
Abstract
Background
The dietary consumption of cuprizone – a copper chelator – has long been known to induce demyelination of specific brain structures and is widely used as model of multiple sclerosis. Despite the extensive use of cuprizone, the mechanism by which it induces demyelination are still unknown. With this review we provide an updated understanding of this model, by showcasing two distinct yet overlapping modes of action for cuprizone-induced demyelination; 1) damage originating from within the oligodendrocyte, caused by mitochondrial dysfunction or reduced myelin protein synthesis. We term this mode of action ‘intrinsic cell damage’. And 2) damage to the oligodendrocyte exerted by inflammatory molecules, brain resident cells, such as oligodendrocytes, astrocytes, and microglia or peripheral immune cells – neutrophils or T-cells. We term this mode of action ‘extrinsic cellular damage’. Lastly, we summarize recent developments in research on different forms of cell death induced by cuprizone, which could add valuable insights into the mechanisms of cuprizone toxicity. With this review we hope to provide a modern understanding of cuprizone-induced demyelination to understand the causes behind the demyelination in MS.
Similar content being viewed by others

Background
Multiple Sclerosis (MS) is a chronic neurological disorder characterized by the loss of myelin or demyelination. Myelin is produced by oligodendrocytes in the central nervous system (CNS) wherein a single oligodendrocyte may myelinate up to 80 axon segments [1] to facilitate axonal signaling [1] and provide metabolic support to the axon [2]. Loss of oligodendrocyte and demyelination is associated with axonal damage [3,4,5,6]. Given the importance of myelin to CNS health, a wide body of MS research focuses on understanding the vulnerability of oligodendrocytes and their associated myelin sheaths to identify novel strategies to improve myelin regeneration, otherwise known as remyelination, and to slow neurodegeneration.
The disease course of MS is variable – different stages of the disease are characterized by distinct cellular mechanisms and inflammatory processes that depend on CNS-resident immune cells such as microglia, and the infiltration of peripheral immune cells, such as T- and B-cells. Primary-progressive MS (PPMS) is characterized by a linear worsening of symptoms from disease onset [7] and given that the blood–brain barrier (BBB) is mostly intact, CNS/cerebrospinal fluid -localized mechanisms likely facilitate demyelination [8, 9]. Relapsing–remitting MS (RRMS) is characterized by new lesions that cause distinct spikes in symptomatic disease that are followed by a recovery from disability [7, 10]. Relapsing stages of MS generally involve BBB breakdown and infiltration of T- and B-cells that then propagate inflammation to induce damage. RRMS often evolves into a progressive state with no symptomatic remission, termed secondary-progressive MS (SPMS) [7, 11]. Due to the different cellular mechanisms that characterize the different MS stages, modeling the disease as a whole is not feasible with one given model. Thus, three types of animal models are widely employed for modeling the different aspects of MS: (1) Virus-induced demyelination, (2) Experimental autoimmune encephalomyelitis (EAE), and (3) toxin provoked demyelination [12, 13]. Viral-induced demyelination is provoked by infection of mice with viruses such as Theiler’s murine encephalomyelitis virus (TMEV) or mouse hepatitis virus. This infection leads to inflammatory demyelination in brain and spinal cord, with a mixture of cluster of differentiation (CD)4+ and CD8+ T-cells, B-cells, microglia, and macrophages contributing to the demyelination [14].
To induce EAE, mice are injected with one or more myelin peptides in combination with an immune-boosting adjuvant and pertussis toxin, myelin reactive primed T-cells, or by expressing an autoimmune T-cell receptor [13]. These stimuli evoke an inflammatory autoimmune response against myelin and axons, facilitated by CNS infiltrating T-cells and monocytes that cause demyelination and disability [15, 16]. EAE is thought to model lesion formation and inflammatory injury characteristic of relapsing–remitting MS. However, the profound immune response and the stochastic appearance of demyelinated lesions make it challenging to decipher CNS intrinsic immune relevance to disease progression and remyelination (see [12, 13] for further reading). For these reasons, researchers may employ toxin-based animal models that limit the stochasticity of lesion formation and peripheral immune cell infiltration [17].
Toxin-based models of demyelination are primarily used to study mechanisms of primary demyelination and subsequent remyelination [18, 19]. Certain toxin-induced models of demyelination are initiated by the injection of toxic compounds such as lysophosphatidylcholine (LPC), lipopolysaccharide (LPS), or ethidium bromide into white matter areas to create a focal lesion at the injection site. An alternative toxin-based model of demyelination is the dietary consumption of cuprizone that causes demyelination of specific brain white matter regions, such as corpus callosum and hippocampus [20]. Among the toxin-based methods to induce demyelination, Bis-cyclohexanone-oxaldihydrazone, colloquially known as cuprizone, was found to be the most commonly used MS model in a recent systematic review and meta-analysis [21]. Cuprizone is a copper chelator that, when fed to rodents, results in a loss of oligodendrocytes and thus, myelin, in specific brain regions, with limited BBB disturbance and infiltration of peripheral immune cells. Despite cuprizone being first described in 1950 and the current widespread use of the toxin as a model to study demyelination and remyelination, little is known about how cuprizone elicits its toxic effects on the oligodendrocyte population.
To this end, there is a need to model demyelination and subsequent remyelination in animal models to enhance our understanding of oligodendrocyte susceptibility and to test potential pro-myelinating treatments that could be moved towards clinical therapies [22]. We argue that an improved understanding of the cuprizone model is vital for the use of this model in pre-clinical studies. To provide a better understanding, we showcase how early cuprizone experiments contributed to our understanding of how it damages oligodendrocytes and myelin. We discuss both cuprizone-induced oligodendrocyte-intrinsic mechanisms of demyelination and oligodendrocyte-extrinsic factors that can induce or exacerbate oligodendrocyte toxicity, with a focus on extrinsic immune-derived factors. We also summarize the diverse forms of oligodendrocyte cell death that occur in the cuprizone model as understanding oligodendrocyte death may illuminate mechanisms of cuprizone toxicity. We believe, those modes of action of cuprizone toxicity can be further categorized into three forms of pathology: (A) Oligodendrocyte cell death as a direct result of the effect of cuprizone on the oligodendrocyte population, which we call ‘primary oligodendrocytopathy’. (B) Oligodendrocyte cell death caused by activated astrocytes and microglia in response to oligodendrocyte damage, which we term ‘toxic innate immunity’ and (C) oligodendrocyte cell death caused by astrocytes and microglia following a direct effect of cuprizone on these cells, which we term ‘primary immunocytopathy’.
Main text
The cuprizone model – past and present
Considering how long and widespread cuprizone is used as a model for demyelination and remyelination [21], it is surprising that so little is understood regarding the mechanisms of its toxicity. The knowledge we do have about the mechanisms of how cuprizone elicits its effect is greatly influenced by findings obtained more than six decades ago. To understand how this still affects the way we think about cuprizone, we must explore how cuprizone was established as a model for MS.
The past – establishment of the cuprizone model of MS
Cuprizone was first described in 1950 by Gustav Nilsson, when he found that cuprizone is a sensitive indicator for copper, producing a colorimetric shift in the presence of copper [23]. In subsequent years, cuprizone was revealed to also produce severe systemic effects on the nervous system and peripheral organs in rodents. William Carlton first fed cuprizone to albino mice in the 1960s and described reduced growth of weanling mice, induced paresis, and terminated pregnancies following dietary consumption [24, 25]. These altercations coincided with brain edema, non-inflammatory demyelination, and astrogliosis in the cerebellum, cerebellar cortex, and medulla [24, 25]. In the late 1960s, others described mitochondrial abnormalities following cuprizone consumption in the mouse liver [26]. Cuprizone administration induces mitochondrial enlargement, severely alters metabolic rates, and impairs oxidative phosphorylation by reducing mitochondrial enzymes that contain copper as a cofactor, such as monoamine oxidase [27] and cytochrome c (Cyt c) oxidase [28, 29] in the brain of mice. Cuprizone treatment impaired hepatocytic mitochondrial function [30], which was thought to be a result of copper deficiency. Whether the consequences of cuprizone are directly related to copper chelation is unclear given that copper supplementation has limited capacity to reduce cuprizone toxicity to the CNS of mice [24].
Although most of the early experiments using cuprizone were conducted in mice, a number of studies demonstrated detrimental effects of cuprizone administration to the brain – similar to those in mice – of other rodents, such as rats [31,32,33,34,35], guinea pigs [31] or hamsters [36, 37]. Interestingly, in experiments using hamsters the cuprizone concentration needed to be increased to 3% or even 5%—compared to 0.2%—0.5% in experiments on mice or rat—to induce brain alterations. One very small study using non-human primates did not find brain demyelination in young cynomolgus macaques even after 18 weeks on a 3% cuprizone diet [38]. However, the lack of demyelination could be because even higher, but untested, doses of cuprizone are needed to induce demyelination in macaques. For this review, outside of these studies, unless stated otherwise, all other cuprizone research mentioned is carried out in mice.
The BBB of mice was found to be largely intact, as confirmed by various assays for leakage of components of the periphery into the CNS, including horseradish peroxidase [39, 40], immunohistochemistry [40], and radioactive tracers [41], which is why it was long believed that the cuprizone-induced demyelination occurs without peripheral involvement. Due to these early findings, metabolic disturbance of the mitochondrion was long thought to be the main reason for oligodendrocyte death and myelin loss. However, the early explorations of cuprizone toxicity used mice of varying strains, sex, and ages, which complicates interpretations of findings between experiments, thus highlighting the need for a more standardized approach.
The present – hallmarks of the modern cuprizone model
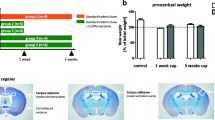
To standardize the cuprizone model, Hiremath and colleagues introduced a protocol for lower-dose cuprizone administration (0.2% w/w) in C57BL/6 mice in 1998 [42, 43] that is still widely used today. Now, cuprizone is commonly administered for 4–6 weeks to study acute demyelination or up to 12 weeks to study chronic demyelination, and mice are occasionally followed after cuprizone withdrawal to study remyelination (Fig. 1A). Interestingly, endogenous repair processes occur despite the consumption of cuprizone [44]. For example, spontaneous remyelination, with up to 50% of myelinated axons and increased numbers of mature oligodendrocytes, can be seen after 6 weeks, despite continued exposure to cuprizone [45, 46]. The histopathological hallmark of the cuprizone model as standardized by Hiremath is oligodendrocyte death and subsequent demyelination in the corpus callosum, superior cerebellar peduncles, hippocampus [42, 46,47,48,49,50,51,52,53,54] and several other areas of the mouse brain (reviewed in [20]). In this model, myelin destabilization and loss begin at 2–3 weeks of cuprizone administration and peaks at week 4–5 [42], myelin levels stay low until at least week 12 [55] with continuous cuprizone feeding. This progressive demyelination is accompanied by robust astrogliosis and microgliosis (Fig. 1B). Astrogliosis begins coincident with demyelination after 2 weeks on cuprizone diet. Astrocyte densities peak around 5–6 weeks and stay high until 12 weeks. Microglia also proliferate and expand after 2 weeks, and their density peaks at 5—6 weeks [45, 55, 56]. Demyelination of brain white matter coincides with behavioral changes and loss of cognitive and motor function [57, 58]. While the cuprizone diet causes widespread demyelination, removal of cuprizone permits remyelination of the corpus callosum and cerebellar peduncles after a seven-week cuprizone diet [59] or even a longer seven-month diet, although remyelination is less efficient with chronic cuprizone consumption [60]. Despite this remyelination, persistent behavioral changes such as poor adaptive motor learning and motor function are apparent 4–7 weeks after cuprizone withdrawal, which is several weeks longer than is required for remyelination [57, 61]. Others found ongoing degeneration of the corpus callosum tracts continues despite robust remyelination [62, 63] suggesting ongoing dysfunction despite myelin repair.

A Representation of commonly used cuprizone exposure protocol. Rodents are fed with concentrations of typically 0.2 – 0.3% (w/w) cuprizone in animal chow for up to 6 weeks to study acute demyelination and 12 weeks to study chronic demyelination. This is sometimes followed by a period of normal chow without cuprizone (withdrawl) to induce remyelination. B Time course of cuprizone intoxication in the corpus callosum. The dashed black line represents myelin integrity, and the colored lines represent cellularity (numbers of cells, unless stated otherwise) of microglia/macrophages in red, astrocytes in blue (here cellularity refers to reactivity), T-cells in teal, and neutrophils in purple. Below the timeline we show different modes of cell death observed in the cuprizone model, and their time of occurrence depicted by the black bar. Apoptosis (green box) occurs between day 2 and 3 weeks. Ferroptosis markers are elevated (yellow box) between day 2 and 4 weeks. Necroptosis (brown box) is seen in the cuprizone model between week 2 and week 5. Inflammasome-mediated cell death/pyroptosis (grey box) occurs between 3 and 5 weeks
Together, the adoption of a standardized cuprizone administration protocol improved inter-study reliability and by using the C57BL/6 inbred mouse strain, provided better options for conducting studies on transgenic mice, many of which are bred to a C57BL/6 background. By combining a standardized cuprizone model with transgenic mice, researchers are able to dissect genes, pathways and cell-specific roles in cuprizone toxicity more intimately.
Cuprizone-induced pathology mimics aspects of MS
Histopathological analysis of post-mortem MS tissue revealed profound lesion heterogeneity both within and between MS patients [64,65,66]. Lucchinetti and colleagues categorized patterns of pathology into distinct lesion types, all of which contain T-cell, plasma cell, and macrophage infiltrates [65, 67]. Lesion types 1 and 2 are thought to be the product of T-cell-mediated autoimmune processes, which are commonly replicated with the EAE model. Lesion types 3 in contrast are displaying features of primary oligodendrogliapathy and heightened oligodendrocyte cell death, comparable to cuprizone-induced demyelination [65, 68]. Other features common to both cuprizone-induced and type 3 MS lesions are mitochondrial stress, selective loss of myelin-associated glycoprotein (MAG), lesions with indistinct borders that are not perivascular, pronounced glial activation, and lymphocyte infiltration [43, 46, 65, 69,70,71]. Type 3 lesions are also often found in the corpus callosum of MS patients, a brain region primarily affected by cuprizone [65].
Like in MS lesions, T-cells are found in cuprizone lesions. Indeed, cuprizone lesions and progressive MS lesions contain comparable, if relatively sparse, T-cell densities [70]. The source of the T-cells in progressive MS lesions is unclear given that the BBB remains largely intact despite the enriched lymphocytic trafficking [72, 73]. These lymphocytes may be remainders from lesions formed earlier in the disease or they may migrate from prominent intrathecal inflammation – a characteristic of progressive MS [74]. With cuprizone exposure, the recruitment of T-cells to the lesion site [70] may result from the decreased BBB integrity induced by cuprizone [75, 76]
Cuprizone toxicity produces profound demyelination in the white and grey matter of the brain that mirrors sites of MS pathology [46]. Corpus callosum demyelination is detectable by histology and magnetic resonance imaging (MRI) in MS patients [65]. Similarly, the medial aspect of the splenium of the corpus callosum and lateral aspect of its body are highly reliable sites of injury during intoxication with 0.2% cuprizone, prone to profound microglia/macrophage accumulation [54]. Thus, the corpus callosum has become a popular region for the study of white matter demyelination. Grey matter demyelination also presents in MS – cortical, and hippocampal atrophy is most severe in the progressive stages of MS and is a prominent source of permanent disability [77, 78]. Cuprizone toxicity is one of the few models of MS that produces grey matter demyelination, specifically in the cortex, hippocampus, and deep grey matter nuclei [79, 80]. Cortical grey matter demyelination is evident after 4 weeks of cuprizone treatment, but microglia/macrophage density in this region remains near baseline levels [54, 81, 82]. Likewise, cuprizone administration results in layer-specific cortical degeneration, with demyelination enriched within layers five and six [54]. Given that cuprizone administration induces both white and grey matter demyelination, it remains a highly relevant and useful model for studying the biological underpinnings of MS lesions.
