
Abstract
Background
Biofuel production from conversion of biomass is indispensable in the portfolio of renewable energies. Complex microbial communities are involved in the anaerobic digestion process of plant material, agricultural residual products and food wastes. Analysis of the genetic potential and microbiology of communities degrading biomass to biofuels is considered to be the key to develop process optimisation strategies. Hence, due to the still incomplete taxonomic and functional characterisation of corresponding communities, new and unknown species are of special interest.
Results
Three mesophilic and one thermophilic production-scale biogas plants (BGPs) were taxonomically profiled using high-throughput 16S rRNA gene amplicon sequencing. All BGPs shared a core microbiome with the thermophilic BGP featuring the lowest diversity. However, the phyla Cloacimonetes and Spirochaetes were unique to BGPs 2 and 3, Fusobacteria were only found in BGP3 and members of the phylum Thermotogae were present only in the thermophilic BGP4. Taxonomic analyses revealed that these distinctive taxa mostly represent so far unknown species. The only exception is the dominant Thermotogae OTU featuring 16S rRNA gene sequence identity to Defluviitoga tunisiensis L3, a sequenced and characterised strain. To further investigate the genetic potential of the biogas communities, corresponding metagenomes were sequenced in a deepness of 347.5 Gbp in total. A combined assembly comprised 80.3 % of all reads and resulted in the prediction of 1.59 million genes on assembled contigs. Genome binning yielded genome bins comprising the prevalent distinctive phyla Cloacimonetes, Spirochaetes, Fusobacteria and Thermotogae. Comparative genome analyses between the most dominant Thermotogae bin and the very closely related Defluviitoga tunisiensis L3 genome originating from the same BGP revealed high genetic similarity. This finding confirmed applicability and reliability of the binning approach. The four highly covered genome bins of the other three distinct phyla showed low or very low genetic similarities to their closest phylogenetic relatives, and therefore indicated their novelty.
Conclusions
In this study, the 16S rRNA gene sequencing approach and a combined metagenome assembly and binning approach were used for the first time on different production-scale biogas plants and revealed insights into the genetic potential and functional role of so far unknown species.
Similar content being viewed by others

Background
The reorientation of the global energy industry towards renewable energy sources is one of the major challenges of this century. The development of techniques using these resources contributes to the reduction of traditional fossil fuel usage [1]. Within the agricultural sector of renewable energy sources, energy generation by decomposition of organic materials has become one of the most important techniques. In particular, the production of biogas represents an economically attractive technology to generate bioenergy [2].
In general, organic material is anaerobically decomposed by complex consortia of microorganisms. The final product of this fermentation process is biogas with methane as the main compound. Several studies investigated and characterised the microbial community composition of agricultural biogas reactors. Among the bacterial community members, those of the classes Clostridia and Bacteroidetes dominate the biogas microbial subcommunities, followed by Proteobacteria, Bacilli, Flavobacteria, Spirochaetes and Erysipelotrichi. Within the domain Archaea, the methanogenic orders Methanomicrobiales, Methanosarcinales and Methanobacteriales were described to be frequently dominant [3–8].
Usually, the anaerobic fermentation process is practised at mesophilic (35–40 °C) or thermophilic (55–60 °C) conditions. Mesophilic biogas plants are typically fed with energy crops in contrast to thermophilic reactors which also convert complex manure mixtures, industrial food residues and organic household wastes [9]. The digestion process under mesophilic conditions requires less process heat and is described to be stable due to the larger diversity of microorganisms, explaining its broader usage [10–12]. In comparison, thermophilic plants show higher methane content of the biogas, faster process turnover rates and a sanitising effect [10]. Depending on the process conditions, such as temperature and fed substrates, differences in the biogas microbiome have been observed [13, 14].
To analyse the structure and function of biogas communities, high-throughput 16S rRNA gene amplicon as well as metagenome sequencing has been applied frequently [3, 4, 7, 15–18]. Still, the complex microbial consortia involved in anaerobic digestion are not fully understood, since many species of the process are unknown and uncharacterised. Culturing of single species is mostly difficult and does not cover the community’s complexity. For culture-independent functional characterisation of microbial species, genome binning from metagenome sequence data has been done for laboratory scale, but not yet on production-scale biogas fermenters [19].
The aim of this study was to compare community structures of different mesophilic and thermophilic production-scale biogas plants for the identification of distinctive taxa and their functional potential. Taxonomic community profiling was achieved by high-throughput 16S rRNA gene amplicon sequencing, whereas ultra-deep metagenome sequencing, assembly of metagenome reads and subsequent binning of obtained contigs resulted in genome bins providing the basis for genome-centred metabolic reconstructions. Genome bins enabled functional predictions for so far unknown and distinctive taxa of the biogas communities analysed.
Methods
Sampling at production-scale biogas plants and DNA extraction
Fermentation samples were taken directly from the main fermenters of four different production-scale biogas plants (BGPs 1, 2, 3 and 4, see Table 1). Before sampling at the sampling devices installed at the BGPs, the reactor content was stirred and dead volumes of the outlet pipelines were discarded. One litre fermentation sludge was then filled into a gas tight bottle, respectively, excess air was removed and the bottle tightly closed with a screw cap. Samples were immediately transferred to the laboratory maintaining the process temperature of the sample and then processed for total community DNA extraction. Whole community DNA was prepared from fermenter samples applying the protocol as follows:
A 26 g aliquot of fermenter sludge was mixed with 50 ml of 1 M phosphate buffered saline solution (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4), centrifuged at 9000×g for 5 min, the supernatant discarded. The pellet was resuspended in 50 ml PBS (4 °C), shaken at 400 rpm, centrifuged at 200×g for 5 min and the supernatant collected. These steps were repeated three times to wash off the microbial biomass off the substrate fibres. The collected supernatant was centrifuged at 9000×g for 5 min, the pellet resuspended in 40 ml PBS and further centrifuged at 5000×g for 15 min, supernatant discarded. For cell disruption, the pellet was resuspended in CTAB containing DNA extraction buffer (DEP, described previously in [20], 5 mg Pronase ε (Serva Electrophoresis GmbH, Heidelberg, Germany) and 2 mg RNAse (Qiagen, Germantown, MD, USA) added and shaken at 180 rpm and 37 °C for 1 h. Afterwards, 30 ml of 10 % SDS solution were added and the suspension incubated in a water bath at 65 °C for 2 h, inverting the suspension every 15 min. After centrifugation at 3900×g for 10 min, the supernatant was filtered through a folded filter (pore size 15–18 µm) and the filtrate mixed 1:1 (v/v) with a 24:1 chloroform/isoamyl alcohol (v/v) mixture, followed by centrifugation at 8000×g and 4 °C for 5 min. The upper phase was taken off, mixed 1:0.7 (v/v) with isopropyl alcohol and left at room temperature for 1 h. The DNA was pelleted by centrifugation at 9000×g and 4 °C for 20 min. From here, the NucleoBond AX-G (Macherey–Nagel, Düren, Germany) ion exchanger columns and solutions were used for DNA purification according to the manufacturer’s instructions, starting with resuspension of DNA in 2 ml N2-Buffer and overnight incubation at 60 °C. In the end, the DNA was resuspended in 100 µl TE buffer (1 m mM Tris, 1 mM EDTA, pH 8.0) and overnight incubated at 4 °C. DNA concentration was measured using the NanoDrop 2000 Spectrophotometer (Thermo Scientific, Waltham, USA). The DNA extraction above was done in quadruplicates and two DNA replicates were sent to the DOE Joint Genome Institute (JGI) in Walnut Creek, California, USA for 16S rRNA gene amplicon and metagenomic sequencing. Samples for (chemical) process parameter measurements were taken and analysed separately and independently by the biogas plants’ operators.
High-throughput 16S rRNA gene amplicon sequencing
To describe and characterise the biogas-producing microbial community composition the high-throughput 16S rRNA gene amplicon sequencing approach was applied as published previously [21]. Library preparation and sequencing were done at the DOE JGI. Briefly, to amplify the hypervariable region V4 of the 16S rRNA gene, the primers 515F 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R 5′-GGACTACHVGGGTWTCTAAT-3′, covering the domains Bacteria and Archaea [21], multiplex identifier (MID) tags and Illumina-specific sequencing adaptor sequences were used. Afterwards, the fragments of expected length (approx. 300 bp) were amplified by PCR. Obtained PCR products were purified with AMPureXP® magnetic beads (Beckman Coulter GmbH, Brea, CA, USA). Qualitative and quantitative analysis of the generated 16S rRNA gene amplicons was performed using the Agilent 2100 Bioanalyzer system (Agilent, Santa Clara, CA, USA) and afterwards pooled together in equimolar amount. Finally, the constructed amplicon libraries were sequenced on the Illumina MiSeq system applying the paired-end protocol.
