
Abstract
Background
In recent years biogas plants in Germany have been supposed to be involved in amplification and dissemination of pathogenic bacteria causing severe infections in humans and animals. In particular, biogas plants are discussed to contribute to the spreading of Escherichia coli infections in humans or chronic botulism in cattle caused by Clostridium botulinum. Metagenome datasets of microbial communities from an agricultural biogas plant as well as from anaerobic lab-scale digesters operating at different temperatures and conditions were analyzed for the presence of putative pathogenic bacteria and virulence determinants by various bioinformatic approaches.
Results
All datasets featured a low abundance of reads that were taxonomically assigned to the genus Escherichia or further selected genera comprising pathogenic species. Higher numbers of reads were taxonomically assigned to the genus Clostridium. However, only very few sequences were predicted to originate from pathogenic clostridial species. Moreover, mapping of metagenome reads to complete genome sequences of selected pathogenic bacteria revealed that not the pathogenic species itself, but only species that are more or less related to pathogenic ones are present in the fermentation samples analyzed. Likewise, known virulence determinants could hardly be detected. Only a marginal number of reads showed similarity to sequences described in the Microbial Virulence Database MvirDB such as those encoding protein toxins, virulence proteins or antibiotic resistance determinants.
Conclusions
Findings of this first study of metagenomic sequence reads of biogas producing microbial communities suggest that the risk of dissemination of pathogenic bacteria by application of digestates from biogas fermentations as fertilizers is low, because obtained results do not indicate the presence of putative pathogenic microorganisms in the samples analyzed.
Similar content being viewed by others

Background
Human pathogenic bacteria causing foodborne or zoonotic diseases are a major healthcare concern even in developed countries [1, 2]. Usage of manure as fertilizer has been discussed as a potential source of infection. Moreover, digestates from anaerobic digesters used as fertilizers were also suspected to transfer human pathogenic bacteria onto vegetables or other crops. The recent outbreak of an enterohemorrhagic Escherichia coli O104:H4 strain in Germany in May 2011 is an example for a foodborne disease having vegetables as source of infection. This outbreak led to the infection of about 3,800 patients suffering from acute gastroenteritis or even the hemolytic-uremic syndrome. Epidemiological and surveillance studies were conducted at the same time by German federal institutions to identify the origin of infection. These studies led to the hypothesis that contaminated vegetables like cucumbers or tomatoes might be involved in spreading of the human pathogenic bacterium [3–5]. Press coverage also hypothesized that digestates from agricultural biogas reactors could have been a source causing these infections. Finally, fenugreek sprouts grown from seeds from Egypt were identified as the most likely source of infection [4].
However, E. coli is not the only relevant potential foodborne pathogen. Examples for other human pathogenic bacteria causing foodborne infections are Listeria monocytogenes, Yersinia enterocolitica or Salmonella species. Moreover, Campylobacter, Vibrio and Clostridium species are also known human pathogens causing foodborne diseases [1, 6]. Particularly the genus Clostridium, which is well known to accomplish the first steps of anaerobic digestion, is widespread in biogas systems. This genus comprises some important pathogens, such as C. botulinum, C. difficile, C. perfringens and C. tetani. For instance, C. botulinum was recently identified in animal feces [7, 8], a potential substrate for agricultural biogas plants. Hence, agricultural biogas plants are also accused to be involved in the spreading of C. botulinum[9] causing chronic botulism [10, 11].
Human pathogenic bacteria are defined as bacteria causing disease in humans [12] while the term ‘virulence’ describes their degree of pathogenicity. It has been proposed that human pathogenic bacteria can enhance their virulence by acquisition of genes encoding virulence factors [12–14]. These factors may facilitate adhesion to and invasion of (specific) host cells. Moreover virulence factors can promote survival of the pathogen in the host tissue by inhibiting the immune response and increase the pathogenicity by encoding toxins. Resistance against antibiotics can also be seen as a virulence factor as it complicates medical treatment of a human pathogenic bacterial infection [14, 15]. As an example, for the E. coli O104:H4 strain causing the outbreak in Germany it is supposed that it evolved from an enteroaggregative ancestor by acquisition of the shiga toxin encoding Stx-phage and a plasmid encoding aggregative adherent fimbriae and further virulence features [3, 4].
A major substrate component used for biogas production besides agricultural plant material is manure from animals such as pigs, cattle or chicken. It is known that manure can contain potential human pathogenic bacteria such as Salmonella sp., Listeria sp., Campylobacter sp. or E. coli. Thus, spreading of manure might contribute to (zoonotic) bacterial infections [1, 6, 16–18]. However, several studies on lab-scale and agricultural anaerobic digesters showed that a reduction of the overall pathogen load is possible even at low temperatures [16–18]. Reduction of pathogens was shown to be very efficient for bacteria belonging to the family of Enterobacteriaceae, while it was less efficient for Listeria, Clostridia and Enterococci[16–18].
Several metagenomes of experimental and agricultural anaerobic digesters have been published recently [19–23]. These data provided insights into the microbial community involved in anaerobic digestion and methane production and into the underlying metabolic pathways.