Despite these similarities, there are some distinct differences between MS and the cuprizone model. For example, grey matter remyelination in MS is more efficient than white matter remyelination [83, 84]. However, upon cuprizone withdrawal, the white matter corpus callosum is remyelinated nearly to control levels within a week. In contrast, the grey matter of the cortex does not efficiently remyelinate in the same period, suggesting that grey matter remyelination may be slower [85]. The slower cortical remyelination may relate to technical challenges in measuring structures with vastly different myelination prior to cuprizone treatment, but this remains to be confirmed. Transgenic models that fate-map new myelin will be essential to better understanding grey matter demyelination and remyelination. Another difference between type 3 and cuprizone lesions is that MS lesions lack complement protein deposition [65], which is a feature of cuprizone toxicity [85]. Despite the striking similarities between cuprizone and MS lesions, an incomplete understanding of the mechanisms of cuprizone limits its full potential as a preclinical model for identifying next-generation therapeutics for MS.
Death from within: how oligodendrocyte intrinsic mechanisms could lead to death during cuprizone treatment
Although MS is generally described as an autoimmune and inflammatory disease of the CNS, some argue that MS may initially reflect a non-inflammatory degenerative disorder that affects myelin and in doing so, begins a cascade that elicits the more commonly reported inflammatory response (for review see [8]). In line with this, researchers explore whether cuprizone acts directly on oligodendrocytes/the myelin sheath. Cuprizone is a copper chelator and considering that dysregulation of copper homeostasis in humans can lead to neurodegenerative diseases such as Menkes disease [86, 87], Wilson’s diseases [88] and is thought to contribute to Alzheimer’s disease and Parkinson’s disease [88, 89], better understanding potential mechanisms underlying demyelination mediated by copper chelation is necessary to better understand cuprizone toxicity.
Copper levels in the brain of cuprizone-treated mice are altered [28, 90, 91], though the extent of this change is unclear, as studies on copper levels in the brains of cuprizone-treated mice are contradictory. Using spectrophotometry Gilles Venturini reported a roughly 40% decrease of copper (Cu2+) in the brain of Swiss mice treated with cuprizone for 3–4 weeks [28], while others reported an increase in copper after 1 week (Cu2+ and Cu+) [90] and 3, 6 and, 9 months (total free and bound) [91] following cuprizone treatment. Similar to the inconclusive findings regarding the copper levels after cuprizone treatment, the actual chemistry of the copper-cuprizone complex is disputed as well. Some studies suggest hydrolysis of cuprizone in aqueous solutions to ‘monohydrazone cuprizone’ and multiple possible copper-cuprizone complexes in solution [23]. For example, Gustav Nilsson’s discovery that cuprizone changes color in copper solutions suggests that it can bind Cu2+ [23]. In later experiments cuprizone was shown to precipitate as a Cu2+-cuprizone complex when in high concentrations of copper. This Cu2+-cuprizone complex could not cross the intestinal epithelial barrier in an ex vivo experiment on mice intestines, which was thought to cause systemic copper deficiency [92]. Messori and colleagues proposed that two monohydrazone cuprizone molecules form a complex with the toxic Cu3+ [93]. A study on the redox properties of copper-cuprizone complexes confirmed the Cu3+-cuprizone complex and furthermore demonstrated that cuprizone can stabilize the toxic Cu3+ [94], which may be a source of oxidative stress.
The unresolved questions surrounding the effect of cuprizone treatment on brain copper levels and complex chemistry make it difficult to judge if the copper chelating property of cuprizone plays a role in cuprizone-induced demyelination. Indeed, there are a few studies suggesting a chelation independent mechanisms of cuprizone. In an in vitro investigation of the cuprizone inhibition of pig-plasma benzylamine oxidase—containing copper and pyridoxal phosphate as cofactor [95]—Lindström and Pettersson found that cuprizone interacts but does not extract copper from the enzymes active site [96]. Cuprizone likely covalently binds the other cofactor pyridoxal phosphate, which prevents enzymatic activity [96]. Taraboletti and colleagues confirmed this cuprizone-pyridoxal phosphate reaction and demonstrated cuprizone inhibits an aminotransferase by this same mechanism [97].
Systematically investigating the copper-cuprizone complex chemistry and copper chelation independent mechanisms may help unravel the mechanisms of cuprizone toxicity. Early after cuprizone initiation and in the context of minimal inflammation, changes in myelin composition and/or lipid polarity have been detected well in advance of oligodendrocyte cell body damage giving weight to the idea that cuprizone may have direct, albeit subtle, effects on myelin [79, 98, 99] – the human relevance of such altered myelin biochemistry prior to overt demyelination has since been confirmed [98]. In fact, the first signs of oligodendrocyte death are seen 2 days after initiation of cuprizone treatment [100, 101], which is several weeks earlier than the accumulation of other cell types at the site of injury [42]. In the following sections, we will review several theories on how cuprizone treatment leads to oligodendrocyte death from within, here defined as intrinsic death.
Mitochondrial dysfunction in oligodendrocytes as cause for oligodendrocyte death
Oligodendrocytes are highly specialized cells that produce myelin sheaths to ensure proper axonal signal transmission [102]. To maintain the myelin sheaths, oligodendrocytes require a high energy supply to synthesize sufficient amounts of the necessary lipids and proteins [103]. Oligodendrocytes also store large quantities of iron within ferritin [104, 105], but have low levels of the important reactive oxygen species (ROS) reducing agent glutathione (GSH) [106], which suggests that oligodendrocytes may be especially vulnerable to changes in metabolic rates and ROS. Therefore, the disturbance of oligodendrocyte mitochondria, the resultant energy shortage, ROS accumulation, and disturbance of lipid and protein synthesis, is discussed as a mode of action of cuprizone toxicity [20, 107, 108].
Cuprizone ingestion alters mitochondria in the CNS of mice [27,28,29, 109]. In vitro, cuprizone lowers the mitochondrial transmembrane potential of oligodendrocytes, but does not affect microglia, astrocytes, and neurons [110] – neither does cuprizone kill rat oligodendrocytes in vitro [111]. In vivo, 3 weeks of cuprizone administration results in morphological disturbances in oligodendrocyte mitochondria in the form of megamitochondria [109]. Enlarged megamitochondria are thought to reflect a compensatory response to elevated levels of ROS, based on evidence in vitro. Treatment of rat hepatocytes with hydrogen peroxide or ROS-inducing chemicals triggers a megamitochondria phenotype [112], which is reversed by buffering with free radical scavengers [113]. Megamitochondria can buffer a certain concentration of ROS, but if levels are kept high for too long, megamitochondria become permeable, swell, and release cyt c and apoptosis-inducing factor-1 (AIF-1) into the cytosol [114,115,116]. Cyt c released from mitochondria binds Apoptotic protease activating factor-1 (APAF-1) and subsequently activates caspase-9 [117] to trigger apoptosis [118,119,120] (Fig. 2A). After 3 weeks on a cuprizone diet, AIF-1—a regulator of Poly (ADP-ribose) polymerase (PARP) mediated cell death [121]—translocates into the nucleus of oligodendrocytes of treated mice, which is a step necessary for apoptosis. Inhibition of PARP prevents oligodendrocyte death and demyelination of the corpus callosum [71]. After 5 weeks of cuprizone diet, mitochondria in the corpus callosum of treated mice swell and release mitochondrial cyt c [122]. It is difficult to know the exact timeline of mitochondria swelling, cyt c release, and AIF-1 nuclear translocation and how this regulates early oligodendrocyte death as these early timepoints have not yet been investigated (Fig. 2A).

Various oligodendrocyte intrinsic mechanisms could lead to cell death of the oligodendrocyte during cuprizone intoxication. A Elevated levels of ROS can lead to permeabilization of mitochondria and subsequent release of cytochrome C and apoptosis-inducing factor-1. Cytosolic cyt c can activate caspase-9 and thus induce apoptosis in the oligodendrocyte, while apoptosis-inducing factor leads to cell death through PARP. B ROS reduces iron stored in ferritin from Fe3+ to Fe2+, which leads to iron release from ferritin. Free Fe2+ and ROS can induce lipid peroxidation, a hallmark of ferroptotic cell death. C Glutamate may reach toxic levels and act via NMDA receptors to permit excessive Ca2 influx. NMDA receptor activity can be modulated by copper. Depending on the concentration of copper present, NMDA receptor channel function is enhanced or inhibited. Also, PrPC can inhibit the NMDA receptor in the presence of copper. Elevated levels of glutamate and changes to copper concentration in the brain as a result of cuprizone administration therefore might increase Ca2+ accumulation in the cell to a toxic level and myelin degradation/oligodendrocyte death
ROS also directly induce oligodendrocyte death (e.g., ferroptosis [101]). Staining the corpus callosum with 8-OhdG reveals that oligodendrocytes contain oxidized nucleotides – an indication of elevated ROS [123] – after 1 week of cuprizone treatment [124]. Oligodendrocytes are particularly sensitive to ROS given their heightened storage of Fe3+ within ferritin. The ROS superoxide reduces Fe3+ to Fe2+ to liberate the iron from ferritin in vitro [125]. This free iron can react via the Fenton reaction to produce hydroxyl radicals to generate toxic 4-Hydroxynonenal (4-HNE) and malondialdehyde (MDA), which are products of lipid peroxidation. Lipid peroxidation is responsible for ferroptotic cell death (Fig. 2B) and may be another mode of oligodendrocyte death (described in more detail below).
In addition to elevated levels of ROS, cuprizone consumption alters mitochondrial enzymes in a way suggestive of metabolic dysfunction. For example, cuprizone administration reduces the mitochondrial enzymes monoamine oxidase (as early as 3 days [27]) and cyt c oxidase [28, 29] in the brain of mice, both of which require copper for catalysis [126]. A recent in vivo study used MRI to describe significantly reduced ATP levels in the brain after 1 week of cuprizone treatment [124], further suggesting metabolic decline. Lowered mitochondrial enzymes could account for the decreased metabolic rates of mitochondria during cuprizone intoxication.
Despite clear mitochondrial and metabolic changes within oligodendrocytes following cuprizone consumption, how these alterations relate to oligodendrocyte death remains unclear. For example, impaired mitochondrial metabolism alone does not induce oligodendrocyte death in vivo [127]. Oligodendrocyte-specific deletion of Cox10, which encodes a crucial component for cyt c oxidase assembly and is necessary for oxidative phosphorylation, failed to cause demyelination or neurodegeneration even at 14-months of age. These COX10-deficient oligodendrocytes maintained axonal thickness without a change in the number of oligodendrocyte progenitor cells (OPC) or astrocytes and did not induce microglial activation, suggesting that oligodendrocytes can function without oxidative phosphorylation. Oligodendrocytes with impaired oxidative phosphorylation instead boost aerobic glycolysis, as indicated by increased lactate levels [127]. Lactate is likely shuttled to the underlying axons as a source of trophic support [128] or used to support myelination [129]. The possibility that aerobic glycolysis can sustain oligodendrocytes without reduced oxidative phosphorylation suggests that other stressors compound mitochondrial function to initiate oligodendrocyte death. Confusingly enough, cuprizone also does not kill oligodendrocytes in vitro [111], suggesting that cuprizone alone might not be enough to induce oligodendrocyte cell death in vivo.
Myelinic glutamate excitotoxicity
Glutamate is the main excitatory neurotransmitter in the brain [130]. Postsynaptic glutamate acts on N-Methyl-D-aspartic acid receptors (NMDAR), which opens a non-selective cation channel on neurons to permit calcium or sodium ion influx [131]. Calcium-dependent downstream signaling of NMDAR is important for modulation of synaptic plasticity and neuronal survival [132]. Despite the importance of glutamate and NMDAR for the healthy brain, overactivation of glutamate receptors leads to toxic levels of intracellular calcium, which can result in cell death [133, 134], for which the term excitotoxicity was coined [135].
In MS, glutamate is elevated prior to the formation of lesions and is correlated to lesion formation [136]. Similar to MS lesions, cuprizone consumption leads to elevated levels of total glutamate in the hippocampus (~ 10% increase based on in vivo 1H magnetic resonance spectroscopy) and corpus callosum (~ 60.8% increase based on glutamate-weighted chemical exchange saturation transfer) of rats after 7 weeks of cuprizone treatment [137, 138], which may result in excitotoxicity. The pathways that elicit elevated glutamate levels remain unclear. It may be that the unitary strength of glutamatergic synapses increases which in turn release more glutamate into synapses. One of the factors influencing synaptic strength is the level of vesicular glutamate transporter (VgluT) 1 [139], which loads synaptic vesicles with glutamate [140, 141]. Cuprizone treatment increases VgluT1 levels after 5 weeks [142] and may lead to increased release of glutamate from the synapse [143]. In contrast, both the NMDAR subunit NR2A and glutamate aspartate transporter (GLAST) – two factors that counteract excitotoxicity – are upregulated in the corpus callosum of cuprizone-fed mice after 5 weeks [144]. NR2A upregulation was correlated to neuroprotection in an in vitro study on NMDA-mediated excitotoxicity in primary rat retinal cells [145]. GLAST is upregulated in astrocytes following cuprizone consumption and likely mediates astrocytic clearance of glutamate from the synaptic cleft [144, 146, 147]. Elevation of GLAST in astrocytes is likely a protective response to increased glutamate levels [148, 149]. Taken together, we know now that glutamate is elevated, but so too are molecules that counteract glutamate excitotoxicity.
Aside from glutamate concentration, the activity of its receptor NMDAR also mediates excitotoxicity. Ions such as magnesium [150], zinc [151], and copper [152] regulate NMDAR activity. For example, copper potentiates the NMDAR current at concentrations < 30 µM while inhibiting it above 30 µM in rat cerebellum granule cells in vitro [152]. Proteins, such as the cellular prion protein (PrPC) also regulate NMDAR activity [153, 154]. PrPC binds copper with high affinity and inhibits NMDAR in the presence of copper [155, 156] to prevent glutamate-mediated excitotoxicity. PrPC is present in myelin and oligodendrocytes [157, 158] and thus may prevent excitotoxicity in the cuprizone model, though there is no direct evidence. In summary, cuprizone-mediated changes in brain copper levels can both increase and decrease NMDAR activity to promote or inhibit excitotoxicity respectively. As mentioned, measurements of brain copper levels following cuprizone treatment were rather inconclusive [28, 90, 91] and therefore it is unclear if copper concentrations decrease sufficiently to promote excitotoxicity via NMDAR. Further, copper levels are often expressed as µgcopper/gdryweightbrain, which makes it difficult to compare changes in copper levels following cuprizone treatment between in vitro and in vivo models. Even though there is evidence that cuprizone could lead to excitotoxicity in oligodendrocytes and neurons, the precise impacts of cuprizone on NMDAR-mediated cell death are not fully understood but could provide a mechanism by which glutamate in the lesion can induce myelin damage (Fig. 2C).
Amino acid starvation results in reduced synthesis of myelin components
Cuprizone consumption may alternatively induce demyelination by reducing myelin protein and lipid synthesis which destabilizes the myelin sheath. For example, Morell and colleagues found that the messenger ribonucleic acid (mRNA) of various myelin proteins, such as MAG and myelin basic protein (MBP), decreases in the first week after cuprizone diet in rats by ~ 75% and ~ 50% respectively [159, 160]. Reduced production of myelin protein mRNA may be due to overall reduction of transcription and protein synthesis as a result of peripheral amino acid starvation. After only 4 days of cuprizone consumption, plasma levels of alanine, glycine, and proline decline. This amino acid starvation may trigger the amino acid response pathway, which decreases overall mRNA expression and protein synthesis [161]. Taraboletti and colleagues also found reduction of several amino acids and related metabolites in the corpus callosum of cuprizone treated animals after 2 weeks [97], which might be a cause for reduced protein synthesis. Ceramide galactosyltransferase (CGT) and β-Hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase are two enzymes that are reduced following cuprizone consumption [159, 160]. CGT is a key enzyme in galactocerebroside synthesis and HMG-CoA reductase in cholesterol synthesis. Galactocerebrosides are the most abundant glycolipid of myelin [162] and cholesterol is an important part of the myelin sheath [163]. Thus, both are important for myelin sheath stability. The reduced production of crucial myelin proteins and proteins involved in myelin lipid synthesis following cuprizone treatment could lead to a lack of those components in the myelin sheath and subsequent destabilization of the myelin sheath. Sheath destabilization can result in vacuole formation within the myelin sheath and the periaxonal space, which could lead to oligodendrocyte and axonal degeneration [164, 165]. However, given that mRNA levels of myelin proteins are reduced at 1 week, when oligodendrocyte death is ongoing, the lower synthesis may be a consequence rather than a driver of oligodendrocyte toxicity (Fig. 3).

