syn internalization triggers endocytosis as well as both clathrin-dependent and clathrin-independent processes. PD-related genes, such as LRRK2, regulate the stages of endocytosis-lysosomal assembly in microglia and might contribute to inflammation and neuronal α-syn pathology
It has been proposed that several cell-surface receptors, including TLR2 and TLR4, are involved in α-syn internalization by clathrin-dependent endocytosis [111, 112]. The uptake of α-syn by clathrin-mediated endocytosis has been demonstrated in primary cultured microglia [113]. However, blocking this pathway cannot fully inhibit α-syn uptake, suggesting that alternative routes are involved in α-syn internalization [114]. It was also found that soluble monomeric α-syn directly translocates across the plasma membrane in a manner not involving endocytic receptors [114, 115].
Intracellular processing of internalized α-syn
Although α-syn aggregates have been observed to be internalized and cleared by microglia, the precise intracellular events responsible for this process is still not fully understood. Studies of familial PD mutation genes have demonstrated that the internalization of α-syn into recipient cells typically leads to its degradation by lysosome vesicles. For example, PD-associated genes carrying causal mutations, such as the LRRK2 mutation identified from patients with autosomal-dominant monogenic familial cases of PD, have been reported to be associated with lysosome processing of α-syn within microglia (Fig. 3). LRRK2 is present on lysosomes [116] and plays a direct role in endocytosis through its interactions with Rab small GTPases, which regulate the fusion of certain endosomes and consequent lysosomal content degradation [117] and synaptic vesicle recycling [118]. Microglial knockout of LRRK2 increases the amount of α-syn taken up and the α-syn clearance efficiency, which is correlated with an increase in the number of Rab5-positive endosomes [31]. Interestingly, LRRK2 has been reported to play a positive role in microglial phagocytosis of neuronal axons and elements [116]. This suggests that LRRK2 strongly controls vesicular trafficking events within endolysosomes and has a wide-ranging influence on endocytosis and the degradation of unfolded/aggregated proteins in microglia. However, the influence of PD-related genes, such as ATP13A2, its common risk variants and GBA, which are linked to lysosomal activity in neurons and astrocytes [112], on the endolysosomal system in microglia is yet to be determined, and thus, further investigation is needed. Another study suggested that after a short incubation time, α-syn selectively induces the expression of p62/SQSTM1 [119], an autophagy-related protein that recognizes ubiquitinated proteins and interacts with these proteins to mediate selective autophagy for clearance [120], through the TLR4/NF-κB pathway. Knocking out p62/SQSTM1 prevents the effective removal of α-syn and aggravates pathology [119].
Signaling pathways involved in α-syn-induced microglial inflammation
Activation of the NF-κB signaling pathway by α-syn
In α-syn-related pathology, the expression level of the microglial surface receptor TLR2 is significantly increased. Oligomeric or fibrillary α-syn activates the microglia surface receptor TLR2, causing intracellular NF-κB pathway activation and the release of inflammatory factors such as TNF-α and IL-6 [111, 121]. The regulation of downstream signals by TLR2 requires its adaptive protein MyD88 [122]. Suppressing the effect of MyD88 on TLR2 can effectively inhibit the activation of NF-κB signaling and alleviate α-syn-induced neuroinflammation and dopaminergic neuron loss [24]. TLR4 regulates microglial phagocytosis of α-syn, activates the intracellular NF-κB signaling pathway, and causes the release of inflammatory factors [123]. The various forms of α-syn have distinct preferences for TLR2 and TLR4, with their effects on these receptors varying depending on the form of α-syn. At low concentrations, monomeric α-syn preferentially acts on TLR4 to induce selective autophagy to clear α-syn, which may be related to its ability to regulate microglial phagocytic activity [124]. In contrast, oligomeric or fibrillary α-syn-activated microglia induce inflammation through cell-surface TLR2. Studies have shown that the α-syn promoter contains NF-κB binding sites, so α-syn-induced NF-κB activation further causes α-syn expression and aggravates α-syn-induced pathology, forming a vicious cycle [125]. A study investigated the consequences of exposure to wild-type α-syn and three α-syn mutants (A53T, A20P, and E46K) on microglial reactivity. It was found that α-syn-induced microglial reactivity appeared to be peptide dependent, and the inflammation caused by the A53T mutant was more substantial than that caused by the wild-type form [126]. Although short-term exposure to the A53T mutant may stimulate MAPK-pERK pathway activation, the long-term inflammatory response elicited by continued exposure to the mutant protein requires further investigation.
α-Syn affects inflammasome formation in microglia
Some reports have suggested the importance of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, a persistent source of microglia-mediated inflammation that drives progressive dopaminergic neuropathology in PD [127, 128]. Activation of the NF-κB pathway promotes NLRP3 inflammasome assembly and IL-1β release and further causes mitochondrial dysfunction, reactive oxygen species (ROS) production, and oxidative stress [129]. RNA-sequencing (RNA-seq) analysis of the substantia nigra of 5-week-old CX3CR1-SNCA mice showed that α-syn aggregation in microglia significantly increased the expression of inflammation-related genes, including the inflammasome components NLRP3 and adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), IL-1β, and NF-κB [75]. This observation indicates that α-syn in microglia activates the NLRP3 inflammasome and induces the release of IL-18 and IL-1β, consistent with previous in vitro studies. Inhibition of NLRP3 inflammasome activity significantly alleviates PD pathology, including neuroinflammation and motor dysfunction [127, 128]. In chronic MPTP-treated NLRP3 knockout mice, NLRP3 deletion was observed to inhibit inflammasome formation and protect against α-synuclein pathology by alleviating autophagy dysfunction [130]. α-Syn-mediated activation of the inflammasome component NLRP3 in microglia promotes ASC release and induces α-syn aggregation in neurons [75].
Research has revealed that the assembly of the NLRP3 inflammasome requires two distinct steps: priming and activation. According to research, α-syn fibrils can initiate intracellular NLRP3 inflammasome assembly or activate the NLRP3 inflammasome through cell surface receptors [131, 132]. These molecules act as priming signals to activate the NLRP3 inflammasome. For instance, α-syn fibrils interact with TLR2, activating downstream signals to activate the NF-κB pathway, which, in turn, upregulates NLRP3, ASC, and pro-caspase-1 [133], and anti-TLR2 antibodies attenuate this response. Scheiblich et al. reported that α-syn monomers and oligomers, but not α-syn fibrils or ribbons, activate the NLRP3 inflammasome via TLR2 and TLR5 under unprimed conditions [134]. In addition, microglial TLR2 and TLR5 neutralization by specific antibodies blocks caspase-1 cleavage and NLRP3 inflammasome activation induced by α-syn monomers and oligomers. These observations indicate that α-syn monomers acting on TLR2 coordinate with α-syn monomers and oligomers acting on TLR5 to contribute to the priming step of NLRP3 inflammasome activation. Another study suggested that α-syn interacting with TLR4 promotes the expression of proinflammatory cytokines and increases mitochondrial ROS (mtROS) levels by damaging mitochondria and triggering NLRP3 inflammasome assembly and activation [135]. An additional study suggested that α-syn fibrils can activate the NLRP3 inflammasome under unprimed conditions; however, it was found that the expression level of IL-1β is lower in the unprimed state than in the primed state but significantly higher than that in untreated microglia [136]. These conflicting results may be due to differences in the concentration and/or quality of the prepared α-syn aggregates and may indicate the existence of alternative pathways involved in NLRP3 inflammasome activation.
For instance, α-syn can bind to CD11b to initiate NLRP3 inflammasome assembly. CD11b is highly expressed on microglia in the CNS and has been reported to regulate microglial phagocytosis and migration [137, 138]. α-Syn fibrils bind to CD11b to cause mtROS production through the RhoA/ROCK pathway to activate the NLRP3 inflammasome [139]. The interaction between oligomeric α-syn and CD11b induces NOX2 activation and the production of hydrogen peroxide, which binds to an intracellular tyrosine-protein kinase known as Lyn, resulting in actin phosphorylation and cytoskeleton rearrangement [140]. This ultimately leads directly to the induction of microglial migration to damaged nerve cells, which are cleared through phagocytosis [141]. Microglial activation may lead to neuronal repair; however, persistent activation of microglia for an extended period may cause indiscriminate phagocytosis of neuronal synapses, disrupting neurotransmission.
