
Abstract
Glutamate is the most abundant free amino acid in the brain and is at the crossroad between multiple metabolic pathways. Considering this, it was a surprise to discover that glutamate has excitatory effects on nerve cells, and that it can excite cells to their death in a process now referred to as “excitotoxicity”. This effect is due to glutamate receptors present on the surface of brain cells. Powerful uptake systems (glutamate transporters) prevent excessive activation of these receptors by continuously removing glutamate from the extracellular fluid in the brain. Further, the blood–brain barrier shields the brain from glutamate in the blood. The highest concentrations of glutamate are found in synaptic vesicles in nerve terminals from where it can be released by exocytosis. In fact, glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. It took, however, a long time to realize that. The present review provides a brief historical description, gives a short overview of glutamate as a transmitter in the healthy brain, and comments on the so-called glutamate–glutamine cycle. The glutamate transporters responsible for the glutamate removal are described in some detail.
Similar content being viewed by others

Introduction
Outside the community of biomedical scientists, glutamate is probably best known as “monosodium glutamate” or “MSG” which is the sodium salt of glutamic acid and a white crystalline solid used as a flavor or taste enhancer in food (food additive number E620). This, however, is not the reason for the enormous scientific interest in glutamate. The main motivation for the ongoing worldwide research on glutamate is that glutamate is the major excitatory transmitter in the brain.
Like other signaling substances, the signaling effect of glutamate is not dependent on the chemical nature of glutamate, but on how cells are programmed to respond when exposed to it. Because the glutamate receptor proteins are expressed on the surface of the cells in such a way that they can only be activated from the outside, it follows that glutamate exerts its neurotransmitter function from the extracellular fluid. Consequently, control of receptor activation is achieved by releasing glutamate to the extracellular fluid and then removing glutamate from it. Because there are no enzymes extracellularly that can degrade glutamate, low extracellular concentrations require cellular uptake. This uptake is catalyzed by a family of transporter proteins located at the cell surface of both astrocytes and neurons (e.g. Danbolt 2001; Grewer and Rauen 2005; Tzingounis and Wadiche 2007; Vandenberg and Ryan 2013).
Because glutamate is the major mediator of excitatory signals as well as of nervous system plasticity, including cell elimination, it follows that glutamate should be present at the right concentrations in the right places at the right time. It further follows that cells should have the correct sensitivity to glutamate and have energy enough to withstand normal stimulation, and that glutamate should be removed with the appropriate rates from the right locations. Both too much glutamate and too little glutamate are harmful. Excessive activation of glutamate receptors may excite nerve cells to their death in a process now referred to as “excitotoxicity”. This toxicity was initially perceived as a paradox like “Dr. Jekyll and Mr. Hyde”, but it is now clear that glutamate is toxic, not in spite of its importance, but because of it. As outlined before (Danbolt 2001), the intensity of glutamatergic stimulation that a given cell can tolerate, depends on several factors. As long as one variable is not extreme, it will be the combination of several factors that will determine the outcome.
It took a long time to realize that glutamate is a neurotransmitter in part because of its abundance in brain tissue and in part because it is at the crossroad of multiple metabolic pathways (e.g. Erecinska and Silver 1990; Broman et al. 2000; McKenna 2007; Hertz 2013). There is 5–15 mmol glutamate per kg brain tissue, depending on the region, more than that of any other amino acid (Schousboe 1981). So although it was noted early on that glutamate plays a central metabolic role in the brain (Krebs 1935), that brain cells have a very high glutamate uptake activity (Stern et al. 1949) and that glutamate has an excitatory effect (Hayashi 1954; Curtis et al. 1959, 1960), the transmitter role was not realized until the early 1980s (for review see Fonnum 1984).
In fact, glutamate metabolism is complex and compartmentalized (Berl et al. 1961, 1962; Van den Berg and Garfinkel 1971; Balcar and Johnston 1975). The important role of glutamate uptake in the control of the excitatory action of glutamate was recognized (Logan and Snyder 1971, 1972; Wofsey et al. 1971; Balcar and Johnston 1972). This became a hot research topic. A number of different glutamate and aspartate analogues were synthesized, and heterogeneity within glutamate uptake was uncovered suggesting more than one uptake mechanism (Ferkany and Coyle 1986; Robinson et al. 1991, 1993; Fletcher and Johnston 1991; Balcar and Li 1992; Rauen et al. 1992).