Cuprizone induced myelin loss could be due to reduced myelin protein production and the subsequent destabilization and vacuolation of the myelin sheath. (1) Cuprizone administration leads to reduced levels of certain amino acids (Glycine, Alanine, and Proline) in the blood plasma. This amino acid starvation could lead directly or indirectly – through the amino acid response pathway – to reduced protein synthesis in the oligodendrocyte. (2) In the cuprizone model several proteins are reduced, that are either part of the myelin sheath or enzymes that produce components of the myelin sheath. For example, MAG and MBP are reduced – both are crucial stabilizers of the myelin sheath. CGT and HMG-CoA reductase are reduced as well. CGT is crucial in producing Galactocerebrosides. HMG-CoA is involved in the cholesterol synthesis pathway. Both Galactocerebrosides and cholesterol are important parts of the myelin sheath membrane. Lack of MAG, MBP, galactocerebrosides, and cholesterol could lead to myelin sheath destabilization, formation of intramyelinic vacuoles, and subsequence myelin and axonal damage degradation
Death from the outside: How oligodendrocyte extrinsic mechanisms could lead to death during cuprizone treatment
Cuprizone treatment also evokes accumulation of various cells of the innate and adaptive immune response, which is accompanied by expression of pro-inflammatory cytokines and other inflammatory modulators. Those cells and molecules create a destructive environment, that can further aggravate or potentially initiate the demyelination process. In the following sections, we will showcase multiple cell types and inflammatory molecules that are known to contribute to oligodendrocyte death and demyelination in the cuprizone model.
The contribution of CNS resident cells to inflammatory demyelination in the cuprizone model
Understanding the CNS resident mechanisms of demyelination is of interest to MS research because not all demyelination in MS is linked to infiltration of peripheral immune cells [8]. Cuprizone treatment elicits a largely CNS-restricted immune response following injury to oligodendrocytes and myelin breakdown, making it an ideal model to study CNS intrinsic immune responses following demyelination.
Oligodendrocyte lineage cells are inflammatory cells
Historically, oligodendrocyte lineage cells that include oligodendrocytes and OPCs were regarded as the victims of inflammatory brain damage. However, oligodendrocyte lineage cells are now recognized to produce immunomodulatory factors that alter CNS inflammation. Oligodendrocyte lineage cells produce many different chemokines, cytokines, antigen presentation complexes, and complement proteins both in vitro and in vivo (reviewed in [166]). In MS lesions and EAE mice, oligodendrocyte lineage cells express immune-modulatory genes such as the major histocompatibility complex (MHC)-II, a protein crucial for antigen presentation [167]. Both oligodendrocytes and OPCs express MHC-II in response to Interferon (INF) and present antigens to regulate the proliferation and cytokine production of CD4+ T-cells [167]. Like EAE, cuprizone may induce immune-modulating properties in oligodendrocytes. The treatment of cultured oligodendrocytes with sodium azide—an inhibitor of the mitochondrial respiration—models altered mitochondrial function during cuprizone treatment and leads to increased mRNA levels of several cytokines and chemokines. These factors include potent inflammatory regulators such as Interleukin (IL)-6, Leukemia inhibitory factor (LIF), C-X-C motif chemokine ligand (CXCL)1, C–C motif chemokine ligand (CCL)5, and Colony stimulating factor (CSF) 1 [168]. Notably, CXCL1, CCL5, LIF, and CSF1 regulate microglia activation, survival, and migration to the site of injury [169,170,171,172] (Fig. 4H). Oligodendrocytes also produce IL-6 in vivo, a roughly fivefold increase in gene expression compared to control, after 2 days of cuprizone treatment [168]. Despite its pro-inflammatory properties, IL-6 protects against mitochondrial damage following bacterial infection [173] and is necessary for the repair of alcohol-induced mitochondrial deoxyribonucleic acid (DNA) damage [174]. The cytokine IL-6 seems to be neuroprotective in MS [175] and over expression of IL-6 in astrocytes preserves oligodendrocytes and reduces the severity of demyelination and numbers of microglia following cuprizone treatment [176, 177]. While oligodendrocytes are now recognized as mediators of CNS inflammation, it remains challenging to understand the consequences of their involvement given that the released factors may have both protective and degenerative functions.

Proinflammatory response of CNS intrinsic cells resulting from cuprizone intoxication. Cuprizone might directly affect oligodendrocytes, astrocytes, and microglia. Cuprizone-induced demyelination immune response involves multiple – in part overlapping – steps. A As oligodendrocytes die, they release various cytokines and chemokines may lead to activation of astrocytes. B Complement is potentially produced or activated by damaged oligodendrocytes or from myelin breakdown. Activated complement produces anaphylatoxin C3a and C5a, which in turn can attract and activate astrocytes and microglia. Complement may also be directly cytotoxic to oligodendrocytes. C Reactive astrocytes may contribute to copper storage and redistribution in the CNS. The high affinity copper transporter CTR1 mediated copper influx into the astrocytes, while ATP7A/B are mediating copper efflux. This may create a locally toxic copper accumulation or release of factors that exacerbate oligodendrocyte death. Reactive astrocytes also produce (D) cytotoxic chemokines such as LTα and TNFα or (E) proinflammatory cytokines that lead to expansion and activation of microglia. Microglia are also activated when oligodendrocytes release (F) myelin debris, (G) by complement and anaphylatoxin, and (H) cytokines released from the injured oligodendrocyte. I Activated microglia require CSF1R stimulation for survival and proliferation. J Microglia are important for phagocytosis of myelin debris, for which TREM2 and MERTK are crucial. K Activated microglia may exhibit direct toxicity towards oligodendrocytes, (L) Other inflammatory mediators that could lead to oligodendrocyte death such as PG E2 via the PGE2-EP2 receptor
Astrocytes are critical for cuprizone-induced demyelination
Astrocytes become reactive following oligodendrocyte damage and release cytokines, chemokines, and other factors that contribute to cuprizone-induced demyelination (Fig. 4). Astrocyte reactivity occurs throughout cuprizone treatment with morphological changes beginning as early as day 5 of cuprizone treatment. Astrocytes become hypertrophic and hyperplastic between weeks 1 and 2 of cuprizone administration, before myelin loss. Astrocyte reactivity is associated with increased activity of oxidative enzymes (glutamate dehydrogenase, lactate dehydrogenase, NAD(P)H dehydrogenase, NADH dehydrogenase) [42, 178, 179] which may contribute to ROS mediated toxicity. By week 3 of cuprizone administration, there is a roughly twofold increase in astrocyte numbers [42, 54] (Fig. 1), which is likely a critical modulator of inflammation given that the number of reactive astrocytes is concomitant with the severity of demyelination [42, 48].
Strategies that limit astrocyte reactivity reduce demyelination for cuprizone-treated animals. For example, knockout mice for receptor tropomyosin receptor kinase B (TrkB) show reduced astrocyte reactivity and in turn, demyelination [180]. TrkB is enriched within reactive astrocytes and stimulating astrocytes with a TrkB ligand in culture promotes the nuclear localization of the prototypical proinflammatory signaling pathway molecule Nuclear factor kappa B (NF-κB). The canonical NF-κB signaling pathway is a critical component of the proinflammatory astrocytic response induced by cuprizone [181]. The canonical NF-κB pathway may be activated by a TrkB ligand [180] but also by other cytokines, such as Tumor necrosis factor (TNF) [182]. In mice deficient in inhibitor kappa B kinase 2 (IKK2) – IKK2 is a subunit of IκB which lies downstream of NF-κB activity [182] – cuprizone treatment elicits less severe astrocyte and microglia reactivity and induces less myelin loss. IKK2 knockout mice also express lower levels of pro-inflammatory cytokines, including IL-1β, TNFα, CCL2, CCL3, and CXCL10, after 5 weeks of cuprizone treatment. Removing IKK2 from non-microglial CNS cells also prevents the loss of oligodendrocytes at weeks 3 and 5 compared to wild-type animals treated with cuprizone, suggesting that NF-κB pathway-derived cytokines aggravate demyelination in the cuprizone model [181]. Similarly, astrocyte-specific inhibition of NF-κB by overexpressing IκBα – the dominant-negative inhibitor of NF-κB – under the glial fibrillary acidic protein (GFAP)-promoter, prevents myelin loss and reduces TNFα, IL-1β, and CCL2 [181]. In summary, the NF-κB pathway mediates proinflammatory responses of astrocytes following cuprizone consumption and may be essential for an astrocytic role in demyelination.
The ablation of astrocytes during cuprizone treatment significantly decreases astrocyte reactivity and microgliosis, while sparing myelin, increasing oligodendrocytes density and improving motor function. Astrocyte ablation during cuprizone treatment also reduces the expression of CXCL10 in mice [183] – an important attractant for microglia during cuprizone treatment [184,185,186]. Astrocytes were also shown to secrete CXCL10 in vitro following stimulation with IFN-γ or TNF-α [185]. This suggests that astrocytes may promote microglial accumulation during cuprizone-induced demyelination. However, CXCL10 is also expressed by oligodendrocytes after 1 week of cuprizone treatment, suggesting that oligodendrocytes themselves may also help attract microglia to the area of demyelination [186]. Loss of CXCL10 does not alter astrocyte reactivity, but instead, reduces demyelination, microglial reactivity, and axonal damage after three weeks of cuprizone diet [186], thus linking this astrocyte-derived chemokine to microglia recruitment and demyelination (Fig. 4E).
Astrocytes are attracted to the site of injury and aggravate demyelination through the production of proinflammatory cytokines and chemokines. Certain astrocyte-released cytokines may induce oligodendrocyte cell death, such as TNFα, while others like CXCL10 can attract microglia to the site of injury. Astrocytes also produce Lymphotoxin α (Ltα) during cuprizone treatment [56], which is directly cytotoxic to oligodendrocytes in vitro [187]. Ltα is elevated in astrocytes, but not microglia, during cuprizone treatment and its astrocytic loss delays cuprizone-induced demyelination, oligodendrocyte loss, and microglia infiltration, suggesting that Ltα is an astrocytic cytokine that potentiates cuprizone toxicity (Fig. 4D).
Astrocytes also regulate copper levels in the brain by transporting and redistributing copper from endothelial cells into the CNS. CNS copper uptake is regulated via the high affinity copper uptake protein 1 (CTR1) whereas copper release from cells is regulated by two copper-transporting P-type ATPases (ATP7A and ATP7B) [188]. Astrocytic expression of CTR1, ATP7A, and ATP7B increases 1 week after cuprizone exposure [180]. Dysregulation of the copper transport system may be potentially toxic in the cuprizone model, as stimulating CTR1 expression in astrocytes along with high copper stimulus in vitro primes astrocytes such that once they return to basal conditions, they release factors—possibly copper—that are toxic to oligodendrocytes [180]. Whether copper or other factors are responsible for oligodendrocyte toxicity remains unclear, as do the consequences of these factors in vivo (Fig. 4C).
Together, astrocyte reactivity and accumulation at the site of injury precedes demyelination. As oligodendrocytes die, astrocytes produce cytokines such as TNFα and LTα that may kill oligodendrocytes. Astrocytes also produce chemokines that promote microglia expansion to the site of injury to further propagate the CNS intrinsic immune response (Fig. 4A-E).
Microglia are critical for cuprizone-induced demyelination
Microglia are highly responsive cells that respond to factors like myelin or oligodendrocyte debris following cuprizone-induced demyelination (Fig. 4F). Microglia also drive cuprizone-induced demyelination [189]. Microglia become reactive as early as 1 week of cuprizone treatment and sustain activity throughout demyelination [189,190,191]. In addition to expressing markers indicative of their reactivity, microglia also expand their numbers into the peak of demyelination observed after 3–5 weeks on a cuprizone diet [42], though, for unknown reasons, microglia numbers decline afterward despite continuing cuprizone diet [55, 56] (Fig. 1B).
One critical role for microglia during demyelination is the clearance of myelin debris, which acts as an inhibitor of remyelination [192,193,194]. Microglia accumulate in response to demyelination to consume myelin debris [195, 196]. Myelin debris is sufficient to induce the expansion and reactivity of microglia [197] (Fig. 4F). Myelin debris clearance during cuprizone consumption is regulated by receptor tyrosine kinases, such as MER proto-oncogene, tyrosine kinase (MERTK) that recognize phosphatidylserine on apoptotic cells [198, 199]. Loss of MERTK does not attenuate demyelination but instead impairs microglia recruitment and remyelination [198]. Similarly, triggering receptor expressed on myeloid cells (TREM) 2, a phospholipid sensing receptor, is expressed by microglia, sustains microglia activity, and mediates myelin debris consumption [200]. In mice lacking TREM2, fewer microglia accumulate in the corpus callosum during demyelination due to reduced proliferation which in turn reduces myelin debris clearance [200,201,202]. Mice lacking TREM2 also experience greater axonal damage after cuprizone treatment. Conversely, TREM2 agonists accelerate myelin clearance in the cuprizone model to facilitate remyelination [202] (Fig. 4J). Together, these findings suggest that TREM2 mediates neuroprotective functions during cuprizone-induced demyelination.
Despite the positive roles of microglia in response to cuprizone, a complete absence of microglia prevents cuprizone toxicity suggesting that these cells are also pathogenic during cuprizone-induced demyelination [189]. Microglia require CSF1 receptor (CSF1R) signaling for ongoing survival [171, 172] (Fig. 4I) and in the CNS, CSF1R is restricted to microglia [203]. The use of CSF1R antagonists ablates microglia, providing a tool to understand the role of microglia during disease [171]. PLX3397 is a CSFR1 inhibitor that ablates microglia with minimal upregulation of inflammatory genes [204], suggesting that inflammation is not overtly induced. Ablation of microglia with PLX3397 during cuprizone treatment reduces the loss of oligodendrocytes and the severity of demyelination in the corpus callosum after 3 weeks and 5 weeks [189, 205]. Pre-treatment of mice with PLX3397 for two weeks before cuprizone treatment to ablate microglia to low levels and continued PLX3397 treatment maintains microglia at very low numbers during cuprizone treatment. The pronounced and prolonged microglia ablation completely prevented cuprizone-induced demyelination with no substantial alterations to myelin ultrastructure [189]. However, it is still unknown how microglia drive cuprizone-mediated demyelination. Surprisingly, promoting the expansion of microglia by treating mice with CSF1 during cuprizone diet also reduced demyelination [172], demonstrating the complicated role of microglia during cuprizone intoxication. Taken together, microglia initiate demyelination potentially through direct toxicity or the expression and secretion of cytokines (Fig. 4K) but may also protect axons and promote remyelination during cuprizone consumption.
The complement system during cuprizone toxicity
In innate immune responses, the complement system acts as a first-line of defense against pathogens, but it also modulates other innate and adaptive immune cells [206]. Given its immunoregulatory functions, the complement system unsurprisingly is involved in cuprizone-induced demyelination. The complement complex C1q is comprised of C1qA, C1qB, and C1qC and facilitates the first step of classical complement pathway activation.[207]. The C1q complex is increased during the first 4 weeks of cuprizone consumption and accumulates in the corpus callosum during demyelination [208]. The gene expression of the component proteins of C1q and other complement proteins, namely C3a receptor and C4, are upregulated at 5 weeks of cuprizone [209]. Coincidentally, endogenous Complement receptor 1-related Gene/Protein y (Crry)—an important inhibitor of complement-mediated injury [210]—is reduced during demyelination [208], suggesting that complement may be disinhibited during cuprizone intoxication. The overexpression of soluble Crry (sCrry) in astrocytes is neuroprotective as it spares myelin in the corpus callosum and decreases microglia infiltration by week 4 after cuprizone exposure [208].