16S rRNA gene sequence processing and quality control
The raw 16S rRNA gene sequencing reads were preprocessed by JGI’s iTagger amplicon analysis pipeline as to perform quality control. It was used to remove contaminants, e.g. PhiX control, sequencing library adapter dimers, etc., deplete sequencing primers and merge read pairs. Further contaminants removal was performed by DUK (v1.05) [22]. Read pairs with at least one read matching against the PhiX genome or the Illumina-specific sequencing artefact library were removed from the library. Adapter trimming was performed through cutadapt (v1.2.1) providing the sequences of the sequencing primers 515F and 806R [23]. Finally, Flash (v1.2.6) was used for iterative read pair merging through consecutive trimming of the read pairs until these could be merged by removing failing read pairs from the library and a final filter with a threshold of 0.3 errors per 100 bp [24]. Before subsequent analysis with the QIIME NGS analysis pipeline [25], the eight libraries were initially merged into a sample tagged QIIME accessible format, including an additional quality control step checking for min base quality of 20 on Phred scale and truncating sequences if necessary.
Operational taxonomic unit (OTU) clustering and taxonomic classification of 16S rRNA gene sequences
Further analysis of the 16S rRNA gene reads was performed within the QIIME analysis pipeline for operational taxonomic unit (OTU) clustering and subsequent taxonomic classification of the OTU representatives. From these OTUs, distinctive taxa per sample have been identified and investigated more closely. The pre-filter +4 step open reference based OTU picking workflow from QIIME v1.9.1 was used in combination with Usearch (v7.0.1090, 64bit) and Greengenes 16S rRNA gene database (v13_08, 97 % identity) as reference dataset [26, 27]. The representative sequences of each OTU were aligned using PyNAST (v0.1) [28], where OTUs with sequences failing to be aligned were removed from the final OTU table. Hence, the community profiles were corrected with CopyRighter (v0.46) [29] to account for different 16S rRNA gene copy numbers within the microbial community. The OTU representatives of the four distinctive phyla were placed into the All-Species Living Tree LTPs123 [30]. Sequences were aligned using the SINA alignment service v.1.2.11 online. The LTPs123 tree and SINA alignments were loaded into ARB [31] and sequences placed into the existing LTP tree using ARB’s parsimony method.
Preparation and metagenome sequencing of total DNA from biogas-producing microbial communities
Library preparation and sequencing from total DNA were done at the DOE JGI. For sequencing purposes, 100 ng of total DNA was sheared to 270 bp fragments using a focused-ultrasonicator (Covaris, Woburn, MA, USA). The DNA fragments were purified and size selected using SPRI beads (Beckman Coulter, Brea, CA, USA). Obtained fragments were blunt-end-repaired, phosphorylated and A-tailed. Subsequently, T-tailed adapters, containing sequences used during cluster formation and Illumina compatible adapters (IDT, San Jose, CA, USA), were ligated to the purified DNA fragments applying the KAPA-Illumina library creation kit (KAPA Biosystems, Wilmington, MA, USA). The prepared sample libraries were quantified applying the KAPA Biosystem’s next-generation sequencing library qPCR kit (KAPA Biosystems, Wilmington, MA, USA) and run on the Roche LightCycler 480 real-time PCR instrument (Roche Basel, Switzerland). Sequencing of the libraries was performed on the Illumina HiSeq 2000 sequencer using the Illumina TruSeq SBS v3-HS kit, following a 2 × 150 indexed high output run protocol.
Metagenome assembly and binning
To reconstruct low-abundance community members and to facilitate downstream genome binning, all sequencing data was combined after quality control (JGI QC pipeline: sequencing artefact removal, removal of reads containing ambiguous (N) bases and filtering based on quality). Ray Meta (v2.3.0) [32] was used for assembly of the pooled sequencing data of all samples, using a k-mer size of 31. To estimate the inclusivity of our metagenome assembly, all sequencing reads were aligned to the assembled contigs with Bowtie 2 (v2.2.4) [33]. SAMtools (v1.0) [34] was used to convert SAM to BAM, sort the alignment file and calculate read mapping statistics.
The gene prediction tool Prodigal v.2.6.0 [35] was used to predict genes on assembled contigs larger than 1 kb. Predicted protein sequences were compared to NCBI’s database using the BLASTP mode of DIAMOND [36]. The resulting output file was loaded into MEGAN5 [37] for taxonomic classification of each gene sequence.
To divide the metagenome assembly into genome bins, MetaBAT (v0.21.3) [38] was used in its very specific mode. Completeness, contamination, and strain heterogeneity were estimated with CheckM (v1.0.4) [39], using sets of clade-specific single-copy marker genes. Genome bins of distinct taxa were identified by (A) counting the aforementioned taxonomic assignments on gene level, and (B) running taxator-tk (v1.2.1; binning-workflow-fasta-blast.sh) [40] to additionally assign a taxon label on contig level. These two approaches were largely in agreement, identifying high-confident genome bins for the taxa of interest. For each taxon, we considered only the most complete and less contaminated genome bin, as estimated by CheckM, for further analyses.
Genome bins were annotated and analysed within the GenDB 2.0 annotation system [41], additionally using the KEGG pathway mapping and BLASTP tool implemented in the system. For gene content comparisons, genome bins and corresponding reference genomes were analysed using the EDGAR 2.0 software [42]. Here, orthologous genes (hereafter referred to as ‘shared genes’) that the compared genomes have in common and singletons (hereafter referred to as ‘unique genes’) that do not have any orthologous counterpart in the respective reference genome were determined based on BLASTP.
Results and discussion
Parameters of the three mesophilic and one thermophilic industrial biogas plants analysed
In this study, four different production-scale biogas plants (BGPs) were compared on taxonomic and functional level, based on high-throughput 16S rDNA amplicon and ultra-deep metagenome sequencing. The BGPs analysed are located in North-Rhine-Westphalia, Germany, and regarding their construction, mainly differ in the number of fermenters, size, process temperature and fed substrates. The main fermenters of the biogas plants BGP1, BGP2 and BGP3 are continuously stirred tank reactors (CSTRs), while in the thermophilic BGP4 mixing of the substrate is achieved by pumping it through the reactor. Regarding the operating temperature, BGP4 is a thermophilic biogas plant (54 °C), whereas the other three BGPs were operated under mesophilic conditions (approx. 40 °C). The substrates of all four biogas plants were based on maize silage with the addition of different manure types. In BGP2 and 4, grass silage was also added and BPG1 is unique due to its fermentation of sugar beet as substrate. All BGPs showed stable biogas production and process parameters around the time of sampling (data not shown).
To interpret microbial compositions within the fermenters, other physico-chemical parameters are of importance. Table 1 summarises all parameters of the four fermenters, measured around the time of sampling, and corresponding optimal parameter ranges as taken from different sources [43–45]. Almost all measured parameters of the four BGPs are in the recommended optimal range. One exception is the total inorganic carbon (TIC) value of BGP2, which is above the recommended level and also the volatile organic acids (VOA) concentration is relatively high, resulting in a VOA/TIC ratio that is within the range of 0.11 to 0.6. This indicates that the system is well buffered [46]. Other characteristics are the acetic acid equivalents (HAC-eq) of BGP2 and 4, which are below the optimal range. However, all other parameters including ammonium/ammonia concentrations of these BGPs are within the optimal range. Only BGP2 and 3 have ammonium/ammonia concentrations in the higher range of the optimum and are considered here as ammonia-stressed. BGP4 shows the highest biogas per kg of organic dry matter (l/kg oDM) output and also shows the highest percentage of methane. Both findings are in accordance with increased methane content of thermophilic biogas plants found in the literature [10, 11, 47] indicating that thermophilic biogas plants generally have higher biogas outputs, due to heat-induced increase of enzymatic activity. This is also supported by the data in this study. The finding that the other biogas outputs are only slightly lower, especially for BGP2, shows that also other process parameters, such as substrate type and pH, have a significant influence on the microbial community and the biogas output, which would be consistent with the literature [10, 12, 48, 49].
Sequencing results for 16S rRNA gene amplicons and metagenomes
To study the taxonomic microbial community compositions of the four studied BGPs, high-throughput 16S rRNA gene sequencing was done in duplicates on total community DNA extracted from reactor samples of each BGP. The microbial taxonomic composition based on the 16S rRNA gene sequencing data was determined using the QIIME software package and additional CopyRighter analysis for gene copy number corrections. 16S rRNA gene amplicon sequencing and quality control (QC) results are summarised in Table 2. To determine the functional potential of the communities, metagenome sequencing was done, of which statistics and QC results are shown in Table 3. In total, approx. 2.3 billion reads (347 Gb; Table 3) were generated, the deepest sequencing of biogas community metagenomes so far.