To evaluate the risk associated with utilization of digestates from biogas plants as fertilizer on fields, the existing metagenome sequence data from different biogas reactor communities were for the first time analyzed for the presence of sequence tags originating from putative pathogenic bacteria and those representing virulence or resistance determinants.
Results
Searching for putative pathogens in taxonomic profiles deduced from metagenome sequence data of biogas-producing microbial communities
Origin and characteristics of metagenome sequence datasets consulted for searching of sequence tags originating from putative pathogenic bacteria are described in Table 1. Metagenomic DNA was isolated from microbial communities residing in agricultural as well as lab-scale biogas reactors at different temperatures. The taxonomic profiles of biogas-producing communities residing in the analyzed biogas reactors were computed by CARMA3 [24] and analyzed for the presence of putative pathogenic bacteria.
In total, CARMA3 classified 2,183,722 environmental gene tags (EGTs), comprising all datasets, while 176,780 of these EGTs were assigned to genus and 16,035 EGTs to 351 species level. Subsequently, the profiles were examined for potentially human pathogenic distinct species (Table 2). One EGT was assigned to C. botulinum. This species is capable to produce the botulinum neurotoxin, which is responsible for the neuroparalytic disease botulism [25]. However, searching for sequences that are similar to the identified EGT in the NCBI non-redundant nucleotide (NT) database revealed that it encodes a part of a 23S rRNA gene of a species rather related to C. haemolyticum or C. ljungahlii (98% similarity) than to C. botulinum. This observation is in accordance with a recent study of methanogenic bioreactors in which pathogenic Clostridia could not be detected [26].
Moreover, a manual BLAST search of the EGTs assigned to other pathogenic species of the genus Clostridium, except for Clostridium clostridioforme, indicated that the majority of these EGTs are highly similar to related species for which pathogenicity has not been described so far. Some of the EGTs assigned to C. clostridioforme are identical to genes encoding hypothetical proteins originating from C. clostridioforme. This species has been reported to be involved in human infections, including bacteremia [27], but it also participates in fermentation of carbohydrates to acetate, lactate and formate [28]. Finally, no EGTs were classified to Clostridium sordelii which is a causative agent of gas gangrene.
Among the order Enterobacteriales, the genera Escherichia, Salmonella and Shigella are present in the taxonomic profiles of all biogas plant samples. No taxonomic assignments on species level were obtained for EGTs classified as Salmonella or Shigella. However, 7 EGTs exhibit a high similarity to genomic fragments originating from Escherichia coli. These EGTs represent a cell division component, a rhamnose-proton symporter and a DNA-damage-inducible protein. No genes encoding toxins were identified for this species.
A detailed analysis of the sequences assigned to Streptococcus species revealed that some EGTs encode DNA recombinases, excisionase protein transposase or hypothetical proteins that are identical in other related species. However, the EGTs assigned to Streptococcus infantarius are identical to the corresponding genome and different from orthologous genes in related species. The identified EGTs encode for example an isoleucyl-tRNA synthetase, N-acetylglucosamine 6-phosphate deacetylase (nagA) and the B subunit of DNA gyrase (gyrB) in S. infantarius, which is associated with various human infections [29].
Mapping of metagenome sequence data to selected reference genomes of relevant pathogens
Sequence reads of the metagenomic datasets were mapped onto published genomes of pathogenic bacteria to reconstruct genomic sequences of putative pathogenic and closely related bacteria within biogas communities. Only a small number of reads of each metagenomic dataset could be mapped to the selected bacteria (Table 3). On average these reads only cover 0.1% of the respective reference genome. In contrast, more than 40% of the Methanoculleus marisnigri JR1 genome could be covered by reads of the U1 dataset [22]. In general, genome sequences of pathogenic strains belonging to the genus Clostridium feature a higher coverage by metagenomic reads than the other species. This reflects the high abundance of Clostridia within the microbial biogas communities [22, 23].
Contigs and corresponding consensus sequences were extracted from the mapping datasets. Subsequently, BLAST-analyses of these sequences against organism-specific databases were performed. Assembled contigs on average are 90% identical to corresponding reference genome sequences, indicating that these biogas-producing communities analyzed only comprise strains that are related to the selected pathogenic bacteria but not identical. Moreover, functional descriptions of corresponding BLAST hits confirm these results since no pathogenicity determinants of the selected pathogenic bacteria could be detected. Most of the BLAST hits correspond to common housekeeping genes. Clostridial species within the biogas communities analyzed mostly are unknown and do not represent well-characterized species covered by database entries. In summary, sequence reads identical or almost identical to genomic sequences of selected pathogenic reference species are not present within the metagenome datasets analyzed in this study. Likewise, virulence determinants of these reference strains could not be detected.
Searching for putative pathogenicity determinants in functional profiles deduced from metagenome sequence data of biogas-producing microbial communities by exploiting Protein Family Database (pfam) assignments
Metagenome sequence reads matching Pfam family entries representing toxins, non-toxic components of toxins and virulence determinants were analyzed. Altogether only a marginal number (0.02 – 0.04%) of the 3,064,324 metagenome sequence reads could be assigned to relevant selected Pfam families (see Table 4).