Anaphylatoxin C3a and C5a—downstream products of the complement pathway [211]—function as chemoattractants for neutrophils [212], macrophages [213], astrocytes [214], and microglia [215,216,217] (Fig. 4B&G). When overexpressed in astrocytes, these factors enhance demyelination and increase microglia numbers after cuprizone exposure. Indeed, treatment of a microglia cell line with C3a and/or C5a in vitro resulted in increased expression of the proinflammatory cytokines and chemokines CCL4, CCL5, CCL11, and IL-6 [218]. During cuprizone-induced demyelination, the complement proteins C3a and C5a increase the numbers of microglia present at the site of injury and might stimulate microglia to express pro-inflammatory cytokines [218]. There is also evidence that complement can be directly cytotoxic to oligodendrocytes in culture [219,220,221]. Given that anaphylatoxins regulate leukocyte phagocytosis [222], they may also alter microglia phagocytosis. Taken together, complement proteins indirectly aggravate demyelination by promoting the accumulation of proinflammatory microglia and astrocytes (Fig. 4B&G) at the lesion site and may directly induce oligodendrocyte death.
Cytokines and chemokines contribute to demyelination
Chemokines and cytokines are critical regulators of cuprizone-induced demyelination. Several inflammatory cytokines and chemokines such as CCL2, CCL3, CCL5, and CXCL10 are increased within the first week of cuprizone treatment, while others are increased later during demyelination such as CCL3, CCL4, C–C motif chemokine receptor (CCR) 5 [100, 186, 223]. Both CXCL10 and CCL2 peak early (~ week one) and decline after, while CCL3 expression increases with intoxication length [100, 186]. TNF expression levels are increased after one week of cuprizone-induced demyelination in vivo [224] and TNFα−/− mice showed a delay in demyelination in the cuprizone model [56], consistent with a role for TNF to exacerbate cuprizone-induced demyelination. Given that TNF directly induces oligodendrocyte death in vitro [225, 226], it may directly participate in cuprizone-induced demyelination in vivo. However, TNF is also required for remyelination [224]. Many other immune factors are involved in cuprizone-induced demyelination. CCL3 knock-out mice have less microglia/macrophage accumulation and less demyelination compared to control mice on the cuprizone diet [223]. Also, loss of CCL10 largely prevented demyelination, oligodendrocyte death, microglia, and astrocyte activation and axonal damage at week 3 on the cuprizone diet [186]. Several other knockout studies showcase the influence of cytokines on demyelination, oligodendrocyte death, astrogliosis, and microgliosis in the cuprizone model, which are listed in Table 1 (modified table from [107]).
Prostaglandin involvement during demyelination
Prostaglandins (PG) are a family of lipids made at the sites of tissue damage that modulate inflammation [252]. PG are derived from arachidonic acid, an unsaturated fatty acid produced from phospholipids in the plasma membrane by phospholipase A2. Subsequently, arachidonic acid is converted to PG via Cyclooxygenase (COX)-1 and COX-2 [253]. During cuprizone consumption, COX-2 expression increases after 1 week and PG levels increase after 5 weeks in the cortex [254]. Indeed, COX-2−/− mice and mice treated with celecoxib—a selective COX-2 inhibitor [255]—experience less oligodendrocyte death and demyelination in the corpus callosum on a cuprizone diet. At the same time, the PG E2 receptor 2 subtype (PGE2-EP2) localizes in apoptotic oligodendrocytes in the cortex, suggesting an involvement of PG in oligodendrocyte death in the cuprizone model. Inhibition of PGE2-EP2 with a selective antagonist also reduces cuprizone-induced demyelination in mice [191]. However, inhibition of PGE2-EP2 only reduces demyelination when started at the same time as cuprizone treatment, not when started during peak demyelination suggesting a role for PG during the initiation of demyelination (Fig. 4L).
The increased presence of PG in areas of cuprizone-induced demyelination underlines the importance of inflammation for cuprizone pathology. PG are also elevated in cerebrospinal fluid of people with MS [256, 257] and COX-2 is upregulated in demyelinating MS plaques [258, 259], suggesting a potential role of those inflammatory mediators in MS.
The contribution of peripheral immune cells to inflammatory demyelination
Apart from inflammatory cells from within the CNS, there is evidence that also peripheral immune cells/components might play a role in cuprizone-induced demyelination. Initially, it was thought that the BBB was intact during cuprizone-induced demyelination, which would limit infiltration of peripheral cells. In contrast, recent research using fluorescently tagged dextran (70 kDa) or Evans Blue tracer injection demonstrates BBB permeability as early as 3 days after cuprizone treatment [75, 76]. Gene expression analysis of corpus callosum tissue shows upregulation of Tnf, Il-1b, Ccl2, and Il-6, cytokines and chemokines involved in BBB breakdown, as early as 5–7 days of cuprizone treatment. Conversely, the expression of genes encoding the tight junction proteins that maintain the BBB, namely claudin-5, occludin, ZO1/tight junction protein-1, cadherin-1, and cadherin-5 decrease initially after 5 days and decreases further by 5 weeks on a cuprizone diet. Microglia likely promote BBB breakdown given that loss of the microglia C-X-C motif chemokine receptor (CXCR) 3 ameliorated both the CNS-wide rise in TNF, IL-1β, and CCL2 and the reduction in tight junction gene expression. Cell-specific expression analysis of Tnf, Il-1β, Il-6, and Ccl2 after 5 days of cuprizone demonstrates that astrocytes are the main source for these cytokines [75]. Together, it is likely that CNS-derived processes alter BBB integrity during cuprizone treatment which may permit the peripheral immune system to exert effects within the CNS.
Another important barrier in the brain exists between the blood and the cerebrospinal fluid, formed by endothelial cells of the choroid plexus (CP). The CP is the major producer of cerebrospinal fluid, and thus an important interface between the blood and cerebrospinal fluid [260]. The CP was shown to be enlarged [261, 262] and a point of T-cell entry into the CNS during neurodegenerative diseases, such as MS or the EAE model [263, 264]. The CP is altered following cuprizone treatment. For example, Fleischer and colleagues found CP enlargement, increased numbers of CP macrophages and T cells in the CP after two weeks of cuprizone treatment [261]. It is still largely unknown however, how the CP or other CNS barrier like the CNS lymphatic or glymphatic system are altered after cuprizone treatment.
Evidence neutrophils contribute to cuprizone demyelination
Neutrophils are innate immune cells that responds to inflammatory stimuli to sense and kill pathogens [265]. In the cuprizone model, neutrophils are only present around week 1 in the corpus callosum (Fig. 1B). Liu and colleagues observed significant CNS localization of neutrophils at day 7 using flow cytometry but did not examine whether neutrophils enter into the CNS parenchyma [238]. Neutrophils are thought to promote cuprizone demyelination as knocking out the neutrophil chemokine receptor, CXCR2, in mice prevents demyelination at week 6 of cuprizone treatment. After 4 weeks of cuprizone treatment, nearly all axons are demyelinated in control mice, while in CXCR2-null mice, almost 60% of axons remain myelinated. Loss of CXCR2 does not affect the loss of MBP and 2’,3’-Cyclic-nucleotide 3’-phosphodiesterase (CNPase) expression within the first 2 weeks of cuprizone treatment, but by weeks 3 and 4 MBP and CNPase expression increases in CXCR2−/− mice, suggesting that loss of CXCR2 protects against a delayed myelin protein loss [238]. Transplanting bone marrow from CXCR2−/− mice into irradiated CXCR2+/+ mice prevents cuprizone-induced demyelination and oligodendrocyte death suggesting that the source of CXCR2 cells producing demyelination is of peripheral origins [238]. However, CXCR2 is not exclusively a marker for neutrophils as it is also expressed on CD4+ T-cells [266] and macrophages [267]. Using flow cytometry, Liu and colleagues find CXCR2 positive cells were Lymphocyte antigen 6 complex locus G6D (Ly6G) positive neutrophils [238]. Depletion of neutrophils using specific GR1 antibodies also reduces the number of apoptotic cells in the corpus callosum by 3 weeks of cuprizone treatment [238], which is a timepoint with sparse neutrophil presence in the CNS. Taken together, CXCR2 plays a role in demyelination, however, it is unclear whether this factor exclusively acts through neutrophils.
T-cells presence during cuprizone
T-cells are a predominant effector of the adaptive immune system. T-cells recognize peptide antigens presented by MHC class I or II. T-cells that express CD4 are known as T helper cells while those expressing CD8 are referred to as cytotoxic T-cells [268]. Historically these cells are not linked to demyelination following cuprizone treatment because the loss of the V(D)J recombination activation gene-1 (RAG-1), which is critical for B- and T- lymphocyte generation [269], does not alter demyelination and T-cell accumulation in the corpus callosum [46, 270]. However, these cells still might have executive functions during cuprizone treatment. Recently, Kaddatz and colleagues found that small numbers of cytotoxic CD8 T-cells, and even fewer CD4 T-cells, accumulate within the corpus callosum of cuprizone treated mice up until week 5 [70] (Fig. 1B). The enrichment of CD8+ T-cells is in contrast to EAE, where CD4 T-cells predominate [271]. The CNS entry of T-cells after cuprizone treatment may be different than EAE given the lack of perivascular cuffs, which is a sign of lymphocyte infiltration [272]. T-cells in the demyelinated corpus callosum often express markers of activation [70]. Even after disruption of the BBB with pertussis toxin, CD4+ and CD8+ T-cells are limited in the corpus callosum of cuprizone animals [273], suggesting that these cells are likely attracted to myelin injury specifically. Recent discoveries found severe atrophy of the thymus and spleen and depletion of CD4+ and CD8+ T-cells due to apoptosis in mice fed with cuprizone [273, 274], which could explain why T-cells are limited in cuprizone lesions.
Peripheral immune cells account only for a small number of all cells that are present at demyelinating sites following cuprizone intoxication and their presence might indicate a role in demyelination that has not been explored yet. Alternatively, T-cells may be responding to demyelination to help restore homeostasis and promote repair.
Cuprizone induced cell death: a mixed bag
In the previous sections, we have outlined several etiological hypotheses for cuprizone-induced demyelination that might lead to cell death of the oligodendrocytes. Studying what forms of cell death occur in the cuprizone model may help establish the mechanism of its toxicity. Despite decades of research demonstrating that cuprizone induces cell death, the modalities of cell death during cuprizone intoxication are still unclear. One of the first studied forms of cell death is apoptosis, with its name being coined in 1972 by Kerr, Wyllie, and Currie [275], and unsurprisingly it is also the most common form of cell death studied in the cuprizone model. More recently, however, new forms of cell death have been discovered. Of the ten non-apoptotic forms of cell death described by the Nomenclature Committee on Cell Death [276], only Ferroptosis, Necroptosis, and Pyroptosis have been studied after cuprizone ingestion and are reviewed in turn below.
Apoptosis
Apoptosis is a programmed cell death induced by both extracellular and intracellular cues. These cues activate caspase 3 by proteolytic cleavage through other caspase isoforms. Once activated, cleaved caspase 3 (CC-3) activates endonucleases and proteases, which ultimately leads to chromatin condensation, reorganization of the cytoskeleton, and cellular disintegration into apoptotic bodies [277].
Oligodendrocytes undergo apoptosis during cuprizone toxicity. Oligodendrocyte apoptosis, based on CC-3 staining, starts as early as day 2 on the cuprizone diet, with extensive depletion of mature oligodendrocytes by day 4 in the corpus callosum [100]. Oligodendrocyte apoptosis reaches its maximum between week 1 and week 3, with the number of apoptotic cells remaining constant thereafter [100, 161, 278]. Hesse and colleagues measured cell death based on nuclear morphology and found that by the end of the first week of cuprizone consumption, dying oligodendrocyte nuclei are almost all condensed, fragmented, and co-label with CC-3 [278]. However, by three weeks of cuprizone consumption, dying oligodendrocytes condense, but are unfragmented, and no longer co-label with CC-3 [278]. The presence of a cell death morphology that lacks CC-3 suggests that apoptosis may shift to a non-CC3 dependent form. Indeed, we found that the condensed nuclear morphology without fragmentation is indicative of a lytic form of cell death [279].
Ferroptosis
Ferroptosis is a caspase-independent form of cell death caused by accumulation of toxic lipid peroxidases and free iron. Free iron can react via the Fenton reaction (with H2O2 from mitochondria), to generate hydroxyl radicals [280] and promote lipid peroxidation [281]. Products of lipid peroxidation, such as MDA and 4-HNE, are highly cytotoxic and serve as common markers of ferroptosis [282, 283].
Iron homeostasis is dysregulated in the liver and brain of mice fed with cuprizone, as several iron metabolism enzymes like hepcidin, transferrin receptor 1 and 2, ferritin heavy chain, and mitochondrial iron transport proteins are dysregulated in the corpus callosum compared to control animals [284, 285]. Dysregulation of these enzymes alters the intracellular iron pool, which can lead to ferroptosis [286]. Cuprizone-mediated reduction in oligodendrocytes between days 1–4 coincides with reduction of the iron storage protein ferritin and an increase of the ferroptosis marker nuclear receptor coactivator 4 (NCOA4). NCOA4 is involved in ferritin breakdown and leads to increased free iron [287]. At the same time, markers of ferroptosis and oxidative stress such as transferrin receptor 1, COX-2, heme oxygenase-1, heat shock protein beta 1, hephaestin, and superoxide dismutase 1 are also dysregulated, suggesting ongoing ferroptosis. However, not all ferroptotic cells are oligodendrocytes as 25% of NCOA4+ cells are not oligodendrocytes during cuprizone intoxication [101].
Signs of ferroptotic oligodendrocyte death are present early after cuprizone ingestion. Cuprizone-induced lipid peroxidation due to free radicals begins at 2 days post-cuprizone and remains high throughout the first 4 weeks. Endogenous free radical scavengers are meant to scavenge free radicals, which are crucial for lipid peroxidation. One important ROS scavenger is the glutathione system, which includes two key enzymes: glutathione peroxidase 4 (GPX4) and cystine/glutamate antiporter system xc− (Sxc−). GPX4 reduces toxic lipid peroxides to harmless lipid alcohols, by oxidation of GSH to glutathione disulfide [288,289,290]. Sxc− is a cysteine/glutamate antiporter, which ensures a constant supply of L-cysteine into the cell for GSH synthesis [291]. Cuprizone diet reduces both GPX4 and Sxc− and thus reduces the scavenging capacity of lipid peroxides between 2 and 7 days after cuprizone ingestion [101]. Daily injection of ferrostatin—a lipid ROS scavenger [286]—markedly reduces oligodendrocyte death and 4-HNE accumulation at two days and two weeks of cuprizone consumption [101]. Ferrostatin and cuprizone treatment for two weeks also limits myelin loss at four weeks compared to mice treated with only cuprizone for two weeks. The fact that blocking of ferroptosis could prevent long-term myelin loss suggests an important role of ferroptosis in early oligodendrocyte loss in the cuprizone model.