Link between α-syn accumulation and microglial inflammation
Some studies suggest that aggregated α-syn promotes M1 microglial polarization [30, 142] and causes monomeric α-syn aggregation, implying that the activation of the microglial immune response might be intrinsically linked to the accumulation of α-syn [131]. Cell-surface Toll-like receptors (TLRs) appear to be key molecules in the signaling pathway that link α-syn phagocytosis with the formation and transport of phagosome cargos into lysosomes for degradation [124, 143]. Evidence has demonstrated that TLR1 [144], TLR2 [145], TLR4 [106], and TLR5 [134], members of the TLR family, functionally interact with α-Syn, with TLR2 and TLR4 playing a more prominent role. Primary microglia exhibit significantly increased expression of TLR1 and TLR2 after exposure to mutant A53T α-syn [145]. Studies indicate that TLR2 can recognize monomeric, oligomeric, and fibrillar forms of α-syn and trigger the intracellular cascade to promote inflammatory activation [125, 146]. Evidence suggests that TLR4 is highly expressed in patients with synucleinopathies and is vital for the uptake of α-syn and the associated inflammatory processes, including the generation of ROS and cytokine release [106]. Upon TLR activation [106], the NF-κB cascade is triggered, leading to the transcription of p62, an autophagy receptor essential for forming a platform for autophagosome formation to eliminate ubiquitinated proteins in microglia [147].
Myeloid cell-triggered receptor II (TREM2), a member of the immunoglobulin superfamily, is abundantly expressed on the surface of microglia in the CNS and plays a critical role in microglial phagocytosis, proliferation, and inflammatory factor production [148, 149]. Studies have revealed that TREM2 is a contributing factor to PD development [150], and genetic research has revealed that the R47H mutation (rs75932628, p.R47H) in TREM2 contributes to the development of both PD and AD [151]. Studies on mouse models of PD induced by MPTP have revealed that TREM2 can reduce pathological changes in the nervous system and inhibit NF-κB, MAPK, and TLR4/TRAF6 signaling during the neuroinflammatory response [152]. Research has demonstrated that overexpressing TREM2 inhibits the neuroinflammatoty response through the PI3K/AKT and NF-κB signaling pathways in LPS-stimulated BV2 cells [153]. Transfection of BV2 microglia with TREM2-siRNA exacerbated the inflammatory responses induced by α-syn and conditioned medium, triggering apoptosis in cultured SH-SY5Y cells [154]. These observations have been confirmed in TREM2 knockout mice with AAV-mediated α-syn overexpression, as it was found that dopaminergic neuron loss is exacerbated in these mice [155]. One clinical study showed that the levels of TREM2 in the CSF of PD patients were elevated compared to those in the CSF of healthy controls and had a positive correlation with total α-syn degradation [156]. These findings demonstrate the potential bidirectional role of TREM2 in processing α-syn on the cell membrane, as well as in inducing microglial inflammation.
The aromatic residues of α-syn can form hydrogen bonds with glycosphingolipids in the cell membrane, forming a round porous oligomeric structure, causing intracellular calcium current disorder, interfering with intracellular homeostasis, and leading to cell degeneration [157]. Oligomers have a higher membrane-binding capacity than monomers, and their binding to lipids is considered the early stage in the development of synucleinopathies [158]. In addition, α-syn aggregation can induce microglia to release the neurotransmitter glutamate, resulting in extracellular glutamate accumulation and CNS excitotoxicity [159]. Metabotropic glutamate receptor 5 (mGluR5) is a G-protein coupled receptor (GPCR) present in neurons and microglia. Stimulating mGluR5 in microglia is a desirable strategy, as it can significantly inhibit the inflammatory response; thus, mGluR5 may play a critical role in microglial homeostasis [160, 161]. mGluR5 agonists can inhibit α-syn-induced inflammatory signals and cytokine secretion in BV2 cells and primary microglia and reduce microglial activation to protect against neurotoxicity. Additionally, mGluR5 was found to colocalize with α-syn in lysosomes. Studies of an α-syn-based PD rat model revealed that α-syn promotes the breakdown of lysosome-dependent degradation of the mGluR5 complex to control neuroinflammation [161].
Microglia mediate α-syn release and transmission between cells
The release of α-syn is associated with exosomes, as exosomes containing α-syn are formed through the fusion of multivesicular bodies with the plasma membrane, a process that is affected by the intracellular Ca2+ concentration [162,163,164]. Upon treatment with α-syn preformed fibrils, exosomes containing α-syn released by donor microglia can induce protein aggregation in recipient neurons [97]. Exosomes containing pathological α-syn may also be internalized by neighboring microglia [165] or other cells by receptor-mediated endocytosis [166]. In an in vitro culture system, it was found that LPS promoted the secretion of soluble factors and TNF-α from activated microglia and increased α-syn levels in the medium. Importantly, TNF-α secreted from LPS-treated microglia promotes cell-to-cell propagation of α-syn between neurons, while soluble factors trigger cellular senescence. Interestingly, the secretion of α-syn was found to be increased in senescent neurons, indicating the acquisition of a senescence-associated secretory phenotype (SASP) in response to TNF-α stimulation. Related morphological studies have confirmed that propagating α-syn aggregates are present in electron-dense lysosome-like compartments, indicating that inflammatory factors can drive cell-to-cell propagation of α-syn by promoting the acquisition of a SASP and the secretion of α-syn [167]. Recently, studies have suggested that microglia can form tunneling nanotubes for microglia-to-microglia spread of α-syn [168]. In addition, α-syn-induced activated microglia promote the release of α-syn-containing exosomes and increase α-syn propagation [97, 166]. The pathways through which α-syn is secreted by microglia may serve as essential mechanisms for amplifying and spreading α-syn pathology to the surrounding milieu.
Recently, lymphocyte activation gene 3 (LAG3) was shown to act as a receptor for pathogenic α-synuclein assemblies in the CNS in PD [169]. Further investigation found that the C-terminus of α-syn binds to LAG3 and amyloid precursor-like protein 1 (APLP1)-positive pockets, facilitating the entry of α-syn into neurons [170]. The phosphorylation of α-syn at S129 in the C-terminus promotes α-syn fibril binding to LAG3 and APLP1, enhancing the propagation of α-syn fibers in neurons [171]. Overexpression of LAG3 increases the phosphorylation of α-syn, thus exacerbating PD pathology, whereas deletion of LAG3 has the opposite effect, inhibiting α-syn neuronal transmission, PFF-induced neuron loss and toxicity both in vitro and in vivo [169]. Increasing evidence suggests that LAG3 may be involved in the endocytosis and intercellular transmission of α-syn [172, 173]. In mouse cortical neurons, deletion of LAG3 can recruit α-syn to early endosomes, where it colocalizes with RAB5, significantly reducing α-syn aggregation; this indicates that LAG3 is involved in processing α-syn during endocytosis [169, 174]. snRNAseq and scRNAseq data suggest that microglia express LAG3, and the administration of an anti-LAG3 monoclonal antibody was found to protect against neuronal impairment [175], indicating a modulatory role for LAG3 in α-syn pathology through microglial pathways.
Peripheral interference with microglial inflammation and α-synucleinopathy
Multiple clinical and preclinical studies have provided evidence that PD pathology can be transmitted from the peripheral to central nervous systems and trigger neuroinflammation through various mechanisms, including the regulation of the gut microbiota and disruption/disturbance of ascending or descending neurotransmission. A study revealed the existence of a bidirectional connection between the CNS and the gastrointestinal system, known as the "brain-gut axis", despite their anatomical separation. Intestinal bacterial colonization regulates immune system maturation and development in the CNS. It has been reported that α-syn-dependent microglial activation may be linked to changes in the microbiome in both animal models and patients [176]. According to these observations, the gut microbiota is involved in the pathogenesis of PD, and its role is mediated by bidirectional regulation of the immune response of microglia in the CNS.
