
Abstract
Motor neuron-extrinsic mechanisms have been shown to participate in the pathogenesis of ALS-SOD1, one familial form of amyotrophic lateral sclerosis (ALS). It remains unclear whether such mechanisms contribute to other familial forms, such as TDP-43 and FUS-associated ALS. Here, we characterize a single-copy mouse model of ALS-FUS that conditionally expresses a disease-relevant truncating FUS mutant from the endogenous murine Fus gene. We show that these mice, but not mice heterozygous for a Fus null allele, develop similar pathology as ALS-FUS patients and a mild motor neuron phenotype. Most importantly, CRE-mediated rescue of the Fus mutation within motor neurons prevented degeneration of motor neuron cell bodies, but only delayed appearance of motor symptoms. Indeed, we observed downregulation of multiple myelin-related genes, and increased numbers of oligodendrocytes in the spinal cord supporting their contribution to behavioral deficits. In all, we show that mutant FUS triggers toxic events in both motor neurons and neighboring cells to elicit motor neuron disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disease clinically characterized by a preferential loss of upper and lower motor neurons, resulting in progressive weakness of skeletal muscles, atrophy, paralysis and death due to respiratory failure [42]. Recent advances in human genetics identified mutations in almost 40 genes associated with ALS, and these familial cases together account for about 10% of ALS cases [62, 79].
Heterozygous mutations in the FUS (Fused in sarcoma) gene, encoding the RNA-binding protein FUS, are the major cause of juvenile forms of ALS [14, 34, 45, 84]. In ALS-FUS patients, the FUS protein accumulates in the cytoplasm in a dimethylated form [19, 75]. FUS is functionally related to TDP-43 (TAR DNA-binding protein 43), the major protein found in ubiquitin-positive inclusions of ALS patients [59], and, like TDP-43, FUS is a nuclear protein involved in multiple steps of gene expression, including mRNA transcription, splicing, transport and translation [49, 56]. In neurons, FUS is found in axons [69], dendrites and at excitatory synapses [24] as well as in RNA transporting granules [4, 10]. Several recent studies demonstrated that the complete loss of FUS protein, either in adult mice or perinatally, was not sufficient to trigger motor neuron degeneration [44, 68, 72, 82]. Contrasting with this, overexpression of FUS, either wild type or mutant, is able to trigger motor neuron degeneration, suggesting that the mutant protein gains a toxic function leading to aggressive neurodegeneration [55, 64, 71–73]. Importantly, the majority of FUS mutations are missense changes clustered in the C-terminal nuclear localization sequence (NLS) or frameshift and stop mutations that truncate the NLS [16]. This impairs the binding of FUS to the nuclear import receptor Transportin, and thus interferes with import of FUS in the nucleus, resulting in the cytoplasmic accumulation of FUS [20]. Consistent with a critical role of nuclear import of FUS, the mutations leading to the most severe forms of ALS are truncating or frameshift mutations in FUS causing the complete deletion of the NLS [3, 11, 16, 87, 88, 96]. These aggressive FUS mutations lead to extensive FUS redistribution to the cytoplasm and age of onset was correlated with the degree of cytosolic mislocalization of FUS [20]. Together, these findings strongly suggest that neurodegeneration is directly related to the altered subcellular localization of FUS.
To study the mechanisms of ALS-FUS in a physiologically relevant manner, we recently generated a conditional knock-in mouse model (Fus ΔNLS mice) in which the NLS of FUS is deleted [68]. We have shown that FUS is completely mislocalized to the cytoplasm in mice homozygous for the Fus ΔNLS mutation [68], leading to motor neuron degeneration in neonates. However, homozygous knock-in mice were lethal at birth, thus precluding the analysis of aging mice homozygous for the Fus mutation. Here, we studied heterozygous Fus ΔNLS/+ mice, as a mouse model carrying a genetic defect that mimics the genetic situation in human ALS-FUS patients. Analysis of these mutant mice revealed progressive motor neuron degeneration and neuropathological changes that faithfully model several key aspects of ALS-FUS, including ubiquitin pathology and cytoplasmic accumulation of dimethylated FUS. Motor neuron death appeared cell autonomous, yet the motor phenotype of these mice was only delayed when the mutation was rescued solely within motor neurons, and axonal defects were still present. Further, we identified alterations in genes involved in myelin structure and function, and showed altered abundance of oligodendrocytes in the spinal cord supporting the contribution of these cells to the disruption of axonal integrity and the motor phenotype. Thus, while expression of mutant FUS within motor neurons is necessary for cell death, motor symptoms are caused by the concerted action of mutant FUS in motor neurons and other cell types, including oligodendrocytes.
Materials and methods
Animal housing and genotyping
Wild type and heterozygous Fus ∆NLS/+ mice and heterozygous Chat-CRE mice, generated as described previously [68], were bred and housed in the central animal facility of the Faculty of Medicine of Strasbourg, with a regular 12-h light and dark cycle (light on at 7:00 am) under constant conditions (21 ± 1 °C; 60% humidity). Standard laboratory rodent food and water were available ad libitum throughout all experiments. Wild type and heterozygous Fus +/− mice, generated as described previously [68], were bred and housed in the animal facility of the Max Planck Institute for Molecular Biomedicine, with a regular 12-h light and dark cycle. Mice were genotyped by PCR of genomic DNA from tail biopsies as described previously [68].
10- to 22-month-old male littermates of each genotype (Fus +/+, Fus ΔNLS/+ and Fus +/−) on a pure genetic background (C57/Bl6) were subjected to behavioral tests and molecular analyses. Behavioral test were done during the light phase of their light/dark cycle.
Compliance with ethical standards
These protocols were approved by the local ethical committees (Cremeas in Strasbourg, LANUV NRW in Muenster), under reference number AL/27/34/02/13; 84-02.04.2011.A100 and 84-02.04.2016.A166.
Subcellular fractionation and western blotting
Nuclear and cytoplasmic fractions were prepared from fresh spinal cord tissue using the NE-PER® Nuclear and Cytoplasmic Extraction reagents (Thermo Scientific) according to the manufacturer. Protein concentration was quantitated using the BCA protein assay kit (Pierce). Equal amounts of protein (10 µg for nuclear and 30 µg for cytoplasmic fraction) were loaded in each well of a gradient 4–20% SDS-PAGE gel, separated and transferred onto a 0.45 µm nitrocellulose membrane (BioRad) using a semi-dry Transblot Turbo transfer system (BioRad). Membranes were saturated with 10% non-fat milk in PBS and were then probed with the following primary antibodies: goat anti-FUS against the N-terminal part of protein (ProteinTech 11570; 1:1000), rabbit anti-FUS against the C-terminal part of protein (Bethyl A300-294A, 1:10000) and rat anti-di-methylated FUS (ADMA, 1:1000) [19, 75] all diluted in 3% non-fat milk in PBS. Blots were incubated with horseradish peroxidase (HRP)-labeled secondary antibodies anti-goat (Sigma A5420), anti-rabbit (P.A.R.I.S. BI2407), anti-sheep (Chemicon AP147) and anti-rat (Rockland 612-1102), all secondary antibodies were diluted 1:5000 in PBS. Antibodies rabbit anti-HDAC1 (Bethyl A300-713A, 1:1000) was used as loading control for nuclear fraction and mouse anti sheep SOD1 (Merk 574597, 1:1000) was used as loading control for cytoplasmic fraction. Blots were analyzed with chemiluminescence (ECL; Luminata Forte Kit, Millipore WBLUF0500) using the Molecular Imager Chemidoc XRS (Biorad) as detection system and total protein as loading controls.
For western blot on protein extracts from spinal cord of Fus +/− and Fus +/+ mice, rabbit anti-FUS (Bethyl A300-294A) was used as a primary antibody in a 1:800 dilution. A mouse monoclonal anti-beta-tubulin antibody (clone E7, DSHB, 1:2000) was used as a loading control. As secondary antibodies, HRP-conjugated anti-rabbit (Promega W4011) and anti-mouse (Promega W4021) antibodies were used in a 1:2500 dilution.
Spinal cord histology
Animals were anesthetized with ketamine (Imalgene 80 mg/kg; Merial, Lyon, France) and xylazine (Rompun 20 mg/kg; Bayer, Lyon, France) and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB), pH 7.4. Spinal cord were dissected and fixed by immersion in 4% paraformaldehyde in 0.1 M phosphate buffer pH 7.4 overnight. The lumbar part of spinal cords (L1–L5) was cryoprotected in 30% sucrose, snap frozen in melting isopentane, and embedded in TissueTek (O.C.T.Compound, SAKURA#4583). Cryosections (Leica CM 3050S) of 16 µm were obtained for histological analysis (10 sections per animal).