Similarly, several families of glutamate receptor proteins were identified with molecular cloning (for review see Niswender and Conn 2010; Traynelis et al. 2010; Nicoletti et al. 2011). The receptors were classified as N-methyl-d-aspartate (NMDA) receptors (Gonda 2012; Bonaccorso et al. 2011; Santangelo et al. 2012), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors (Rogawski 2013), kainate receptors (Lerma and Marques 2013) and metabotropic receptors (Gregory et al. 2013). Most, if not all, cells in the nervous system express at least one type of glutamate receptor (Steinhauser and Gallo 1996; Vernadakis 1996; Forsythe and Barnes-Davies 1997; Wenthold and Roche 1998; Petralia et al. 1999; Conti et al. 1999; Shelton and McCarthy 1999; Bergles et al. 2000). The locations and functional properties of each type are beyond the scope of this review.
Medicinal chemists continued to synthesize new compounds and it is now possible to differentiate pretty well between the various receptors and transporters. Considering the relatively large number of proteins with ability to bind glutamate, it may seem strange that it is possible to find compounds that can distinguish between them. The reason is the high flexibility of the glutamate molecule which permits several conformations that are only minimally less favorable energetically at body temperature than the lowest energy conformation (Bridges et al. 1991). This implies that glutamate can take many shapes and explains, in part, why the various glutamate binding proteins (transporters, receptors, enzymes) can have quite different binding sites and still be able to bind glutamate. A large number of compounds are now available, and there are a number of excellent reviews on the topic (e.g. Bräuner-Osborne et al. 1997; Jensen and Bräuner-Osborne 2004; Shigeri et al. 2004; Ritzen et al. 2005; Thompson et al. 2005; Bridges and Esslinger 2005; Shimamoto 2008; Bridges et al. 2012a, b; Gregory et al. 2013; Gonda 2012; Bonaccorso et al. 2011).
Identification of plasma membrane glutamate transporters
A glutamate transporter, now known as EAAT2 (GLT-1; slc1a2; Pines et al. 1992), was purified in an active form from rat brain by employing reconstitution of transport as the assay to monitor the purification process (Danbolt et al. 1990). The purification was based on solubilization of rat brain membranes with a detergent and fractionation by conventional chromatographic techniques. This resulted in a 30-fold increase in specific activity, but due to inactivation, the purification ratio was closer to 100-fold. It was hard to convince ourselves that this moderate enrichment was sufficient to yield a pure preparation, and it was even harder to convince others. The fact that the protein tends to give wide bands in electrophoresis gels did not make the task any easier (see Danbolt 1994). Nevertheless, this was a pure preparation (Levy et al. 1993; Lehre and Danbolt 1998). Antibodies were raised to the purified protein and used to localize it in the brain (Danbolt et al. 1992; Levy et al. 1993) and to screen expression libraries. The sequence of the isolated cDNA predicted correctly a protein of 573 amino acids (Pines et al. 1992). Simultaneously, but independently of each other, three other research teams succeeded in cloning another two glutamate transporters using completely different approaches. Storck et al. (1992) were purifying a galactosyltransferase from rat brain and observed that a 66 kDa hydrophobic glycoprotein copurified with this protein. The purified protein was subjected to limited proteolysis. Partial amino acid sequences were obtained and used for synthesizing degenerate oligonucleotide probes for screening of a rat brain cDNA library. This resulted in the identification of 543 amino acid residues long protein now referred to as EAAT1 (GLAST; slc1a3; Storck et al. 1992). EAAT3 (EAAC1; slc1a1) was isolated from a rabbit jejunum by Xenopus laevis oocyte expression cloning (Kanai and Hediger 1992). The cDNA sequence contains an open reading frame coding for a protein of 524 amino acids. The rat brain equivalent is 89.9 % identical and 523 amino acids long (Kanai et al. 1993; Bjørås et al. 1996). The three human counterparts were quickly identified and named excitatory amino acid transporter (EAAT)1–3 (Arriza et al. 1994). Another two glutamate transporters were found later: EAAT4 (Fairman et al. 1995) and EAAT5 (Arriza et al. 1997). All the EAATs catalyze coupled transport of 1H+, 3Na+, and 1K+ with one substrate molecule (Klöckner et al. 1993; Zerangue and Kavanaugh 1996a; Levy et al. 1998; Owe et al. 2006). l-Glutamate and dl-aspartate are transported with similar affinities while d-glutamate is not. It is important to note that the transporters are performing exchange in addition to net uptake. Exchange is a process whereby the transporters exchange external and internal substrate molecules in a 1:1 relationship (see Fig. 5 in Danbolt 2001). Thus, when transportable uptake inhibitors are added to cell cultures, the inhibitors induce glutamate release from the cells (e.g. Volterra et al. 1996; Danbolt 2001) Table 1.