The microbiomes of one thermophilic and three mesophilic production-scale biogas plants determined by high-throughput 16S rRNA gene amplicon sequencing
Similarities and differences of the microbiomes prevailing in the four analysed BGPs
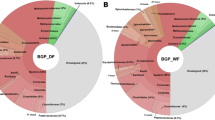
To analyse and interpret the taxonomic structure of the microbial communities residing in the three mesophilic and one thermophilic biogas plants, OTU clustering of the 16S rRNA gene sequence data was done for two biological replicates of each BGP. OTUs were clustered on taxonomic ranks from phylum to genus level, calculating their respective percentage share within the respective sample. Figure 1 shows the microbial taxonomic profile for each replicate, with percentage shares for each phylum. The vast majority of taxa prevailing in all four BGPs was assigned to the bacterial superkingdom with between 97.37 % in biogas plant 4 (replicate 1) and 99.19 % (replicate 2), while Archaea have a share of between 0.36 % (BGP1) and 2.25 % (BGP4). These results are in accordance with other 16S rRNA gene sequencing studies addressing microbial communities of anaerobic methane-producing reactor systems [8, 50, 51]. Compared with BGP1, 2 and 3, BGP4 shows the lowest diversity among bacterial and archaeal taxa based on the Shannon index of 7.5, 7.4, 6.8 and 5.7, respectively. This is most likely due to the higher process temperature of BGP4. For similar thermophilic systems, it has been shown that the temperature has the main influence on microbial community structures [52–54]. Regarding temperature differences between the three mesophilic and the only thermophilic BGPs, community profiles in general reflect previous findings obtained for similar anaerobic reactor systems [50, 55, 56].

Taxonomic profiles of the four biogas plants (BGPs) on phylum level, based on 16S rRNA gene amplicon sequencing. The respective relative abundances of the replicates for each BGP are shown. Four taxa, distinctly and abundantly present in one or two of the BGPs, were identified: Thermotogae, Fusobacteria, Spirochaetes and Cloacimonetes, highlighted with asterisks
Within the bacterial superkingdom, ten phyla were identified in all BGPs, namely Firmicutes, Spirochaetes, Bacteroidetes, Proteobacteria, Tenericutes, Actinobacteria, Synergistetes, Fibrobacteres, Chloroflexi and OP9 division (Fig. 1). The proportions of these phyla differ considerably between the four BGPs. Sequences assigned to the phylum Firmicutes are less abundant in BGP2 and 3 in comparison to BGP1 and 4, with BGP1 having the highest share (Fig. 1). Their dominance in biogas and other fermentation reactors with cellulose-rich substrates was found frequently, underlining their importance and specific adaptation abilities [3, 5, 56–58]. In BGPs 1, 3 and 4 members of the phylum Bacteroidetes are the second most abundant bacterial group, while in BGP2 they feature a lower relative abundance, due to a slightly higher proportion of Spirochaetes. The relatively high abundance of Bacteroidetes in BGP4 is surprising, as it has been shown that members of this phylum are sensitive to high temperatures [57, 59, 60].
The archaeal superkingdom in all samples exclusively comprises the phylum Euryarchaeota (Fig. 1) that on class level, is represented by Methanomicrobia (0.31–1.84 %), Methanobacteria (0.01–0.34 %) and Thermoplasmata (0.03–0.08 %) (data not shown). In all four BGPs, the order Methanomicrobiales is the most abundant, with Methanoculleus being the dominant genus accounting for 0.3 % (BGP1) to 1.8 % (BGP4) of all assigned sequences (not shown). This genus was found dominant in several other mesophilic and thermophilic biogas-producing communities and may outcompete other Archaea due to a broad temperature optimum spectrum (20–55 °C) and higher growth rate [4, 8, 18, 46, 50, 51, 61, 62]. However, the genus Methanothermobacter (order Methanobacteriales) is present only in the thermophilic BGP4, with a share of 0.29 % (not shown). Members of this genus are known to be thermophilic and often are dominant in thermophilic methane-producing microbial consortia [52, 60, 61]. In general, the mostly hydrogenotrophic genera dominated methanogenic communities indicate that they are mostly based on CO2 and formate as electron acceptors and H2 as electron donors for methanogenesis in all four BGPs. High affinities towards hydrogen and a better adaptation to lower hydrogen pressures may explain the strict dominance of the hydrogenotrophic metabolism [61, 62]. It is also possible that aceticlastic methanogens have been inhibited by elevated ammonium/ammonia concentrations in the reactors BGP2 and 3 as they all are in the upper part of the optimal range regarding this process parameter (Table 1). In case of the thermophilic BGP4, the operating temperature also drives methanogenesis towards the hydrogenotrophic mode, as it is thermodynamically more favourable and it has also been reported that hydrogenotrophic methanogenesis is the dominant pathway in thermophilic methane-producing reactors [45, 51, 54, 59, 60].
When comparing the phyla percentages of the four BGPs, it is noticeable that members of the Thermotogae are present in only one, Fusobacteria and Cloacimonetes members in only two and Spirochaetes members are present in only three of four BGPs, the latter being highly abundant in only two BGPs (Fig. 1). To further investigate these distinct taxonomic features, community profiles were followed down towards deeper taxonomic levels.
The phylum Thermotogae and its members are present only in the thermophilic BGP4
The taxonomic profile of BGP4 features a high share of Thermotogae (approx. 7.5 %), which are absent (BGP2/3) or present only in very low abundances (approx. 0.02 %, BGP1) in the mesophilic BGPs (Fig. 1). All 16S rRNA gene sequences assigned to the phylum Thermotogae were classified as class Thermotogae, order Thermotogales and family Thermotogaceae (see Add. File 1).
Members of the phylum Thermotogae were also found to be present in similar reactors operated under mesophilic conditions (31–41 °C) [18, 54, 63, 64], but were identified more frequently and in higher abundances in anaerobic reactors operated at thermophilic temperatures (50–60 °C) [54, 59, 64]. The results of this study reflect previous findings, which can be explained by the temperature optimum for Thermotogae members being mostly in the range of approx. 50–60 °C. The existence of mesophilic (‘mesotoga’) and hyperthermophilic members of the phylum were also described and at least for the latter group, several genome sequences are available in the literature [65–68]. However, the prevailing temperature of 54 °C in BGP4 meets the preferred temperature demands of thermophilic Thermotogae species, explaining their presence in this reactor. Moreover, their high share and the lower abundances of other bacterial phyla (i.e. Proteobacteria, Tenericutes, Cloacimonetes, see Fig. 1) is most likely due to the adaptation-based outcompeting effect at the expense of those community members that are not adapted to high temperatures. In anaerobic fermentation of biomass, Thermotogae members are involved in the degradation of cellulose and highly complex polysaccharides, producing acetate, carbon dioxide and hydrogen, and therefore are involved in hydrolysis and acetogenesis [64, 67, 69]. Additionally, members of the phylum Thermotogae are thought to be syntrophically associated with methanogenic Archaea, and therefore are essential for the maintenance of methane production [65, 69, 70]. It can be assumed that Thermotogae species within BGP4 have a similar metabolism and syntrophic character compared to reference species.
The phylum Fusobacteria and its members are solely present in the mesophilic BGP3
The taxonomic profile of BGP3 is distinct due to the presence of Fusobacteria, having an average share of 8.3 % of all rRNA gene sequences in the replicates, compared to 0–0.02 % in the communities of the other BGPs (Fig. 1). All Fusobacteria sequences from BGP3 were assigned to the class Fusobacteria and the order Fusobacteriales (see Additional file 1).
Fusobacteria were also found in other studies focusing on microbial communities in biogas-producing reactors, but with a share below 2 % [6, 18]. Naturally, these anaerobic, mesophilic bacteria are found, e.g. in the mouth and gastrointestinal (GI) tract of humans, rats, cattle, sheep and chicken, were they can cause severe diseases [71–73]. Therefore, it can be assumed that Fusobacteria in BGP3 originated from the fed cattle manure. Frequently, the Fusobacterium species F. necrophorum and F. nucleatum are associated with infections in humans and animals [71], but neither of these species was identified within BGP3.
Since the focus of research is more on the pathogenic Fusobacteria species, nothing is known about the role of Fusobacteria within methane-producing biogas plants. Hence, the lack of reference genomes of species playing a role in these systems hampered a taxonomic classification on lower ranks. Their absence in BGP4 can be explained by the high temperature, since these bacteria most likely are adapted to the mesophilic body temperatures of their hosts. One explanation for their absence in BGPs 1 and 2, despite added manure, could be the higher pH values of corresponding fermentation samples. It is known that Fusobacteria prevailing in the microbial communities of human oral biofilms live at fluctuating pH values of 6.3–7.0 [74]. BGP3 had the lowest pH value (7.53) of all BGPs analysed. However, due to the lack of information on the phylum Fusobacteria in the context of biomass fermentation, it cannot be understood clearly why these bacteria are present only in BGP3.