Necroptosis
Necroptosis is a regulated form of necrosis triggered by intra- and extracellular signals and is initiated by chemokine receptors such as FAS [292], by pathogen recognition receptors such as Toll-like receptor (TLR)3, TLR4, and Z-DNA binding protein (ZBP)1 [293, 294], and by the TNF receptor (TNFR)1 [295]. Activation of TNFR1 by TNFα leads to the eventual activation of receptor interacting serine/threonine kinase (RIPK)3 via RIPK1. RIPK3 phosphorylates mixed lineage kinase domain-like protein (MLKL) to initiate the oligomerization and translocation of a RIPK3, RIPK1, MLKL complex to the cell membrane where it permeabilizes the cell membrane [296, 297]. Necroptosis is inhibited by caspase-8, a key enzyme for apoptosis, and thus the absence of caspase-8 is required for RIPK3-mediated necroptosis [298]. Given that oligodendrocytes express low levels of caspase-8 [299], they are more susceptible to necroptotic cell death. In the corpus callosum of cuprizone-fed mice, overall RIPK1 levels and RIPK1+ oligodendrocytes increase until week 5 of treatment. The administration of 7 N-1, an analog of necrostatin-1 which inhibits RIPK1 [300], reduces the density of RIPK1+ oligodendrocytes [301]. 7 N-1 treatment of cuprizone-fed mice also reduces oligodendrocytes loss and increases motor function in animals after 5 weeks. Administration of 7 N-1 three weeks after beginning of cuprizone treatment rescues some of the motor defects, thus linking demyelination and motor outcomes [301]. Similarly, mice lacking RIPK3 display less corpus callosum demyelination compared to WT [301]. However, necroptosis inhibition with RIPA-56, an alternative RIPK1 inhibitor [302], does not prevent cuprizone-induced demyelination but does block the progression of EAE [303]. Inhibition of RIPK1 decreases demyelination only when inhibited by 7 N-1 but not with RIPA-56, which both prevent necroptosis [300, 304], suggesting that RIPK1 might have multiple functions during cuprizone-induced demyelination.
Necroptosis requires the permeability of the cell membrane, which currently, is only indirectly measured by assessing RIPK1/RIPK3/MLKL3 oligomerization. However, these necroptotic proteins may have functions outside of traditional necroptosis. One alternative downstream effect of RIPK1/RIPK3 is the PGAM5-dependent activation of dynamin-related protein 1 (Drp1) [305, 306], a crucial protein for mitochondrial fission [307]. Drp1 hyperactivation induces a variety of cellular damage and is involved in several forms of cell death, including apoptosis [308, 309], necrosis [305, 310], and autophagic cell death [311, 312]. Cuprizone induces DRP1 translocation to the mitochondria, indicative of its hyperactivation, after one week on the diet, which coincides with the peak of oligodendrocyte apoptosis. Treatment of cultured oligodendrocytes with H2O2 and TNFα induces mitochondrial translocation of Drp-1 and mitochondrial fragmentation [305]. Pharmacological inhibition of Drp-1 in the cuprizone model mitigates MBP and oligodendrocyte loss [306]. Despite the downstream activation of Drp1 by RIPK1/RIPK3/MLKL, necroptosis does not require activation of Drp1 [313,314,315], suggesting that Drp1-mediated mitochondrial deficiency is a byproduct, and not a direct cause, of necroptosis.
Inflammasome mediated cytokine release/pyroptosis
The canonical inflammasome pathway is activated by cytosolic inflammasome sensors such as LRR- and pyrin domain-containing protein (NLRP)3. NLRP3 (reviewed in [316]) detects pathogen associated molecular patterns (PAMPs) or factors released from damaged/dying cells [317,318,319,320] and activates caspase-1 (casp-1), the executing caspase in the canonical inflammasome pathway [320]. Activated casp-1 forms a heterodimer complex that catalyzes the maturation of IL-1β and IL-18 [321]. At the same time, casp-1 mediates pyroptotic cell death by cleavage of Gasdermin-D [322]. Cleavage of Gasdermin-D by casp-1 leads to the release of its N-terminal “death domain” [322, 323]. This death domain oligomerizes with components of the cell membrane, forming a lytic pore that results in the release of cellular content [324]. The lytic complex formed by cleaved Gasdermin-D does not necessarily lead to cell lysis but is required to release the cytokines IL-1β and IL-18 [325, 326]. Additionally, Gasdermin-D also cleaves pro-IL-1α to permit the release of mature IL-1α [327]. In the absence of Gasdermin-D, casp-1 activation leads to apoptosis, which can be followed by secondary pyroptosis. In vitro experiments suggest that this secondary pyroptosis is mediated by Gasdermin-E, instead of Gasdermin-D [328]; Brie and colleagues found casp-1 and cleaved Gasdermin-D mediated pyroptosis of oligodendrocytes and microglia in the EAE model and within brain lesions of people with MS [329]. In the cuprizone model, the role of Gasdermin-D has not been explored yet.
In the cuprizone model, NLRP3 expression increases by week 3 and remains high through to week 5 [244]. NLRP3 and NLR Family CARD Domain Containing (NLRC)4, another inflammasome sensor [330], knockout reduces microglia and astrocyte numbers and attenuates demyelination during cuprizone treatment [244, 331, 332]. NLRC4 is expressed highly in astrocytes and microglia and is necessary for the secretion of IL-1β in both cell types after acute demyelination with LPC [332] and may be important for cuprizone-mediated demyelination as well. The activated inflammasome triggers oligodendrocyte death, demyelination, microglia infiltration, and astrocyte activation in the cuprizone model; knockout of IL-18, NLRP3, NLRC4, and Casp-1 in mice yields less severe demyelination after cuprizone treatment [244, 332]. Saito and colleagues recently described elevation of casp-1 expression levels alongside NLRP3 in the cerebellum of mice fed cuprizone for 6 weeks [333]. Inhibition of casp-1 with VX‐765 reduces the severity of inflammation and behavioral deficits in the EAE model [329]. Casp-1 inhibition with VX‐765 in cuprizone-fed mice significantly reduces the number of Iba-1 positive cells and prevents loss of MBP. Cuprizone-fed mice treated with VX‐765 also have greater levels of spared oligodendrocytes compared to animals not treated with VX-765 [333]. Taken together, inflammasome activation executes critical roles for cuprizone-induced demyelination, though it remains unclear how prominent inflammasome activation results in pyroptotic cells in the cuprizone model.
Cause of cuprizone-induced demyelination
The cuprizone model is in many ways an ideal system to understand innate immunity in the context of demyelination. Astrocytes and microglia collaborate through the release of cytokines, chemokines, and other, yet to be identified factors, to drive demyelination [79, 107], preventing the release of specific cytokines or chemokines, restricting astrocyte reactivity, or ablating microglia all reduce cuprizone-induced demyelination [107, 181, 189, 243]. Yet many questions remain. For example, what are the contributions of cuprizone itself or its copper chelation in the brain to the ongoing demyelination? Alternatively, are chelation independent mechanisms or cuprizone metabolites the cause for oligodendrocyte death. Does cuprizone act as a toxic factor by altering metabolism and ROS production to trigger oligodendrocyte pathology?
Early after cuprizone administration, ROS production increases coincident with mitochondrial and metabolic dysfunction that could directly cause oligodendrocyte death [28, 29, 101, 243]. We term this initial trigger of cuprizone-induced oligodendrocyte death as a primary ‘oligodendrocytopathy’ (Fig. 5A). Following this initial oligodendrocyte destruction, an immune response mediated primarily by astrocytes and microglia seems to amplify the demyelination. We call this cascade of destructive, innate inflammation involving microglia and astrocytes ‘toxic innate immunity’ (Fig. 5B). It may also be possible that cuprizone induces an innate immune response where cuprizone intake or the subsequent metabolic processes activate innate immune cells and direct them to induce oligodendrocyte toxicity, which we term ‘primary immunocytopathy’ (Fig. 5C). If and how cuprizone affects astrocytes and microglia directly is still largely unexplored. Importantly, those are not mutually exclusive—on the contrary, we think it highly likely that these mechanisms occur simultaneously.

Hypothesis on different pathologies in the cuprizone model: A Primary ‘oligodendrocytopathy’, where the effects of cuprizone on the oligodendrocyte population induces cell death. B ‘Toxic innate immunity’, where innate immune cells of the CNS take on a cytotoxic phenotype towards the oligodendrocyte population as a reaction to an initial damage to oligodendrocytes. C ‘Primary immunocytopathy’, in which the effect of cuprizone on CNS innate immune cells triggers a cytotoxic phenotype of those cells towards oligodendrocytes, without prior damage to the oligodendrocyte population
Conclusion
Cuprizone intoxication leads to myelin loss and selective oligodendrocyte death in several brain regions with demyelination pathology that displays a striking resemblance to lesions from progressive stages of MS. This demyelination is accompanied by expansion and transition of CNS-derived microglia and astrocytes to become more reactive. Cuprizone intake also results in BBB breakdown and a limited infiltration of peripheral immune cells. Although cuprizone-induced oligodendrocyte loss was historically attributed solely to primary oligodendrocyte death, we now know that CNS inflammation either causes or aggravates the demyelination. We argue that two distinct, yet overlapping and sequential, modes of action within the CNS propagate demyelination. The first mode of action stems from oligodendrocyte intrinsic mechanisms, which we have termed a ‘primary oligodendrogliapathy’, wherein cuprizone alters mitochondrial metabolism and the synthesis of myelin components which eventually leads to cell death. The second mode of action stems from oligodendrocyte extrinsic inflammatory mechanisms within the CNS by both resident and peripherally-derived immune cells. These cells adopt a pro-inflammatory phenotype, either as a direct response to cuprizone, which we have termed a ‘primary inmmunocytopathy’, or by molecules released from dying oligodendrocytes, refered as ‘toxic innate immunity’. In the latter case, reactive astrocytes produce cytokines and chemokines that are directly cytotoxic to oligodendrocytes or attract and stimulate microglia. Reactive microglia are vital for phagocytosis of myelin debris, which inhibits remyelination, but are also necessary for demyelination as their ablation prevents demyelination after cuprizone intoxication. Although oligodendrocytes are primarily the victims of demyelination, they might themselves contribute to the proinflammatory environment by release/secretion of cytokines and chemokines and/or the expression of MHC-II molecules for antigen presentation and further amplified immune cell activation. Other parts of the innate immune response, such as the complement system may have direct cytotoxic effects on oligodendrocytes and attract microglia and astrocytes. Peripheral immune cells such as T-cells are present at the site of demyelination, but they are likely not promoting demyelination.
Cuprizone intake causes the loss of oligodendrocytes by a variety of different cell death forms. Apoptosis is the most commonly studied type of cell death in the cuprizone model as it is well understood and easy to assess. However, we know now that other forms of cell death, such as necroptosis, ferroptosis, and inflammasome mediated cell death/pyroptosis lead to oligodendrocyte death in the cuprizone model and occur at different time points after intoxication. For example, apoptosis and ferroptosis occur up until week 2 – 3 of cuprizone intoxication while other types of cell death seem to contribute to oligodendrocyte loss starting at week 2 – 3, concomitant with myelin loss and accumulation of astrocytes and microglia. A shift from apoptotic/ferroptotic cell death to other forms might also indicate a shift in the underlying mechanisms of cell death. To better understand the complexity of the cuprizone model, it is critical to explore the earliest time points of cuprizone treatment, particularly when microglia and astrocyte activities are impaired. We believe that resolving the time-dependent pathways that underlie the various types of cell death will be vital to better understanding the mechanisms of cuprizone toxicity.
As early loss of oligodendrocytes appears sufficient to drive late-stage myelin loss [101], it is critical to understand what factors drive oligodendrocyte loss in the first week of cuprizone treatment. Although we continue to better understand the mechanisms of cuprizone toxicity, there are still many unresolved questions surrounding this model. Is astrocyte and microglia reactivity secondary to an initial oligodendrocyte insult or does cuprizone per se induce a toxic pro-inflammatory phenotype in those cells? How much do oligodendrocytes themselves contribute to their downfall? Also, regarding the interaction of cuprizone with cellular components many questions remain. How widespread is the copper-chelation independent protein inhibition and could this be a reason for oligodendrocyte death? To date, it is unclear if cuprizone is metabolized in the body and if a potential metabolites cause oligodendrocyte death. By rigorously identifying how cuprizone induces demyelination, we may find new parallels with human white matter diseases, such as MS, and be able to use this model more efficiently in preclinical studies.
Availability of data and materials
Not applicable.
Abbreviations
- 4-HNE:
-
4-Hydroxynonenal
- AIF-1:
-
Apoptosis-inducing factor-1
- APAF-1:
-
Apoptotic protease activating factor-1
- ATP7:
-
Copper-transporting P-type ATPase
- BBB:
-
Blood–brain-barrier
- Casp-1:
-
Caspase-1
- CC-3:
-
Cleaved caspase 3
- CCL:
-
C–C motif chemokine ligand
- CCR:
-
C–C motif chemokine receptor
- CD:
-
Cluster of differentiation
- CGT:
-
Ceramide galactosyltransferase
- CNS:
-
Central nervous system
- CNPase:
-
2’,3’-Cyclic-nucleotide 3’-phosphodiesterase
- COX:
-
Cyclooxygenase
- CP:
-
Choroid plexus
- Crry:
-
Complement receptor 1-related Gene/Protein y
- CSF:
-
Colony stimulating factor
- CSF1R:
-
Colony stimulating factor 1 receptor
- CTR1:
-
High affinity copper uptake protein 1
- CXCL:
-
C-X-C motif chemokine ligand
- CXCR:
-
C-X-C motif chemokine receptor
- Cyt c:
-
Cytochrome c
- DNA:
-
Deoxyribonucleic acid
- Drp1:
-
Dynamin-related protein 1
- EAE:
-
Experimental autoimmune encephalomyelitis
- GFAP:
-
Glial fibrillary acidic protein
- GLAST:
-
Glutamate aspartate transporter
- GPX4:
-
Glutathione peroxidase 4
- GSH:
-
Glutathione
- HMG-CoA:
-
β-Hydroxy β-methylglutaryl-CoA
- IKK2:
-
Inhibitor kappa B kinase 2
- IL:
-
Interleukin
- INF:
-
Interferon
- IRF-8:
-
Interferon regulatory factor 8
- LIF:
-
Leukemia inhibitory factor
- LPC:
-
Lysophosphatidylcholine
- LPS:
-
Lipopolysaccharide
- Ltα:
-
Lymphotoxin α
- MAG:
-
Myelin-associated glycoprotein
- MBP:
-
Myelin basic protein
- MDA:
-
Malondialdehyde
- MERTK:
-
MER proto-oncogene, tyrosine kinase
- MHC:
-
Major histocompatibility complex
- MLKL:
-
Mixed lineage kinase domain-like protein
- MRI:
-
Magnetic resonance imaging
- mRNA:
-
Messenger ribonucleic acid
- MS:
-
Multiple Sclerosis
- NCOA4:
-
Nuclear receptor coactivator 4
- NF-κB:
-
Nuclear factor kappa B
- NLRC:
-
NLR Family CARD domain containing
- NLRP:
-
LRR- and pyrin domain-containing protein
- NMDAR:
-
N-Methyl-D-aspartic acid receptors
- OPC:
-
Oligodendrocyte progenitor cell
- OSM:
-
Oncostatin M
- P75NTR:
-
P75 neurotrophin receptor
- PAMPs:
-
Pathogen associated molecular patterns
- PARP:
-
Poly (ADP-ribose) polymerase
- PDGF:
-
Platelet-derived growth factor
- PG:
-
Prostaglandins
- PGE2-EP2:
-
PG E2 receptor 2 subtype
- PPMS:
-
Primary-progressive MS
- PrPC:
-
Cellular prion protein
- RAG-1:
-
Recombination activation gene-1
- RIPK:
-
Receptor interacting serine/threonine kinase
- ROS:
-
Reactive oxygen species
- RRMS:
-
Relapsing–remitting MS
- sCrry:
-
Soluble Crry
- SPMS:
-
Secondary-progressive MS
- Sxc-:
-
Cystine/glutamate antiporter system xc −
- TLR:
-
Toll-like receptor
- TMEV:
-
Theiler’s murine encephalomyelitis virus
- TNF:
-
Tumor necrosis factor
- TNFR:
-
TNF receptor
- TREM:
-
Triggering receptor expressed on myeloid cells
- TrkB:
-
Tropomyosin receptor kinase B
- VgluT:
-
Vesicular glutamate transporter
- ZBP:
-
Z-DNA binding protein
References
Snaidero N, Simons M. Myelination at a glance. J Cell Sci. 2014;127(Pt 14):2999–3004.
Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest. 2017;127(9):3271–80.
Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, Riethmacher D, Franklin RJ, Rudin M, Suter U. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci. 2011;31(3):1069–80.