A series of well-designed experiments demonstrated that α-syn-overexpressing mice harboring a complex microbiota have worse motor function and constipation, accompanied by marked microglial neuroinflammatory responses and α-syn aggregation in the brain, than α-syn-overexpressing germ-free (GF) mice (born and raised in sterile environments). Gut microbiota transplantation from PD patients into GF α-syn-overexpressing mice was found to exacerbate motor dysfunction [177]. Intriguingly, using antibiotics to eliminate the microbiota in PD model mice was shown to alleviate motor dysfunction and pathological features [176]. Increasing the gut microbiota diversity in PD model mice improves motor function and ameliorates pathological features [176]. GF mice display global microglial defects, such as altered cell proportions, defects in microglial maturation, and impaired innate immune responses. However, another critical study highlighted the substantial contribution of host-dwelling bacteria to microglial homeostasis. In GF mice, microglia in the brain are globally defective, exhibiting altered proportions and maturation. The expression of the microglial surface molecules CSF1R, F4/80, and CD31, which gradually decreases during cell maturation in the brain, was found to be increased. These findings indicate that host bacteria are vital for microglial development and function, and that microglial impairment can be at least partially rectified by a complex microbiota [178, 179]. Gut microbiota supplementation, especially with short-chain fatty acids (SCFAs) and microbial metabolites, promotes the maturation of microglia in GF mice, suggesting that the gut microbiota regulates the development and growth of microglia. Indeed, intensive studies have shown the potential effects of the gut microbiota on the pathogenic features of PD in various models, including Drosophila melanogaster [180, 181], mouse [176, 182, 183] and nonhuman primate [184, 185] models. Additionally, clinical studies have shown gut microbiota changes in PD patients [186, 187]. Akkermansia, Bifidobacterium, and Lactobacillus, bacteria commonly considered beneficial, are increased in abundance in PD [188], but the mechanisms underlying this change are unknown.
Accumulating evidence indicates that inflammation caused by gut dysbiosis involves α-syn transmission from the gut to the brain and/or α-syn aggregation during PD progression. The transmission of α-syn from the gut to the brain, accompanied by the degeneration of dopaminergic neurons in the substantia nigra and PD-like motor and nonmotor symptoms, has been shown to occur in rodent models [182, 183]. Aged rats receiving gastrointestinal injections of aggregated α-syn show more efficient gut-to-brain spreading than young rats, suggesting that aging is a critical risk factor for α-syn transmission via this pathway [189, 190]. Studies have shown that there are alterations in gut microbiome transmission and the effects of microglia-related neuroinflammation within the brain, and studies on intestinal microbiome perturbations have also shown the relationship between intestinal hyperinflammation and local α-syn aggregation. For example, marmosets with colitis show increased phosphorylated α-syn accumulation in the colonic myenteric plexus [185]. In addition, transgenic mice overexpressing α-syn show gut microbiota alterations [191]. Therefore, more studies are needed to clarify the molecular signaling pathways involved in the influence of the microbiota on microglial activation.
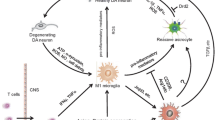
Various potential pathways, such as bidirectional endocrine and immune pathways involving the brain-gut-microbiota axis, might be involved in the pathological processes of PD [192]. The blood‒brain barrier (BBB) can prevent harmful substances from entering the brain through the blood, and it plays an essential role in maintaining the normal physiological state of the CNS and preventing pathogens from invading the brain [193]. PD affects the permeability of the BBB and causes abnormally deposited proteins, such as α-syn, in the CNS to diffuse into the intestine through the BBB, affecting the gut microbiota balance. Nevertheless, studies have suggested that gut microbiota impairment affects the permeability of the BBB, thus allowing intestinal microbial metabolites and intestinal inflammatory factors to enter the CNS [194]. In addition to microglia, peripheral immune cells and activators can cross the BBB under pathological conditions [193]. For instance, microglia-specific expression of α-syn induces the release of several chemokines, such as CCL1, CCL2, and CXCL10 [75]. These chemokines increase inflammation and recruit peripheral immune cells, including CD4+ and CD8+ T cells, to the brain, thus exacerbating inflammation [75, 195]. T helper 17 (Th17) cells, a subtype of CD4+ T cells, enter the brain through the CXCR4-CXCL12 pathway and interact with microglia to interact with α-syn and participate in IL-17A-regulated neuronal injury [196]. Furthermore, GF mice show increased BBB permeability [197], and BBB permeability can be decreased by SCFA-producing bacteria and oral gavage with solidum butyrate [198].
Evidence of some important neuroanatomical pathways in PD has led to the proposal of the "dual-hit" hypothesis to explain the disease's peripheral onset [199]. Hyposmia and constipation are common nonmotor symptoms of PD that can appear years, or even decades, before diagnosis, and Lewy-type pathology can be detected in peripheral tissues up to twenty years before diagnosis. Truncal vagotomy disrupts α-syn transmission from the gut to the brain in animals [183] and substantially reduces PD risk in patients [200]. Animal and clinical studies have demonstrated that LB-like aggregates are deposited in the enteric nervous system (ENS) and dorsal motor nucleus of the vagus nerve (DMV) at an early stage and are linked to the severity of motor and gastrointestinal symptoms [201, 202]. Consistent with these findings, injection of α-syn into intestinal tissues can induce α-syn pathology in the vagus nerve and brainstem [182]. However, this evidence has not been confirmed clinically, as there is a lack of postmortem patients with isolated gastrointestinal α-syn pathology without accompanying CNS pathology. Despite the controversy concerning the peripheral origin of diseases, this theory provides an intriguing perspective and significant directions for the development of treatments for PD.
Therapeutic strategies targeting α-syn aggregation and neuroinflammation
α-Syn aggregation and transmission and neuroinflammation play crucial roles in the pathogenesis of PD, and unsurprisingly, reducing α-syn aggregation and inhibiting inflammation have become fundamental goals of PD therapy. To this end, clinical and preclinical trials of approaches for preventing α-syn aggregation and inflammation have been carried out or are underway.
The ability of various approaches targeting different points in the life cycle of α-syn to prevent or lessen its harmful effects in PD is being investigated. Immunotherapies targeting pathological α-syn are currently being developed as primary therapeutic approaches and tested in clinical trials [203]. In addition to immunotherapies, including those that suppress α-syn (NTP200-11) [204], reduce α-syn transcription [205, 206] or translation [207], eliminate pathological aggregates [208,209,210,211], and enhance α-syn clearance and degradation [212,213,214], have been tested as disease-modifying treatments for α-synucleinopathies. In addition to blocking the release of α-syn from donor cells in the CNS, preventing α-syn transmission from the peripheral nervous system has recently been identified as an effective therapeutic strategy [183]. An alternative approach is to target other genes or proteins associated with or implicated in PD that contribute to the processing or aggregation of α-syn, such as Gcase, LRRK2, NMDAR, and AMPAP [215,216,217,218]. The proteolytic system is an efficient mechanism for eliminating abnormally deposited proteins within cells, and regulating protease expression and enzymatic activity is important for facilitating α-syn clearance [219]. For example, the expression of the protease cathepsin D, which is a component of the proteolytic system, is reduced during α-syn aggregation, and this protease protects against α-syn-induced cell death in vitro and in C. elegans models [220]. These findings suggest that the proteolytic system efficiently eliminates abnormally accumulated proteins within cells, highlighting its potential as a crucial target for PD treatment.