The protein families PF05588 (C. botulinum HA-17 protein) as well as PF05105 (Holin family) were identified within all biogas samples (Table 4). PF05588 consists of hemagglutinin (HA) subcomponents, which are part of the L toxin, a progenitor toxin of C. botulinum type D strain 4947 [30]. The Pfam Holin family (PF05105) comprises TcdE/UtxA, which is involved in toxin secretion in C. difficile[31], but also other proteins, which are involved in bacterial lysis and virus dissemination. Interestingly, both protein families were clearly increased (PF05588, 74 EGTs, PF05105, 27 EGTs) within the hyperthermophilic digestate sample derived from the two-phase biogas system at 70°C (S70, Table 4) indicating that sanitation effect commonly assumed as consequence of increased temperatures was ineffective at least as far as clostridial species in general are concerned. Moreover, the protein family PF03496 (ADP-ribosyltransferase exoenzyme), including the ADP-ribosylating function of actin leading to lethal and dermonecrotic reactions in mammals [32], was particularly identified within the hyperthermophilic biogas samples (S70, Table 4). All other samples derived from mesophilic (38°C, 41°C) or thermophilic (55°C, 65°C) biogas reactors or batch fermentations showed a reduced number of EGTs for PF05588 and PF05105 and hardly any assignment to PF03496 (Table 4).
Beside these clostridial toxin-associated protein families, toxins derived from other bacteria (see Table 5) were not identified. For instance, the heat-labile enterotoxins (PF01375, PF01376) as well as the heat-stable enterotoxins (PF02048, PF08090) of E. coli were not detected within these biogas samples.
Searching for putative virulence determinants in metagenome sequence data implementing BLAST searches vs. the Microbial virulence database MvirDB
To identify possible virulence determinants within metagenome datasets of biogas-producing communities, BLAST analyses vs. the Microbial virulence Database MvirDB were accomplished. Metagenomic reads of each dataset were annotated based on BLASTn analyses against nucleotide sequences of the MvirDB database to identify putative virulence and resistance determinants. In total about 3.7% of all reads generated hits against sequences within the MvirDB, while about 2% of these reads featured hits against reference sequences classified as ‘virulence factor’ (Table 6). Most matching metagenomic reads were annotated as ‘virulence proteins’. Further but fewer hits corresponded to the categories ‘antibiotic resistance’, ‘transcription factor’, ‘protein toxin’ and ‘differential gene regulation’ with about 0.03 to 0.18% of all reads (Table 6). Reads annotated as ‘antibiotic resistance’, ‘protein toxin’ or ’virulence protein’ were further classified regarding their predicted function.
Protein toxins
Among the total number of metagenome sequence reads obtained for the different biogas reactors, only about 0.02 to 0.08% represent genes encoding different protein toxins (Table 7). A total of 67 different protein toxins were identified within the datasets by sequence similarity. Most of the detected protein toxins were assigned to the group of exotoxins and within this subgroup subtilisins, hyaluronidases, hemolysins and RTX toxins were annotated.
Within these exotoxins, 37 different subtilisins and subtlilisin-like serine proteases were detected by sequence similarity and accordingly constitute the most prominent subgroup within the detected protein toxins. Corresponding proteases are present in microorganisms and even in higher eukaryotes [33]. Some subtilisins function as scavengers for nutrients [34, 35] or their proteolytic properties are activated during pathogenesis in plants [36]. Risk assessment by the Toxic Substances Control Act of B. subtilis, one of the main producers of subtilisin, revealed that the protease only shows very low toxigenic properties. However, subtilisin is able to cause allergic reactions. The fact, that subtilisins are commonly used in different detergents may be interpreted in a way that subtilisin production by biogas community members does not pose an imponderable hazard to the environment or human health.
The second subgroup of exotoxins detected in every biogas sample comprises RTX toxins. The number of reads assigned to corresponding protein toxins varies between 13 and 63 representing only three different RTX genes. RTX toxins contribute to pathogenicity by interacting with the host’s immune system [37]. The gene products of the three different RTX genes detected are involved in the transport of the corresponding exotoxins, which were not verifiably within any sample.
In four of the biogas reactors, hyaluronidase genes probably originating from the species C. perfringens were detected. This species is a ubiquitous environmental organism [38] and a common human and livestock pathogen, causing gastroenteritis and gas gangrene in humans [39]. The number of detected sequences assigned to this gene family is relatively low and only ranges between 1 and 5 hits.
Altogether four different hemolysin genes were traceable in a low amount within each sample. Hemolysins are cytotoxic proteins that destroy the integrity of the host cell membrane by different mechanisms. The function of these hemolysin toxins is aimed at nutrient acquisition mostly by lysing leukocytes of the host [40]. Among the hemolysin genes identified in the datasets analyzed, the gene hlyC is present as deduced from sequence similarity analyses. The hlyC gene product activates the pore forming hemolysin HlyA in an unknown way [41]. However, hlyA-like genes were not detectable in the metagenome data. Additionally remaining possible and pore-forming hemolysins were not identified within the present data.
Only one to two reads per metagenome dataset were assigned to other exotoxin genes. Moreover, four different genes predicted to be involved in lipopolysaccharide (LPS) synthesis from the human stomach pathogen Helicobacter pylori were detected within six datasets. LPS originating from this pathogen mimics human glycan structures and contributes to the virulence by modulation of the immune system [42].