Ghosh A, Manrique-Hoyos N, Voigt A, Schulz JB, Kreutzfeldt M, Merkler D, Simons M. Targeted ablation of oligodendrocytes triggers axonal damage. PLoS ONE. 2011;6(7): e22735.
Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, Merson TD. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci. 2012;32(24):8317–30.
Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Myelin damage and repair in pathologic CNS: challenges and prospects. Front Mol Neurosci. 2015;8:35.
Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–31.
Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13(7):507–14.
Dobson R, Giovannoni G. Multiple sclerosis - a review. Eur J Neurol. 2019;26(1):27–40.
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–68.
Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, Hoffman B, Morgan BP. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50(5):646–57.
Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15(8):1074–7.
Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 2017;133(2):223–44.
Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17(1):174–207.
Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol. 2014;122:173–89.
Skundric DS. Experimental models of relapsing-remitting multiple sclerosis: current concepts and perspective. Curr Neurovasc Res. 2005;2(4):349–62.
Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol. 2005;58(6):939–45.
Zendedel A, Beyer C, Kipp M. Cuprizone-induced demyelination as a tool to study remyelination and axonal protection. J Mol Neurosci. 2013;51(2):567–72.
Blakemore WF, Franklin RJ. Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol. 2008;318:193–212.
Vega-Riquer JM, Mendez-Victoriano G, Morales-Luckie RA, Gonzalez-Perez O. Five decades of cuprizone, an updated model to replicate demyelinating diseases. Curr Neuropharmacol. 2019;17(2):129–41.
Hooijmans CR, Hlavica M, Schuler FAF, Good N, Good A, Baumgartner L, Galeno G, Schneider MP, Jung T, de Vries R, et al. Remyelination promoting therapies in multiple sclerosis animal models: a systematic review and meta-analysis. Sci Rep. 2019;9(1):822.
Plemel JR, Liu WQ, Yong VW. Remyelination therapies: a new direction and challenge in multiple sclerosis. Nat Rev Drug Discov. 2017;16(9):617–34.
Nilsson G, Erdtman H, Lindstedt G, Kinell P. A new colour reaction on copper and certain carbonyl compounds. Acta Chem Scand. 1950;4:205–205.
Carlton WW. Studies on the induction of hydrocephalus and spongy degeneration by cuprizone feeding and attempts to antidote the toxicity. Life Sci. 1967;6(1):11–9.
Carlton WW. Response of mice to the chelating agents sodium diethyldithiocarbamate, alpha-benzoinoxime, and biscyclohexanone oxaldihydrazone. Toxicol Appl Pharmacol. 1966;8(3):512–21.
Suzuki K. Giant hepatic mitochondria: production in mice fed with cuprizone. Science. 1969;163(3862):81–2.
Kesterson JW, Carlton WW. Monoamine oxidase inhibition and the activity of other oxidative enzymes in the brains of mice fed cuprizone. Toxicol Appl Pharmacol. 1971;20(3):386–95.
Venturini G. Enzymic activities and sodium, potassium and copper concentrations in mouse brain and liver after cuprizone treatment in vivo. J Neurochem. 1973;21(5):1147–51.
Acs P, Selak MA, Komoly S, Kalman B. Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. J Neuroimmunol. 2013;262(1–2):128–31.
Hoppel CL, Tandler B. Biochemical effects of cuprizone on mouse liver and heart mitochondria. Biochem Pharmacol. 1973;22(18):2311–8.
Carlton WW. Spongiform encephalopathy induced in rats and guinea pigs by cuprizone. Exp Mol Pathol. 1969;10(3):274–87.
Silvestroff L, Bartucci S, Pasquini J, Franco P. Cuprizone-induced demyelination in the rat cerebral cortex and thyroid hormone effects on cortical remyelination. Exp Neurol. 2012;235(1):357–67.
Oakden W, Bock NA, Al-Ebraheem A, Farquharson MJ, Stanisz GJ. Early regional cuprizone-induced demyelination in a rat model revealed with MRI. NMR Biomed. 2017;30(9):e3743.
Adamo AM, Paez PM, Escobar Cabrera OE, Wolfson M, Franco PG, Pasquini JM, Soto EF. Remyelination after cuprizone-induced demyelination in the rat is stimulated by apotransferrin. Exp Neurol. 2006;198(2):519–29.
Kanno T, Sasaki S, Yamada N, Kawasako K, Tsuchitani M. Hexachlorophene and cuprizone induce the spongy change of the developing rat brain by different mechanisms: the role of 2’, 3’-cyclic nucleotide 3’-phosphodiesterase (CNPase). J Vet Med Sci. 2012;74(7):837–43.
Buyukmihci N, Goehring-Harmon F, Marsh RF. Retinal degeneration during clinical scrapie encephalopathy in hamsters. J Comp Neurol. 1982;205(2):153–60.
Kimberlin RH, Collis SC, Walker CA. Profiles of brain glycosidase activity in cuprizone-fed Syrian hamsters and in scrapie-affected mice, rats, Chinese hamsters and Syrian hamsters. J Comp Pathol. 1976;86(1):135–42.
Chen Z, Chen JT, Johnson M, Gossman ZC, Hendrickson M, Sakaie K, Martinez-Rubio C, Gale JT, Trapp BD. Cuprizone does not induce CNS demyelination in nonhuman primates. Ann Clin Transl Neurol. 2015;2(2):208–13.
Kondo A, Nakano T, Suzuki K. Blood-brain barrier permeability to horseradish peroxidase in twitcher and cuprizone-intoxicated mice. Brain Res. 1987;425(1):186–90.
Bakker DA, Ludwin SK. Blood-brain barrier permeability during Cuprizone-induced demyelination. Implications for the pathogenesis of immune-mediated demyelinating diseases. J Neurol Sci. 1987;78(2):125–37.
Sansom BF, Pattison IH, Jebbett JN. Permeability of blood vessels in mice affected with scrapie or fed with cuprizone. J Comp Pathol. 1973;83(4):461–6.
Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92(1–2):38–49.
Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. 2008;188:72–6.
Xing YL, Roth PT, Stratton JA, Chuang BH, Danne J, Ellis SL, Ng SW, Kilpatrick TJ, Merson TD. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J Neurosci. 2014;34(42):14128–46.
Berghoff SA, Gerndt N, Winchenbach J, Stumpf SK, Hosang L, Odoardi F, Ruhwedel T, Bohler C, Barrette B, Stassart R, et al. Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat Commun. 2017;8:14241.
Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11(1):107–16.
Blakemore WF. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J Neurol Sci. 1973;20(1):63–72.
Ludwin SK. Central nervous system demyelination and remyelination in the mouse: an ultrastructural study of cuprizone toxicity. Lab Invest. 1978;39(6):597–612.
Jurevics H, Largent C, Hostettler J, Sammond DW, Matsushima GK, Kleindienst A, Toews AD, Morell P. Alterations in metabolism and gene expression in brain regions during cuprizone-induced demyelination and remyelination. J Neurochem. 2002;82(1):126–36.
Silvestroff L, Bartucci S, Soto E, Gallo V, Pasquini J, Franco P. Cuprizone-induced demyelination in CNP::GFP transgenic mice. J Comp Neurol. 2010;518(12):2261–83.
Norkute A, Hieble A, Braun A, Johann S, Clarner T, Baumgartner W, Beyer C, Kipp M. Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J Neurosci Res. 2009;87(6):1343–55.
Yang HJ, Wang H, Zhang Y, Xiao L, Clough RW, Browning R, Li XM, Xu H. Region-specific susceptibilities to cuprizone-induced lesions in the mouse forebrain: Implications for the pathophysiology of schizophrenia. Brain Res. 2009;1270:121–30.
Hoffmann K, Lindner M, Groticke I, Stangel M, Loscher W. Epileptic seizures and hippocampal damage after cuprizone-induced demyelination in C57BL/6 mice. Exp Neurol. 2008;210(2):308–21.
Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res. 2009;1283:127–38.
Lindner M, Fokuhl J, Linsmeier F, Trebst C, Stangel M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci Lett. 2009;453(2):120–5.
Plant SR, Arnett HA, Ting JP. Astroglial-derived lymphotoxin-alpha exacerbates inflammation and demyelination, but not remyelination. Glia. 2005;49(1):1–14.
Franco-Pons N, Torrente M, Colomina MT, Vilella E. Behavioral deficits in the cuprizone-induced murine model of demyelination/remyelination. Toxicol Lett. 2007;169(3):205–13.
Hibbits N, Pannu R, Wu TJ, Armstrong RC. Cuprizone demyelination of the corpus callosum in mice correlates with altered social interaction and impaired bilateral sensorimotor coordination. ASN Neuro. 2009;1(3):e00013.
Blakemore WF. Remyelination of the superior cerebellar peduncle in the mouse following demyelination induced by feeding cuprizone. J Neurol Sci. 1973;20(1):73–83.
Ludwin SK. Chronic demyelination inhibits remyelination in the central nervous system. An analysis of contributing factors. Lab Invest. 1980;43(4):382–7.
Liebetanz D, Merkler D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp Neurol. 2006;202(1):217–24.
Manrique-Hoyos N, Jurgens T, Gronborg M, Kreutzfeldt M, Schedensack M, Kuhlmann T, Schrick C, Bruck W, Urlaub H, Simons M, et al. Late motor decline after accomplished remyelination: impact for progressive multiple sclerosis. Ann Neurol. 2012;71(2):227–44.
Morgan ML, Kaushik DK, Stys PK, Caprariello AV. Autofluorescence spectroscopy as a proxy for chronic white matter pathology. Mult Scler. 2021;27(7):1046–56.
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain. 1999;122(Pt 12):2279–95.
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–17.
Metz I, Gavrilova RH, Weigand SD, Frischer JM, Popescu BF, Guo Y, Gloth M, Tobin WO, Zalewski NL, Lassmann H, et al. magnetic resonance imaging correlates of multiple sclerosis immunopathological patterns. Ann Neurol. 2021;90(3):440–54.
Metz I, Weigand SD, Popescu BF, Frischer JM, Parisi JE, Guo Y, Lassmann H, Bruck W, Lucchinetti CF. Pathologic heterogeneity persists in early active multiple sclerosis lesions. Ann Neurol. 2014;75(5):728–38.
Torkildsen O, Brunborg LA, Milde AM, Mork SJ, Myhr KM, Bo L. A salmon based diet protects mice from behavioural changes in the cuprizone model for demyelination. Clin Nutr. 2009;28(1):83–7.
Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131(Pt 7):1722–35.
Kaddatz H, Joost S, Nedelcu J, Chrzanowski U, Schmitz C, Gingele S, Gudi V, Stangel M, Zhan J, Santrau E, et al. Cuprizone-induced demyelination triggers a CD8-pronounced T cell recruitment. Glia. 2021;69(4):925–42.
Veto S, Acs P, Bauer J, Lassmann H, Berente Z, Setalo G Jr, Borgulya G, Sumegi B, Komoly S, Gallyas F Jr, et al. Inhibiting poly(ADP-ribose) polymerase: a potential therapy against oligodendrocyte death. Brain. 2010;133(Pt 3):822–34.
Hochmeister S, Grundtner R, Bauer J, Engelhardt B, Lyck R, Gordon G, Korosec T, Kutzelnigg A, Berger JJ, Bradl M, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol. 2006;65(9):855–65.
Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18(12):905–22.
Milstein JL, Barbour CR, Jackson K, Kosa P, Bielekova B. Intrathecal, not systemic inflammation is correlated with multiple sclerosis severity, especially in progressive multiple sclerosis. Front Neurol. 2019;10:1232.
Berghoff SA, Duking T, Spieth L, Winchenbach J, Stumpf SK, Gerndt N, Kusch K, Ruhwedel T, Mobius W, Saher G. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol Commun. 2017;5(1):94.
Shelestak J, Singhal N, Frankle L, Tomor R, Sternbach S, McDonough J, Freeman E, Clements R. Increased blood-brain barrier hyperpermeability coincides with mast cell activation early under cuprizone administration. PLoS ONE. 2020;15(6):e0234001.
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(Pt 11):2705–12.
Calabrese M, Rinaldi F, Grossi P, Gallo P. Cortical pathology and cognitive impairment in multiple sclerosis. Expert Rev Neurother. 2011;11(3):425–32.
Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118(6):723–36.
Zhang Y, Cai L, Fan K, Fan B, Li N, Gao W, Yang X, Ma J. The spatial and temporal characters of demyelination and remyelination in the cuprizone animal model. Anat Rec (Hoboken). 2019;302(11):2020–9.
Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7(9):841–51.
Carassiti D, Altmann DR, Petrova N, Pakkenberg B, Scaravilli F, Schmierer K. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol Appl Neurobiol. 2018;44(4):377–90.
Strijbis EMM, Kooi EJ, van der Valk P, Geurts JJG. Cortical remyelination is heterogeneous in multiple sclerosis. J Neuropathol Exp Neurol. 2017;76(5):390–401.
Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM, Yong VW, Fox RJ, Kidd GJ, Trapp BD. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol. 2012;72(6):918–26.
Wergeland S, Torkildsen O, Myhr KM, Mork SJ, Bo L. The cuprizone model: regional heterogeneity of pathology. APMIS. 2012;120(8):648–57.
Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605–14.
Tumer Z, Moller LB. Menkes disease. Eur J Hum Genet. 2010;18(5):511–8.
Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14(1):103–13.
Gromadzka G, Tarnacka B, Flaga A, Adamczyk A. Copper dyshomeostasis in neurodegenerative diseases-therapeutic implications. Int J Mol Sci. 2020;21(23):9259.
Tezuka T, Tamura M, Kondo MA, Sakaue M, Okada K, Takemoto K, Fukunari A, Miwa K, Ohzeki H, Kano S, et al. Cuprizone short-term exposure: astrocytic IL-6 activation and behavioral changes relevant to psychosis. Neurobiol Dis. 2013;59:63–8.
Zatta P, Raso M, Zambenedetti P, Wittkowski W, Messori L, Piccioli F, Mauri PL, Beltramini M. Copper and zinc dismetabolism in the mouse brain upon chronic cuprizone treatment. Cell Mol Life Sci. 2005;62(13):1502–13.
Benetti F, Ventura M, Salmini B, Ceola S, Carbonera D, Mammi S, Zitolo A, D’Angelo P, Urso E, Maffia M, et al. Cuprizone neurotoxicity, copper deficiency and neurodegeneration. Neurotoxicology. 2010;31(5):509–17.
Messori L, Casini A, Gabbiani C, Sorace L, Muniz-Miranda M, Zatta P. Unravelling the chemical nature of copper cuprizone. Dalton Trans. 2007;21:2112–4.
Yamamoto N, Kuwata K. DFT studies on redox properties of copper-chelating cuprizone: Unusually high-valent copper(III) state. J Mol Struct (Thoechem). 2009;895(1):52–6.
Kapeller-Adler R. Amine oxidases and methods for their study. Wiley-Interscience; 1970.
Lindstrom A, Pettersson G. The mechanism of inhibition of pig-plasma benzylamine oxidase by the copper-chelating reagent cuprizone. Eur J Biochem. 1974;48(1):229–36.
Taraboletti A, Walker T, Avila R, Huang H, Caporoso J, Manandhar E, Leeper TC, Modarelli DA, Medicetty S, Shriver LP. Cuprizone intoxication induces cell intrinsic alterations in oligodendrocyte metabolism independent of copper chelation. Biochemistry. 2017;56(10):1518–28.
Teo W, Caprariello AV, Morgan ML, Luchicchi A, Schenk GJ, Joseph JT, et al. Nile Red fluorescence spectroscopy reports early physicochemical changes in myelin with high sensitivity. Proc Natl Acad Sci U S A. 2021;118(8):e2016897118. https://doi.org/10.1073/pnas.2016897118.
Caprariello AV, Rogers JA, Morgan ML, Hoghooghi V, Plemel JR, Koebel A, Tsutsui S, Dunn JF, Kotra LP, Ousman SS, et al. Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci U S A. 2018;115(21):5528–33.
Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J Mol Neurosci. 2012;48(1):66–76.