Targeting inflammation has emerged as a potential therapeutic approach for PD, as it may help to slow or halt the progression of the disease. Inflammation damages nerve cells and promotes the expression of α-syn, further aggravating PD pathology [125]. Studies have highlighted innate and adaptive immune response abnormalities in PD patients, including increased proinflammatory cytokine levels and altered immune cell populations such as monocytes and their precursors [221]. Neuroinflammatory features have been observed in clinical studies and experimental models of PD, thus establishing a link between inflammation and the pathogenesis of this condition [222, 223]. TLR plays a crucial role in activating microglia-induced neuroinflammation mediated by α-syn. Treatment with an anti-TLR2 antibody was shown to significantly decrease α-syn deposition and neuroinflammation in animal models of PD, leading to alleviation of movement disorders [224]. Additionally, CU-CPT22, a small molecule inhibitor targeting TLR1/2, attenuates oligomeric α-syn-induced inflammation in primary microglia [121]. Upregulation of NLRP3 in microglia has been demonstrated to significantly upregulate inflammatory cytokines, transcription factors, and critical components of the inflammasome pathway, contributing to dopaminergic cell loss in PD mouse models [225]. MCC950, a small molecule inhibitor targeting the NLRP3 inflammasome, has been demonstrated to suppress neuronal damage, attenuate α-syn deposition and inflammation, and alleviate dyskinesia in various PD models, including PFF- and 6-OHDA-treated and α-syn A53T mutant animals [127, 128].
Autoantigens, such as pathological α-syn and inflammatory factors, which can be presented on the surface or secreted into the extracellular environment by neurons, activated microglia, and macrophages in the brain and CSF [226, 227], can increase BBB permeability and thus allow T-cell entry, resulting in detrimental effects on neurons [221, 228, 229]. Sargramostim, an FDA-approved recombinant form of human granulocyte–macrophage colony-stimulating factor (GM-CSF), has been demonstrated to enhance regulatory T-cell function and improve motor outcomes. Due to its antineuroinflammatory properties, it is currently being investigated as a potential therapeutic option for PD [230]. Epidemiological evidence suggests that the secretion of TNF-α by activated microglia in patients is associated with an elevated risk of developing PD [231], while anti-TNF therapy has been linked to a significant reduction in PD incidence and promotes the survival of dopaminergic neurons [232, 233]. Consistent with these observations, anti-TNF antibodies prevent the death of dopaminergic neurons in mice [234, 235]. The transcription factor PPARγ, expressed in neurons and glia, is a molecular link between glucose metabolism and the regulation of microglial inflammation [236]. The PPARγ agonist pioglitazone has been used in the clinic for T2DM treatment, and its effectiveness was recently evaluated in PD animal models [237]. The PPARγ agonist rosiglitazone effectively inhibits LPS-induced microglial activation, whereas the antagonist T0070907 induces a shift of microglia from an inflammatory phenotype to a homeostasis-restoring phenotype [238]. The PPARγ agonist exerts inhibitory effects on microglial activation, leading to a reduction in the production of proinflammatory factors and protecting dopaminergic neurons by modulating multiple signaling pathways, including the JUN, NK-κb, and NF-AT pathways.
The involvement of the gut microbiota in chronic inflammation and α-synuclein aggregation in the enteric nervous system presents new therapeutic opportunities that are largely unexplored. Notably, an ongoing clinical trial (NCT03958708) is investigating the effects of rifaximin, an antibiotic, in reducing systemic inflammation and α-synuclein aggregation by targeting the gut microbiota in individuals with PD. Another study (NCT03808389) is exploring the potential benefits of fecal transplantation in alleviating gut inflammation in PD patients. While most of these trials are primarily assessing clinical motor endpoints or the ability of the treatments to act on their targets, evaluating immune-related endpoints and outcomes is crucial in preclinical and clinical studies owing to the close relationship between synucleinopathies and neuroinflammation.
Conclusions, limitations, and perspectives
This review emphasizes the importance of microglia, which are located in the inflammatory environment within the CNS, in establishing a connection between neuroinflammation and α-synucleinopathies associated with PD. The exact mechanism by which disruption of microglial homeostasis contributes to α-synucleinopathy is still under investigation, but several findings suggest that microglia may act as regulators of this process. Despite the wealth of information presented in this review regarding microglia-related pathological changes, there remains significant uncertainty surrounding the specific states of microglia and their role in the pathogenesis of PD. Additionally, recent research has highlighted distinct functional variations among microglial phenotypes across different brain regions, potentially contributing to the unique patterns of microglial-mediated inflammation in PD [239]. The complexity of cellular phenotypes extends beyond conventional classifications, suggesting that further investigation is imperative for elucidating inflammation signatures associated with neurodegenerative diseases [240]. Therefore, developing enhanced methods such as scRNA-seq or spatial transcriptomics for characterizing microglial signatures in humans or disease models would be highly advantageous to the field.
Several large-scale epidemiological and clinical studies have provided limited evidence of a relationship between intestinal diseases, gut-targeted interventions, microbiome changes, microglial homeostasis, and α-synucleinopathies [241]. Inflammation in the gut contributes to disease development through systemic mechanisms such as increased cytokine production, disruption of the blood brain barrier, migration of inflammatory cells into the brain, and activation of microglia according to studies of PD patient biopsies and fecal samples. Identifying gut microbes and metabolites that cause the disease is extremely challenging [242], as they can act independently or exert enhancing or counteracting effects within the microbial community. However, recent advances in technical and computational tools used to investigate the composition and function of the microbiome could facilitate the analysis of variations in the influence of host-associated microbial communities [243] and may provide clues for understanding the communication between the microbiome and microglia in the progression of α-synucleinopathies.
The current understanding of microglial states and their involvement in α-synucleinopathies is derived from studies utilizing diverse animal models, cell culture systems, and human samples. Researchers have made significant progress in the field by reprogramming primary microglia from fresh postmortem brains of individuals with disease or stem cells obtained from human or animal models to become microglia-like cells, enabling access to more rapid and physiological findings [244]. Recent advances in the use of human induced pluripotent stem cell (iPSC)-derived microglia-like cells (iMGLs) have allowed successful recapitulation of disease phenotypes, providing a better understanding of the pathological roles of microglia in neurological diseases [245, 246]. Alternatively, the monocyte-derived microglia-like cell (MDMi) model is another in vitro culture system that both recapitulates the genetic background of the humans from which the cells are derived [247] and allows for rapid large-scale cultivation. This system may be beneficial for exploring the interaction between the disruption of microglial homeostasis and disease progression [248, 249]. When investigating the disruption of microglial homeostasis and α-syn, model selection should be contingent on the context, with the model cells being cultured either alone or in combination with other cells, to obtain the most robust findings that reveal pertinent disease pathways. Furthermore, these findings should be cross-validated in other systems according to downstream applications to evaluate potential treatment methods.
In summary, microglia play a pivotal role in central inflammation, and the interaction of these cells with α-syn may contribute to the development of PD pathogenesis; thus, microglia are indispensable targets for therapeutic interventions.
Availability of data and materials
Not applicable.
Abbreviations
- LBs:
-
Lewy bodies
- LNs:
-
Lewy neurites
- α-syn:
-
α-synuclein
- PD:
-
Parkinson's disease
- SNpc:
-
Substantia nigra pars compacta
- GWAS:
-
Genome-wide association study
- DLB:
-
Dementia with Lewy bodies
- MSA:
-
Multiple system atrophy
- LPS:
-
Lipopolysaccharide
- CNS:
-
Central nervous system
- NAC:
-
Nonamyloid component of plaques
- TNF-α:
-
Tumor necrosis factor α
- IL-1β:
-
Interleukin 1
- IL-6:
-
Interleukin 6
- MHC:
-
Major histocompatibility complex
- IL-4:
-
Interleukin 4
- IL-10:
-
Interleukin 10
- ARG-1:
-
Arginase-1
- mGluR5:
-
Metabotropic glutamate receptor 5
- GPCR:
-
G-protein coupled receptor
- KLK6:
-
Kallikrein
- EVs:
-
Extracellular vesicles
- PTMs:
-
Posttranslational modifications
- L-DOPA:
-
Levodopa
- LAG3:
-
Lymphocyte activation gene 3
- APLP1:
-
Amyloid precursor-like protein 1
- NF-κB:
-
Nuclear factor kappa-B
- TLR4:
-
Toll-like receptor 4
- CXCL1:
-
Chemokine ligand 1
- iNOS:
-
Inducible nitric oxide synthase
- COX-2:
-
Cyclooxygenase-2
- TLR2:
-
Toll-like receptor 2
- Myd88:
-
Myeloid differentiation factor 88
- NLRP3:
-
NLR family pyrin domain containing 3
- ROS:
-
Reactive oxygen species
- ASC:
-
Adaptor protein apoptosis-associated speck-like protein containing a CARD
- H2O2 :
-
Hydrogen peroxide
- BBB:
-
Blood‒brain barrier
- GF:
-
Germ-free
- LCMV:
-
Lymphocytic choriomeningitis virus
- SCFAs:
-
Short-chain fatty acids
- NMDAR:
-
N-methyl-D-aspartate receptor
- AMPAP:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- TSPO:
-
Translocator protein
- GLP1:
-
Glucagon-like peptide 1
- T2DM:
-
Type 2 diabetes mellitus
- iMGLs:
-
IPSC-derived microglia-like cells
- MDMi:
-
Monocyte-derived microglia-like cells
References
Reich SG, Savitt JM. Parkinson’s Disease. Med Clin North Am. 2019;103:337–50.