Spinal cord sections were stained using rabbit anti-FUS antibody against the FUS N-terminal part (ProteinTech 11570, 1:100), goat anti-ChAT (Millipore AB144-P, diluted 1:50), and Hoechst (Sigma 33342, 1:1000) followed by fluorescently labeled secondary antibodies donkey anti-rabbit Alexa 488 (Jackson A21206), goat anti-rabbit Alexa 488 (Invitrogen A11008), goat anti-mouse Alexa 594 (Invitrogen A11005) and donkey anti-goat Alexa 594 (Molecular Probes A11058) diluted 1:500.
Other antibodies used for spinal cord staining included rat anti-di-methylated FUS (ADMA, 1:100), rabbit anti UBIQUITIN (Abcam ab179434, 1:100), mouse anti-ubiquitin (Millipore MAB1510, 1:100), guinea pig anti-P62 (Progen GP-62C, 1:100), rabbit anti-FUS (Bethyl A300-302A, 1:150), mouse anti-NeuN (clone A60, Millipore MAB377, 1:500), mouse anti-CNPase (Sigma C-5922, 1:100) and rabbit anti-carbonic anhydrase II (kind gift of Dr S. Ghandour, 1:200 for IF and 1:1000 for DAB staining) [13, 35, 74].
Imaging
Single-layer images were acquired using a laser-scanning microscope (confocal Leica SP5 Leica Microsystems CMS GmbH) equipped with ×63 oil objective (NA1.4). Excitation rays are sequential: Hoechst 33342 was excited using diode 405 nm, Alexa 488 by the argon laser 488 nm, Alexa 594 by diode 561 nm and Alexa 647 by the Helium Neon laser 633 nm. Emission bandwidths were 410–470 nm for Hoechst 33342, 520–550 nm for Alexa 488, 570–620 nm for Alexa 594, and 650–690 nm for Alexa 647. Intensity of FUS fluorescent staining was measured using the software Nis Elements version 4.0.
Motor coordination and muscle strength analysis
Mice were followed weekly for general health, neurological symptoms, body weight, grip test and accelerating rotarod performances starting from weaning (4 weeks of age) until 22 months of age as described previously [39]. Briefly, mouse motor performance was assessed using rotarod (Ugobasile model 7650). Each session consisted of three tests of 300 s with an acceleration period (4–20 rpm during 150 s) followed by 150 s at constant speed. To evaluate muscle strength, we used a grip strength meter test (Bioseb, ALG01; France). The muscle force (in Newton) was measured three times per mouse. Results are presented as one measurement point per month.
Inverted grid test
The four limbs hang test uses a wire grid system to non-invasively measure the ability of mice to use sustained limb tension to oppose their gravitational force. The procedure measures 4 limbs hang time in seconds as well as the minimal holding impulse. Each mouse was placed at the simple cage grid and was allowed to accommodate to this environment for 3–5 s before the grid was inverted and held approximately 35 cm over a mouse cage containing 5–6 cm of bedding (wood chips). Each of these holding periods began with all four paws of the mouse grasping the grid. The wire grid hanging time (or “hang time”) was defined as the amount of time that it takes the mouse to fall down from the inverted grid and was measured visually with a stop watch. In each session, the procedure was repeated three times with approximately 10 min between each assessment of holding time. The mouse body weight was obtained shortly before the test. The physical impulse (holding impulse) is the hanging time multiplied by the gravitational force of the mouse [body mass (g) × 0.00980665 N/g × hanging time (s)]. This parameter represents the minimal total sustained force that was exerted to oppose the gravitational force [12].
Gait analysis
Gait parameters of freely moving mice were measured using the CatWalk gait analysis system (Noldus Information Technology, The Netherlands). The CatWalk instrument consists of a hardware system of a long, enclosed glass walkway plate, illuminated with green light, a high-speed video camera, and a software package for quantitative assessment of animal footprints. A green light emitted by a fluorescent lamp positioned underneath the glass plate is reflected within the glass plate except at points where the mouse paws made contact with the glass plate. It scatters and illuminates the contact area. The intensity of the area of illumination, which is proportional to the exerted pressure, is digitally captured by the video camera connected to a computer that runs the CatWalk software 7.1.
The recordings were carried out when the room was completely dark, except for computer screen. Each mouse was placed individually in the CatWalk walkway and allowed to walk freely, in an unforced manner and traverse from side to side the walkway glass plate. Mouse tracks that were straight without any interruption or hesitation were treated as successful runs. Runs with any wall climbing, grooming, and staying on the walkway were not analyzed. An average number of 3 replicate crossings made by each mouse were recorded. The CatWalk software was used to analyze crossings that had at least five cycles of complete steps. The software automatically labeled all areas containing pixels above the set threshold. These areas were identified and assigned to the respective paws. Analysis of the recording generated a wide range of parameters from which the following gait and coordination parameters were analyzed: Stride length (distance between two consecutive paw placements of the same paw in pixel), swing speed (distance between two consecutive paw placements of the same paw per second), body speed (distance that the animal walks per second) and body speed variation (regularity of body speed, in %) [2, 54, 89].
Electromyography
Electromyography was performed as previously described [21, 22]. Mice at 10 and 22 month of age were anesthetized with a solution of ketamine/xylazine (100 mg/kg; 5 mg/kg) and electrical activity was recorded using a monopolar needle electrode (diameter 0.3 mm; 9013R0312; Medtronic, Minneapolis, MN, USA) inserted into the tail of the mouse (grounding electrode). Recordings were made with a concentric needle electrode (diameter 0.3 mm; 9013S0011; Medtronic). Electrical activity was monitored in both GA and TA on both legs for at least 2 min. Spontaneous activity was differentiated from voluntary activity by visual inspection. Results were scored as described previously [21, 22].
Compound muscle action potentials (CMAP) were recorded in gastrocnemius muscle as described previously [61]. Briefly, CMAPs were elicited by supramaximal square pulses, of 0.2 ms duration, delivered with a monopolar needle electrode to the sciatic nerve at the sciatic notch level. CMAPs were measured by a monopolar needle electrode inserted in the gastrocnemius, and the system was grounded by subcutaneously inserted monopolar needle electrodes in the back and the tail of the animal. Amplitudes (mV) from the left and right muscle-evoked responses were measured and averaged, resulting in one average CMAP amplitude per animal, which was used for statistical analysis. The latency was measured as the time from the given electrical stimulus to the appearance of a muscle response—the initial CMAP deflection from the baseline.
Spinal cord motor neurons quantifications
To quantify lower motor neurons, spinal cord cryostat sections of 16 µm were stained with 0.1% Cresyl violet acetate (Certistain®, MERK#5235) and anti-ChAT (Millipore, AB144-P; diluted 1:50) followed by biotinylated donkey anti-goat IgG (Jackson, 705-066-147; 1:250) as secondary antibody. The staining was revealed using the ABC kit (Vektor, PK7200; 1:4000), by the avidin–biotin complex immunoperoxidase technique.
Counting of motor neurons was performed in L1–L3 ventral horn in every tenth section for ten sections in total per animal. Total number of motor neurons was counted using ImageJ freeware (http://rsbweb.nih.gov/ij/) after image acquisition at ×20 magnification under the same exposition parameters with a digital camera (Nikon Digital Sight DS-U3). The observer was blinded to the genotype of studied mice.
RNAseq
Total RNA from spinal cords (including dorsal and ventral roots) of Fus ΔNLS/+ (n = 4) and their control littermates (n = 4) were extracted with TRIzol (Invitrogen). RNA quality was measured using the Agilent Bioanalyzer system or RNA screen Tape (Agilent technologies) according to the manufacturer’s recommendations, and processed using the Illumina TruSeq Stranded mRNA Sample Preparation Kit according to the manufacturer’s protocol. Generated cDNA libraries were sequenced using an Illumina HiSeq 2000 sequencer with 4 biological replicates sequenced per condition using single read, 50 cycle runs. Quality of sequencing reads was assessed using FastQC (Babraham Bioinformatics) and then aligned to a mouse reference genome (mm9, UCSC Genome Browser) using TopHat (version v2.0.10). Sequencing yielded, on average, 39 million non-redundant reads per sample with a 55–65% mapping rate. Cufflinks (version v2.1.1) was used to generate transcript abundance for each annotated protein-coding gene as Fragments Per Kilobase of transcript per Million mapped reads (FPKM), and statistical analysis and comparison of FPKM values was calculated using Cuffdiff (version v2.1.1).