The phylum Spirochaetes and its members are most abundant only in the mesophilic plants BGP2 and BGP3
The biogas plants 2 and 3 both have the highest proportion of Spirochaetes, with average shares of 10.6 and 4.7 % of all 16S rRNA gene sequence reads, respectively, while in BGP1 the average share of Spirochaetes is 0.92 % and BGP4 almost completely lacks this phylum (0.01 %) (Fig. 1). All Spirochaetes sequences from BGP3 and the majority in BGP2 were assigned to the class Spirochaetes and the order Spirochaetales (see Additional file 1).
Not much is known about non-pathogenic Spirochaetes since most members of this phylum are described to be human or animal pathogens [75]. Non-pathogenic Spirochaetes, especially Treponema species, can be found in termite guts, where they form a symbiosis with their hosts [76, 77]. In the context of anaerobic digestion of municipal and/or agricultural wastes, representatives of this phylum were mostly found in mesophilic anaerobic reactors fed with swine manure and cellulose-rich substrates. It is assumed that within these environments Spirochaetes are involved in cellulose degradation [56, 78, 79]. In BGP2 and 3, swine manure and the cellulose-rich substrates maize and grass silage were fed, which may explain the high abundance of Spirochaetes in the respective microbial communities. Although BGP4 is fed with pig manure, the phylum Spirochaetes is underrepresented which may be explained by the thermophilic conditions prevailing in BGP4. Spirochaetes are probably not adapted to higher temperatures, and therefore, their function was adopted by other community members such as for example Thermotogae species.
The candidate phylum Cloacimonetes (WWE1) was only identified in the mesophilic biogas plants BGP2 and BGP3
In the taxonomic profiles of the BGPs 2 and 3, an average share of 3.7 and 4.5 % of all 16S rRNA gene sequences was assigned to the candidate phylum Cloacimonetes (WWE1), respectively, while in the BGP1 and 4, this phylum is almost absent with an average of max. 0.05 % (Fig. 1). All Cloacimonetes (WWE1) sequences from BGP3 and the majority in BGP2 were assigned to the class Cloacamonae and the order Cloacamonales (see Additional file 1). Since there is very little information about the phylum Cloacimonetes and its lower taxonomic levels, there are no alternatives to this class and order that were proposed by the RDP classifier used to classify the 16S rRNA gene sequence data.
The phylum was identified in 2005, named WWE1 (for waste water for Evry 1) and later renamed candidate phylum Cloacimonetes (WWE1) [80, 81]. Recently, it has been characterised as a separate phylum, belonging to the Fibrobacteres-Chlorobi-Bacteroidetes (FCB) superphylum and proposed to be a sister group of the phylum Spirochaetes [81]. Due to its novelty, missing reference genomes most probably hampered a deeper taxonomic classification of Cloacimonetes (WWE1) species residing in BGPs 2 and 3. Members of this phylum are mostly present in anaerobic habitats, such as biogas plants and the porcine digestive tract. It is assumed, and evidence is increasing, that corresponding bacteria are involved in mostly cellulose or sugar degradation, directly derived from cellulose and produce primarily acetate and hydrogen, which are further metabolised by their syntrophic partners, methanogenic Archaea [3, 78–84].
No reports exist for the presence of candidate phylum Cloacimonetes (WWE1) members in thermophilic environments. This supports the assumption that the temperatures in BGP4 exceed the optimum for species belonging to this candidate phylum. In BGPs 2 and 3, a lower share of Firmicutes, also being involved in hydrolysis, compared to the other two BGPs, can be observed. Possibly, Cloacimonetes (WWE1) species partly complemented the function of Firmicutes species. Recently, Cloacimonetes species were shown to increase in their abundance when the ammonium/ammonia concentration was high. This indicates that they can adapt to this condition and even seem to benefit from it when the system is ammonium/ammonia adapted [82]. Regarding the addition of swine manure, this may also explain the presence of Cloacimonetes species in BGPs 2 and 3, as these show the highest ammonia concentrations, especially BGP3, whose values are in the higher range of the optimum.
Most of the dominant distinctive taxa are not represented by closely related reference species in databases
Taxonomic profiling of the community structure revealed the presence of distinctive taxa that are present only in one or two of the four BGPs. This observation raises the question concerning the function of these distinctive taxa within the trophic network of the biogas plant’s microbial communities. To determine the closest relatives of distinctive taxa, corresponding dominant OTUs deduced from clustering of 16S rRNA gene sequence data were placed in a phylogenetic tree. Figure 2 shows condensed phylogenetic trees considering only the dominant OTUs of the taxa Thermotogae (OTU_ 777316, Fig. 2a) Spirochaetes (OTU_1139645), Fusobacteria (OTU_4357841) and Cloacimonetes (OTU_575765 and OTU_543067) (Fig. 2b). The complete phylogenetic tree can be found in the supplementary material (see Additional file 2).

Partial phylogenetic trees of all available type strains with operational taxonomic units (OTUs) of Thermotogae (a), Fusobacteria, Spirochaetes and Cloacimonetes (b) taxa of the studied biogas plants and their closest non-type strain relatives embedded. Type strains are in black, OTUs are in red, their closest relatives in purple, their closest sequenced relatives in blue and their closest type strain relatives in green. 16S rRNA sequence tree construction was done using the ARB software [31]
Of the four distinctive phyla, only the dominant OTU of the phylum Thermotogae shows close relatedness to an characterised bacterium (purple), followed by the type strain Defluviitoga tunisiensis SulfLac1T (green, [85]), and the recently characterised and sequenced non-type strain Defluviitoga tunisiensis L3 (blue, [86]) .
The Fusobacteria OTU derived from 16S rRNA gene sequence data of BGP3 was placed within a sister group of the Leptotrichiaceae. However, its closest relative is an uncultured bacterium belonging to the genus Fusobacterium (purple). Interestingly, the closest type strain relative belongs to the genus Psychrilyobacter (green), while the closest characterised and sequenced non-type strain relative is Fusobacterium varium ATCC 27725 (blue) (accession number NZ_ACIE00000000.2).
The representative OTU 16S rRNA gene sequence from BGP2 belonging to the phylum Spirochaetes was placed within the group of the genus Treponema of the family Spirochaetaceae. For this OTU, the most closely related sequence belongs to the uncultured type strain Treponema lecithinolyticum OMZ 684T (green, [87]) that is not further characterised. When comparing the query OTU to all 16S rRNA gene sequences available in the NCBI database, it appeared that the closest non-type strain relative is the genome sequenced bacterium Treponema brennaborense (blue, [88]). This indicates that the dominant OTU represents a new species of the genus Treponema that needs further characterisation.
The two most dominant representative OTU 16S rRNA gene sequences belonging to the phylum Cloacimonetes (WWE1) originate from BGP2 and 3, respectively, and are closely related to each other. They were placed within the phylum Spirochaetes, which is inconsistent regarding the newer literature, since Cloacimonetes has recently been classified as an autonomous sister group of the Spirochaetes [81]. The closest relative of the Cloacimonetes OTUs is an uncultured uncharacterised bacterium (purple) and the only characterised and closest relative is Candidatus Cloacamonas acidaminovorans (blue) [89].
In summary, with the exception of the Thermotogae OTU, no clear classification of the dominant OTUs representing the distinctive taxa could be achieved. The lack of suitable reference genomes hampers the classification of these OTUs and accordingly, information on the functional role of these taxa within the biogas process is currently not available. These issues cannot be solved by 16S rRNA gene sequence comparison. Exploration of the corresponding metagenome sequence datasets of the four biogas plants is needed to uncover the functional role of the identified distinctive taxa. This can be achieved by binning of metagenome contigs representing dominant species of the four distinctive taxa, their annotation and comparative analyses using sequenced and characterised relatives (blue, Fig. 2) identified in this section.
Reconstruction of genomes representing distinctive taxa applying metagenome assemblies and binning
Metagenome assembly and binning results
A total of 1.49 Gbp metagenomic data were assembled (Table 4) and contigs were sorted into 532 genome bins, five of which belong to the taxa of interest and met the stringent quality requirements as defined in the “Methods” section: One Thermotogae genome bin (206_Thermotogae), one Fusobacteria bin (175_Fusobacteria), one Spirochaetes bin (128_Spirochaetes) and two Cloacimonetes (WWE1) bins (120_Cloacimonetes; 244_Cloacimonetes) were chosen. The estimated amount of contamination is largely negligible and mostly due to strain heterogeneity (Table 5).
To deduce the metabolism of all five genome bins, they were annotated and analysed in the annotation platform GenDB 2.0 [41]. Encoded enzymes were mapped on KEGG pathways within GenDB to enable metabolic pathway reconstructions. An example of this analysis is given in Additional file 3, showing the coverage of the ‘Alanine, Aspartate and Glutamate metabolism’ KEGG pathway by enzymes encoded in the genome bin assigned to the phylum Fusobacteria.