Overall only a low number of reads feature similarity to sequences categorized as ‘protein toxin’. Moreover, reference proteins encoded by these sequences are known to possess a low degree of toxicity.
Virulence proteins
The assignments of MvirDB entries classified as ‘virulence protein’ show a great diversity regarding their function. However, some of these annotations were present at high abundance in all datasets (see Table 8). Among these some may play a role in stress response (endopeptidase Clp ATP-binding chain C, ATP-dependent Clp protease ATP-binding subunit ClpX, ClpB protein, DNA mismatch repair protein, chaperonin GroEL) [43, 44], sugar and energy metabolism (pyruvate kinase, GTP pyrophosphokinase, UDP-N-acetylglucosamine 2-epimerase) or are thought to have further functions not directly related to virulence (carbamoyl-phosphate synthase large chain, putative lysil-tRNA synthetase LysU). At first view, corresponding genes mediate general features of microorganisms and do not pose a potential risk regarding virulence. However, some of these genes are described to be involved in virulence of certain bacteria. For example the Clp ATPase and proteases are involved in quality control of proteins and their structure [44] in non-stress as well as in stress situations and are needed for cellular differentiation. Hence, these enzymes most probably also ensure the survival of cells in pathogenic interactions [44]. Moreover, they regulate the expression of further virulence determinants.
Accordingly, presence of metagenomic reads sharing similarity to those genes described to be involved in bacterial virulence does not allow drawing the conclusion that virulent bacteria reside in microbial communities of the samples analyzed because a read based analysis per se cannot take into account the genomic context of a bacterium harboring a putative virulence determinant. Certainly, a putative virulence gene in a pathogenic organism might be more severe than the same gene in an otherwise harmless bacterium.
Antibiotic resistance determinants
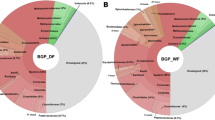
About 0.09% (B55) to 0.22% (S70) of metagenome sequence reads were annotated to have a predicted function in the context of resistance to antimicrobial drugs. Corresponding annotations mainly represent eight groups of antimicrobial compounds for which resistance determinants were identified (Figure 1). These groups comprise vancomycin, macrolide, tetracycline, polypeptide (bacitracin, polymyxin), β-lactam, streptogramin and aminoglycoside (kasugamycin, streptomycin, kanamycin, spectinomycin) resistance determinants as well as multidrug exporter components. Further refer to resistances against a number of additional antibiotics (Figure 1). No clear differences concerning the abundance of specific resistance types can be observed between the samples (Figure 1). Moreover, annotated resistances are based on different mechanisms [45] including enzymatic inactivation of the drug (beta-lactames, amidoglycosides), mutational alteration of the target protein (fluoroquinolones), acquisition of genes encoding gene products that are less susceptible to the antibiotic (trimethoprim), bypassing the target of antimicrobial action (vancomycin) or by prevention of drug access to the target (multidrug efflux pumps). Especially for the last four resistance mechanisms the approach to predict the existence of resistance determinants by means of similarity searches in curated databases such as MvirDB has limitations because reliable functional conclusions cannot be drawn. For example, reads annotated as multidrug exporters might encode pumps for the transport of compounds that do not act as antibiotics or reads annotated as products less susceptible to a drug might encode a drug sensitive target. Surprisingly, a high number of reads were annotated to have a predicted function in vancomycin resistance. Vancomycin binds to the D-Ala-D-Ala termini of peptidoglycan intermediates and inhibits the crosslinking of the peptidoglycan layer [46]. Some bacteria (such as Enterococci or Leuconostoc mesenteroides) are resistant to vancomycin because their cell wall does not contain the D-Ala-D-Ala but D-Ala-D-Lactate termini instead. Enzymes involved in the formation of each type of termini are closely related ligases [46, 47] which may again lead to the annotation of reads encoding D-Ala-D-Ala ligases as vancomycin resistance determinants. These intrinsic limitations might cause an overestimation of reads involved in antibiotic resistance.

Relative abundances of reads annotated to have a predicted function in the context of resistance to antimicrobial drugs. Annotations by means of BLASTn analyses of metagenomic reads against the MvirDB identified about 0.09% (B55) to 0.22% (S70) of metagenome sequence reads to confer resistances against groups of or specific antibiotics or to encode putative multidrug exporters.
Overall a variety of putative antibiotic resistance determinants was identified. However, their abundance within each metagenome dataset is quite low.
Discussion
Biogas plants are discussed to contribute to the proliferation and dissemination of pathogenic bacteria and pathogenicity/virulence determinants in the environment since digestates from biogas reactors are applied as fertilizer on fields. This practice bears the risk that pathogens residing in digestates contaminate crops and vegetables that serve as food for animals and humans thus abetting zoonotic diseases. To our knowledge, in this study metagenome sequence data were analyzed for the presence of sequence tags indicative for the occurrence of pathogens or pathogenicity/virulence determinants for the first time. The sensitivity and resolution of this kind of approach should be very high since it is based on nucleotide sequence data. Moreover, this approach is less biased compared to methods based on PCR for detection of pathogenicity determinants or cultivation of putative pathogens.