RT-PCR analysis
Spinal cord were harvested, rapidly frozen in liquid nitrogen and stored at −80 °C until analysis. For RT-qPCR, frozen tissues were placed into tubes containing a 5-mm stainless steel bead (Qiagen, Courtaboeuf, France) and 1 ml of Trizol reagent (Invitrogen, Paisley, UK) and homogenized using a Tissue Lyser (Qiagen). RNA was prepared from tissue homogenates following the Trizol manufacturer’s instructions. RNA reverse transcription and SYBR Green real-time PCR assays were performed using the Bio-Rad (Biorad, Marnes la Coquette, France) iCycler kits and protocols. PCR conditions were 3 min at 94 °C, followed by 40 cycles of 45 s at 94 °C and 10 s at 60 °C. Three standard genes: 18S (18S Ribosomal RNA), Pol2 (Polr2 polymerase RNA 2 DNA directed polypeptide A) and Tbp (TATA-box binding protein) were used to compute a normalization factor using Genorm software v3.5 [85]. Primer sequences are provided in Supplementary Table 1.
Toluidine blue staining
The L4 ventral root sections were stained with toluidine blue to investigate the axon diameter, the degree of demyelination and the myelin pathology among the different groups of animals. Five animals per genotype were analyzed. The ventral roots at the level of L4 were removed, treated with 1% osmium tetroxide, and embedded in Araldite Epon mixture. Semi-thin sections (1.5 µm) were cut, placed on the slides, and oven dried. The slides were stained with 1% toluidine blue solution for 1 min, rinsed with water, dehydrated and mounted. Internal diameter of myelinated axons was measured, and divided by the external diameter to calculate g-ratios. The following abnormalities in myelin were quantified: onion bulbs (as a sign of demyelination and remyelination), demyelinated axons, and abnormal myelin structures including complex or abnormal myelin outfoldings [1, 32]. These quantifications were performed by an observer blinded to the genotype.
Statistical analysis
For the animal experiments with two groups, the values from each animal were averaged for each genotype group and analyzed by unpaired Student’s t test, two-tailed. Comparison of three or four groups was performed using one-way ANOVA and Tukey post hoc test. Data were analyzed by using the Graphics Prism Program (Graph Pad Software, San Diego, CA) and expressed as mean ± SEM (standard error of the mean) and differences were considered significant when p ≤ 0.05.
Results
Partial cytoplasmic mislocalization of FUS in Fus ΔNLS/+ mice
Fus ΔNLS/+ mice represent the first animal model with a heterozygous mutation in the endogenous Fus gene, a similar genetic situation as in ALS-FUS patients. Fus mRNA levels were modestly increased in spinal cord of Fus ΔNLS/+ mice suggesting disruption of the normal autoregulatory loop controlling FUS levels (Supplementary Fig. 1a, b), while expression of Taf15 and Ewsr1, the two other FET family members, as well as that of Tardbp (encoding TDP-43) was not significantly changed in Fus ΔNLS/+ mice (Supplementary Fig. 1b). Subcellular fractionation of protein extracts from spinal cord followed by western blotting with an antibody recognizing both wild type and ∆NLS FUS protein (N-ter 1) yielded a robust FUS signal in cytoplasmic fractions from Fus ΔNLS/+ spinal cord, but not from wild-type littermate spinal cord (Fig. 1a, b). Contrastingly, western blotting using an antibody specific for the C-terminal NLS of FUS, and thus unable to recognize the mutant FUS protein, did not show increased cytoplasmic levels (Fig. 1c), demonstrating that FUS protein produced from the wild-type allele remains mostly nuclear. Nuclear FUS levels were not altered on immunoblots using either of the two antibodies (Fig. 1a–c). Consistently, increased cytoplasmic FUS staining was observed in Fus ΔNLS/+ motor neurons using double immunofluorescence with FUS and choline acetyltransferase (ChAT) antibodies (Fig. 1d). Quantification of fluorescence signals in individual motor neurons showed that the cytoplasmic staining of FUS was substantially elevated in Fus ΔNLS/+ motor neurons (Fig. 1e). The large majority (85%) of Fus +/+ motor neurons demonstrated an exclusive localization of FUS in the nucleus, while most (74%) Fus ΔNLS/+ motor neurons showed mixed cytoplasmic and nuclear localization. Notably, 21% of motor neurons showed exclusively cytoplasmic FUS accumulation accompanied by complete nuclear clearance of FUS protein (Fig. 1f). We did not observe large cytoplasmic FUS aggregates associated with FUS nuclear clearance.

FUS localization in Fus ΔNLS/+ mice. a Immunoblot analysis of FUS protein subcellular localization in spinal cord of 2 Fus +/+ and 2 Fus ΔNLS/+ 4-month-old mice using two different antibodies targeting either the N-terminal part (N-ter. 1) of FUS or the C-terminal (C-ter. 1) NLS. Molecular weight markers are shown on the left. SOD1 and HDAC1 are used as loading controls for cytoplasmic and nuclear protein extracts fractions, respectively. b, c Quantification of FUS protein levels in cytoplasmic and nuclear fractions from immunoblots for Fus +/+ (blue bars) and Fus ΔNLS/+ (red bars). N = 6. *p < 0.05, ***p < 0.01 by Student’s unpaired t test. d Double immunostaining for the motoneuronal marker ChAT and FUS (N-terminal part) in the spinal cord ventral horn at 22 months of age. Note the cytoplasmic redistribution of truncated FUS in Fus ΔNLS/+ mice. Scale bar 7.5 µm. e Quantification of FUS (N-terminal part) staining intensity in different cellular compartments of motor neuron. N = 70 Fus +/+, N = 68 Fus ΔNLS/+. ***p < 0.01 by Student’s unpaired t test. f Distribution of FUS cytoplasmic/nuclear localization in motor neurons. N = 70 Fus +/+, N = 68 Fus ΔNLS/+
Fus ΔNLS/+ mice recapitulate pathological hallmarks of ALS-FUS
In human ALS-FUS patients, FUS is asymmetrically dimethylated at arginine residues (ADMA), and this modified form of FUS is found in FUS-positive inclusions [19, 75, 81]. In contrast, unmethylated FUS and monomethylated FUS, but not ADMA-FUS, accumulates in cytoplasmic inclusions of FTLD-FUS patients [19, 75]. ADMA-FUS can be readily identified using an antibody specific to the ADMA RGG3 domain of FUS [19]. Interestingly, ADMA-FUS was strongly increased in both nuclear and cytoplasmic fractions of Fus ΔNLS/+ spinal cord (Fig. 2a, b). Furthermore, triple immunolabeling using antibodies against FUS, ADMA-FUS and ChAT revealed a pattern of ADMA-FUS subcellular distribution similar to truncated FUS (Fig. 2c). ALS-FUS patients also develop ubiquitin and p62 pathology [43]. While we did not observe p62 inclusions (Supplementary Fig. 3), we observed cytoplasmic and nuclear ubiquitin pathology in motor neurons of Fus ΔNLS/+ mice (Fig. 2d). Cytoplasmic ubiquitin pathology was also observed using a K63 linkage-specific ubiquitin antibody (Supplementary Fig. 2). Ubiquitin inclusions were occasionally present in motor neurons with mixed nuclear/cytoplasmic FUS localization and systematically present in motor neurons showing complete nuclear FUS clearance (Fig. 2d). Importantly, FUS and ubiquitin stainings did not systematically overlap, suggesting that Fus ΔNLS/+ motor neurons do not develop ubiquitin-positive FUS inclusions. Thus, Fus ΔNLS/+ mice and ALS-FUS patients develop partially similar pathology, with cytoplasmic accumulation of methylated FUS and ubiquitin pathology but no large FUS aggregates.

Subcellular redistribution of asymmetrically arginine dimethylated (ADMA) FUS. a Representative immunoblots on cytoplasmic and nuclear fractions of protein extracts from spinal cord of Fus +/+ and Fus ΔNLS/+ mice, using an antibody recognizing asymmetrically arginine dimethylated FUS (ADMA-FUS). HDAC1 is used as a loading control for nuclear fractions and SOD1 for cytoplasmic fractions. Molecular weight markers are shown on the left. b Quantification of ADMA-FUS protein levels in cytoplasmic and nuclear fractions from immunoblots for Fus +/+ (blue bars) and Fus ΔNLS/+ (red bars). N = 6. *p < 0.05, ***p < 0.01 by Student’s unpaired t test. c Triple immunostaining for the motoneuronal marker ChAT (green), for FUS (N-terminal part) (cyan) and for ADMA-FUS (red) in the spinal cord ventral horn. Scale bar 10 µm. d Triple immunostaining with antibodies to the N terminus of FUS (green), ChAT (red) and pan-Ubiquitin (cyan), showing diffuse cytoplasmic and nuclear punctate aggregates within motor neurons with relocated FUS. Scale bar 10 μm
Cytoplasmic mislocalization of FUS leads to a mild motor deficit in Fus ΔNLS/+ mice
We next investigated whether cytoplasmic accumulation of mutant FUS triggers ALS-like motor symptoms during the lifespan of heterozygous knock-in animals. To this aim, we longitudinally followed Fus ΔNLS/+ male mice and their Fus +/+ wild-type littermates until 2 years of age. Animals were weekly monitored for general health, neurological symptoms, body weight, grip strength and accelerating rotarod performance. Until 22 months of age, when mice were killed, expression of mutant FUS was neither associated with important weight loss nor with development of paralysis (Supplementary Fig. 4a–c). Although grip test and rotarod are among the most commonly used tests to assess motor function in mice [70], they often lack the sensitivity needed to detect subtle alterations in the motor system and should be complemented by additional tests to evaluate motor function in Fus ΔNLS/+mice.