The substrate selectivities are not reviewed here. We will only point out (a) that the commonly used uptake inhibitor dihydrokainate (DHK; CAS 52497-36-6) blocks EAAT2 with high selectivity over the other EAATs (Arriza et al. 1994; Bridges et al. 1999), and (b) that dl-threo-β-benzyloxyaspartate (TBOA; CAS 205309-81-5) and its variants (e.g. PMB-TBOA and TFB-TBOA) block all the five EAATs (Bridges et al. 1999; Shimamoto 2008). These compounds are competitive inhibitors that are not transportable. This implies that they block both uptake and exchange (for a detailed explanation, see sect. 6.5 in Danbolt 2001). For more information, we recommend the outstanding review by Bridges et al. (1999) as an introduction and more recent reviews for the last updates (e.g. Jensen and Bräuner-Osborne 2004; Shigeri et al. 2004; Bridges and Esslinger 2005; Shimamoto and Shigeri 2006; Shimamoto 2008; Sagot et al. 2008).
The EAAT-type of transporters also functions as chloride channels (Fairman et al. 1995; Zerangue and Kavanaugh 1996a; Wadiche et al. 1995a, b; Ryan and Mindell 2007; Takayasu et al. 2009). EAAT4 and EAAT5 have the largest chloride conductance (Mim et al. 2005; Gameiro et al. 2011), and may function more as inhibitory glutamate receptors than as transporters (Dehnes et al. 1998; Veruki et al. 2006; Schneider et al. 2014). Arachidonic acid elicits a substrate-gated proton current associated with the glutamate transporter EAAT4 (Fairman et al. 1998; Tzingounis et al. 1998). In addition, a general feature of sodium coupled transport appears to be transport of water (MacAulay et al. 2001, 2004).
Even though the mammalian transporters have not yet been crystallized, we already know quite a lot about their complex structure (Kanner 2007; Gouaux 2009; Kanner 2013; Vandenberg and Ryan 2013). The EAAT2 and EAAT3 proteins are believed to be homotrimers where the subunits are non-covalently connected (Haugeto et al. 1996). This is in agreement with studies of glutamate transporters from Bacillus Caldotenax and Bacillus stearothermophilus (Yernool et al. 2003) although crosslinking studies of the mammalian transporters indicate that there may be differences between the EAAT subtypes (Dehnes et al. 1998). These proteins are integral membrane proteins and they depend on the lipid environment, and are influenced by fatty acids such as arachidonic acid (Barbour et al. 1989; Trotti et al. 1995; Zerangue et al. 1995) and by oxidation (Trotti et al. 1996; Trotti et al. 1998). The recent determination of the crystall structure of a glutamate transporter homologue (GltPh) from Pyrococcus horikoshii (Yernool et al. 2004) and other transporters (Penmatsa and Gouaux 2013) implies a milestone similar to the cloning of the first transporters in the early 1990s and the generation of knockout mice in the late 1990s. GltPh appear to be a bowl-shaped trimer with a solvent-filled extracellular basin extending halfway across the membrane bilayer. At the bottom of the basin are three independent binding sites (Yernool et al. 2004). This structure is, as uncovered recently, ideal to facilitate rapid transport (Leary et al. 2011).