Inspection of taxonomic profiles deduced from metagenome sequence data and mapping results on pathogenic reference genomes does not elucidate strong evidence for the presence of pathogens within fermentation samples of biogas reactors. Sequence tags originating from pathogenic members of the family Enterobacteriaceae could hardly be detected within the metagenome data analyzed which is in accordance with earlier studies based on microbiological and molecular genetic methods applied for detection of species belonging to this group of pathogens [16–18]. Hence, survival of enterobacterial species seems to be drastically reduced in biogas fermentations. Sanitation under thermophilic conditions might occur. However, this effect is not visible from our data, since even mesophilic conditions in fermentation seem to be non-permissive for Enterobacteria. Likewise, clostridial pathogens are absent in the samples analyzed in this study which also is line with previous results obtained for experimental methanogenic bioreactors [26]. The authors of the latter study concluded that neither pathogenic Clostridium species nor Clostridia closely related to pathogenic ones could be detected in their samples [26]. Occurrence of pathogens such as Clostridium clostridioforme and Streptococcus infantarius in biogas fermentation samples should specifically be addressed in future studies since few identical EGTs were identified in the metagenome datasets analyzed here. C. clostridioforme appeared to be associated with serious or invasive human infections including bacteremia [27] whereas S. infantarius can be isolated from traditionally fermented dairy and plant products and holds a potential health risk for animals and humans [29]. Regarding the latter species, a residual risk remains when applying digestates as fertilizer. It should also be noted here that the metagenomes of this study were not sequenced to saturation. Accordingly, rare pathogens might not have been detected due to low coverage of their genomes within the metagenome sequence datasets. Moreover, it has to be considered that some of the reactors sampled in this study were not continuously fed with manure. Hence, the pathogenic load in reactors regularly fed with manure – especially pig manure – could be higher. In future studies regarding detection of pathogens in biogas fermentation samples, gene-centered approaches applying high-throughput sequencing would be appropriate to identify specific rare pathogens. In this context, 16S rRNA gene amplicon sequencing or PCR-based analysis of pathogen-specific signature genes should be considered. It should also be taken into account that genomic traces identified in this study might not be in an active state anymore. Hence, in situ analyses along with metagenome analysis might further improve detection of pathogens in this context.
Concerning identification of sequence tags representing bacterial toxins and virulence determinants, it has to be taken into account that the genomic context of organisms encoding these determinants is of importance. For example, virulence determinants that are present in a non-pathogenic species most probably are harmless, whereas these genes may enhance virulence of pathogens. Metagenome studies intrinsically do not allow drawing any reliable conclusions regarding the genomic context of particular determinants. Accordingly, possible detection of toxin genes and virulence determinants only allows for very vague assessments concerning the presence of pathogens. However, antibiotic resistance determinants may be released with digestates and hence spread in the environment. Since in most biogas reactors manure from cattle or pigs is used as substrate, antibiotic resistant bacteria selected by application of antimicrobial treatments will end up in biogas plants where resistance determinants located on mobile genetic elements potentially can be transferred to biogas community members and finally be released with digestates into the environment. It cannot be excluded that bacteria harboring resistance determinants occasionally get incorporated by humans. However, prediction of resistance determinants by sequence similarity based methods clearly leads to an overestimation of resistance determinants since in principle functionality of these determinants remains unclear. It should also be noted here that direct application of manure from cattle or pigs as fertilizer on fields is a commonly accepted agricultural practice.
In summary, detection of putative pathogenic bacteria exploiting metagenome sequence data currently is the most reliable approach addressing this issue. However, the informative value of the method clearly depends on careful selection of pathogen-indicative determinants. Results of this study revealed that the risk of unintended proliferation of pathogens in biogas fermentations and their dissemination in the environment is rather low.
Methods
Datasets
Seven metagenomic datasets from different experimental and agricultural biogas reactors were analyzed for the presence of putative pathogenic bacteria (Table 1). The samples B55, S55, S65 and S70 were taken from an experimental two-phase leach-bed biogas reactor. This system consisted of a leach-bed reactor, a leachate reservoir and an anaerobic filter reactor, which was described recently [19]. The reactor was inoculated with manure after it was brought into service. Since then it has been fed with rye silage and straw. The samples S55, S65 and S70 derived from the digestate of the leach-bed reactor at 55°C, 65°C and 70°C, respectively, whereas B55 was taken from a packing of the anaerobic filter reactor at 55°C. The samples G5 and G30 derived from a 30-day anaerobic digestion batch test (37°C) with recalcitrant substrate taken at day 5 and day 30. Here, digestates of an anaerobic digester supplied with maize and manure was mixed with low amounts of straw and hay. Finally, the sample U1 derived from a mesophilic (41°C) agricultural biogas plant supplied with maize silage, green rye and low amounts of chicken manure [21].
The libraries, which were created from the isolated metagenomic DNAs, were sequenced on the Genome Sequencer (GS) FLX platform applying the FLX Titanium sequencing chemistry (Roche Applied Science). Raw data were processed by means of the analysis pipeline for whole genome shotgun sequence reads applying the GS FLX System Software (version 2.6).