The genome bin representing a Thermotogae species from the thermophilic biogas plant 4
Analyses of 16S rRNA gene sequences revealed a close relatedness of the dominant Thermotogae OTU from the thermophilic BGP4 to the strain D. tunisiensis L3 (Fig. 2a). Compilation of a Thermotogae genome bin derived from metagenomic contigs of BGP4 and comparison with the D. tunisiensis L3 genome was conducted to further determine the degree of similarity and evaluate the binning approach itself. The yielded genome bin assigned to the class Thermotogae (Table 5) is covered by 4.45 % of all BGP4 metagenome reads. In addition to the annotation and functional interpretation in GenDB, the genome bin was examined for the presence of genes encoding proteins involved in energy generation of D. tunisiensis [90]. Enzymes encoded in the Thermotogae bin and predicted to be involved in sugar utilisation and fermentation metabolism are listed in Additional file 4. These analyses indicated that the Thermotogae species represented by the bin is able to utilise xylose, glucose, mannose, galactose, lactose, maltose, fructose, ribose and l-lactate with acetate, CO2 and H2 as end products of the fermentation.
Results of the comparative analysis of the annotated Thermotogae genome bin and the reference sequence of D. tunisiensis L3 using EDGAR were visualized in a Venn diagram (see Fig. 3a, 206_Thermotogae). The vast majority of their genes are shared corroborating that the D. tunisiensis and the bin genome are closely related. Deeper analysis of the core gene set indicated that both species are anaerobic bacteria featuring a metabolism based on sugar fermentation (see Additional file 4). However, the unique gene set of D. tunisiensis L3 indicates that it can utilise a broader spectrum of carbohydrates, since it encodes genes for the import and fermentation of sugars that are missing in the genome bin. Analyses on the Thermotogae genome bin’s unique gene set showed that the vast majority (approx. 60 %) could not be functionally classified and the remaining ones do not provide any further information on this strain’s metabolism.

Venn diagrams showing the number of unique and shared genes between the five genome bins and their respective reference strains. Diagrams are shown for genome bins assigned to the phyla Thermotogae (206_Thermotogae, a), Fusobacteria (175_Fusobacteria, b), Spirochaetes (138_Spirochaetes, c) and Cloacimonetes (120_Cloacimonetes, 244_Cloacimonetes; d). Venn diagrams were redrawn manually, based on the original EDGAR output, with their areas drawn to scale
All above mentioned analyses indicate that the species represented by the bin has a metabolism based on sugar fermentation, with acetate, CO2 and H2 as end products of this process, which has also been predicted for the reference strain D. tunisiensis L3. It therefore can be assumed that Defluviitoga strains contribute to acetogenesis of biomass digestion within the biogas fermenter. Furthermore, obtained comparison results also support the idea that the analysed Defluviitoga species may be syntrophically associated with methanogenic Archaea that can utilise CO2 and H2 for methanogenesis [65, 69, 70]. Additionally, comparative analyses between the genome bin und the highly related reference genome to evaluate the binning approach were done with the outcome that the approach proved valuable and reliable.
The genome bin representing a Fusobacteria species from the mesophilic biogas plant 3
To analyse the most dominant Fusobacteria species of BGP3 on the genomic level, genome bins assigned to corresponding taxa were extracted. The most dominant Fusobacteria bin featuring the highest completeness and lowest contamination (Table 5) is covered by 1.4 % of all BGP3 metagenome reads. Since the representative Fusobacterium OTU (Fig. 2b) is moderately related to the reference strain Fusobacterium varium (see Fig. 2b, blue), the corresponding genome bin was compared to this sequenced and annotated reference genome (NZ_ACIE00000000.2).
The Fusobacteria genome bin was annotated, analysed and compared with the F. varium genome in GenDB. Results of these analyses suggest that the Fusobacteria species represented by the genome bin is an acidogenic bacterium, whose metabolism is mainly based on amino acids as energy and carbon source. This is also known for most Fusobacteria species, including the reference strain F. varium [91–94], suggesting that they take part in acidogenesis having a metabolism based on sugars and amino acids. Genome bin analysis also indicates that corresponding pathways lead to the production of the end products CO2, NH3, H2, acetate and lactate that can be partly further metabolised by methanogenic Archaea. Results of the comparative analysis of the annotated Fusobacteria genome bin and the reference sequence of F. varium using EDGAR were visualized in a Venn diagram (see Fig. 3b, 175_Fusobacteria). It shows that they share a comparatively low number of genes. Further analyses on the unique genes of the Fusobacteria bin did not result in additional metabolic information, as for most of them no functional prediction could be obtained (76.03 %). This suggests that the genome bin strain is highly different from other known Fusobacteria species and specific genome features remain to be determined.
In conclusion, the analysed Fusobacteria species represented by the genome bin most likely is involved in amino acid fermentation and produces CO2, NH3, H2, acetate and lactate, making it an acidogenic bacterium. It may be syntrophically associated with hydrogenotrophic methanogens. Due to a high number of unique unknown genes, this new and uncharacterised species requires further analyses.
The genome bin representing a Spirochaetes species from the mesophilic biogas plant 3
To analyse the most dominant Spirochaetes species of BGP3 on the genomic level, genome bins assigned to corresponding taxa were extracted. The largest and most complete Spirochaetes (Table 5) genome bin is covered with 0.2 % of all BGP3 metagenome reads and was chosen for further analyses. Since the representative Spirochaetes OTU (Fig. 2b) is moderately related to the reference strain T. brennaborense (see Fig. 2b, blue), the corresponding genome bin was compared to this sequenced and annotated reference genome [NC_015500].
Genome annotation and metabolic reconstruction in GenDB revealed that the corresponding Spirochaetes species is a hydrolytic bacterium. The presence of a large number of transporters and a number of genes encoding enzymes involved in sugar utilisation indicates that it primarily uses sugars for energy generation, namely glucose, mannose, fructose, rhamnose, xylose, melibiose, stachyose, raffinose and additionally L-lactate. End products of this fermentation were predicted to be acetate, CO2 and hydrogen, which can be directly used by methanogenic Archaea. Since some Treponema species are found in termite guts producing acetate from CO2 and H2, the presence of the gene (fhs) for this reaction’s key enzyme, formyl tetrahydrofolate synthetase [76], was searched by BLAST analysis implemented in GenDB. Its absence indicated that this species is not able to perform homoacetogenesis. Interestingly, no motility genes were found, although it is known that Spirochaetes species possess flagella and are motile [95].
To enable a comparison with the reference strain T. brennaborense, the EDGAR software was used, resulting in a Venn diagram (Fig. 3c, 138_Spirochaetes). It shows that the genomes share a relatively low number of genes, of which 91.54 % were functionally annotated, most being housekeeping genes and others predicted to be involved in sugar metabolism. In contrast, 47.35 % of the unique bin genes are uncharacterised or hypothetical, while 52.65 % were characterised. Of these, many encode for ABC transporters, sugar transporters and enzymes involved in sugar utilisation, underlining the assumption that the corresponding species is particularly dependent on sugars. This and the comparatively low number of shared genes highly suggest that the genome bin represents a new Spirochaetes species.
In conclusion, the analysed Spirochaetes species represented by the genome bin most likely ferments sugars and produces acetate, CO2 and H2. It was predicted to utilise a wide range of carbohydrates.
The genome bins representing Cloacimonetes species from mesophilic biogas plants 2 and 3
To deduce the functional role of dominant Cloacimonetes species from BGP2 and 3, the two genome bins representing species, assigned to this phylum with the highest completeness and lowest contamination, were analysed. The Cloacimonetes genome bin 1 and 2 are covered by 0.08 % BGP3 reads and 0.23 % BGP2 reads, respectively. The only sequenced species of the phylum Cloacimonetes, Candidatus Cloacamonas acidaminovorans (Fig. 2b), is larger in size and also has a significantly lower GC-content of 37.9 % [89]. Candidatus Cloacamonas acidaminovorans, whose genome was reconstructed from metagenomic data, may produce hydrogen, and therefore, most likely is syntrophically associated with hydrogenotrophic methanogens [89].
Gene prediction, annotation and interpretation of genome bin 1 in GenDB showed that it lacks genes encoding enzymes involved in the synthesis of eleven amino acids, namely arginine, cysteine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, tryptophane, tyrosine and valine. Different from what was proposed for species of the candidate phylum Cloacimonetes (WWE1) [78], the analysed bin does not have the genetic potential to degrade cellulose or cellobiose. Genome bin 1 possesses genes encoding enzymes for energy generation from glucose via glycolysis, but in addition to this also has the potential to generate energy by degrading the amino acids proline, alanine, aspartate, glutamate, lysine and asparagine with CO2 and H2 as products. Additionally, corresponding species may be able to produce energy via proton and sodium pumps in combination with hydrogenases. For gaining additional information, proteins involved in fermentation, and energy metabolism compiled in a corresponding study on the reference strain [89] were compared to those encoded by the genome bins (see Additional file 5). It appeared that bin 1 encodes all listed proteins, suggesting that this species can shortly tolerate small amounts of oxygen, but is adapted to an anaerobic lifestyle. Additionally, this species presumably generates energy by Fe-hydrogenases, with H2 as end product. Analyses on Cloacimonetes bin 2 showed that it was predicted to feature the same metabolism as compared to Cloacimonetes bin 1 regarding amino acid fermentation metabolism yielding CO2 and H2 as products.