Indeed, despite normal performance on rotarod and grip strength, Fus ΔNLS/+ mice displayed a significantly shorter hanging time in an inverted grid test (Fig. 3a), both at 10 and 22 months of age. Holding impulse, which represents the total sustained force exerted by the mouse to oppose the gravitational force [12], was also significantly decreased in Fus ΔNLS/+ mice (Fig. 3b). The evaluation of gait performance by CatWalk analysis further confirmed this motor defect. Fus ΔNLS/+ mice demonstrated an irregular walking pattern characterized by phases of fast walking interrupted with stance phases (Fig. 3c). We also observed a reduction of hind limb stride length (Fig. 3d) associated with an increase in body speed variation (i.e., the variation in the speed of the walking mouse) for both ages compared to the control mice (Fig. 3g). In addition, 22-month-old Fus ΔNLS/+ mice showed impaired swing and body speed (Fig. 3e–f). Thus, expression of mutant FUS at a physiological level is associated with partial cytoplasmic mislocalization of the protein and a mild motor deficit in mice.

Fus ΔNLS/+ mice display a mild motor deficit. Age-dependent changes in the mean hanging time (s) (a) and holding impulse (N s) (b) in the four-limb wire inverted grid test in Fus +/+ and Fus ΔNLS/+ mice. N = 7 for 10 months; N = 5 for 22 months. *p < 0.05, ***p < 0.01 as compared to Fus +/+ by Student’s unpaired t test. c Representative gait patterns of Fus +/+and Fus ΔNLS/+ mice at 10 months (left panels) and 22 months (right panels) of age. The panels show the digitized prints with colorful phase lags representing the stance phase duration of each individual paw in a single-step cycle. d–g Gait changes and variability in Fus ΔNLS/+mice. Stride length (d, distance between successive placements of the same paw in cm); swing speed (e, distance traveled by one paw per second), body speed (f, distance traveled by the animal per second, in cm/s), and body speed variation (g, regularity of body speed in %), are shown. N = 3 for 10 months; N = 5 for 22 months. All graphs show the overall sample means and standard errors at various ages (10 months; 22 months) for Fus +/+(blue bars) and Fus ΔNLS/+ (red bars) mice. *p < 0.05, ***p < 0.01 as compared to Fus+/+ by Student’s unpaired t test
Cytoplasmic mislocalization of FUS drives age-dependent, progressive motor neuron degeneration
To determine whether this mild motor phenotype could be due to an underlying motor neuron disease, we performed electromyography analysis (EMG) on Fus +/+ and Fus ΔNLS/+ mice at 10 and 22 months of age. We did not observe stereotypical denervation-related electrical activities in gastrocnemius (GA) or tibialis anterior (TA) muscle of 10-month-old Fus +/+ and Fus ΔNLS/+ mice. However, 22-month-old Fus ΔNLS/+ mice showed typical fibrillation and fasciculation potentials in both muscles (Fig. 4a). Consistent with qualitative observations, a quantitative analysis of the EMG recordings demonstrated a significantly increased frequency of abnormal potentials in 22-month-old Fus ΔNLS/+ mice, but not at 10 months of age (Fig. 4b) and the amplitude of compound muscle action potentials (CMAP) was decreased in 18- to 22-month-old Fus ∆NLS/+ mice (Fig. 4d).

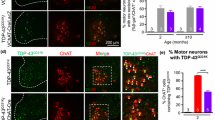
Muscle denervation and progressive degeneration of spinal motor neurons in Fus ΔNLS/+ mice. a Representative electromyograms of Fus ΔNLS/+ mice in gastrocnemius and tibialis anterior muscles. Note the presence of typical spontaneous denervation activities (fibrillation potentials) in Fus ΔNLS/+ mice. Scale bars 50 ms and 50 µV. b Graph showing EMG scores for Fus +/+ (blue bars) and Fus ΔNLS/+ (red bars) mice. Note that a significant difference was only detected for 22-month-old animals. ***p < 0.01 as compared to Fus +/+; N = 7 for 10 months; N = 9 for 22 months; Student’s unpaired t test. c Representative images of Nissl and ChAT staining of spinal cord ventral horn of 10-month-old (left panels) and 22-month-old (right panels) Fus +/+ and Fus ΔNLS/+ animals. In the 22-month-old Fus ΔNLS/+ mice degenerative changes (shrinking, chromatolysis) and loss of motor neurons occur. Scale bars 35 μm. d Compound muscle action potential (CMAP) amplitude. *p < 0.05 as compared to Fus +/+; N = 10 Fus +/+, N = 9 Fus ΔNLS/+. Student’s unpaired t test. Bar graphs showing means and standard errors of Nissl+ (e) and ChAT+ (f) motor neuron number in the ventral horn of the spinal cord at 10 months and 22 months for Fus +/+ (blue bars) and Fus ΔNLS/+ (red bars) mice. ***p < 0.01 as compared to Fus +/+; N = 3 for 10 months; N = 6 for 22 months; Student’s unpaired t test
Importantly, the abnormal electrical activity in Fus ΔNLS/+ mice was accompanied by degeneration of motor neurons in the lumbar spinal cord of 22-month-old Fus ΔNLS/+ mice, as evaluated using either Nissl staining or immunostaining for ChAT (Fig. 4c). Quantitative analysis revealed that the number of motor neurons was reduced by ~30% in 22-month-old Fus ΔNLS/+ mice as compared to Fus +/+ mice (Fig. 4e–f). Importantly, the number of lumbar spinal cord motor neurons was not altered at 10 months of age, indicating that the pathological process is progressive. Thus, partial cytoplasmic mislocalization of FUS triggers late-onset progressive motor neuron loss associated with a mild motor deficit.
Reduced levels of FUS do not lead to motor neuron degeneration
We next investigated whether partial loss of nuclear FUS function might contribute to the observed phenotypes of Fus ΔNLS/+ mice. To determine whether a mild loss of FUS function could be sufficient to trigger motor neuron degeneration, we longitudinally followed Fus +/− mice [68]. At the age of 23 months, Fus transcript levels were reduced by 25% in spinal cord of Fus +/− mice (Fig. 5a). Consistently, western blot analysis revealed reduced FUS protein levels in the spinal cord of these mice (Fig. 5b, c). These reduced FUS expression levels did not alter the subcellular localization of FUS in the spinal cord (Fig. 5d). Up to the age of 23 months, the performance of Fus +/− mice in the inverted grid test (Fig. 5e) was not different from Fus +/+ control mice, as well as their grip strength and body weight (data not shown). Consistently, Fus +/− mice did neither show denervation potentials in EMG nor decreased CMAP (Fig. 5f). Finally, normal numbers of spinal motor neurons were found at 23 months of age (Fig. 5g, h). Thus, Fus reduction is not sufficient to trigger the motor neuron defects observed in Fus ΔNLS/+ mice.

Lack of motor neuron disease in Fus +/− mice. a Expression levels of Fus mRNA in spinal cord. Fus mRNA levels were significantly reduced in Fus +/− mice as revealed by quantitative real-time PCR analysis. N = 7 Fus +/+, N = 9 Fus +/−. *p < 0.05 by Student’s unpaired t test. b Quantification of FUS protein levels from immunoblots showed a lower amount of FUS in spinal cord of Fus +/− mice. N = 7 Fus +/+, N = 8 Fus +/−. *p < 0.05 by Student’s unpaired t test. c Representative immunoblot for FUS on protein extracts from spinal cord of 100-week-old mice. TUBULIN was used as loading control. d Immunostaining for the neuronal marker NeuN and FUS on the spinal cord ventral horn of 100-week-old Fus +/+ and Fus +/− mice. Note preserved nuclear localization of FUS in Fus +/− mice. e Mean hanging time in the four-limb wire inverted grid test of Fus +/+ and Fus +/− mice. N = 4 Fus +/+, N = 3 Fus +/−. p = not significant (NS) by Student’s unpaired t test. f Bar graphs showing means and standard errors for compound muscle action potential (CMAP) amplitude. No difference was observed between groups. N = 6 Fus +/+, N = 5 Fus +/−. p = NS by Student’s unpaired t test. g Representative images of ChAT immunostaining on spinal cord ventral horn. Scale bar 35 μm. h Quantification of the number of motor neurons per spinal cord ventral horn in Fus +/+ and Fus +/− mice. The number of ChAT+ motor neurons was not altered in Fus +/− mice. N = 6 Fus +/+, N = 5 Fus +/−. p = NS by Student’s unpaired t test
Cre-dependent reversal of the Fus mutation in motor neurons largely prevents FUS mislocalization
We then sought to determine whether motor neuron degeneration in Fus ΔNLS/+ mice was dependent on the mislocalization of FUS in motor neurons themselves. To this aim, we took advantage of the presence of loxP sites on the ∆NLS allele, allowing us to restore a normal protein in specific cell types upon CRE recombination [68]. Fus ΔNLS/+ mice were bred with mice expressing the CRE recombinase from the endogenous Chat locus, which leads to CRE recombinase activity in virtually all cholinergic neurons [65, 67]. In double transgenic Fus ΔNLS/+/Chat-CRE mice, the cytoplasmic accumulation of FUS was rescued, although not completely (Supplementary Fig. 5a), and the proportion of motor neurons with complete nuclear FUS clearance was reduced to similar levels as in Fus +/+ mice (Supplementary Fig. 5b–d). Consistently, immunoreactivity for ADMA-FUS was barely detectable in the cytoplasm of motor neurons of Fus ΔNLS/+/Chat-CRE mice (Supplementary Fig. 6). Thus, motor neuron-selective reversal of the Fus mutation rescues FUS mislocalization in motor neurons of Fus ΔNLS/+ mice.