Taxonomic profiles
The metagenome sequences obtained from the different samples were classified using the BLASTx-approach of CARMA3 [24] in order to determine the prevalence of potentially human pathogenic bacteria. For this purpose, CARMA3 was executed using standard settings. Afterwards, the profile was evaluated for the presence of selected species that are associated with infections in humans (species of the genera Escherichia, Streptococcus, Vibrio, Clostridium, Salmonella and Shigella). Finally, identified environmental gene tags (EGTs) were manually searched for homologue matches in the NCBI non-redundant nucleotide (NT) database using standard BLAST settings [48].
Genome mappings
The metagenomic reads of the different datasets were aligned to chromosomal sequences of selected pathogenic bacteria (Table 9) by means of the gsMapper program (Roche Genome Analyzer Data Analysis Software Package, version 2.6) in order to confirm the presence of their virulence determinants. Default settings of the gsMapper (90% sequence identity, 40 bp overlap) were used to also map reads originating from closely related species. Multiple contigs and corresponding consensus sequences were generated from the mapped reads. To identify virulence determinants of selected reference strains in the metagenomic datasets, a BLAST-analysis of the resulting contigs was performed. The BLAST-analysis was done with rather relaxed settings (E-value: 1*10-4, sequence identity: 80%), but with organism-specific BLAST-database. The results were then analyzed for the presence of known pathogenic determinants.
Identification of reads with similarity to toxin protein families
The Pfam database encompasses altogether 13,672 protein families also including toxic protein families and virulence determinants, which are all represented by multiple sequence alignments and hidden Markov models [49]. Different protein families relevant for toxicity of Clostridium sp., E. coli, Streptococcus sp., Staphylococcus sp., Shigella sp. and Vibrio sp. were identified (Table 5) using the Pfam database version 26.0 [49]. Seed sequences matching these Pfam domains were extracted from the Pfam database. The metagenomes were then screened for the presence of these factors based on a BLASTx analysis (e-value cutoff: 1*10-20) and annotated according to their best hit. The results were then checked for the Pfam accessions of interest. The stringent cutoff was applied because metagenomic reads typically represent gene fragments which is due to short read lengths. Therefore it is important to apply stringent cutoffs to avoid false positive assignments caused by conserved domains. The length of a query sequence often does not suffice to distinguish between hits to conserved domains (false positives) and full-length gene alignments. Thus, a more stringent cutoff is required when analysing short reads compared to analyses involving full-length genes. Additionally, the sequence database applied is a comparatively small database which also requires a more stringent cutoff to exclude false positive hits.
BLASTn vs. the microbial virulence database MvirDB
Metagenomic sequences were screened for the presence of gene fragments encoding putative virulence factors based on a BLAST search versus the MvirDB database [14] using an e-value cutoff of 1*10-20 and annotated with the best hit to analyze the presence of further and previously not selected putative virulence determinants. Only those hits against database entries categorized as “virulence factor” were used for further analysis. Hits against database entries classified as “protein toxin”, “antibiotic resistance” or “virulence protein” were further classified regarding their function.
Abbreviations
- MvirDB:
-
Microbial Virulence Database
- BLAST:
-
Basic Local Alignment Search Tool
- RTX:
-
Repeats in Toxin
- PCR:
-
Polymerase Chain Reaction
- Stx:
-
Shiga toxin
- EGT:
-
environmental gene tag
- NT:
-
NCBI non-redundant nucleotide database
- LPS:
-
lipopolysaccharide
- GS:
-
Genome Sequencer
- Pfam:
-
Protein families
- CSTR:
-
Continuously stirred tank reactor.
References
Newell DG, Koopmans M, Verhoef L, Duizer E, Aidara-Kane A, Sprong H, Opsteegh M, Langelaar M, Threfall J, Scheutz F, van der Giessen J, Kruse H: Food-borne diseases — The challenges of 20 years ago still persist while new ones continue to emerge. Future Challenges to Microbial Food Safety Contributions resulting from a conference held in Wolfheze, the Netherlands. Int J Food Microbiol 2010,139(S1):3-15.
Cork SC: Epidemiology of Pathogens in the Food Supply. In Zoonotic pathogens in the food chain. Edited by: Krause DO, Hendrick S. Wallingford, Oxfordshire: CABI; 2011:21-58.
Brzuszkiewicz E, Thuermer A, Schuldes J, Leimbach A, Liesegang H, Meyer F, Boelter J, Petersen H, Gottschalk G, Daniel R: Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: Entero-Aggregative-Haemorrhagic Escherichia coli (EAHEC). Arch Microbiol 2011, 12: 883-891.
Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H: Prospective genomic characterization of the german enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid Next Generation Sequencing technology. PLoS One 2011, 7: e22751.
Altmann M, Wadl M, Altmann D, Benzler J, Eckmanns T, Krause G, Spode A, Ander Heiden M: Timeliness of surveillance during outbreak of shiga toxin-producing Escherichia coli infection, Germany, 2011. Emerg Infect Dis 2011, 10: 1906-1909.
Milinovich GJ, Klieve AV: Manure as a Source of Zoonotic Pathogens. In Zoonotic pathogens in the food chain. Edited by: Krause DO, Hendrick S. Wallingford, Oxfordshire: CABI; 2011:59-83.