To enable further comparison with the reference strain Candidatus Cloacamonas acidaminovorans (see Fig. 2b), the EDGAR software was used. To further analyse the degree of genetic similarity between both annotated Cloacimonetes genome bins and Candidatus Cloacamonas acidaminovorans, the number of all shared and unique genes was determined. A resulting Venn diagram (Fig. 3d, 120_Cloacimonetes, 244_Cloacimonetes) shows that all three genomes share the vast majority of genes. Further analyses on the unique genes of Cloacimonetes bin 1 and 2 did not result in additional information, since for only 17 and 20 % of these genes, functional prediction could be obtained. These comparative analyses indicate that corresponding microorganisms share a similar metabolism based on utilisation of certain amino acids. However, specific genome features remain to be determined.
In conclusion, the two analysed Cloacimonetes species represented by genome bins are most likely amino acid fermenting, CO2 and H2 producing anaerobes that might be syntrophically associated with hydrogenotrophic methanogens.
Conclusions
To understand, evaluate and optimise the production process in biogas fermenters, it is crucial to study their microbial communities with all their members and interactions, which are diverse and highly dependent on different process parameters. In this study, three different mesophilic and one thermophilic production-scale biogas plant (BGP) were comparatively characterised. For taxonomic investigation and comparison, the approach of high-throughput 16S rRNA gene sequencing was used. Results showed that microbial communities of biogas plants are taxonomically complex and the process temperature is an important parameter shaping biogas consortia. Still, a core microbiome seems to be present in all BGPs, including taxa belonging to the phyla Firmicutes and Bacteroidetes and with lower abundances to the Euryarchaeota. These are commonly found in BGPs, especially the former two in those fed with cellulose-rich substrates, as they are responsible for the hydrolysis and acetogenesis/acidogenesis steps of anaerobic digestion, while members of the Euryarchaeota are involved in methanogenesis. Differences in taxonomic profiles between the BGPs, most probably, are due to adaptations of particular community members to prevailing process parameters, especially when comparing temperature, as the overall diversity is lower within the thermophilic BGP. However, the identification of four highly distinctive phyla characteristic for one (Thermotogae, Fusobacteria) or two (Cloacimonetes, Spirochaetes) of the biogas plants was notable and represented the main differences between the BGPs. They showed a high prevalence within their respective reactor environment and seemed to be mostly dominated by only a small number of genera.
To additionally uncover the genetic potential of the four studied BGPs, ultra-deep Illumina HiSeq metagenome sequencing was done. In contrast to read-based approaches on microbial metagenomes in the past, a combined assembly of all metagenomes was done in our study. It resulted in a high number of taxonomically and functionally characterised contigs enabling context-based community analyses. Based on the contigs, a genome binning approach was applied successfully. Comparative analyses of the genome bin representing a dominant species belonging to the Thermotogae with its closest relative Defluviitoga tunisiensis L3 were done. They showed a high similarity and with this confirmed the applicability and reliability of the binning approach. Further exploitation of genome bin information also enabled evaluation of so far unknown biogas species belonging to the phyla Fusobacteria, Spirochaetes and Cloacimonetes. Insights into their genetic potential and putative roles within the biogas fermentation process were obtained. In the past, this was achieved only by cultivation and subsequent genome sequencing or single cell sequencing, which can be difficult especially for taxonomically diverse communities. The assembly based genome binning approach therefore can be seen as an alternative regarding the identification and genetic evaluation of unknown species circumventing the limitations of the methods mentioned before.
This is the first study to use the genome binning approach on deeply sequenced metagenome data originating from different production-scale biogas plants. The combined assembly based binning strategy enabled the identification of five high quality genome bins, representing dominant but mostly unknown species within the complex biogas microbiome. In combination with the taxonomic evaluation of the community by 16S rRNA gene amplicon sequencing and its relation to prevailing process parameters, it allows deep insights into the members’ functional roles and genetic potentials. The next step will be to characterise binned genomes by elucidating their actual transcriptional activity. In this aspect, metatranscriptome analyses will enable identification of predominantly transcribed genes which are believed to encode important functions within the biogas production process with respect to prevailing fermentation conditions. Accordingly, integrative analyses of deeply sequenced metagenome and metatranscriptome data will provide cultivation-independent insights into the performance of so far uncharacterized biogas community members.
Abbreviations
- BGP:
-
biogas plant
- CSTR:
-
continuously stirred tank reactor
- JGI:
-
U.S. Department of Energy Joint Genome Institute
- HAC-eq:
-
acetic acid equivalents
- KEGG:
-
Kyoto encyclopaedia of genes and genomes
- oDM:
-
organic dry matter
- OTU:
-
operational taxonomic unit
- QC:
-
quality control
- TIC:
-
total inorganic carbon
- VOA:
-
volatile organic acids
References
Ho DP, Ngo HH, Guo W. A mini review on renewable sources for biofuel. Bioresour Technol. 2014;169:742–9.
Dewil R, Appels L, Baeyens J. Energy use of biogas hampered by the presence of siloxanes. Energy Convers Manag. 2006;47:1711–22.
Stolze Y, Zakrzewski M, Maus I, Eikmeyer F, Jaenicke S, Rottmann N, Siebner C, et al. Comparative metagenomics of biogas-producing microbial communities from production-scale biogas plants operating under wet or dry fermentation conditions. Biotechnol Biofuels. 2015;8:14.
Zakrzewski M, Goesmann A, Jaenicke S, Jünemann S, Eikmeyer F, Szczepanowski R, Al-Soud WA, et al. Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J Biotechnol. 2012;158:248–58.
Kröber M, Bekel T, Diaz NN, Goesmann A, Jaenicke S, Krause L, Miller D, et al. Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J Biotechnol. 2009;142:38–49.
Wirth R, Kovács E, Maróti G, Bagi Z, Rákhely G, Kovács KL. Characterization of a biogas-producing microbial community by short-read next generation DNA sequencing. Biotechnol Biofuels. 2012;5:41.
Jaenicke S, Ander C, Bekel T, Bisdorf R, Dröge M, Gartemann K, Jünemann S, et al. Comparative and joint analysis of two metagenomic datasets from a biogas fermenter obtained by 454-pyrosequencing. PLoS One. 2011;6(1):e14519.
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH, Sørensen SJ, et al. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol. 2013;85:612–26.
Weiss A, Jérôme V, Freitag R, Mayer HK. Diversity of the resident microbiota in a thermophilic municipal biogas plant. Appl Microbiol Biotechnol. 2008;81:163–73.
Ruile S, Schmitz S, Mönch-Tegeder M, Oechsner H. Degradation efficiency of agricultural biogas plants–a full-scale study. Bioresour Technol. 2015;178:341–9.
Kardos L. Comparing of mesophilic and thermophilic anaerobic fermented sewage sludge based on chemical and biochemical tests. Appl Ecol Env Res. 2011;9:293–302.
Weiland P. Biogas production: current state and perspectives. Appl Microbiol Biotechnol. 2010;85:849–60.
Ziembińska-Buczyńska A, Banach A, Bacza T, Pieczykolan M. Diversity and variability of methanogens during the shift from mesophilic to thermohilic conditions while biogas production. World J Microbiol Biotechnol. 2014;30:3047–53.
Lee S, Kang H, Lee YH, Lee TJ, Han K, Choi Y, Park H. Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J Environ Monit. 2012;14:1893–905.
Bremges A, Maus I, Belmann P, Eikmeyer F, Winkler A, Albersmeier A, Pühler A, et al. Deeply sequenced metagenome and metatranscriptome of a biogas-producing microbial community from an agricultural production-scale biogas plant. GigaSci. 2015;4:849.
Abendroth C, Vilanova C, Günther T, Luschnig O, Porcar M. Eubacteria and archaea communities in seven mesophile anaerobic digester plants in Germany. Biotechnol Biofuels. 2015;8:447.
Eikmeyer FG, Rademacher A, Hanreich A, Hennig M, Jaenicke S, Maus I, Wibberg D, et al. Detailed analysis of metagenome datasets obtained from biogas-producing microbial communities residing in biogas reactors does not indicate the presence of putative pathogenic microorganisms. Biotechnol Biofuels. 2013;6:49.
Li A, Chu Y, Wang X, Ren L, Yu J, Liu X, Yan J, et al. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol Biofuels. 2013;6:3.