Jhelum P, Santos-Nogueira E, Teo W, Haumont A, Lenoel I, Stys PK, David S. Ferroptosis mediates cuprizone-induced loss of oligodendrocytes and demyelination. J Neurosci. 2020;40(48):9327–41.
Raine CS. Morphology of Myelin and Myelination. In: Myelin. edn. Edited by Morell P. Boston: Springer US; 1984: p. 1–50.
McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107(1):1–19.
Cheepsunthorn P, Palmer C, Connor JR. Cellular distribution of ferritin subunits in postnatal rat brain. J Comp Neurol. 1998;400(1):73–86.
Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17(2):83–93.
Thorburne SK, Juurlink BH. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem. 1996;67(3):1014–22.
Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev. 2014;47:485–505.
Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci. 2014;8:73.
Acs P, Komoly S. Selective ultrastructural vulnerability in the cuprizone-induced experimental demyelination. Ideggyogy Sz. 2012;65(7–8):266–70.
Benardais K, Kotsiari A, Skuljec J, Koutsoudaki PN, Gudi V, Singh V, Vulinovic F, Skripuletz T, Stangel M. Cuprizone [bis(cyclohexylidenehydrazide)] is selectively toxic for mature oligodendrocytes. Neurotox Res. 2013;24(2):244–50.
Pasquini LA, Calatayud CA, Bertone Una AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem Res. 2007;32(2):279–92.
Karbowski M, Kurono C, Nishizawa Y, Horie Y, Soji T, Wakabayashi T. Induction of megamitochondria by some chemicals inducing oxidative stress in primary cultured rat hepatocytes. Biochim Biophys Acta. 1997;1349(3):242–50.
Wakabayashi T, Adachi K, Matsuhashi T, Wozniak M, Antosiewicz J, Karbowsky M. Suppression of the formation of megamitochondria by scavengers for free radicals. Mol Aspects Med. 1997;18(Suppl):S51-61.
Wakabayashi T. Megamitochondria formation - physiology and pathology. J Cell Mol Med. 2002;6(4):497–538.
Wakabayashi T. Structural changes of mitochondria related to apoptosis: swelling and megamitochondria formation. Acta Biochim Pol. 1999;46(2):223–37.
Tiwari BS, Belenghi B, Levine A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002;128(4):1271–81.
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144(2):281–92.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–57.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–32.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–6.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–63.
Faizi M, Salimi A, Seydi E, Naserzadeh P, Kouhnavard M, Rahimi A, Pourahmad J. Toxicity of cuprizone a Cu(2+) chelating agent on isolated mouse brain mitochondria: a justification for demyelination and subsequent behavioral dysfunction. Toxicol Mech Methods. 2016;26(4):276–83.
Floyd RA, Watson JJ, Wong PK, Altmiller DH, Rickard RC. Hydroxyl free radical adduct of deoxyguanosine: sensitive detection and mechanisms of formation. Free Radic Res Commun. 1986;1(3):163–72.
Luo M, Deng M, Yu Z, Zhang Y, Xu S, Hu S, Xu H. Differential Susceptibility and Vulnerability of Brain Cells in C57BL/6 Mouse to Mitochondrial Dysfunction Induced by Short-Term Cuprizone Exposure. Front Neuroanat. 2020;14:30.
Biemond P, Swaak AJ, van Eijk HG, Koster JF. Superoxide dependent iron release from ferritin in inflammatory diseases. Free Radic Biol Med. 1988;4(3):185–98.
Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life. 2008;60(7):421–9.
Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485(7399):517–21.
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487(7408):443–8.
Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci. 2011;31(2):538–48.
Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna). 2014;121(8):799–817.
McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74(3):723–60.
Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1041–8.
Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58(3):293–7.
Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23(9):1261–76.
Olney JW. Inciting excitotoxic cytocide among central neurons. Adv Exp Med Biol. 1986;203:631–45.
Klauser AM, Wiebenga OT, Eijlers AJ, Schoonheim MM, Uitdehaag BM, Barkhof F, Pouwels PJ, Geurts JJ. Metabolites predict lesion formation and severity in relapsing-remitting multiple sclerosis. Mult Scler. 2018;24(4):491–500.
Lee DW, Kwon JI, Woo CW, Heo H, Kim KW, Woo DC, Kim JK, Lee DH. In vivo measurement of neurochemical abnormalities in the hippocampus in a rat model of cuprizone-induced demyelination. Diagnostics (Basel). 2020;11(1):45.
Lee DW, Heo H, Woo CW, Woo DC, Kim JK, Kim KW, Lee DH. Temporal changes in in vivo glutamate signal during demyelination and remyelination in the corpus callosum: a glutamate-weighted chemical exchange saturation transfer imaging study. Int J Mol Sci. 2020;21(24):9468.
Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101(18):7158–63.
Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407(6801):189–94.
Bellocchio EE, Reimer RJ, Fremeau RT Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289(5481):957–60.
Hoflich KM, Beyer C, Clarner T, Schmitz C, Nyamoya S, Kipp M, Hochstrasser T. Acute axonal damage in three different murine models of multiple sclerosis: a comparative approach. Brain Res. 2016;1650:125–33.
Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25(26):6221–34.
Azami Tameh A, Clarner T, Beyer C, Atlasi MA, Hassanzadeh G, Naderian H. Regional regulation of glutamate signaling during cuprizone-induced demyelination in the brain. Ann Anat. 2013;195(5):415–23.
Matteucci A, Cammarota R, Paradisi S, Varano M, Balduzzi M, Leo L, Bellenchi GC, De Nuccio C, Carnovale-Scalzo G, Scorcia G, et al. Curcumin protects against NMDA-induced toxicity: a possible role for NR2A subunit. Invest Ophthalmol Vis Sci. 2011;52(2):1070–7.
Peterson AR, Binder DK. Astrocyte Glutamate Uptake and Signaling as Novel Targets for Antiepileptogenic Therapy. Front Neurol. 2020;11:1006.
Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105.
Vallejo-Illarramendi A, Domercq M, Perez-Cerda F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21(1):154–64.
Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7(4):494–506.
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307(5950):462–5.
Peters S, Koh J, Choi DW. Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science. 1987;236(4801):589–93.
Marchetti C, Baranowska-Bosiacka I, Gavazzo P. Multiple effects of copper on NMDA receptor currents. Brain Res. 2014;1542:20–31.
Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Gen Physiol. 2008;131(6):i5.
You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109(5):1737–42.
Stys PK, You H, Zamponi GW. Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J Physiol. 2012;590(6):1357–68.
Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181(3):551–65.
Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005;25(19):4879–88.
Bribian A, Fontana X, Llorens F, Gavin R, Reina M, Garcia-Verdugo JM, Torres JM, de Castro F, del Rio JA. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS ONE. 2012;7(4):e33872.
Morell P, Barrett CV, Mason JL, Toews AD, Hostettler JD, Knapp GW, Matsushima GK. Gene expression in brain during cuprizone-induced demyelination and remyelination. Mol Cell Neurosci. 1998;12(4–5):220–7.
Jurevics H, Hostettler J, Muse ED, Sammond DW, Matsushima GK, Toews AD, Morell P. Cerebroside synthesis as a measure of the rate of remyelination following cuprizone-induced demyelination in brain. J Neurochem. 2001;77(4):1067–76.
Goldberg J, Daniel M, van Heuvel Y, Victor M, Beyer C, Clarner T, Kipp M. Short-term cuprizone feeding induces selective amino acid deprivation with concomitant activation of an integrated stress response in oligodendrocytes. Cell Mol Neurobiol. 2013;33(8):1087–98.
Podbielska M, Levery SB, Hogan EL. The structural and functional role of myelin fast-migrating cerebrosides: pathological importance in multiple sclerosis. Clin Lipidol. 2011;6(2):159–79.
Saher G, Quintes S, Nave KA. Cholesterol: a novel regulatory role in myelin formation. Neuroscientist. 2011;17(1):79–93.
Hemm RD, Carlton WW, Welser JR. Ultrastructural changes of cuprizone encephalopathy in mice. Toxicol Appl Pharmacol. 1971;18(4):869–82.
Blakemore WF. The response of oligodendrocytes to chemical injury. Acta Neurol Scand Suppl. 1984;100:33–8.
Zeis T, Enz L, Schaeren-Wiemers N. The immunomodulatory oligodendrocyte. Brain Res. 2016;1641(Pt A):139–48.
Falcao AM, van Bruggen D, Marques S, Meijer M, Jakel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24(12):1837–44. https://doi.org/10.1038/s41591-018-0236-y.
Scheld M, Fragoulis A, Nyamoya S, Zendedel A, Denecke B, Krauspe B, Teske N, Kipp M, Beyer C, Clarner T. mitochondrial impairment in oligodendroglial cells induces cytokine expression and signaling. J Mol Neurosci. 2019;67(2):265–75.
Skuljec J, Sun H, Pul R, Benardais K, Ragancokova D, Moharregh-Khiabani D, Kotsiari A, Trebst C, Stangel M. CCL5 induces a pro-inflammatory profile in microglia in vitro. Cell Immunol. 2011;270(2):164–71.
Holmberg KH, Patterson PH. Leukemia inhibitory factor is a key regulator of astrocytic, microglial and neuronal responses in a low-dose pilocarpine injury model. Brain Res. 2006;1075(1):26–35.
Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, Green KN. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139.
Laflamme N, Cisbani G, Prefontaine P, Srour Y, Bernier J, St-Pierre MK, Tremblay ME, Rivest S. mCSF-Induced microglial activation prevents myelin loss and promotes its repair in a mouse model of multiple sclerosis. Front Cell Neurosci. 2018;12:178.
Maiti AK, Sharba S, Navabi N, Forsman H, Fernandez HR, Linden SK. IL-4 Protects the mitochondria against tnfalpha and ifngamma induced insult during clearance of infection with citrobacter rodentium and escherichia coli. Sci Rep. 2015;5:15434.
Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology. 2010;52(6):2137–47.
Schonrock LM, Gawlowski G, Bruck W. Interleukin-6 expression in human multiple sclerosis lesions. Neurosci Lett. 2000;294(1):45–8.
Petkovic F, Campbell IL, Gonzalez B, Castellano B. Astrocyte-targeted production of interleukin-6 reduces astroglial and microglial activation in the cuprizone demyelination model: Implications for myelin clearance and oligodendrocyte maturation. Glia. 2016;64(12):2104–19.
Petkovic F, Campbell IL, Gonzalez B, Castellano B. Reduced cuprizone-induced cerebellar demyelination in mice with astrocyte-targeted production of IL-6 is associated with chronically activated, but less responsive microglia. J Neuroimmunol. 2017;310:97–102.
Kesterson JW, Carlton WW. Histopathologic and enzyme histochemical observations of the cuprizone-induced brain edema. Exp Mol Pathol. 1971;15(1):82–96.
Blakemore WF. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J Neurocytol. 1972;1(4):413–26.
Colombo E, Triolo D, Bassani C, Bedogni F, Di Dario M, Dina G, Fredrickx E, Fermo I, Martinelli V, Newcombe J, et al. Dysregulated copper transport in multiple sclerosis may cause demyelination via astrocytes. Proc Natl Acad Sci U S A. 2021;118(27):e2025804118.
Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, Knobeloch KP, Bethea JR, Waisman A, Knust M, et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain. 2011;134(Pt 4):1184–98.
Zhang H, Sun SC. NF-kappaB in inflammation and renal diseases. Cell Biosci. 2015;5:63.
Madadi S, Pasbakhsh P, Tahmasebi F, Mortezaee K, Khanehzad M, Boroujeni FB, Noorzehi G, Kashani IR. Astrocyte ablation induced by La-aminoadipate (L-AAA) potentiates remyelination in a cuprizone demyelinating mouse model. Metab Brain Dis. 2019;34(2):593–603.
Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, et al. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24(39):8500–9.
Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M, Baumgartner W, Stangel M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 2013;136(Pt 1):147–67.
Clarner T, Janssen K, Nellessen L, Stangel M, Skripuletz T, Krauspe B, Hess FM, Denecke B, Beutner C, Linnartz-Gerlach B, et al. CXCL10 triggers early microglial activation in the cuprizone model. J Immunol. 2015;194(7):3400–13.
Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes Apoptosis induced by lymphotoxin. J Immunol. 1991;147(5):1522–9.
Zheng W, Monnot AD. Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol Ther. 2012;133(2):177–88.
Marzan DE, Brugger-Verdon V, West BL, Liddelow S, Samanta J, Salzer JL. Activated microglia drive demyelination via CSF1R signaling. Glia. 2021;69(6):1583–604.
Greter M, Lelios I, Croxford AL. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front Immunol. 2015;6:249.
Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F. The cyclooxygenase-2 pathway via the PGE(2) EP2 receptor contributes to oligodendrocytes apoptosis in cuprizone-induced demyelination. J Neurochem. 2012;121(3):418–27.
Plemel JR, Manesh SB, Sparling JS, Tetzlaff W. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia. 2013;61(9):1471–87.
Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, Franklin RJ, Altmann F, Lubec G, Kotter MR. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. 2009;132(Pt 2):465–81.
Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006;26(1):328–32.
Plemel JR, Stratton JA, Michaels NJ, Rawji KS, Zhang E, Sinha S, Baaklini CS, Dong Y, Ho M, Thorburn K, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv. 2020;6(3):eaay6324.
Rawji KS, Young AMH, Ghosh T, Michaels NJ, Mirzaei R, Kappen J, Kolehmainen KL, Alaeiilkhchi N, Lozinski B, Mishra MK, et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol. 2020;139(5):893–909.
Clarner T, Diederichs F, Berger K, Denecke B, Gan L, van der Valk P, Beyer C, Amor S, Kipp M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia. 2012;60(10):1468–80.
Shen K, Reichelt M, Kyauk RV, Ngu H, Shen YA, Foreman O, Modrusan Z, Friedman BA, Sheng M, Yuen TJ. Multiple sclerosis risk gene Mertk is required for microglial activation and subsequent remyelination. Cell Rep. 2021;34(10):108835.
Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11):a009076.
Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129(3):429–47.
Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125(5):2161–70.
Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R, Manis M, Ibrahim A, Deng L, Benitez BA, et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020;140(4):513–34.
Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, Liu Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017;37(6):2224–36.
Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, Wang J, Zhao L, Liang YX, Wu T, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–40.
Tahmasebi F, Pasbakhsh P, Barati S, Madadi S, Kashani IR. The effect of microglial ablation and mesenchymal stem cell transplantation on a cuprizone-induced demyelination model. J Cell Physiol. 2021;236(5):3552–64.
Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50.
Kishore U, Reid KB. Modular organization of proteins containing C1q-like globular domain. Immunopharmacology. 1999;42(1–3):15–21.
Briggs DT, Martin CB, Ingersoll SA, Barnum SR, Martin BK. Astrocyte-specific expression of a soluble form of the murine complement control protein Crry confers demyelination protection in the cuprizone model. Glia. 2007;55(14):1405–15.
Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JP. Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. J Neurosci. 2003;23(30):9824–32.
Bao L, Wang Y, Chang A, Minto AW, Zhou J, Kang H, Haas M, Quigg RJ. Unrestricted C3 activation occurs in Crry-deficient kidneys and rapidly leads to chronic renal failure. J Am Soc Nephrol. 2007;18(3):811–22.
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97.
Ehrengruber MU, Geiser T, Deranleau DA. Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett. 1994;346(2–3):181–4.
van Lookeren CM, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol. 2007;9(9):2095–102.
Armstrong RC, Harvath L, Dubois-Dalcq ME. Type 1 astrocytes and oligodendrocyte-type 2 astrocyte glial progenitors migrate toward distinct molecules. J Neurosci Res. 1990;27(3):400–7.
Crehan H, Hardy J, Pocock J. Microglia, Alzheimer’s disease, and complement. Int J Alzheimers Dis. 2012;2012:983640.
Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–49.
Nolte C, Moller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73(4):1091–107.
Ingersoll SA, Martin CB, Barnum SR, Martin BK. CNS-specific expression of C3a and C5a exacerbate demyelination severity in the cuprizone model. Mol Immunol. 2010;48(1–3):219–30.