Dahlenborg M, Borch E, Radstrom P: Development of a combined selection and enrichment PCR procedure for Clostridium botulinum types B, E, and F and its use to determine prevalence in fecal samples from slaughtered pigs. Appl Environ Microbiol 2001, 10: 4781-4788.
Dahlenborg M, Borch E, Radstrom P: Prevalence of Clostridium botulinum types B, E and F in faecal samples from Swedish cattle. Int J Food Microbiol 2003, 2: 105-110.
Boll C: Chronischer Botulismus. Tod aus der Biogasanlage. Wild und Hund 2011, 10: 14-19.
Rodloff AC, Krüger M: Chronic Clostridium botulinum infections in farmers. Anaerobe 2012, 2: 226-228.
Krüger M, Große-Herrenthey A, Schrödl W, Gerlach A, Rodloff A: Visceral botulism at dairy farms in Schleswig Holstein, Germany: prevalence of Clostridium botulinum in feces of cows, in animal feeds, in feces of the farmers, and in house dust. Anaerobe 2012, 2: 221-223.
Pallen MJ, Wren BW: Bacterial pathogenomics. Nature 2007, 7164: 835-842.
Dobrindt U, Hochhut B, Hentschel U, Hacker J: Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2004, 5: 414-424.
Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T: MvirDB - a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res 2007,35(supl. 1):D391-D394.
Liu B, Pop M: ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res 2009,37(supl. 1):D443-447. Database issue
Goberna M, Podmirseg SM, Waldhuber S, Knapp BA, García C, Insam H: Pathogenic bacteria and mineral N in soils following the land spreading of biogas digestates and fresh manure. Appl Soil Ecol 2011, 49: 18-25.
Massé D, Gilbert Y, Topp E: Pathogen removal in farm-scale psychrophilic anaerobic digesters processing swine manure. Bioresource Technol 2011, 2: 641-646.
Watcharasukarn M, Kaparaju P, Steyer J, Krogfelt KA, Angelidaki I: Screening Escherichia coli , Enterococcus faecalis , and Clostridium perfringens as indicator organisms in evaluating pathogen-reducing capacity in biogas plants. Microb Ecol 2009, 2: 221-230.
Rademacher A, Zakrzewski M, Schlüter A, Schoenberg M, Szczepanowski R, Goesmann A, Pühler A, Klocke M: Characterization of microbial biofilms in a thermophilic biogas system by high-throughput metagenome sequencing. FEMS Microbiol Ecol 2012, 3: 785-799.
Hanreich A, Schimpf U, Zakrzewski M, Schlüter A, Benndorf D, Heyer R, Rapp E, Pühler A, Reichl U, Klocke M: Metagenome and metaproteome analyses of microbial communities in mesophilic biogas producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst Appl Microbiol 2013. in Press
Krause L, Diaz NN, Edwards RA, Gartemann K, Kroemeke H, Neuweger H, Pühler A, Runte KJ, Schlüter A, Stoye J, Szczepanowski R, Tauch A, Goesmann A: Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol 2008, 1–2: 91-101.
Jaenicke S, Ander C, Bekel T, Bisdorf R, Droege M, Gartemann K, Juenemann S, Kaiser O, Krause L, Tille F, Zakrzewski M, Pühler A, Schlüter A, Goesmann A: Comparative and joint analysis of two metagenomic datasets from a biogas fermenter obtained by 454-pyrosequencing. PLoS One 2011, 1: e14519.
Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann K, Krahn I, Krause L, Krömeke H, Kruse O, Mussgnug JH, Neuweger H, Niehaus K, Pühler A, Runte KJ, Szczepanowski R, Tauch A, Tilker A, Viehöver P, Goesmann A: The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol 2008, 1–2: 77-90.
Gerlach W, Stoye J: Taxonomic classification of metagenomic shotgun sequences with CARMA3. Nucleic Acids Res 2011, 14: e91.
Popoff MR, Bouvet P: Clostridial toxins. Future Microbiol 2009, 8: 1021-1064.
Dohrmann AB, Baumert S, Klingebiel L, Weiland P, Tebbe CC: Bacterial community structure in experimental methanogenic bioreactors and search for pathogenic clostridia as community members. Appl Microbiol Biotechnol 2011, 6: 1991-2004.
Finegold SM, Song Y, Liu C, Hecht DW, Summanen P, Kononen E, Allen SD: Clostridium clostridioforme : a mixture of three clinically important species. Eur J Clin Microbiol 2005, 5: 319-324.
Deublein D, Steinhauser A: Biogas from waste and renewable resources. An introduction. Weinheim, Chichester: Wiley-VCH; 2010.
Jans C, Follador R, Lacroix C, Meile L, Stevens MJA: Complete genome sequence of the african dairy isolate Streptococcus infantarius subsp infantarius strain CJ18. J Bacteriol 2012, 8: 2105-2106.
Kouguchi H, Watanabe T, Sagane Y, Sunagawa H, Ohyama T: In vitro reconstitution of the Clostridium botulinum type D progenitor toxin. J Biol Chem 2002, 4: 2650-2656.
Tan KS, Wee BY, Song KP: Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile . J Med Microbiol 2001, 7: 613-619.
Tsuge H, Nagahama M, Nishimura H, Hisatsune J, Sakaguchi Y, Itogawa Y, Katunuma N, Sakurai J: Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J Mol Biol 2003, 3: 471-483.