The glutamate-cystine exchanger
Another transporter that has got quite a lot of attention lately is the so called glutamine-cystine exchanger (xCT; slc7a11). This transporter was first described in human fibroblasts as an electroneutral 1:1 cystine-glutamate exchanger that carries cystine into the cell in exchange for internal glutamate (Bannai 1986). Thus, the physiological role of this transporter is to act as a cystine transporter that uses the transmembrane gradient of glutamate as driving force. It follows from this that extracellular glutamate inhibits uptake of cystine and that uptake of cystine causes glutamate release. The transporter responsible for this uptake has been identified by molecular cloning (Sato et al. 1999). It is a heterooligomer consisting of two different subunits: the 4F2hc surface antigen (slc3a2) the xCT protein (slc7a11). The substrate selectivities are excellently reviewed by Bridges et al. (2012a, b).
There are several reasons why xCT has become a hot topic (Conrad and Sato 2012; Lewerenz et al. 2013; Bridges et al. 2012a, b). The first important observation was that glioma express high levels of xCT and low levels of EAATs suggesting that they release glutamate and that glutamate toxicity may be a mechanism facilitating their invasion of normal tissue (e.g. Ye et al. 1999; Sontheimer 2004; Takeuchi et al. 2013). Another reason for the interest is that cystine is a source of cysteine needed for synthesis of glutathione (Dringen 2000). There are, however, a number of transporters that can transport cysteine. These comprise EAAT3 (Zerangue and Kavanaugh 1996b), the two alanine-serine-cysteine transporters (Arriza et al. 1993; Shafqat et al. 1993; Hofmann et al. 1994), ASCT1 (slc1a4) and ASCT2 (slc1a5) as well as several others (Bröer 2008). So if cystine is reduced to cysteine at the cell surface, then cysteine can be taken up independently of xCT. Nevertheless, xCT-deficient mice display redox imbalance suggesting that xCT does play a role in glutathione production (Sato et al. 2005). A third reason for the interest in xCT is that xCT has been suggested to be a major source of extracellular glutamate (Baker et al. 2002). This has been highly controversial, but a recent paper based on the xCT-deficient mice is supporting the idea (De Bundel et al. 2011). There are, however, a number of unresolved issues. The distribution of xCT in the brain has not yet been definitively determined, and available data suggest low levels (Sato et al. 2002). If the above observations are due to direct actions of xCT, then there must be enough xCT molecules present to perform the proposed functions. Thus, both the expression levels and the speed which xCT operates (translocation cycles per second per xCT molecule) are important to determine. As xCT is highly inducible (Sato et al. 2001, 2004), it is should be kept in mind that expression levels may change in stressful situations. Finally, if xCT exchanges glutamate and cystine in a 1:1 relationship, then a massive glutamate release can only be mediated by xCT if there is a similar transport of cystine (or another substrate) in the other direction. In conclusion, more work is needed before we fully understand the roles that xCT plays.
Intracellular glutamate carriers
When glutamate enters the cytoplasm, it may undergo further redistribution to mitochondria or synaptic vesicles (Erecinska and Silver 1990; Nicholls 1993). (A) Mitochondrial glutamate transport: Several of the enzymes for which glutamate is a substrate are located in mitochondria. In agreement, mitochondria possess mechanisms for glutamate translocation. In fact, there are four different carriers: AGC1 (Slc25a12; aralar1; del Arco and Satrustegui 1998), AGC2 (Slc25a13; citrin; aralar2; Kobayashi et al. 1999; Yasuda et al. 2000), GC1 (Slc25a22; Fiermonte et al. 2002) and GC2 (Slc25a18; Fiermonte et al. 2002).
These transporters are very different from the glutamate transporters in the plasma membranes and will not be discussed further here (for review see Palmieri 2013). (B) Glutamate transporters in synaptic vesicles: In glutamatergic nerve terminals, glutamate is carried into synaptic vesicles by means of the so called vesicular glutamate transporters (VGLUTs). These are also very different from those in the plasma membrane (for review see El Mestikawy et al. 2011; Omote et al. 2011) by being independent of sodium and potassium, and by having lower affinity (km around 1 mM). There are three different isoforms: VGLUT1 (Slc17a7; Ni et al. 1994; Bellocchio et al. 1998, 2000; Takamori et al. 2000), VGLUT2 (Slc17a6; DNPI; Aihara et al. 2000) and VGLUT3 (Slc17a8; Takamori et al. 2002).