Campanaro S, Treu L, Kougias PG, de Francisci D, Valle G, Angelidaki I. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol Biofuels. 2016;9:5478.
Henne A, Daniel R, Schmitz RA, Gottschalk G. Construction of environmental DNA libraries in Escherichia coli and screening for the presence of genes conferring utilization of 4-hydroxybutyrate. Appl Environ Microbiol. 1999;65:3901–7.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4516–22.
The Berkeley Lab Publications System. DUK—a fast and efficient Kmer based sequence matching tool. https://publications.lbl.gov/islandora/object/ir%3A155200. Accessed 09 Mar 2016.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–7.
Angly FE, Dennis PG, Skarshewski A, Vanwonterghem I, Hugenholtz P, Tyson GW. CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome. 2014;2:11.
Munoz R, Yarza P, Ludwig W, Euzéby J, Amann R, Schleifer K, Oliver Glöckner F, Rosselló-Móra R. Release LTPs104 of the all-species living tree. Syst Appl Microbiol. 2011;34:169–70.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Boisvert S, Raymond F, Godzaridis É, Laviolette F, Corbeil J. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012;13:R122.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics. 2012;28:2223–30.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2014;12:59–60.
Huson DH, Mitra S, Ruscheweyh H, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–60.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer J. 2015;3:e1165.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Dröge J, Gregor I, McHardy AC. Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics. 2015;31:817–24.
Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Pühler A. GenDB - an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–95.
Blom J, Kreis J, Spänig S, Juhre T, Bertelli C, Ernst C, Goesmann A. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016;44:W22–8.
Kovács E, Wirth R, Maróti G, Bagi Z, Rákhely G, Kovács KL. Biogas production from protein-rich biomass: fed-batch anaerobic fermentation of casein and of pig blood and associated changes in microbial community composition. PLoS One. 2013;8:e77265.
Mao C, Feng Y, Wang X, Ren G. Review on research achievements of biogas from anaerobic digestion. Renew Sustain Energy Rev. 2015;45:540–55.
Ziganshin AM, Ziganshina EE, Kleinsteuber S, Pröter J, Ilinskaya ON. Methanogenic Community Dynamics during Anaerobic Utilization of Agricultural Wastes. Acta Naturae. 2012;4:91–7.
Munk B, Lebuhn M. Process diagnosis using methanogenic Archaea in maize-fed, trace element depleted fermenters. Anaerobe. 2014;29:22–8.
Tuan NN, Chang Y, Yu C, Huang S. Multiple approaches to characterize the microbial community in a thermophilic anaerobic digester running on swine manure: a case study. Microbiol Res. 2014;169:717–24.
Angelidaki I, Karakashev D, Batstone DJ, Plugge CM. Stams, Alfons J M. Biomethanation and its potential. Methods Enzymol. 2011;494:327–51.
Lebuhn M, Munk B, Effenberger M. Agricultural biogas production in Germany - from practice to microbiology basics. Energ Sustain Soc. 2014;4:10.
Tang Y, Ji P, Hayashi J, Koike Y, Wu X, Kida K. Characteristic microbial community of a dry thermophilic methanogenic digester: its long-term stability and change with feeding. Appl Microbiol Biotechnol. 2011;91:1447–61.
Ziganshin AM, Liebetrau J, Pröter J, Kleinsteuber S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl Microbiol Biotechnol. 2013;97:5161–74.
Yu D, Kurola JM, Lähde K, Kymäläinen M, Sinkkonen A, Romantschuk M. Biogas production and methanogenic archaeal community in mesophilic and thermophilic anaerobic co-digestion processes. J Environ Manage. 2014;143:54–60.
Rademacher A, Zakrzewski M, Schlüter A, Schönberg M, Szczepanowski R, Goesmann A. Pühler, Klocke M. Characterization of microbial biofilms in a thermophilic biogas system by high-throughput metagenome sequencing. FEMS Microbiol Ecol. 2012;79:785–99.
Levén L. Eriksson, Anders R B, Schnürer A. Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic household waste. FEMS Microbiol Ecol. 2007;59:683–93.
Krause L, Diaz NN, Edwards RA, Gartemann K, Krömeke H, Neuweger H, Pühler A, Runte KJ, Schlüter A, Stoye J, Szczepanowski R, Tauch A, Goesmann A. Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol. 2008;136:91–101.
Liu FH, Wang SB, Zhang JS, Zhang J, Yan X, Zhou HK, Zhao GP, et al. The structure of the bacterial and archaeal community in a biogas digester as revealed by denaturing gradient gel electrophoresis and 16S rDNA sequencing analysis. J Appl Microbiol. 2009;106:952–66.
Luo G, de Francisci D, Kougias PG, Laura T, Zhu X, Angelidaki I. New steady-state microbial community compositions and process performances in biogas reactors induced by temperature disturbances. Biotechnol Biofuels. 2015;8:3.
Tracy BP, Jones SW, Fast AG, Indurthi DC, Papoutsakis ET. Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr Opin Biotechnol. 2012;23:364–81.
Pap B, Györkei Á, Boboescu IZ, Nagy IK, Bíró T, Kondorosi É, Maróti G. Temperature-dependent transformation of biogas-producing microbial communities points to the increased importance of hydrogenotrophic methanogenesis under thermophilic operation. Bioresour Technol. 2015;177:375–80.
Vanwonterghem I, Jensen PD, Rabaey K, Tyson GW. Temperature and solids retention time control microbial population dynamics and volatile fatty acid production in replicated anaerobic digesters. Sci Rep. 2015;5:8496.
Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–89.
Stantscheff R, Kuever J, Rabenstein A, Seyfarth K, Dröge S, König H. Isolation and differentiation of methanogenic Archaea from mesophilic corn-fed on-farm biogas plants with special emphasis on the genus Methanobacterium. Appl Microbiol Biotechnol. 2014;98:5719–35.
Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann K, Krahn I, et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol. 2008;136:77–90.
Pervin HM, Dennis PG, Lim HJ, Tyson GW, Batstone DJ, Bond PL. Drivers of microbial community composition in mesophilic and thermophilic temperature-phased anaerobic digestion pre-treatment reactors. Water Res. 2013;47:7098–108.
Johnson MR, Conners SB, Montero CI, Chou CJ, Shockley KR, Kelly RM. The Thermotoga maritima phenotype is impacted by syntrophic interaction with Methanococcus jannaschii in hyperthermophilic coculture. Appl Environ Microbiol. 2006;72:811–8.
Nesbø CL, Dlutek M, Zhaxybayeva O, Doolittle WF. Evidence for existence of “mesotogas”, members of the order Thermotogales adapted to low-temperature environments. Appl Environ Microbiol. 2006;72:5061–8.
Gupta RS, Bhandari V. Phylogeny and molecular signatures for the phylum Thermotogae and its subgroups. Antonie Van Leeuwenhoek. 2011;100:1–34.
Green AG, Swithers KS, Gogarten JF, Gogarten JP. Reconstruction of ancestral 16S rRNA reveals mutation bias in the evolution of optimal growth temperature in the Thermotogae phylum. Mol Biol Evol. 2013;30:2463–74.
Röske I, Sabra W, Nacke H, Daniel R, Zeng A, Antranikian G, Sahm K. Microbial community composition and dynamics in high-temperature biogas reactors using industrial bioethanol waste as substrate. Appl Microbiol Biotechnol. 2014;98:9095–106.
Lykidis A, Chen C, Tringe SG, McHardy AC, Copeland A, Kyrpides NC, Hugenholtz P, Macarie H, Olmos A, Monroy O, Liu WT. Multiple syntrophic interactions in a terephthalate-degrading methanogenic consortium. ISME J. 2011;5:122–30.
Moore C, Addison D, Wilson JM, Zeluff B. First case of Fusobacterium necrophorum endocarditis to have presented after the 2nd decade of life. Tex Heart Inst J. 2013;40:449–52.
Rehman HU, Vahjen W, Awad WA, Zentek J. Indigenous bacteria and bacterial metabolic products in the gastrointestinal tract of broiler chickens. Arch Anim Nutr. 2007;61:319–35.
Suchodolski JS. Companion animals symposium: microbes and gastrointestinal health of dogs and cats. J Anim Sci. 2011;89:1520–30.
Kolenbrander PE. Oral Microbial Communities: biofilms, Interactions, and Genetic Systems 1. Annu Rev Microbiol. 2000;54:413–37.
Hampson DJ, Ahmed N. Spirochaetes as intestinal pathogens: lessons from a Brachyspira genome. Gut Pathog. 2009;1:10.
Pester M, Brune A. Expression profiles of fhs (FTHFS) genes support the hypothesis that spirochaetes dominate reductive acetogenesis in the hindgut of lower termites. Environ Microbiol. 2006;8:1261–70.