Reversal of the Fus mutation in motor neurons prevents motor neuron degeneration and delays motor deficits
The presence of a Chat-CRE allele in Fus ΔNLS/+ mice was sufficient to fully restore motor neuron counts to similar numbers as in Fus +/+ mice at 22 months of age (Fig. 6a–c). Consistently, Fus ΔNLS/+/Chat-CRE mice did not develop the EMG abnormalities found in their Fus ΔNLS/+ littermates and their EMG score did not significantly differ from Fus +/+ mice (Fig. 6d–e). While littermate Fus ΔNLS/+ mice displayed a significantly shorter hanging time and shorter holding impulse than Fus +/+ mice in the inverted grid test at both 10 and 22 months of age (Figs. 3a, b, 6f, g), Fus ΔNLS/+/Chat-CRE mice displayed motor performance similar to Fus +/+ mice at 10 months of age (Fig. 6f, g). At 22 months of age, however, Fus ΔNLS/+/Chat-CRE mice displayed significantly impaired motor performance as compared to Fus +/+ mice, leading to a similar motor deficit as Fus ΔNLS/+ mice. Thus, the selective reversal of the Fus mutation in motor neurons is sufficient to fully rescue motor neuron degeneration, even at older ages, while the motor deficits are delayed but not prevented.

Motor neuron-selective reversal of the Fus ΔNLS allele to wild type delays but does not prevent Fus ΔNLS/+ motor phenotypes. a Representative images of spinal cord ventral horn of Fus +/+/ChAT-CRE, Fus ΔNLS/+/− and Fus ΔNLS/+/ChAT-CRE mice at 22 months stained with Nissl (left panels) or anti-choline acetyl transferase (ChAT, right panels). b, c Quantification of motor neurons per spinal cord ventral horn. The number of Nissl+ (b) and ChAT+ (c) motor neurons is rescued in Fus ΔNLS/+/ChAT-CRE mice while significantly reduced in Fus ΔNLS/+/− mice. N = 5 per genotype; ***p < 0.01 versus Fus +/+/ChAT-CRE, ### p < 0.01 versus Fus ΔNLS/+/ChAT-CRE; one-way ANOVA followed by Tukey post hoc test. In all graphs genotypes are represented as Fus +/+/ChAT-CRE (blue bars), Fus ΔNLS/+/− (red bars) and Fus ΔNLS/+/ChAT-CRE (green bars). d EMG recording traces in gastrocnemius muscle of 22-month-old animals. Note the absence of typical spontaneous denervation activities in Fus ΔNLS/+/ChAT-CRE versus Fus ΔNLS/+/− mice. Scale bars 50 ms and 50 µV. e EMG score showing significantly decreased spontaneous activity in Fus ΔNLS/+/ChAT-CRE as compared to Fus ΔNLS/+/− in 22-month-old animals. N = 5 Fus +/+/ChAT-CRE, N = 7 Fus ΔNLS/+/− and N = 6 Fus ΔNLS/+/ChAT-CRE. ***p < 0.01 versus Fus +/+/ChAT-CRE; # p < 0.05 versus Fus ΔNLS/+/ChAT-CRE; one-way ANOVA followed by Tukey post hoc test. Inverted grid test mean hanging time (f) and holding impulse (g). N = 7–8 for 10 months; N = 5–7 for 22 months. *p < 0.05, ***p < 0.01 versus Fus +/+/ChAT-CRE; # p < 0.05 versus Fus ΔNLS/+/ChAT-CRE; one-way ANOVA followed by Tukey post hoc test
Oligodendrocytic alterations in Fus ΔNLS/+ spinal cord
To evaluate the molecular mechanisms underlying the phenotypes in Fus ΔNLS/+ mice, we performed RNAseq on spinal cord RNA extracts from 22-month-old Fus ΔNLS/+ and control littermates. Among the genes showing differential expression, several of them encoded for proteins related to myelination (Fig. 7a). Indeed, mRNA levels of myocilin, Ncmap, Pmp2, Pmp22, Cldn19 and Prx, were all downregulated in RNAseq from Fus ΔNLS/+ mice (Fig. 7a), and this was confirmed using RT-qPCR on samples obtained from an independent cohort of mice (Fig. 7b). For instance, Myocilin is required for peripheral myelination [46], as is Pmp2 [94] and Prx [25]. Pmp22 [33] and Prx [15] are involved in the morphology of myelinating Schwann cells. Interestingly, we did not observe altered expression of other major myelin genes such as Mbp, Plp1 or Abca1 (Supplementary Fig. 7). Increased cytoplasmic FUS staining was observed in Fus ΔNLS/+ oligodendrocytes using double immunofluorescence for FUS and oligodendrocyte specific markers CNPase and carbonic anhydrase II (Fig. 7c; supplementary Fig. 8). Oligodendrocytes were more numerous in ventral horn white matter of Fus ΔNLS/+ mice as compared to Fus +/+ mice (Fig. 7d, e), and this was not reverted in Fus ΔNLS/+/Chat-CRE mice. Together with RNAseq results, these data suggest that FUS mislocalization leads to defective oligodendrocyte physiology, independently of motor neuron involvement. To understand whether these defects could translate into abnormal myelination of motor axons, we studied the morphology of the ventral roots that collect all motor axons exiting from the spinal cord at 22 months of age. Consistent with a defect in myelination, g-ratio of ventral root axons were modestly decreased for the smaller axonal calibers (Supplementary Fig. 9a). Furthermore, motor axons of Fus ΔNLS/+ mice showed a distribution shifted towards smaller caliber (Supplementary Fig. 9b), and this axonal defect was not rescued in Fus ΔNLS/+/Chat-CRE mice (Supplementary Fig. 9b, c). Moreover, there were fewer axons showing normal myelination in ventral roots of both Fus ΔNLS/+ and Fus ΔNLS/+/Chat-CRE mice as compared to control littermates (Supplementary Fig. 9d), and thus a higher frequency of typical features of myelination defects in both Fus ΔNLS/+ groups. Last, consistent with a myelin defect, the latency of CMAP was increased in Fus ΔNLS/+ muscles (Supplementary Fig. 9e). These results indicate that the function of myelinating cells, both in spinal cord and in the periphery, is altered in Fus ΔNLS/+ mice, what may contribute to the observed motor deficits in aged mice.

Axonal and myelin abnormalities in Fus ΔNLS/+ mice. a, b Normalized expression of myelin-related genes (Myocilin, NcMap, Pmp22, Pmp2, Cldn19, Prx, Dhh) in Fus ΔNLS/+ compared to their control littermates based on FPKM from RNAseq (a, n = 4) or RT quantitative PCR (b, n = 6–7). Error bars represent SEM. *p < 0.05, ***p < 0.01; Student’s t test. c Double immunolabeling for the oligodendrocyte marker carbonic anhydrase II (CAII) (green) and FUS (N-terminal part) (red) in the spinal cord. Scale bar 5 µm. d Representative images of spinal cord ventral horn of Fus +/+/ChAT-CRE, Fus ΔNLS/+ and Fus ΔNLS/+/ChAT-CRE mice at 22 months stained with anti-carbonic anhydrase II showing increased numbers of oligodendrocytes. Scale bars: left column 70 µm; right column 15 µm. e Oligodendrocyte density in whole spinal cord ventral quadrant, and separately in white and grey matter. Note increased numbers in white matter in 22-month-old Fus ΔNLS/+/−, not rescued in Fus ΔNLS/+/ChAT-CRE mice. N = 4 animals per genotype. ***p < 0.01 versus Fus +/+/ChAT-CRE; one-way ANOVA followed by Tukey post hoc test
Discussion
In this study we show that a heterozygous mutation in the endogenous murine Fus gene, that is similar to the most severe mutations in juvenile ALS, partially recapitulates ALS-FUS pathology and triggers mild progressive ALS-like symptoms. We demonstrate that this Fus mutation is associated with motor neuron degeneration through FUS mislocalization in motor neurons, while axonal damage and demyelination occur independent of mutant FUS expression in motor neurons.