Release of glutamate
Glutamate is continuously being released to the extracellular fluid, and inhibition of glutamate uptake leads to extracellular buildups of glutamate within seconds (Jabaudon et al. 1999). Although most of the focus has been on synaptic release of glutamate from nerve terminals by exocytosis of synaptic vesicles, this is not the only mechanism able to supply the extracellular fluid with glutamate (Danbolt 2001). In fact, there are several different non-vesicular (non-exocytotic) mechanisms that appear to be important. One is through anion channels (Kimelberg et al. 1990; Kimelberg and Mongin 1998; Wang et al. 2013) and another is via reversed operation of the glutamate transporting proteins at the plasma membrane (e.g. Levi and Raiteri 1993; Longuemare and Swanson 1995; Roettger and Lipton 1996; Jensen et al. 2000; Rossi et al. 2000; Jabaudon et al. 2000; Sontheimer 2008). A third is via xCT as explained above. A fourth mechanism that has been vividly debated over the last decade is whether mature brain astrocytes in situ also have the ability to release glutamate by exocytosis (Bezzi et al. 2004). The differences in opinions can to some extent be explained by the use of different model systems. For instance, primary astrocytes in culture differ from mature astrocytes in the brain (Cahoy et al. 2008) so observations from cultures are not necessarily valid for the intact living brain. Nevertheless, it seems likely that also mature astrocytes in situ may release glutamate (Malarkey and Parpura 2008; Nedergaard and Verkhratsky 2012; Wang et al. 2013), but exocytosis of vesicles similar to those in nerve terminals is questionable (Hamilton and Attwell 2010). In fact, a recent paper refutes the notion that astrocytes express vesicular glutamate transporters (Li et al. 2013). Thus, this does not entirely rule out the concept of gliotransmitters because glutamate may be released via other mechanisms as explained above, but it does suggest critical evaluation of the literature.
Regulation of the EAAT-type of transporters
Considering the importance of the glutamate transporters, pharmacological manipulation of transporter function may prove to be highly interesting from a therapeutic point of view (Sheldon and Robinson 2007). Although there are several examples where dysregulation of transporters contributes to the pathogenetic process, there are few examples of transporters being the primary cause (e.g. Danbolt 2001; Sattler and Rothstein 2006; Lauriat and Mcinnes 2007; Bröer and Palacin 2011). For instance, it is clear that complete absence of EAAT2 results in spontaneous epilepsy (Tanaka et al. 1997) and increased extracellular glutamate (Mitani and Tanaka 2003; Takasaki et al. 2008), but studies of humans with epilepsy have not uncovered any direct link to glutamate transporter expression (Tessler et al. 1999; Akbar et al. 1997; Bjørnsen et al. 2007). Nevertheless, studies from knockout mice and from humans with mutated transporters show links to disease (for a recent short update see Zhou and Danbolt 2013). Consequently, uncovering regulatory mechanisms is something that has been a hot topic and has interested a large number of researchers. A full account is beyond the scope of this review. Here we only mention a few points.
The first observation revealing regulation of glial glutamate transporter expression came from lesion experiments (Levy et al. 1995). Unilateral ablation of the neocortex in young adult rats resulted in ipsilateral down regulation of EAAT1 and EAAT2 in the striatum. The lesions did not penetrate the corpus callosum so striatum was not directly affected. However, the neocortical lesion eliminated the cell bodies that are responsible for the corticostriatal axons resulting in a loss of glutamatergic terminals in the striatum. Because astrocytes reduced their levels of EAAT1 and EAAT2 in response to the removal of these terminals, it was assumed that neuro-glia interactions were important in the regulation of transporter expression (Levy et al. 1995). This was followed up in cell cultures. Astrocytes cultured in the absence of neurons hardly expressed EAAT2 at all, while addition of neuron conditioned medium turned on EAAT2 expression (e.g., Gegelashvili et al. 1996, 1997, 2000, 2001; Plachez et al. 2000). This regulation turned out to be via several different pathways. Further, glutamate transporters are regulated by protein kinase C (Casado et al. 1993; reviewed by: Gonzalez and Robinson 2004; Vandenberg and Ryan 2013), by zinc (Vandenberg et al. 1998; Mitrovic et al. 2001; Vandenberg and Ryan 2013), and by arachidonic acid as mentioned above. In fact, there is regulation on more or less all levels from transcription to posttranslational modification and trafficking (for review see Seal and Amara 1999; Bergles et al. 1999; Hediger 1999; Kullmann 1999; Sims and Robinson 1999; Danbolt 2001; Robinson 2006; Sattler and Rothstein 2006). The most exciting discovery so far from a drug development point of view is the finding that beta-lactam antibiotics, e.g. Ceftriaxone, increase EAAT2 expression (Rothstein et al. 2005; Berry et al. 2013). Another team has also started high-throughput screening in order to identify translational activators of glial glutamate transporter EAAT2 (Colton et al. 2010) and have identified some pyridazine derivatives that may serve as lead compounds for drug development (Xing et al. 2011). Another interesting finding is a spider toxin that enhances EAAT2 transport activity (Fontana et al. 2007), but the compound responsible has not yet been identified.