Leadbetter JR. Acetogenesis from H2 Plus CO2 by Spirochetes from termite guts. Science. 1999;283:686–9.
Limam RD, Chouari R, Mazéas L, Wu T, Li T, Grossin-Debattista J, Guerquin-Kern J, et al. Members of the uncultured bacterial candidate division WWE1 are implicated in anaerobic digestion of cellulose. Microbiologyopen. 2014;3:157–67.
Rivière D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J. 2009;3:700–14.
Chouari R, Le Paslier D, Dauga C, Daegelen P, Weissenbach J, Sghir A. Novel major bacterial candidate division within a municipal anaerobic sludge digester. Appl Environ Microbiol. 2005;71:2145–53.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7.
Su H, Liu L, Wang S, Wang Q, Jiang Y, Hou X, Tan T. Semi-continuous anaerobic digestion for biogas production: influence of ammonium acetate supplement and structure of the microbial community. Biotechnol Biofuels. 2015;8:13.
Gies EA, Konwar KM, Beatty JT, Hallam SJ. Illuminating microbial dark matter in meromictic Sakinaw Lake. Appl Environ Microbiol. 2014;80:6807–18.
Solli L, Håvelsrud OE, Horn SJ, Rike AG. A metagenomic study of the microbial communities in four parallel biogas reactors. Biotechnol Biofuels. 2014;7:146.
Ben Hania W, Godbane R, Postec A, Hamdi M, Ollivier B, Fardeau M. Defluviitoga tunisiensis gen. nov., sp. nov., a thermophilic bacterium isolated from a mesothermic and anaerobic whey digester. Int J Syst Evol Microbiol. 2012;62:1377–82.
Maus I, Cibis KG, Wibberg D, Winkler A, Stolze Y, König H, Pühler A, Schlüter A. Complete genome sequence of the strain Defluviitoga tunisiensis L3, isolated from a thermophilic, production-scale biogas plant. J Biotechnol. 2015;203:17–8.
Wyss C, Choi B, Schupbach P, Moter A, Guggenheim B, Gobel UB. Treponema lecithinolyticum sp. nov., a small saccharolytic spirochaete with phospholipase A and C activities associated with periodontal diseases. Int J Syst Bacteriol. 1999;49:1329–39.
Schrank K, Choi BK, Grund S, Moter A, Heuner K, Nattermann H, Göbel UB. Treponema brennaborense sp. nov., a novel spirochaete isolated from a dairy cow suffering from digital dermatitis. Int J Syst Bacteriol. 1999;49(Pt 1):43–50.
Pelletier E, Kreimeyer A, Bocs S, Rouy Z, Gyapay G, Chouari R, Rivière D, Ganesan A, Daegelen P, Sghir A, Cohen GN, Médigue C, Weissenbach J, Le Paslier D. “Candidatus Cloacamonas acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol. 2008;190:2572–9.
Maus I, Cibis KG, Bremges A, Stolze Y, Wibberg D, Tomazetto G, Blom J, Sczyrba A, König H, Pühler A, Schlüter A. Genomic characterization of Defluviitoga tunisiensis L3, a key hydrolytic bacterium in a thermophilic biogas plant and its abundance as determined by metagenome fragment recruitment. J Biotechnol. 2016. doi:10.1016/j.jbiotec.2016.05.001.
Resmer KL, White RL. Metabolic footprinting of the anaerobic bacterium Fusobacterium varium using 1H NMR spectroscopy. Mol BioSyst. 2011;7:2220.
Kapatral V, Anderson I, Ivanova N, Reznik G, Los T, Lykidis A, Bhattacharyya A, Bartman A, Gardner W, Grechkin G, Zhu L, Vasieva O, Chu L, Kogan Y, Chaga O, Goltsman E, Bernal A, Larsen N, D’Souza M, Walunas T, Pusch G, Haselkorn R, Fonstein M, Kyrpides N, Overbeek R. Genome sequence and analysis of the oral bacterium Fusobacterium nucleatum strain ATCC 25586. J Bacteriol. 2002;184:2005–18.
Pikis A, Immel S, Robrish SA, Thompson J. Metabolism of sucrose and its five isomers by Fusobacterium mortiferum. Microbiology (Reading, England). 2002;148:843–52.
Robrish SA, Oliver C, Thompson J. Sugar metabolism by fusobacteria: regulation of transport, phosphorylation, and polymer formation by Fusobacterium mortiferum ATCC 25557. Infect Immun. 1991;59:4547–54.
Charon NW, Greenberg EP, Koopman M, Limberger RJ. Spirochete chemotaxis, motility, and the structure of the spirochetal periplasmic flagella. Res Microbiol. 1992;143:597–603.
Authors’ contributions
YS performed the laboratory work, biological data analysis, coordinated drafting and drafted the manuscript. AB conceived and performed bioinformatic metagenome analysis and participated in manuscript drafting. MR carried out 16S rRNA amplicon analysis and participated in manuscript drafting. CH participated in metagenome data processing and revised the manuscript. IM participated in data analyses and manuscript drafting. AScz co-designed the study, supervised all bioinformatic analyses and participated in manuscript drafting. ASch and AP conceived the study, participated in manuscript coordination, supervised all biological analyses and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We greatly acknowledge the work conducted by the US Department of Energy Joint Genome Institute (DOE JGI).
We also acknowledge the funding by the Bundesministerium für Ernährung und Landwirtschaft (BMEL), the Fachagentur für nachwachsende Rohstoffe (FNR) and the Projektträger Jülich (PTJ) and the bioinformatics support of the BMBF-funded project ‘Bielefeld-Gießen Center for Microbial Bioinformatics – BiGi’ within the German Network for Bioinformatics Infrastructure (de.NBI).
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
The 16S data supporting the conclusions of this article is included within the article’s additional files (BIOM file). The datasets supporting the conclusions of this article are available in the short read archive (SRA): http://www.ncbi.nlm.nih.gov/sra/?term=SRA357211, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357213, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357208, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357209, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357214, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357221, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357222, http://www.ncbi.nlm.nih.gov/sra/?term=SRA357223.
Funding
The sequencing work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231.
AB and IM are supported by a fellowship from the CLIB Graduate Cluster Industrial Biotechnology. MR is funded (and AB is partially funded) by the International DFG Research Training Group GRK 1906/1.
The Bundesministerium für Ernährung und Landwirtschaft (BMEL) and the Fachagentur für Nachwachsende Rohstoffe (FNR) projects, Grant numbers 22006712 and 22404015, supported the work of AS and YS; the Projektträger Jülich (PTJ), grant number 03SF0440C, supported the work of AS and IM.
The work of AP is supported by the BMBF-funded project ‘Bielefeld-Gießen Center for Microbial Bioinformatics—BiGi (Grant number 031A533)’ within the German Network for Bioinformatics Infrastructure (de.NBI).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yvonne Stolze and Andreas Bremges contributed equally to this work
Additional files
13068_2016_565_MOESM1_ESM.biom
Additional file 1: BIOM-File S1. Final operational taxonomic unit (OTU) table in BIOM format, format specification 1.0.0. Contained are clustered OTUs with specific OTU IDs, their supporting 16S rRNA gene sequence counts per sample, and as metadata taxonomic classification per OTU, descriptive labels per sample.
13068_2016_565_MOESM2_ESM.pdf
Additional file 2: Figure S1. Phylogenetic tree of all available type strains with operational taxonomic units (OTUs) of Thermotogae, Fusobacteria, Spirochaetes and Cloacimonetes taxa of the studied biogas plants and their closest non-type strain relatives embedded. Type strains are in black, OTUs are in red, their closest relatives in purple, their closest sequenced relatives in blue and their closest type strain relatives in green. 16S rRNA gene sequence tree construction was done using the ARB [31] software.
13068_2016_565_MOESM3_ESM.png
Additional file 3: Figure S2. ‘Alanine, Aspartate and Glutamate Metabolism’ with proteins encoded in the Fusobacteria bin marked in green and red. Violet marked proteins are uncertain. The analysis was done with the KEGG-based protein mapping tool implemented within the annotation system GenDB 2.0 [41].
13068_2016_565_MOESM4_ESM.pdf
Additional file 4: Table S1. Genes encoding proteins involved in energy generation found in the reference genome of Defluviitoga tunisiensis L3 [90] and their presence or absence within the Thermotogae genome bin.
13068_2016_565_MOESM5_ESM.pdf
Additional file 5: Table S2. Genes encoding proteins involved in life in the presence of oxygen, anaerobic lifestyle and energy generation found in the reference genome of Candidatus Cloacamonas acidaminovorans [89] and presence or absence of these within the Cloacimonetes genome bin.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Stolze, Y., Bremges, A., Rumming, M. et al. Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production-scale biogas plants. Biotechnol Biofuels 9, 156 (2016). https://doi.org/10.1186/s13068-016-0565-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-016-0565-3