Obeso JA, Rodriguez-Oroz M, Marin C, Alonso F, Zamarbide I, Lanciego JL, Rodriguez-Diaz M. The origin of motor fluctuations in Parkinson’s disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 2004;62:S17-30.
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castano-Diez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–109.
Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–72.
Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397:2284–303.
Shadrina MI, Slominsky PA. Genetic Architecture of Parkinson’s Disease. Biochemistry (Mosc). 2023;88:417–33.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–102.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12.
Henderson MX, Trojanowski JQ, Lee VM. alpha-Synuclein pathology in Parkinson’s disease and related alpha-synucleinopathies. Neurosci Lett. 2019;709:134316.
Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018;109:249–57.
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–8.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–73.
Gai WP, Power JH, Blumbergs PC, Blessing WW. Multiple-system atrophy: a new alpha-synuclein disease? Lancet. 1998;352:547–8.
Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature. 2018;557:558–63.
Laferriere F, Claverol S, Bezard E, De Giorgi F, Ichas F. Similar neuronal imprint and no cross-seeded fibrils in alpha-synuclein aggregates from MSA and Parkinson’s disease. NPJ Parkinsons Dis. 2022;8:10.
Kaur C, Rathnasamy G, Ling EA. Biology of Microglia in the Developing Brain. J Neuropathol Exp Neurol. 2017;76:736–53.
Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–83.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97.
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65.
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–98.
Dutta D, Jana M, Majumder M, Mondal S, Roy A, Pahan K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat Commun. 2021;12:5382.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–42.
Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Koshimori Y, Ko JH, Mizrahi R, Rusjan P, Mabrouk R, Jacobs MF, Christopher L, Hamani C, Lang AE, Wilson AA, et al. Imaging Striatal Microglial Activation in Patients with Parkinson’s Disease. PLoS One. 2015;10:e0138721.
Koller EJ, Brooks MM, Golde TE, Giasson BI, Chakrabarty P. Inflammatory pre-conditioning restricts the seeded induction of alpha-synuclein pathology in wild type mice. Mol Neurodegener. 2017;12:1.
Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56:1215–23.
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–42.
Maekawa T, Sasaoka T, Azuma S, Ichikawa T, Melrose HL, Farrer MJ, Obata F. Leucine-rich repeat kinase 2 (LRRK2) regulates alpha-synuclein clearance in microglia. BMC Neurosci. 2016;17:77.
Nash Y, Schmukler E, Trudler D, Pinkas-Kramarski R, Frenkel D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. 2017;143:584–94.
Iseki T, Imai Y, Hattori N. Is Glial Dysfunction the Key Pathogenesis of LRRK2-Linked Parkinson’s Disease? Biomolecules. 2023;13:178.
Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK. alpha-synuclein aggregation and its modulation. Int J Biol Macromol. 2017;100:37–54.
Kim CY, Alcalay RN. Genetic Forms of Parkinson’s Disease. Semin Neurol. 2017;37:135–46.
Durante V, de Iure A, Loffredo V, Vaikath N, De Risi M, Paciotti S, Quiroga-Varela A, Chiasserini D, Mellone M, Mazzocchetti P, et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain. 2019;142:1365–85.
Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun JC, Schneggenburger R, Knott GW, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–69.
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7.
Akintade DD, Chaudhuri B. The effect of copy number on alpha-synuclein’s toxicity and its protective role in Bax-induced apoptosis, in yeast. Biosci Rep. 2020;40:BSR20201912.
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–9.
Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–54.
Eschbach J, Danzer KM. alpha-Synuclein in Parkinson’s disease: pathogenic function and translation into animal models. Neurodegener Dis. 2014;14:1–17.
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–6.
Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J Am Chem Soc. 2008;130:12856–7.
Vasili E, Dominguez-Meijide A, Outeiro TF. Spreading of alpha-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front Mol Neurosci. 2019;12:107.
Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94.
Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13.
Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–20.
Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7.
El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 1998;440:67–70.
Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry. 2001;40:11604–13.
Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–902.
Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–6.
Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, et al. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–25.
Sode K, Ochiai S, Kobayashi N, Usuzaka E. Effect of reparation of repeat sequences in the human alpha-synuclein on fibrillation ability. Int J Biol Sci. 2006;3:1–7.
Han H, Weinreb PH, Lansbury PT Jr. The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol. 1995;2:163–9.
Iwai A, Yoshimoto M, Masliah E, Saitoh T. Non-A beta component of Alzheimer’s disease amyloid (NAC) is amyloidogenic. Biochemistry. 1995;34:10139–45.
Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–9.
Nielsen MS, Vorum H, Lindersson E, Jensen PH. Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J Biol Chem. 2001;276:22680–4.
Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, Lee VM. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep. 2016;16:3373–87.
Cheng F, Vivacqua G, Yu S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. 2011;42:242–8.
Kim TD, Paik SR, Yang CH. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry. 2002;41:13782–90.
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–4.
Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–9.
Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, et al. Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc Natl Acad Sci U S A. 2015;112:E1994-2003.
Sanchez SE, Whiten DR, Meisl G, Ruggeri FS, Hidari E, Klenerman D. Alpha Synuclein only Forms Fibrils In Vitro when Larger than its Critical Size of 70 Monomers. ChemBioChem. 2021;22:2867–71.
Tofaris GK. Initiation and progression of alpha-synuclein pathology in Parkinson’s disease. Cell Mol Life Sci. 2022;79:210.
Li D, Liu C. Conformational strains of pathogenic amyloid proteins in neurodegenerative diseases. Nat Rev Neurosci. 2022;23:523–34.
Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53:1181–94.
Joers V, Tansey MG, Mulas G, Carta AR. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol. 2017;155:57–75.
Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, Amit I, Audinat E, Bechmann I, Bennett M, et al. Microglia states and nomenclature: A field at its crossroads. Neuron. 2022;110:3458–83.
Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. 2020;329:113310.
Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, Kiechle M, Feiler MS, Freischmidt A, Brenner D, et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016;131:379–91.
Rabenstein M, Besong Agbo D, Wolf E, Dams J, Nicolai M, Roeder A, Bacher M, Dodel RC, Noelker C. Effect of naturally occurring alpha-synuclein-antibodies on toxic alpha-synuclein-fragments. Neurosci Lett. 2019;704:181–8.
Bido S, Muggeo S, Massimino L, Marzi MJ, Giannelli SG, Melacini E, Nannoni M, Gambare D, Bellini E, Ordazzo G, et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat Commun. 2021;12:6237.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42.
Li N, Stewart T, Sheng L, Shi M, Cilento EM, Wu Y, Hong JS, Zhang J. Immunoregulation of microglial polarization: an unrecognized physiological function of alpha-synuclein. J Neuroinflammation. 2020;17:272.
Balusu S, Praschberger R, Lauwers E, De Strooper B, Verstreken P. Neurodegeneration cell per cell. Neuron. 2023;111:767–86.
Smajic S, Prada-Medina CA, Landoulsi Z, Ghelfi J, Delcambre S, Dietrich C, Jarazo J, Henck J, Balachandran S, Pachchek S, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145:964–78.