The generation of faithful animal models of neurodegenerative diseases is a long-standing aim of the scientific community. Recently, heterozygous knock-in mouse models of Alzheimer’s disease were shown to display mild but significant behavioral abnormalities, overcoming most of the artificial phenotypes observed in classical transgenic AD mouse models [66]. Similar artificial phenotypes are also confounding analysis in most currently used transgenic mouse models of ALS [5, 23, 30]. Indeed, the vast majority of described ALS mice expressing either mutant SOD1 [29], mutant TDP-43 [90] or mutant FUS [64, 73], are multi-copy transgenic lines, with poorly documented sites of transgene insertion. Moreover, overexpression of the wild type forms of SOD1 [26], TDP-43 [78, 91, 92] or FUS [55] generally leads to similar, if not exacerbated, symptoms as compared to expression of the ALS-linked mutations, casting doubts on their relevance as faithful animal models of the disease. Recently, Sharma and collaborators generated mice with targeted expression of wild type or mutant FUS from the Tau locus [72]. These mice express physiological levels of FUS, yet under the control of the Tau promoter which is more active in neurons than in other cell types of the CNS. In Fus ΔNLS/+ mice, the endogenous murine Fus gene carries the mutation on a single allele thus providing the unique opportunity to study the effects of an ALS-like mutation in an authentic genetic context. The genetic construct used here features a poly-adenylation cassette that precludes the inclusion of regulatory elements present in the endogenous 3′UTR of the Fus mRNA. Since the 3′UTR of Fus mRNA has been found important for FUS autoregulation [17], this could in principle result in altered autoregulation of FUS. However, exon 7 and adjacent introns that are primarily involved in Fus autoregulation are conserved in this model [95].
Fus ΔNLS/+ mice replicated a number of the pathological hallmarks observed in ALS-FUS patients. Similar to ALS-FUS patients or iPSC-derived cells of these patients, the FUS protein was partially mislocalized to the cytoplasm [36, 38, 45, 48, 51] in a dimethylated form [19, 75]. Consistent with the heterogeneity observed in human cases, Fus ΔNLS/+ motor neurons displayed various stages of FUS mislocalization [51, 52], and a subset of them showed complete nuclear FUS clearance. Importantly, not all pathological features of ALS-FUS were reproduced, suggesting that Fus ΔNLS/+ mice recapitulate early stages of disease. For instance, we did not observe neuronal or glial large cytoplasmic inclusions of FUS [43, 51], even in cells showing complete nuclear clearance of FUS. This is similar to mice expressing mutant FUS from the TAU locus [72]. Although FUS does not spontaneously form large pathological aggregates, we cannot exclude that the biochemical properties of FUS could be altered in these mice. For instance, a proportion of the FUS protein could become insoluble and/or its repertoire of binding partners could be modified [6, 86]. Further work in Fus ΔNLS/+ mice should clarify the biochemical consequences of FUS truncation and mislocalization.
There was significant ubiquitin pathology but no p62 pathology. Such an early pathological stage is consistent with the observed slowly progressive motor neuron loss, mild motor symptoms, and a presumably unaltered mouse lifespan at least until 22 months of age. Several factors could explain why Fus ΔNLS/+ mice did not progress to the full-blown pathological and behavioral features of ALS-FUS. First, the genetic background of mice influences ALS-related disease course [31, 53]. Second, it cannot be excluded that additional hits, either environmental or genetic, are necessary for the progression of ALS-FUS, as previously postulated [18]. Third, the life span of mice might simply be too short to develop full-blown ALS, as in humans the disease becomes symptomatic after several decades.
Both ALS-FUS patients and Fus ΔNLS/+ mice carry one mutant copy of the FUS gene, and comparing Fus ΔNLS/+ mice with Fus +/− mice allowed us to provide definitive evidence that gain of function is required to cause ALS-FUS. Indeed, Fus +/− mice did not show motor neuron loss or motor symptoms in contrast to Fus ΔNLS/+ mice. This is consistent with the absence of motor phenotypes in Fus knock-out mice, showing that even the complete absence of the FUS protein is not sufficient to trigger motor neuron degeneration [44, 68, 72]. Our results are consistent with recently published studies documenting that cytoplasmic FUS accumulation is the only necessary toxic event to trigger motor neuron loss [72, 73] and provide evidence that endogenous levels of mutant FUS protein are sufficient in animal models carrying identical gene dosage as in ALS-FUS patients.
The observed analogies between Fus ΔNLS/+ mice and ALS-FUS indicate that Fus ΔNLS/+ mice could be a model of choice to elucidate the cellular and molecular basis of ALS-FUS. In this respect, our study suggests that both cell-autonomous and non-cell autonomous toxicity contributes to trigger the motor phenotype in ALS-FUS. A previous study by Sharma and collaborators has demonstrated that motoneuronal expression of a FUS mutation is sufficient to trigger motor symptoms [72]. However, the expression of mutant FUS from the mostly neuronal TAU locus did not allow them to rigorously test the contribution of other FUS-expressing cell types. Here we show that loss of motor neuron cell bodies is completely rescued by reversal of the mutation in motor neurons. However, Fus ΔNLS/+/Chat-CRE mice still developed motor symptoms after 10 months of age, and axonal damage appeared similar, independent of cytoplasmic FUS expression by motor neurons. In this knock-in Chat-CRE mouse strain, recombination occurs as early as E12 in motor neurons, and is extremely efficient [67]. Thus, both motor neuron autonomous and non-autonomous mechanisms contribute to motor neuron disease in Fus ΔNLS/+ mice. These results are consistent with previous findings obtained in mutant SOD1 mice [8, 9, 93] as well as with results obtained in conditional transgenic TDP-43 mice (Da Cruz and Cleveland, personal communication). Hence, non-cell autonomous toxicity to motor neurons likely represents a ubiquitous mechanism of ALS.
Motor defects that are not rescued in Fus ΔNLS/+/Chat-CRE mice could be due to defects in myelinating cells. Indeed, FUS was mislocalized in spinal cord oligodendrocytes of Fus ΔNLS/+ mice and this mislocalization was maintained in Fus ΔNLS/+/Chat-CRE mice. Consistent with a defect in oligodendrocytes, RNAseq uncovered decreased spinal cord expression of a number of genes that have been previously involved in morphology and function of myelin. Several of them are mutated in human demyelinating neuropathies, and loss of their function in mice leads to profound biochemical and morphological abnormalities of myelin. For instance, decreases in PMP2, whose mutations can cause demyelinating Charcot–Marie–Tooth disease (CMT) [57] is sufficient to modify the lipidome of peripheral myelin [94], while heterozygous loss of Pmp22, causing human peripheral neuropathy [60], disrupts myelin junctions in mice [28, 33]. Furthermore, loss of periaxin, causing CMT4F in humans [7, 27], is sufficient to profoundly alter the morphology of myelinating Schwann cells [15]. Importantly, we observed an increased number of oligodendrocytes in the ventral spinal cord white matter, consistent with altered function of this cell type, and this increased number was not dependent upon expression of the mutation in motor neurons as it persisted in Fus ΔNLS/+/Chat-CRE mice. These results provide further evidence for the involvement of oligodendrocytes in ALS, and are consistent with results obtained in transgenic mice expressing mutant SOD1. In SOD1(G93A) mice, generation of oligodendrocytes from NG2+ cells is increased [40], and axons are abnormally myelinated in the grey matter [41, 63]. Interestingly, however, oligodendrocyte numbers appear increased in Fus ΔNLS/+ mice, while they were reported to be unchanged in SOD1(G93A) mice. Such a difference might be due to the very different disease courses of both mouse strains. Oligodendrocytic expression of mutant SOD1 appears to play a critical role in mutant SOD1-ALS [41, 47, 63], and our current study provides suggestive, but not conclusive, evidence for a critical role of oligodendrocytes in ALS-FUS, and it does not identify pathogenic mechanisms elicited by mutant FUS in myelinating cells. Further work, using relevant CRE expressing lines and/or cell-specific gene expression profiling [76] is mandatory to explore this hypothesis.
Besides oligodendrocytes, we also provide evidence of defects in Schwann cells of Fus ΔNLS/+ mice. There were myelin defects in ventral roots, and this was accompanied by increased CMAP latencies, characteristic for peripheral demyelination. Defects in Schwann cells have been recently observed in multiple models of ALS [37, 80], but expression of mutant SOD1 in Schwann cells appeared neither necessary [50] nor sufficient [83] for the disease triggered by this specific ALS-linked mutation, and the role of Schwann cells in ALS remains uncertain.