Liu Y, Harlow DE, Given KS, Owens GP, Macklin WB, Bennett JL. Variable sensitivity to complement-dependent cytotoxicity in murine models of neuromyelitis optica. J Neuroinflammation. 2016;13(1):301.
Scolding NJ, Morgan BP, Campbell AK, Compston DA. Complement mediated serum cytotoxicity against oligodendrocytes: a comparison with other cells of the oligodendrocyte-type 2 astrocyte lineage. J Neurol Sci. 1990;97(2–3):155–62.
Tradtrantip L, Yao X, Su T, Smith AJ, Verkman AS. Bystander mechanism for complement-initiated early oligodendrocyte injury in neuromyelitis optica. Acta Neuropathol. 2017;134(1):35–44.
Vandendriessche S, Cambier S, Proost P, Marques PE. Complement receptors and their role in leukocyte recruitment and phagocytosis. Front Cell Dev Biol. 2021;9:624025.
McMahon EJ, Cook DN, Suzuki K, Matsushima GK. Absence of macrophage-inflammatory protein-1alpha delays central nervous system demyelination in the presence of an intact blood-brain barrier. J Immunol. 2001;167(5):2964–71.
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4(11):1116–22.
Cammer W. Apoptosis of oligodendrocytes in secondary cultures from neonatal rat brains. Neurosci Lett. 2002;327(2):123–7.
Jurewicz A, Matysiak M, Tybor K, Selmaj K. TNF-induced death of adult human oligodendrocytes is mediated by c-jun NH2-terminal kinase-3. Brain. 2003;126(Pt 6):1358–70.
Janssens K, Maheshwari A, Van den Haute C, Baekelandt V, Stinissen P, Hendriks JJ, Slaets H, Hellings N. Oncostatin M protects against demyelination by inducing a protective microglial phenotype. Glia. 2015;63(10):1729–37.
Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25(2):252–62.
Zimmermann J, Emrich M, Krauthausen M, Saxe S, Nitsch L, Heneka MT, Campbell IL, Muller M. IL-17A promotes granulocyte infiltration, myelin loss, microglia activation, and behavioral deficits during cuprizone-induced demyelination. Mol Neurobiol. 2018;55(2):946–57.
Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129(Pt 5):1306–18.
Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117(2):448–56.
Gao X, Gillig TA, Ye P, D’Ercole AJ, Matsushima GK, Popko B. Interferon-gamma protects against cuprizone-induced demyelination. Mol Cell Neurosci. 2000;16(4):338–49.
Pons V, Laflamme N, Prefontaine P, Rivest S. Role of macrophage colony-stimulating factor receptor on the proliferation and survival of microglia following systemic nerve and cuprizone-induced injuries. Front Immunol. 2020;11:47.
Janssen K, Rickert M, Clarner T, Beyer C, Kipp M. Absence of CCL2 and CCL3 ameliorates central nervous system grey matter but not white matter demyelination in the presence of an intact blood-brain barrier. Mol Neurobiol. 2016;53(3):1551–64.
Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, Yong VW, Stys PK, Tremblay ME, Rivest S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015;212(4):481–95.
Remington LT, Babcock AA, Zehntner SP, Owens T. Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol. 2007;170(5):1713–24.
Lindner M, Trebst C, Heine S, Skripuletz T, Koutsoudaki PN, Stangel M. The chemokine receptor CXCR2 is differentially regulated on glial cells in vivo but is not required for successful remyelination after cuprizone-induced demyelination. Glia. 2008;56(10):1104–13.
Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010;13(3):319–26.
Krauthausen M, Saxe S, Zimmermann J, Emrich M, Heneka MT, Muller M. CXCR3 modulates glial accumulation and activation in cuprizone-induced demyelination of the central nervous system. J Neuroinflammation. 2014;11:109.
Trebst C, Heine S, Lienenklaus S, Lindner M, Baumgartner W, Weiss S, Stangel M. Lack of interferon-beta leads to accelerated remyelination in a toxic model of central nervous system demyelination. Acta Neuropathol. 2007;114(6):587–96.
Schmidt H, Raasch J, Merkler D, Klinker F, Krauss S, Bruck W, Prinz M. Type I interferon receptor signalling is induced during demyelination while its function for myelin damage and repair is redundant. Exp Neurol. 2009;216(2):306–11.
Mana P, Linares D, Fordham S, Staykova M, Willenborg D. Deleterious role of IFNgamma in a toxic model of central nervous system demyelination. Am J Pathol. 2006;168(5):1464–73.
Kang Z, Liu L, Spangler R, Spear C, Wang C, Gulen MF, Veenstra M, Ouyang W, Ransohoff RM, Li X. IL-17-induced Act1-mediated signaling is critical for cuprizone-induced demyelination. J Neurosci. 2012;32(24):8284–92.
Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30(47):15811–20.
Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21(18):7046–52.
Horiuchi M, Wakayama K, Itoh A, Kawai K, Pleasure D, Ozato K, Itoh T. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J Neuroinflammation. 2012;9:227.
Marriott MP, Emery B, Cate HS, Binder MD, Kemper D, Wu Q, Kolbe S, Gordon IR, Wang H, Egan G, et al. Leukemia inhibitory factor signaling modulates both central nervous system demyelination and myelin repair. Glia. 2008;56(6):686–98.
Plant SR, Iocca HA, Wang Y, Thrash JC, O’Connor BP, Arnett HA, Fu YX, Carson MJ, Ting JP. Lymphotoxin beta receptor (Lt betaR): dual roles in demyelination and remyelination and successful therapeutic intervention using Lt betaR-Ig protein. J Neurosci. 2007;27(28):7429–37.
Copray JC, Kust BM, Mantingh-Otter I, Boddeke HW. p75NTR independent oligodendrocyte death in cuprizone-induced demyelination in C57BL/6 mice. Neuropathol Appl Neurobiol. 2005;31(6):600–9.
Iocca HA, Plant SR, Wang Y, Runkel L, O’Connor BP, Lundsmith ET, Hahm K, van Deventer HW, Burkly LC, Ting JP. TNF superfamily member TWEAK exacerbates inflammation and demyelination in the cuprizone-induced model. J Neuroimmunol. 2008;194(1–2):97–106.
Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19(1–2):171–82.
Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000.
Fitzpatrick FA. Cyclooxygenase enzymes: regulation and function. Curr Pharm Des. 2004;10(6):577–88.
Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F. Time-dependent changes in the brain arachidonic acid cascade during cuprizone-induced demyelination and remyelination. Prostaglandins Leukot Essent Fatty Acids. 2011;85(1):29–35.
Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet. 2000;38(3):225–42.
Dore-Duffy P, Donaldson JO, Koff T, Longo M, Perry W. Prostaglandin release in multiple sclerosis: correlation with disease activity. Neurology. 1986;36(12):1587–90.
Dore-Duffy P, Ho SY, Donovan C. Cerebrospinal fluid eicosanoid levels: endogenous PGD2 and LTC4 synthesis by antigen-presenting cells that migrate to the central nervous system. Neurology. 1991;41(2 ( Pt 1)):322–4.
Rose JW, Hill KE, Watt HE, Carlson NG. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J Neuroimmunol. 2004;149(1–2):40–9.
Carlson NG, Hill KE, Tsunoda I, Fujinami RS, Rose JW. The pathologic role for COX-2 in apoptotic oligodendrocytes in virus induced demyelinating disease: implications for multiple sclerosis. J Neuroimmunol. 2006;174(1–2):21–31.
Liddelow SA. Development of the choroid plexus and blood-CSF barrier. Front Neurosci. 2015;9:32.
Fleischer V, Gonzalez-Escamilla G, Ciolac D, Albrecht P, Kury P, Gruchot J, Dietrich M, Hecker C, Muntefering T, Bock S, et al. Translational value of choroid plexus imaging for tracking neuroinflammation in mice and humans. Proc Natl Acad Sci U S A. 2021;118(36):e2025000118.
Ricigliano VAG, Morena E, Colombi A, Tonietto M, Hamzaoui M, Poirion E, Bottlaender M, Gervais P, Louapre C, Bodini B, et al. Choroid plexus enlargement in inflammatory multiple sclerosis: 3.0-T MRI and Translocator Protein PET Evaluation. Radiology. 2021;301(1):166–77.
Solar P, Zamani A, Kubickova L, Dubovy P, Joukal M. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17(1):35.
Greiner T, Kipp M. What guides peripheral immune cells into the central nervous system? Cells. 2021;10(8):2041.
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–75.
Khaw YM, Tierney A, Cunningham C, Soto-Diaz K, Kang E, Steelman AJ, Inoue M. Astrocytes lure CXCR2-expressing CD4(+) T cells to gray matter via TAK1-mediated chemokine production in a mouse model of multiple sclerosis. Proc Natl Acad Sci U S A. 2021;118(8).
Sun B, Li F, Lai S, Zhang X, Wang H, Li Y, Wang W, Chen Y, Liu B, Zheng Y. Inhibition of CXCR2 alleviates the development of abdominal aortic aneurysm in Apo E-/- mice. Acta Cir Bras. 2021;36(1):e360105.
Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S3–23.
Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–77.
Hiremath MM, Chen VS, Suzuki K, Ting JP, Matsushima GK. MHC class II exacerbates demyelination in vivo independently of T cells. J Neuroimmunol. 2008;203(1):23–32.
Duarte J, Carrie N, Oliveira VG, Almeida C, Agua-Doce A, Rodrigues L, Simas JP, Mars LT, Graca L. T cell apoptosis and induction of Foxp3+ regulatory T cells underlie the therapeutic efficacy of CD4 blockade in experimental autoimmune encephalomyelitis. J Immunol. 2012;189(4):1680–8.
Roggendorf W, Sasaki S, Ludwig H. Light microscope and immunohistological investigations on the brain of Borna disease virus-infected rabbits. Neuropathol Appl Neurobiol. 1983;9(4):287–96.
Sen MK, Almuslehi MSM, Gyengesi E, Myers SJ, Shortland PJ, Mahns DA, Coorssen JR. Suppression of the peripheral immune system limits the central immune response following cuprizone-feeding: relevance to modelling multiple sclerosis. Cells. 2019;8(11):1314.
Solti I, Kvell K, Talaber G, Veto S, Acs P, Gallyas F Jr, Illes Z, Fekete K, Zalan P, Szanto A, et al. Thymic Atrophy and Apoptosis of CD4+CD8+ Thymocytes in the Cuprizone Model of Multiple Sclerosis. PLoS ONE. 2015;10(6):e0129217.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
Hesse A, Wagner M, Held J, Bruck W, Salinas-Riester G, Hao Z, Waisman A, Kuhlmann T. In toxic demyelination oligodendroglial cell death occurs early and is FAS independent. Neurobiol Dis. 2010;37(2):362–9.
Plemel JR, Caprariello AV, Keough MB, Henry TJ, Tsutsui S, Chu TH, Schenk GJ, Klaver R, Yong VW, Stys PK. Unique spectral signatures of the nucleic acid dye acridine orange can distinguish cell death by apoptosis and necroptosis. J Cell Biol. 2017;216(4):1163–81.
Connor JR, Menzies SL, Burdo JR, Boyer PJ. Iron and iron management proteins in neurobiology. Pediatr Neurol. 2001;25(2):118–29.
Tien M, Svingen BA, Aust SD. Superoxide dependent lipid peroxidation. Fed Proc. 1981;40(2):179–82.
Benedetti A, Comporti M, Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta. 1980;620(2):281–96.
Yau TM. Mutagenicity and cytotoxicity of malonaldehyde in mammalian cells. Mech Ageing Dev. 1979;11(2):137–44.
Pandur E, Pap R, Varga E, Janosa G, Komoly S, Forizs J, Sipos K. Relationship of iron metabolism and short-term cuprizone treatment of C57BL/6 Mice. Int J Mol Sci. 2019;20(9):2257.
Varga E, Pandur E, Abraham H, Horvath A, Acs P, Komoly S, Miseta A, Sipos K. Cuprizone administration alters the iron metabolism in the mouse model of multiple sclerosis. Cell Mol Neurobiol. 2018;38(5):1081–97.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–9.
Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta. 1985;839(1):62–70.
Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710(2):197–211.
Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics. 2019;19(18):e1800311.
Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165(1):20–34.
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919–30.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–7.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79.
Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–46.
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–7.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–47.
Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437.
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10(11):1836–49.
Ren Y, Su Y, Sun L, He S, Meng L, Liao D, Liu X, Ma Y, Liu C, Li S, et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of Receptor-Interacting Protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J Med Chem. 2017;60(3):972–86.
Zhang S, Su Y, Ying Z, Guo D, Pan C, Guo J, Zou Z, Wang L, Zhang Z, Jiang Z, et al. RIP1 kinase inhibitor halts the progression of an immune-induced demyelination disease at the stage of monocyte elevation. Proc Natl Acad Sci U S A. 2019;116(12):5675–80.
Xie Y, Chen H, Luo D, Yang X, Yao J, Zhang C, Lv L, Guo Z, Deng C, Li Y, et al. Inhibiting necroptosis of spermatogonial stem cell as a novel strategy for male fertility preservation. Stem Cells Dev. 2020;29(8):475–87.
Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1–2):228–43.
Luo F, Herrup K, Qi X, Yang Y. Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis. Exp Neurol. 2017;292:21–34.
Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–9.
Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14(6):1086–94.
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14(2):193–204.
Guo X, Sesaki H, Qi X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem J. 2014;461(1):137–46.
Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet. 2013;22(22):4545–61.
Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology. 2014;86:103–15.
Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. 2016;196(1):407–15.
Moujalled DM, Cook WD, Murphy JM, Vaux DL. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014;5:e1086.
Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5(4):878–85.
Prochnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res 2016, 5.
Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–86.
Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015;10:395–424.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–87.
Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: two cytokine substrates for ICE (caspase-1). J Clin Immunol. 1999;19(1):1–11.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–98.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–6.
Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, Holley CL, Boucher D, Shakespear MR, Kapetanovic R, et al. Interleukin-1beta maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 2018;24(6):1425–33.
Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48(1):35-44 e36.
Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, Suda T. Gasdermin D mediates the maturation and release of IL-1alpha downstream of inflammasomes. Cell Rep. 2021;34(12):108887.
Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Le Manh T, Hori O, Mahib MR, Yamaguchi Y, Miura M, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10(1):2091.
McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, Lu JQ, Branton WG, Power C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. 2018;115(26):E6065–74.
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600.
Liu Y, Fan H, Li X, Liu J, Qu X, Wu X, Liu M, Liu Z, Yao R. Trpv4 regulates Nlrp3 inflammasome via SIRT1/PGC-1alpha pathway in a cuprizone-induced mouse model of demyelination. Exp Neurol. 2021;337:113593.
Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351–70.
Saito LB, Fernandes JP, Smith MJ, Doan MAL, Branton WG, Schmitt LM, Wuest M, Monaco MC, Major EO, Wuest F, et al. Intranasal anti-caspase-1 therapy preserves myelin and glucose metabolism in a model of progressive multiple sclerosis. Glia. 2021;69(1):216–29.
Acknowledgements
The authors thank Brady Hammond for critically reviewing this manuscript. Figures: Created with BioRender.com
Funding
P.A. was supported by a Marie Skłodowska-Curie Individual Fellowship (MSCA-IF) and is currently supported by a Banting Postdoctoral Fellowship (Canadian Institute for Health Research; CIHR). This work was funded by operating grants held by JRP from CIHR, NSERC, and the University of Alberta. This project has been made possible by Brain Canada Foundation through the Canada Brain Research fund, with financial support of Health Canada and the Azrieli Foundation.
Author information
Authors and Affiliations
Contributions
MZ prepared the manuscript with contributions from PA, AS, AVC and JRP. MZ designed the figures with contributions from PA. PA, AVC and JRP reviewed the manuscript. All authors approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zirngibl, M., Assinck, P., Sizov, A. et al. Oligodendrocyte death and myelin loss in the cuprizone model: an updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol Neurodegeneration 17, 34 (2022). https://doi.org/10.1186/s13024-022-00538-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00538-8