Reynolds RH, Botia J, Nalls MA. International Parkinson’s Disease Genomics C, System Genomics of Parkinson’s D, Hardy J, Gagliano Taliun SA, Ryten M: Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinsons Dis. 2019;5:6.
Ma SX, Lim SB. Single-Cell RNA Sequencing in Parkinson’s Disease. Biomedicines. 2021;9:368.
Bachiller S, Jimenez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, Boza-Serrano A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front Cell Neurosci. 2018;12:488.
De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang HY, Liu QR, Shen H, Xi ZX, et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron. 2017;95(341–356):e346.
Wang L, Gong X, Liu Y, Du T, Zhang Z, Zhang T, Wang X. CD200 maintains the region-specific phenotype of microglia in the midbrain and its role in Parkinson’s disease. Glia. 2020;68:1874–90.
Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–16.
Badanjak K, Fixemer S, Smajic S, Skupin A, Grunewald A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int J Mol Sci. 2021;22(9):4676.
Liu WW, Wei SZ, Huang GD, Liu LB, Gu C, Shen Y, Wang XH, Xia ST, Xie AM, Hu LF, et al. BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J. 2020;34:6570–81.
Ghiglieri V, Calabrese V, Calabresi P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front Neurol. 2018;9:295.
Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci Lett. 2000;287:65–7.
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17:1945–7.
Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–24.
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–7.
Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M, Miura E, Tanaka N, Tamai K, Furukawa K, et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PLoS One. 2011;6:e29460.
Peng C, Trojanowski JQ, Lee VM. Protein transmission in neurodegenerative disease. Nat Rev Neurol. 2020;16:199–212.
Li Z, Wang X, Wang X, Yi X, Wong YK, Wu J, Xie F, Hu D, Wang Q, Wang J, Zhong T. Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl Neurodegener. 2023;12:43.
Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, Kou L, Yin S, Liu L, Huang J, et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019;10:174.
Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, Cui M, Tieu K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain. 2020;143:1476–97.
Fan RZ, Guo M, Luo S, Cui M, Tieu K. Exosome release and neuropathology induced by alpha-synuclein: new insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol Commun. 2019;7:184.
George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, Pierce S, Burmeister AR, Madaj Z, Steiner JA, et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener. 2019;14:34.
Gordon S. Phagocytosis: An Immunobiologic Process. Immunity. 2016;44:463–75.
Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112.
Tremblay ME, Cookson MR, Civiero L. Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener. 2019;14:16.
Austin SA, Floden AM, Murphy EJ, Combs CK. Alpha-synuclein expression modulates microglial activation phenotype. J Neurosci. 2006;26:10558–63.
Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol. 2011;179:954–63.
Depboylu C, Stricker S, Ghobril JP, Oertel WH, Priller J, Hoglinger GU. Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Exp Neurol. 2012;238:183–91.
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–60.
Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019;568:187–92.
Choi YR, Kang SJ, Kim JM, Lee SJ, Jou I, Joe EH, Park SM. FcgammaRIIB mediates the inhibitory effect of aggregated alpha-synuclein on microglial phagocytosis. Neurobiol Dis. 2015;83:90–9.
Park JY, Kim KS, Lee SB, Ryu JS, Chung KC, Choo YK, Jou I, Kim J, Park SM. On the mechanism of internalization of alpha-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J Neurochem. 2009;110:400–11.
Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, Miller DS, Chen B, Zhang W, McGeer PL, et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia. 2007;55:1178–88.
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.
Filippini A, Gennarelli M, Russo I. alpha-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System? Cell Mol Neurobiol. 2019;39:161–8.
Liu J, Zhou Y, Wang Y, Fong H, Murray TM, Zhang J. Identification of proteins involved in microglial endocytosis of alpha-synuclein. J Proteome Res. 2007;6:3614–27.
Ahn KJ, Paik SR, Chung KC, Kim J. Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J Neurochem. 2006;97:265–79.
Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–49.
Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation. 2012;9:261.
Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener. 2016;11:73.
Inoshita T, Arano T, Hosaka Y, Meng H, Umezaki Y, Kosugi S, Morimoto T, Koike M, Chang HY, Imai Y, Hattori N. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum Mol Genet. 2017;26:2933–48.
Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, Zhang B, Yue Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386.
Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–90.
Daniele SG, Beraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. 2015;8:ra45.
Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, et al. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med. 2004;199:81–90.
Cui B, Guo X, You Y, Fu R. Farrerol attenuates MPP(+) -induced inflammatory response by TLR4 signaling in a microglia cell line. Phytother Res. 2019;33:1134–41.
Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, Zhang B, Yue Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386.
Dutta D, Jana M, Majumder M, Mondal S, Roy A, Pahan K. Selective targeting of the TLR2/MyD88/NF-kappaB pathway reduces alpha-synuclein spreading in vitro and in vivo. Nat Commun. 2021;12:5382.
Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, Grandbarbe L, Heuschling P, Heurtaux T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS One. 2016;11:e0162717.
Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe DB, O’Neill LA, et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. 2018;10:eaah4066.
Grotemeyer A, Fischer JF, Koprich JB, Brotchie JM, Blum R, Volkmann J, Ip CW. Inflammasome inhibition protects dopaminergic neurons from alpha-synuclein pathology in a model of progressive Parkinson’s disease. J Neuroinflammation. 2023;20:79.
Wu AG, Zhou XG, Qiao G, Yu L, Tang Y, Yan L, Qiu WQ, Pan R, Yu CL, Law BY, et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev. 2021;65:101202.
Ou Z, Zhou Y, Wang L, Xue L, Zheng J, Chen L, Tong Q. NLRP3 Inflammasome Inhibition Prevents alpha-Synuclein Pathology by Relieving Autophagy Dysfunction in Chronic MPTP-Treated NLRP3 Knockout Mice. Mol Neurobiol. 2021;58:1303–11.
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One. 2013;8:e55375.
Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L, Sun Y, Wu J, Zhou Q, Huang J, et al. Targeting Microglial alpha-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front Immunol. 2021;12:719807.
Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, Saminathan H, Langley M, Charli A, Samidurai M, et al. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. 2019;216:1411–30.
Scheiblich H, Bousset L, Schwartz S, Griep A, Latz E, Melki R, Heneka MT. Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 Ligation by Distinct alpha-Synuclein Assemblies. J Immunol. 2021;207:2143–54.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203.
Pike AF, Varanita T, Herrebout MAC, Plug BC, Kole J, Musters RJP, Teunissen CE, Hoozemans JJM, Bubacco L, Veerhuis R. alpha-Synuclein evokes NLRP3 inflammasome-mediated IL-1beta secretion from primary human microglia. Glia. 2021;69:1413–28.
Erdei A, Lukácsi S, Mácsik-Valent B, Nagy-Baló Z, Kurucz I, Bajtay Z. Non-identical twins: Different faces of CR3 and CR4 in myeloid and lymphoid cells of mice and men. Semin Cell Dev Biol. 2019;85:110–21.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–6.
Hou L, Bao X, Zang C, Yang H, Sun F, Che Y, Wu X, Li S, Zhang D, Wang Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018;14:600–8.
Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, Guo M, Wilson B, Hong JS, Zhang J. alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci U S A. 2015;112:E1926-1935.
Butler CA, Popescu AS, Kitchener EJA, Allendorf DH, Puigdellivol M, Brown GC. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J Neurochem. 2021;158:621–39.
Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J, Winkler J, Xiang W, Schlachetzki JCM. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun. 2016;479:881–6.
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7.
Sanchez K, Maguire-Zeiss K. MMP13 Expression Is Increased Following Mutant alpha-Synuclein Exposure and Promotes Inflammatory Responses in Microglia. Front Neurosci. 2020;14:585544.
Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, Fu Y, Halliday GM. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133:303–19.
La Vitola P, Balducci C, Cerovic M, Santamaria G, Brandi E, Grandi F, Caldinelli L, Colombo L, Morgese MG, Trabace L, et al. Alpha-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain Behav Immun. 2018;69:591–602.