To our knowledge, myelin ultrastructure has not been systematically studied in ALS-FUS patients, yet several studies observed loss of myelin in the cortico-spinal tract of ALS-FUS patients [43, 84]. Moreover, FUS cytoplasmic aggregates have been observed in oligodendrocytes of ALS-FUS patients [51, 77] as well as in FTD patients with FUS pathology [58]. In non-FUS ALS patients, myelin loss has been observed in sporadic ALS patients and inclusions of TDP-43 are frequent in oligodendrocytes [41, 63]. Besides glial cells, other cell types, such as skeletal muscle, could contribute to the neuromuscular phenotypes, and the conditionality of Fus mutation in Fus ΔNLS/+ mice will allow to investigate the role of these cells using appropriate CRE lines.
In conclusion, we characterize here a heterozygous knock-in mouse model of ALS and demonstrate that mutations in FUS result in a toxic gain of function leading to motor neuron disease through cell autonomous and non-cell autonomous mechanisms. Fus ΔNLS/+ mice will be instrumental in deciphering the molecular derailments elicited by mutant FUS and could be useful for preclinical testing of therapeutic strategies.
References
Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U (1997) Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating to maculous neuropathy. J Neurosci 17:4662–4671
Angeby-Moller K, Berge OG, Hamers FP (2008) Using the CatWalk method to assess weight-bearing and pain behaviour in walking rats with ankle joint monoarthritis induced by carrageenan: effects of morphine and rofecoxib. J Neurosci Methods 174:1–9
Baumer D, Hilton D, Paine SM, Turner MR, Lowe J, Talbot K, Ansorge O (2016) Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 75:611–618
Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y (2005) Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett 379:152–157
Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Rehnmark A, Marklund SL (2006) Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci 26:4147–4154
Blokhuis AM, Koppers M, Groen EJ, van den Heuvel DM, Dini Modigliani S, Anink JJ, Fumoto K, van Diggelen F, Snelting A, Sodaar P, Verheijen BM, Demmers JA, Veldink JH, Aronica E, Bozzoni I, den Hertog J, van den Berg LH, Pasterkamp RJ (2016) Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol 132:175–196
Boerkoel CF, Takashima H, Stankiewicz P, Garcia CA, Leber SM, Rhee-Morris L, Lupski JR (2001) Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet 68:325–333
Boillee S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Bowden HA, Dormann D (2016) Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem 138(Suppl 1):112–133
Calvo A, Moglia C, Canosa A, Brunetti M, Barberis M, Traynor BJ, Carrara G, Valentini C, Restagno G, Chio A (2014) De novo nonsense mutation of the FUS gene in an apparently familial amyotrophic lateral sclerosis case. Neurobiol Aging 35(1513):e1517
Carlson CG, Rutter J, Bledsoe C, Singh R, Hoff H, Bruemmer K, Sesti J, Gatti F, Berge J, McCarthy L (2010) A simple protocol for assessing inter-trial and inter-examiner reliability for two noninvasive measures of limb muscle strength. J Neurosci Methods 186:226–230
Cerghet M, Skoff RP, Bessert D, Zhang Z, Mullins C, Ghandour MS (2006) Proliferation and death of oligodendrocytes and myelin proteins are differentially regulated in male and female rodents. J Neurosci 26:1439–1447
Conte A, Lattante S, Zollino M, Marangi G, Luigetti M, Del Grande A, Servidei S, Trombetta F, Sabatelli M (2012) P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscular disorders: NMD 22:73–75
Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ (2004) Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature 431:191–195
Deng H, Gao K, Jankovic J (2014) The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10:337–348
Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I (2014) An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun 5:4335
Dormann D, Haass C (2011) TDP-43 and FUS: a nuclear affair. Trends Neurosci 34:339–348
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, Haass C (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31:4258–4275
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Dupuis L, Fergani A, Braunstein KE, Eschbach J, Holl N, Rene F, Gonzalez De Aguilar JL, Zoerner B, Schwalenstocker B, Ludolph AC, Loeffler JP (2009) Mice with a mutation in the dynein heavy chain 1 gene display sensory neuropathy but lack motor neuron disease. Exp Neurol 215:146–152
Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C, Schaeffer L, Bouillaud F, Loeffler JP (2009) Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One 4:e5390
Esmaeili MA, Panahi M, Yadav S, Hennings L, Kiaei M (2013) Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int J Exp Pathol 94:56–64
Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593
Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR, Smith A, Brophy PJ (2000) Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 26:523–531
Graffmo KS, Forsberg K, Bergh J, Birve A, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T (2013) Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet 22:51–60
Guilbot A, Williams A, Ravise N, Verny C, Brice A, Sherman DL, Brophy PJ, LeGuern E, Delague V, Bareil C, Megarbane A, Claustres M (2001) A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet 10:415–421
Guo J, Wang L, Zhang Y, Wu J, Arpag S, Hu B, Imhof BA, Tian X, Carter BD, Suter U, Li J (2014) Abnormal junctions and permeability of myelin in PMP22-deficient nerves. Ann Neurol 75:255–265
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Hatzipetros T, Bogdanik LP, Tassinari VR, Kidd JD, Moreno AJ, Davis C, Osborne M, Austin A, Vieira FG, Lutz C, Perrin S (2014) C57BL/6J congenic Prp-TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Res 1584:59–72
Heiman-Patterson TD, Sher RB, Blankenhorn EA, Alexander G, Deitch JS, Kunst CB, Maragakis N, Cox G (2011) Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: a window of opportunity in the search for genetic modifiers. Amyotroph Lateral Scler 12:79–86
Horn M, Baumann R, Pereira JA, Sidiropoulos PN, Somandin C, Welzl H, Stendel C, Luhmann T, Wessig C, Toyka KV, Relvas JB, Senderek J, Suter U (2012) Myelin is dependent on the Charcot-Marie-Tooth Type 4H disease culprit protein FRABIN/FGD4 in Schwann cells. Brain 135:3567–3583
Hu B, Arpag S, Zhang X, Mobius W, Werner H, Sosinsky G, Ellisman M, Zhang Y, Hamilton A, Chernoff J, Li J (2016) Tuning PAK activity to rescue abnormal myelin permeability in HNPP. PLoS Genet 12:e1006290
Hubers A, Just W, Rosenbohm A, Muller K, Marroquin N, Goebel I, Hogel J, Thiele H, Altmuller J, Nurnberg P, Weishaupt JH, Kubisch C, Ludolph AC, Volk AE (2015) De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging 36(3117):e3111–e3116
Hussain R, Ghoumari AM, Bielecki B, Steibel J, Boehm N, Liere P, Macklin WB, Kumar N, Habert R, Mhaouty-Kodja S, Tronche F, Sitruk-Ware R, Schumacher M, Ghandour MS (2013) The neural androgen receptor: a therapeutic target for myelin repair in chronic demyelination. Brain 136:132–146
Ichiyanagi N, Fujimori K, Yano M, Ishihara-Fujisaki C, Sone T, Akiyama T, Okada Y, Akamatsu W, Matsumoto T, Ishikawa M, Nishimoto Y, Ishihara Y, Sakuma T, Yamamoto T, Tsuiji H, Suzuki N, Warita H, Aoki M, Okano H (2016) Establishment of in vitro FUS-associated familial amyotrophic lateral sclerosis model using human induced pluripotent stem cells. Stem Cell Reports 6:496–510
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353:603–608
Japtok J, Lojewski X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, Putz S, Demestre M, Boeckers TM, Ludolph AC, Liebau S, Storch A, Hermann A (2015) Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 82:420–429
Kabashi E, El Oussini H, Bercier V, Gros-Louis F, Valdmanis PN, McDearmid JR, Mejier IA, Dion PA, Dupre N, Hollinger D, Sinniger J, Dirrig-Grosch S, Camu W, Meininger V, Loeffler JP, Rene F, Drapeau P, Rouleau GA, Dupuis L (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22:2350–2360
Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE (2010) NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68:668–681
Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955
King A, Troakes C, Smith B, Nolan M, Curran O, Vance C, Shaw CE, Al-Sarraj S (2015) ALS-FUS pathology revisited: singleton FUS mutations and an unusual case with both a FUS and TARDBP mutation. Acta Neuropathol Commun 3:62
Kino Y, Washizu C, Kurosawa M, Yamada M, Miyazaki H, Akagi T, Hashikawa T, Doi H, Takumi T, Hicks GG, Hattori N, Shimogori T, Nukina N (2015) FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol Commun 3:24
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Kwon HS, Johnson TV, Joe MK, Abu-Asab M, Zhang J, Chan CC, Tomarev SI (2013) Myocilin mediates myelination in the peripheral nervous system through ErbB2/3 signaling. J Biol Chem 288:26357–26371
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448
Lim SM, Choi WJ, Oh KW, Xue Y, Choi JY, Kim SH, Nahm M, Kim YE, Lee J, Noh MY, Lee S, Hwang S, Ki CS, Fu XD, Kim SH (2016) Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol Neurodegener 11:8
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438
Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW (2009) Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci USA 106:4465–4470
Mackenzie IR, Ansorge O, Strong M, Bilbao J, Zinman L, Ang LC, Baker M, Stewart H, Eisen A, Rademakers R, Neumann M (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol 122:87–98
Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M (2011) Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121:207–218
Marino M, Papa S, Crippa V, Nardo G, Peviani M, Cheroni C, Trolese MC, Lauranzano E, Bonetto V, Poletti A, DeBiasi S, Ferraiuolo L, Shaw PJ, Bendotti C (2015) Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 36:492–504
Masocha W, Parvathy SS (2009) Assessment of weight bearing changes and pharmacological antinociception in mice with LPS-induced monoarthritis using the Catwalk gait analysis system. Life Sci 85:462–469
Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CC, Cooper JD, Greensmith L, Shaw CE (2013) Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol 125:273–288
Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E, Bozzoni I (2012) FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J 31:4502–4510
Motley WW, Palaima P, Yum SW, Gonzalez MA, Tao F, Wanschitz JV, Strickland AV, Loscher WN, De Vriendt E, Koppi S, Medne L, Janecke AR, Jordanova A, Zuchner S, Scherer SS (2016) De novo PMP2 mutations in families with type 1 Charcot-Marie-Tooth disease. Brain 139:1649–1656
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nicholson GA, Valentijn LJ, Cherryson AK, Kennerson ML, Bragg TL, DeKroon RM, Ross DA, Pollard JD, McLeod JG, Bolhuis PA et al (1994) A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet 6:263–266
Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 28:131–138
Peters OM, Ghasemi M, Brown RH Jr (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125:2548
Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Kusters B, Van Den Bosch L, Van Damme P, Richardson WD, Robberecht W (2013) Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136:471–482
Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, Wang H, Shorter J, Filiano AJ, Roberson ED, Tourtellotte WG, Chen B, Tsai LH, Huang EJ (2014) ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124:981–999
Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13:195–204
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17:661–663
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96
Scekic-Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, Wagner M, Dieterle S, Sinniger J, Dirrig-Grosch S, Drenner K, Birling MC, Qiu J, Zhou Y, Li H, Fu XD, Rouaux C, Shelkovnikova T, Witting A, Ludolph AC, Kiefer F, Storkebaum E, Lagier-Tourenne C, Dupuis L (2016) Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J 35:1077–1097
Schoen M, Reichel JM, Demestre M, Putz S, Deshpande D, Proepper C, Liebau S, Schmeisser MJ, Ludolph AC, Michaelis J, Boeckers TM (2015) Super-resolution microscopy reveals presynaptic localization of the ALS/FTD related protein FUS in hippocampal neurons. Front Cell Neurosci 9:496
Sedelis M, Schwarting RK, Huston JP (2001) Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav Brain Res 125:109–125
Sephton CF, Tang AA, Kulkarni A, West J, Brooks M, Stubblefield JJ, Liu Y, Zhang MQ, Green CB, Huber KM, Huang EJ, Herz J, Yu G (2014) Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc Natl Acad Sci USA 111:E4769–E4778
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA (2016) ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7:10465
Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, Suzuki N (2016) Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 139:2380–2394
Skoff RP, Ghandour MS (1995) Oligodendrocytes in female carriers of the jimpy gene make more myelin than normal oligodendrocytes. J Comp Neurol 355:124–133
Suarez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Gobl C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp MD, Mackenzie IR, Ansorge O, Dormann D, Haass C (2016) Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol 131:587–604
Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S, Kopelevich A, Kaspar BK, Lagier-Tourenne C, Cleveland DW (2015) Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci USA 112:E6993–E7002
Suzuki N, Kato S, Kato M, Warita H, Mizuno H, Kato M, Shimakura N, Akiyama H, Kobayashi Z, Konno H, Aoki M (2012) FUS/TLS-immunoreactive neuronal and glial cell inclusions increase with disease duration in familial amyotrophic lateral sclerosis with an R521C FUS/TLS mutation. J Neuropathol Exp Neurol 71:779–788
Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, Julien JP (2011) Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 134:2610–2626
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670
Tian F, Yang W, Mordes DA, Wang JY, Salameh JS, Mok J, Chew J, Sharma A, Leno-Duran E, Suzuki-Uematsu S, Suzuki N, Han SS, Lu FK, Ji M, Zhang R, Liu Y, Strominger J, Shneider NA, Petrucelli L, Xie XS, Eggan K (2016) Monitoring peripheral nerve degeneration in ALS by label-free stimulated Raman scattering imaging. Nat Commun 7:13283
Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S (2012) Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet 21:136–149
Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, Sobue G, Yamanaka K (2013) Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5:221–234
Turner BJ, Ackerley S, Davies KE, Talbot K (2010) Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum Mol Genet 19:815–824
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034.0031–0034.0011
Verbeeck C, Deng Q, Dejesus-Hernandez M, Taylor G, Ceballos-Diaz C, Kocerha J, Golde T, Das P, Rademakers R, Dickson DW, Kukar T (2012) Expression of fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol Neurodegener 7:53
Waibel S, Neumann M, Rabe M, Meyer T, Ludolph AC (2010) Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology 75:815–817
Waibel S, Neumann M, Rosenbohm A, Birve A, Volk AE, Weishaupt JH, Meyer T, Muller U, Andersen PM, Ludolph AC (2013) Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur J Neurol 20:540–546
Wang XH, Lu G, Hu X, Tsang KS, Kwong WH, Wu FX, Meng HW, Jiang S, Liu SW, Ng HK, Poon WS (2012) Quantitative assessment of gait and neurochemical correlation in a classical murine model of Parkinson’s disease. BMC Neurosci 13:142
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 106:18809–18814
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 107:3858–3863
Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L (2010) Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci 30:10851–10859
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253
Zenker J, Stettner M, Ruskamo S, Domenech-Estevez E, Baloui H, Medard JJ, Verheijen MH, Brouwers JF, Kursula P, Kieseier BC, Chrast R (2014) A role of peripheral myelin protein 2 in lipid homeostasis of myelinating Schwann cells. Glia 62:1502–1512
Zhou Y, Liu S, Liu G, Ozturk A, Hicks GG (2013) ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet 9:e1003895
Zou ZY, Cui LY, Sun Q, Li XG, Liu MS, Xu Y, Zhou Y, Yang XZ (2013) De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol Aging 34(1312):e1311–e1318
Acknowledgements
Open access funding provided by Max Planck Society. We thank Dr Fabien Alpy (Igbmc, Illkirch, France) and Dr. Said Ghandour (Strasbourg, France) for their kind gift of CNPase and carbonic anhydrase antibodies. This work was supported by ALS Association Investigator Initiated Award (Grants 2235, 3209 and 8075; to LD and CLT); the Frick Foundation (award 2010 to ES, award 2013 to LD and CLT); Association Française contre les Myopathies (Grant #18280; to LD, ES and CLT); Virtual Helmholtz Institute “RNA dysmetabolism in ALS and FTD” (WP2, to LD and ACL) and agence nationale de la recherche (ToFU, EpiFUS, to LD). Work in our laboratories is supported by fondation «recherche sur le cerveau» (call 2015, to LD), Fondation Thierry Latran (SpastALS, to LD), ARSla (call 2014 and 2016 to LD, call 2015 to FR), the Max Planck Society (to ES), the Minna-James-Heinemann-Stiftung (to ES), the Muscular Dystrophy Association (MDA) (to ES), the European Research Council under the European Union’s Seventh Framework Program (FP7/2007–2013) ERC Grant Agreement No. 321366-Amyloid (to CH), the Deutsche Forschungsgemeinschaft (German Research Foundation, to CH and DD) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy) and the Emmy Noether program (DO 1804/1-1, to DD).
Authors contributions
JSZ, CLT, ES, and LD designed research; JSZ, HEO, SM, KD, MW, KA, SD, JS, SDG, FR and ES performed research; SY, DD, CH, and ACL contributed reagents and data analysis; JSZ, CLT, ES and LD analyzed data and made figures; and JSZ and LD wrote the manuscript. CLT supervised RNAseq and its analysis; ES supervised phenotypic analysis of Fus +/− mice; LD supervised phenotypic and pathological analysis of Fus ΔNLS/+ mice and reversal of the mutation by Chat-CRE.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Scekic-Zahirovic, J., Oussini, H.E., Mersmann, S. et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol 133, 887–906 (2017). https://doi.org/10.1007/s00401-017-1687-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1687-9