Approaches used to localize glutamate transporters
Early attempt to localize glutamate uptake sites were done using autoradiography in combination with tissue slices or synaptosome preparations (e.g. Minchin and Beart 1975; McLennan 1976; Beart 1976; Storm-Mathisen 1981; Storm-Mathisen and Wold 1981). To obtain higher resolution, thinner sections were needed. By using dry mount autoradiography (Young and Kuhar 1979; Danbolt et al. 1993) in combination with “sodium-dependent binding” of excitatory amino acids the uptake sites (for references see Danbolt 1994), higher resolution seemed to be within reach. However, heteroexchange complicated the interpretations as the amount of retained radioactively labeled ligand was dependent on both the number of transporter molecules and by the amount of endogenous dicarboxylic amino acid trapped within the membranes (Danbolt and Storm-Mathisen 1986a, b; Danbolt 1994).
From the early days of glutamate research, it was believed that glutamate is taken up by glutamatergic nerve terminals (Fonnum 1984), but the finding that glial glutamate transporters are down-regulated after glutamatergic denervation (Levy et al. 1995), weakened the evidence (for a discussion, see sect. 4.2 in Danbolt 2001). By incubating tissue slices in d-aspartate and fixing the slices, it was possible to detect fixed d-aspartate with antibodies. With this technique, uptake in both astrocytes and nerve terminals was demonstrated at the electron microscopic level (Gundersen et al. 1993). d-aspartate is often used instead of l-glutamate as a probe for glutamate uptake because it is slowly metabolized in brain tissue (Davies and Johnston 1976).
After the protein sequences of the transporters were known, synthetic peptides could be used to generate antibodies to the transporters themselves (Danbolt et al. 1998) rather than to the substrates. This led to an explosion in the use of antibodies to transporters, but, unfortunately, not all investigators validated their antibodies and procedures well enough (for detailed discussion see Holmseth et al. 2005, 2006, 2012a). The most difficult part is to obtain good negative controls. Antibodies may react with seemingly unrelated proteins (Holmseth et al. 2005; Zhou et al. 2014). In fact, antibody binding can always be achieved (see for instance Fig. 3 in: Holmseth et al. 2005). This is just a question of adjusting the assay conditions. Without a good negative control (e.g. tissue from knockout mice processed in parallel with tissue from wild-type mice), it is not possible to prove that the binding is to the antigen of interest. Therefore, antibody binding does not in itself prove that a given antigen is present. In this context it should be noted that the so called pre-adsorption test can easily give a false impression of specificity (Holmseth et al. 2012a). Whenever possible, it is a good idea to use additional methods, including in situ hybridization and Western blotting in combination with immunocytochemistry. TaqMan Real Time PCR is an excellent method for getting a first approximation of expression levels (e.g. Lehre et al. 2011; Zhou et al. 2012a). Another approach is to search available transcriptome and proteome datasets. For instance, proteome data from rat proximal tubules (http://dir.nhlbi.nih.gov/papers/lkem/pttr/) confirms the presence of EAAT3, but does not confirm expression of any of the other EAATs. Similarly, EAAT2 is in liver, but the other EAATs were not detected (http://141.61.102.16/), and neither the EAATs nor the VGLUTs were detected by proteome analysis of mouse pancreas (Zhou et al. 2014). Together, these data cast doubt over a large number of immunocytochemistry reports. The reason is obvious. Labeling with antibodies can always be obtained, and without good negative controls, it is not possible to tell if the labeling represents the antigen of interest or artifacts (see Holmseth et al. 2012a). Further, rapid post mortem proteolysis represents and additional challenge when studying human samples (Beckstrøm et al. 1999; Tessler et al. 1999; Li et al. 2012). Also note that water soluble proteins present in the samples may inhibit binding of transporters to the blotting membranes (Zhou et al. 2012b). Thus, strong upregulation of