Kageyama S, Gudmundsson SR, Sou YS, Ichimura Y, Tamura N, Kazuno S, Ueno T, Miura Y, Noshiro D, Abe M, et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat Commun. 2021;12:16.
Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Pina-Crespo JC, Zhang M, et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron. 2018;97(1023–1031):e1027.
Li XX, Zhang F. Targeting TREM2 for Parkinson’s Disease: Where to Go? Front Immunol. 2021;12:795036.
Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, Chen Z, Li K, Liu J. Convergent Genetic and Expression Datasets Highlight TREM2 in Parkinson’s Disease Susceptibility. Mol Neurobiol. 2016;53:4931–8.
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19.
Ren M, Guo Y, Wei X, Yan S, Qin Y, Zhang X, Jiang F, Lou H. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp Neurol. 2018;302:205–13.
Li C, Zhao B, Lin C, Gong Z, An X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-kappaB signaling. Cell Biol Int. 2019;43:360–72.
Guo Y, Wei X, Yan H, Qin Y, Yan S, Liu J, Zhao Y, Jiang F, Lou H. TREM2 deficiency aggravates alpha-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 2019;33:12164–74.
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–56.
Peng G, Qiu J, Liu H, Zhou M, Huang S, Guo W, Lin Y, Chen X, Li Z, Li G, et al. Analysis of Cerebrospinal Fluid Soluble TREM2 and Polymorphisms in Sporadic Parkinson’s Disease in a Chinese Population. J Mol Neurosci. 2020;70:294–301.
Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science. 2017;358:1440–3.
Killinger BA, Melki R, Brundin P, Kordower JH. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: let’s talk about the lipids in the room. NPJ Parkinsons Dis. 2019;5:23.
Dos-Santos-Pereira M, Acuna L, Hamadat S, Rocca J, Gonzalez-Lizarraga F, Chehin R, Sepulveda-Diaz J, Del-Bel E, Raisman-Vozari R, Michel PP. Microglial glutamate release evoked by alpha-synuclein aggregates is prevented by dopamine. Glia. 2018;66:2353–65.
Gonzalez-Lozano MA, Wortel J, van der Loo RJ, van Weering JRT, Smit AB, Li KW. Reduced mGluR5 Activity Modulates Mitochondrial Function. Cells. 2021;10:1375.
Zhang YN, Fan JK, Gu L, Yang HM, Zhan SQ, Zhang H. Metabotropic glutamate receptor 5 inhibits alpha-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J Neuroinflammation. 2021;18:23.
Bras IC, Outeiro TF. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells. 2021;10:375.
Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Thery C, Raposo G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci. 2013;126:5553–65.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51.
Pandey N, Schmidt RE, Galvin JE. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp Neurol. 2006;197:515–20.
Xia Y, Zhang G, Kou L, Yin S, Han C, Hu J, Wan F, Sun Y, Wu J, Li Y, et al. Reactive microglia enhance the transmission of exosomal alpha-synuclein via toll-like receptor 2. Brain. 2021;144:2024–37.
Bae EJ, Choi M, Kim JT, Kim DK, Jung MK, Kim C, Kim TK, Lee JS, Jung BC, Shin SJ, et al. TNF-alpha promotes alpha-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp Mol Med. 2022;54:788–800.
Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, Eikens F, Odainic A, Spitzer J, Griep A, et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. 2021;184(5089–5106):e5021.
Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353:aah3374.
Mao X, Gu H, Kim D, Kimura Y, Wang N, Xu E, Wang H, Chen C, Zhang S, Jia C, et al. Aplp1 and the Aplp1-Lag3 Complex facilitates transmission of pathologic α-synuclein. bioRxiv. 2021;2001:442157.
Zhang S, Liu YQ, Jia C, Lim YJ, Feng G, Xu E, Long H, Kimura Y, Tao Y, Zhao C, et al. Mechanistic basis for receptor-mediated pathological alpha-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc Natl Acad Sci U S A. 2021;118:e2011196118.
Angelopoulou E, Paudel YN, Villa C, Shaikh MF, Piperi C. Lymphocyte-Activation Gene 3 (LAG3) Protein as a Possible Therapeutic Target for Parkinson’s Disease: Molecular Mechanisms Connecting Neuroinflammation to alpha-Synuclein Spreading Pathology. Biology (Basel). 2020;9::86.
Zhang Q, Duan Q, Gao Y, He P, Huang R, Huang H, Li Y, Ma G, Zhang Y, Nie K, Wang L. Cerebral Microvascular Injury Induced by Lag3-Dependent alpha-Synuclein Fibril Endocytosis Exacerbates Cognitive Impairment in a Mouse Model of alpha-Synucleinopathies. Adv Sci (Weinh). 2023;10:e2301903.
Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J Biol Chem. 2001;276:27441–8.
Emmenegger M, De Cecco E, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, Tantardini E, de Rossi P, Bacioglu M, Langston RG, et al. LAG3 is not expressed in human and murine neurons and does not modulate alpha-synucleinopathies. EMBO Mol Med. 2021;13:e14745.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell. 2016;167(1469–1480):e1412.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell. 2016;167:1469-1480.e1412.
Erny D, de HraběAngelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–77.
Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:8670.
Xu Y, Xie M, Xue J, Xiang L, Li Y, Xiao J, Xiao G, Wang HL. EGCG ameliorates neuronal and behavioral defects by remodeling gut microbiota and TotM expression in Drosophila models of Parkinson’s disease. FASEB J. 2020;34:5931–50.
Parker-Character J, Hager DR, Call TB, Pickup ZS, Turnbull SA, Marshman EM, Korch SB, Chaston JM, Call GB. An altered microbiome in a Parkinson’s disease model Drosophila melanogaster has a negative effect on development. Sci Rep. 2021;11:23635.
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–20.
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron. 2019;103(627–641):e627.
Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, Thiolat ML, Tasselli M, Aubert P, Kruse N, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. 2020;143:1462–75.
Resnikoff H, Metzger JM, Lopez M, Bondarenko V, Mejia A, Simmons HA, Emborg ME. Colonic inflammation affects myenteric alpha-synuclein in nonhuman primates. J Inflamm Res. 2019;12:113–26.
Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, Mutlu E, Shannon KM. Colonic bacterial composition in Parkinson’s disease. Mov Disord. 2015;30:1351–60.
Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord. 2015;30:350–8.
Tan AH, Hor JW, Chong CW, Lim SY. Probiotics for Parkinson’s disease: Current evidence and future directions. JGH Open. 2021;5:414–9.
Van Den Berge N, Ferreira N, Mikkelsen TW, Alstrup AKO, Tamguney G, Karlsson P, Terkelsen AJ, Nyengaard JR, Jensen PH, Borghammer P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain. 2021;144:1853–68.
Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA, Gradinaru V. Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. 2020;23:327–36.
Singh Y, El-Hadidi M, Admard J, Wassouf Z, Schulze-Hentrich JM, Kohlhofer U, Quintanilla-Martinez L, Huson D, Riess O, Casadei N. Enriched Environmental Conditions Modify the Gut Microbiome Composition and Fecal Markers of Inflammation in Parkinson’s Disease. Front Neurosci. 2019;13:1032.
Dong S, Sun M, He C, Cheng H. Brain-gut-microbiota axis in Parkinson’s disease: A historical review and future perspective. Brain Res Bull. 2022;183:84–93.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163:1064–78.
Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22:586–97.
Subbarayan MS, Hudson C, Moss LD, Nash KR, Bickford PC. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an alpha-synuclein rat model of Parkinson’s disease. J Neuroinflammation. 2020;17:242.
Gate D, Tapp E, Leventhal O, Shahid M, Nonninger TJ, Yang AC, Strempfl K, Unger MS, Fehlmann T, Oh H, et al. CD4(+) T cells contribute to neurodegeneration in Lewy body dementia. Science. 2021;374:868–74.
Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, Korecka A, Bakocevic N, Ng LG, Kundu P, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158.
Michel L, Prat A. One more role for the gut: microbiota and blood brain barrier. Ann Transl Med. 2016;4:15.
Borghammer P. How does parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov Disord. 2018;33:48–57.