Siezen RJ, Leunissen JA: Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 1997, 3: 501-523.
Valbuzzi A, Ferrari E, Albertini AM: A novel member of the subtilisin-like protease family from Bacillus subtilis . Microbiol (Reading Engl 1999, 145: 3121-3127.
Graycar T: Proteolytic cleavage, reaction mechanism. In Encyclopedia of bioprocess technology. Fermentation, biocatalysis, and bioseparation. Edited by: Flickinger MC. New York, NY: Wiley; 1999.
Tornero P, Conejero V, Vera P: Identification of a new pathogen-induced member of the subtilisin-like processing protease family from plants. J Biol Chem 1997, 22: 14412-14419.
Welch RA: RTX toxin structure and function: A story of numerous anomalies and few analogies in toxin biology. Curr Top Microbiol 2001, 257: 85-111. 10.1007/978-3-642-56508-3_5
Ficko-Blean E, Boraston AB: Cloning, recombinant production, crystallization and preliminary X-ray diffraction studies of a family 84 glycoside hydrolase from Clostridium perfringens . Acta Crystallogr F 2005, 61: 834-836. 10.1107/S1744309105024012
Adams JJ, Gregg K, Bayer EA, Boraston AB, Smith SP: Structural basis of Clostridium perfringens toxin complex formation. Proc Natl Acad Sci USA 2008, 34: 12194-12199.
Robertson KP, Smith CJ, Am G, Rocha ER: Characterization of Bacteroides fragilis hemolysins and regulation and synergistic interactions of HlyA and HlyB. Infect Immun 2006, 4: 2304-2316.
Welch RA, Pellett S: Transcriptional organization of the Escherichia coli hemolysin genes. J Bacteriol 1988, 4: 1622-1630.
Hug I, Couturier MR, Rooker MM, Taylor DE, Stein M, Feldman MF: Helicobacter pylori lipopolysaccharide is synthesized via a novel pathway with an evolutionary connection to protein N-glycosylation. PLoS Pathog 2010,6(3):e1000819. 10.1371/journal.ppat.1000819
Horwich AL, Fenton WA, Chapman E, Farr GW: Two families of chaperonin: physiology and mechanism. Annu Rev Cell Dev Biol 2007, 23: 115-145. 10.1146/annurev.cellbio.23.090506.123555
Frees D, Savijoki K, Varmanen P, Ingmer H: Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol 2007, 5: 1285-1295.
Nikaido H: Multidrug resistance in bacteria. Annu Rev Biochem 2009, 78: 119-146. 10.1146/annurev.biochem.78.082907.145923
Kuzin AP, Sun T, Jorczak-Baillass J, Healy VL, Walsh CT: Knox, JR: Enzymes of vancomycin resistance: the structure of D-alanine-D-lactate ligase of naturally resistant Leuconostoc mesenteroides . Struct Fold Des 2000, 5: 463-470.
Roper DI, Huyton T, Vagin A, Dodson G: The molecular basis of vancomycin resistance in clinically relevant Enterococci : crystal structure of D-alanyl-D-lactate ligase (VanA). Proc Natl Acad Sci USA 2000, 16: 8921-8925.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 3: 403-410.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD: The Pfam protein families database. Nucleic Acids Res 2012, 40: D290-301. Database issue 10.1093/nar/gkr1065
Acknowledgements
FGE, MZ, IM and DW acknowledge the receipt of a scholarship from the CLIB-Graduate Cluster Industrial Biotechnology co-financed by the Ministry of Innovation of North Rhine-Westphalia. AS gratefully acknowledges the METAEXPLORE grant from the European Community (KBBE 222625). SJ acknowledges funding from the BMBF grant GenoMik-Transfer (0315599A & 0315599B). AR and AH were supported by grants provided as part of the BioEnergie2021 program of the German Federal Ministry of Education and Research (BMBF) coordinated by the Project Management Jülich (PTJ) (grant number 03SF0349C and 03SF0346B). We acknowledge support for the Article Processing Charge by the Deutsche Forschungsgemeinschaft and the Open Access Publication Funds of Bielefeld University Library.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FGE participated in the search of putative virulence determinants in the metagenome sequence data and helped to draft the manuscript. AR and AH searched for putative pathogenicity determinants in functional profiles by exploiting Pfam assignments. MH and IM were involved in the search of putative virulence and resistance determinants in the metagenome sequence data. SJ and MZ managed metagenome sequence data, initiated computational analyses and performed bioinformatic analyses by means of BLAST, MetaSAMS and CARMA3. MZ deduced taxonomic profiles from metagenome sequence data and searched for putative pathogens. DW mapped metagenome sequence data on selected reference genomes. AP, MK and AS conceived of the study, coordinated analyses, and formed the paper concept. All authors contributed to writing of the manuscript, read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Eikmeyer, F.G., Rademacher, A., Hanreich, A. et al. Detailed analysis of metagenome datasets obtained from biogas-producing microbial communities residing in biogas reactors does not indicate the presence of putative pathogenic microorganisms. Biotechnol Biofuels 6, 49 (2013). https://doi.org/10.1186/1754-6834-6-49
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-6-49