Svensson E, Horvath-Puho E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sorensen HT. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78:522–9.
Cersosimo MG, Raina GB, Pecci C, Pellene A, Calandra CR, Gutierrez C, Micheli FE, Benarroch EE. Gastrointestinal manifestations in Parkinson’s disease: prevalence and occurrence before motor symptoms. J Neurol. 2013;260:1332–8.
Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener. 2018;13:21.
McFarthing K, Simuni T. Clinical Trial Highlights: Targetting Alpha-Synuclein. J Parkinsons Dis. 2019;9:5–16.
Price DL, Koike MA, Khan A, Wrasidlo W, Rockenstein E, Masliah E, Bonhaus D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci Rep. 2018;8:16165.
Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science. 2017;357:891–8.
Gronich N, Abernethy DR, Auriel E, Lavi I, Rennert G, Saliba W. beta2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov Disord. 2018;33:1465–71.
Walczak-Nowicka LJ, Herbet M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. Int J Mol Sci. 2021;22:9290.
Hauser RA, Sutherland D, Madrid JA, Rol MA, Frucht S, Isaacson S, Pagan F, Maddux BN, Li G, Tse W, et al. Targeting neurons in the gastrointestinal tract to treat Parkinson’s disease. Clin Park Relat Disord. 2019;1:2–7.
Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, Heller GT, Meisl G, Chen SW, Kumita JR, et al. Multistep Inhibition of alpha-Synuclein Aggregation and Toxicity in Vitro and in Vivo by Trodusquemine. ACS Chem Biol. 2018;13:2308–19.
Grolez G, Moreau C, Sablonniere B, Garcon G, Devedjian JC, Meguig S, Gele P, Delmaire C, Bordet R, Defebvre L, et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 2015;15:74.
Martin-Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C, Patel MC, Spino M, Connelly J, Tricta F, et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci Rep. 2017;7:1398.
Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, Wilmarth BM, Howard H, Dunn C, Carlson A, et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J Parkinsons Dis. 2016;6:503–17.
Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, Yusuf N, Starr NJ, Arellano J, Howard HH, et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol Res Perspect. 2019;7:e00470.
Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, Lafontant DE, Matthews H, Wyse RK, Brundin P, et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021;78:312–20.
Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, Hallqvist J, Toffoli M, Streeter A, Hosking J, et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized Noncontrolled Trial. JAMA Neurol. 2020;77:427–34.
Hur EM, Lee BD. LRRK2 at the Crossroad of Aging and Parkinson’s Disease. Genes (Basel). 2021;12:505.
Rascol O, Fabbri M, Poewe W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021;20:1048–56.
Ueda J, Uemura N, Sawamura M, Taguchi T, Ikuno M, Kaji S, Taruno Y, Matsuzawa S, Yamakado H, Takahashi R. Perampanel Inhibits alpha-Synuclein Transmission in Parkinson’s Disease Models. Mov Disord. 2021;36:1554–64.
Sampognaro PJ, Arya S, Knudsen GM, Gunderson EL, Sandoval-Perez A, Hodul M, Bowles K, Craik CS, Jacobson MP, Kao AW. Mutations in alpha-synuclein, TDP-43 and tau prolong protein half-life through diminished degradation by lysosomal proteases. Mol Neurodegener. 2023;18:29.
Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17.
Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J. Parkinson disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol. 2020;16:303–18.
Heidari A, Yazdanpanah N, Rezaei N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J Neuroinflammation. 2022;19:135.
Kouli A, Camacho M, Allinson K, Williams-Gray CH. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun. 2020;8:211.
Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, Fields JA, Masliah D, Iba M, Lee HJ, et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol Neurodegener. 2018;13:43.
Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu JW. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019;26:213–28.
Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33:9592–600.
Williams GP, Schonhoff AM, Jurkuvenaite A, Thome AD, Standaert DG, Harms AS. Targeting of the class II transactivator attenuates inflammation and neurodegeneration in an alpha-synuclein model of Parkinson’s disease. J Neuroinflammation. 2018;15:244.
Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009381.
Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, Liong C, McMurtrey C, Hildebrand WH, Mao X, et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature. 2017;546:656–61.
Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, Shetty BLD, Lu Y, Estes KA, Standaert DG, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinsons Dis. 2017;3:10.
Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, Wang A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol. 2018;75:939–46.
McCoy MK, Ruhn KA, Blesch A, Tansey MG. TNF: a key neuroinflammatory mediator of neurotoxicity and neurodegeneration in models of Parkinson’s disease. Adv Exp Med Biol. 2011;691:539–40.
Wenker SD, Farias MI, Gradaschi V, Garcia C, Beauquis J, Leal MC, Ferrari C, Zeng X, Pitossi FJ. Microglia-secreted TNF-alpha affects differentiation efficiency and viability of pluripotent stem cell-derived human dopaminergic precursors. PLoS ONE. 2023;18:e0263021.
Barnum CJ, Chen X, Chung J, Chang J, Williams M, Grigoryan N, Tesi RJ, Tansey MG. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro(R)1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J Parkinsons Dis. 2014;4:349–60.
McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci. 2006;26:9365–75.
Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta. 2007;1771:1031–45.
Machado MMF, Bassani TB, Coppola-Segovia V, Moura ELR, Zanata SM, Andreatini R, Vital M. PPAR-gamma agonist pioglitazone reduces microglial proliferation and NF-kappaB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol Rep. 2019;71:556–64.
Ji J, Xue TF, Guo XD, Yang J, Guo RB, Wang J, Huang JY, Zhao XJ, Sun XL. Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell. 2018;17:e12774.
Basurco L, Abellanas MA, Ayerra L, Conde E, Vinueza-Gavilanes R, Luquin E, Vales A, Vilas A, Martin-Uriz PS, Tamayo I, et al. Microglia and astrocyte activation is region-dependent in the alpha-synuclein mouse model of Parkinson’s disease. Glia. 2023;71:571–87.
Javanmehr N, Saleki K, Alijanizadeh P, Rezaei N. Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J Neuroinflammation. 2022;19:273.
Tan AH, Lim SY, Lang AE. The microbiome-gut-brain axis in Parkinson disease - from basic research to the clinic. Nat Rev Neurol. 2022;18:476–95.
Walter J, Armet AM, Finlay BB, Shanahan F. Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell. 2020;180:221–32.
Laukens D, Brinkman BM, Raes J, De Vos M, Vandenabeele P. Heterogeneity of the gut microbiome in mice: guidelines for optimizing experimental design. FEMS Microbiol Rev. 2016;40:117–32.
Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, Jaenisch R. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 2016;22:1358–67.
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 2017;94(278–293):e279.
Drager NM, Sattler SM, Huang CT, Teter OM, Leng K, Hashemi SH, Hong J, Aviles G, Clelland CD, Zhan L, et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat Neurosci. 2022;25:1149–62.
Ryan KJ, White CC, Patel K, Xu J, Olah M, Replogle JM, Frangieh M, Cimpean M, Winn P, McHenry A, et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med. 2017;9:eaai7635.
Smit T, Ormel PR, Sluijs JA, Hulshof LA, Middeldorp J, de Witte LD, Hol EM, Donega V. Transcriptomic and functional analysis of Abeta(1–42) oligomer-stimulated human monocyte-derived microglia-like cells. Brain Behav Immun. 2022;100:219–30.
Sargeant TJ, Fourrier C. Human monocyte-derived microglia-like cell models: A review of the benefits, limitations and recommendations. Brain Behav Immun. 2023;107:98–109.
Acknowledgements
We would like to thank AJE (https://www.aje.cn) for English language editing.
Funding
This work was supported by a special launch fund from Soochow University, the Science and Technology Program of Suzhou (2022SS02), and the Natural Science Foundation of China (NSFC) (82171414).
Author information
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miao, Y., Meng, H. The involvement of α-synucleinopathy in the disruption of microglial homeostasis contributes to the pathogenesis of Parkinson’s disease. Cell Commun Signal 22, 31 (2024). https://doi.org/10.1186/s12964-023-01402-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01402-y