other proteins should be considered a potential source of error when estimating transporter levels by immunoblotting. Electron microscopy in combination with pre-embedding immunocytochemistry without detergents on unfrozen tissue is ideal for identification of labeled cell types, but is not ideal for subcellular distribution as the peroxidase reaction product diffuses some distance before precipitating. (Depending on the strength of the reaction, the reaction product may diffuse a couple of hundred nanometers.) In contrast, post-embedding immunogold is better for collecting semi-quantitative data and gives better intracellular resolution, but when cell membranes are labeled and cells are close to each other as they typically are in the brain, then immunogold cannot tell which membrane labeling belongs to (for description of these methods, see Danbolt et al. 1998; Amiry-Moghaddam and Ottersen 2013). Another problem with post-embedding immunogold is that there must be a sufficient number of target molecules in the plane of the section. This is case for EAAT1, EAAT2 and EAAT4. These proteins are present at very high concentrations (Dehnes et al. 1998; Lehre and Danbolt 1998) making them ideal targets for immunogold investigations. This explains, in part, why our early localization studies were so successful (Chaudhry et al. 1995; Dehnes et al. 1998). In contrast, EAAT3 is expressed at lower levels resulting in too few molecules per micrometer plasma membrane length to distinguish real labeling from background noise (Holmseth et al. 2012b). It should be recalled that the tissue sections used for electron microscopy are thin (40–60 nm) and thereby only slightly thicker than the outer diameter of synaptic vesicles (40 nm), and only two/three times thicker than the width of the synaptic cleft (20 nm). The antibodies do not penetrate well into the sections. To maximize labeling, the section may be mounted so that they can be labeled on both sides. Thus, the sensitivity of the post-embedding immunogold technique is limited by the number of proteins in the exact section plane. Another challenge follows from the vulnerability of the sections and thereby also the labeling. These sections are easily damaged during processing. Consequently, there is variability and this leads to another challenge: avoiding sampling error. This challenge comes in addition to those mentioned above (specificity, proteolysis, etc.).
It is also important to consider if any detected proteins are expressed at physiologically relevant levels. The number of molecules needed to accomplish a given task depends on what that task is. This consideration is particularly relevant for neurotransmitter transporters because the transport process is fairly slow. The cycling time of EAAT2 and EAAT3 are in the order of 30 glutamate molecules per second at Vmax (Otis and Jahr 1998; Otis and Kavanaugh 2000; Bergles et al. 2002; Grewer and Rauen 2005) and EAAT5 is even slower (Gameiro et al. 2011). The cycling time of the GABA transporters appear to be comparable to those of the EAATs (Mager et al. 1993; Sacher et al. 2002; Karakossian et al. 2005; Gonzales et al. 2007). This means that the number of transporters must be high. There is a rapid extracellular turnover of glutamate (Jabaudon et al. 1999), and despite this, the resting levels of extracellular glutamate in normal brains are low, possibly as low as 25 nm (Herman and Jahr 2007). Because the km-values are about 1,000 times higher (Danbolt 2001), maintenance of such low extracellular levels implies a vast excess of transporter proteins (Bergles and Jahr 1997; Dehnes et al. 1998; Lehre and Danbolt 1998; Otis and Kavanaugh 2000).
Cellular and subcellular distribution of glutamate transporters in normal mature brain tissue
A large number of papers on transporter distributions have been published, and it is not easy to navigate in the literature as many of the statements are corrected in later publications. Figure 1 is a schematic illustration of the distributions of EAAT1, EAAT2 and EAAT3 in the forebrain.

A schematic illustration of glutamate transporter distributions around synapses close to a blood vessel in the hippocampus. Four glutamatergic nerve terminals (T) are shown forming synapses onto dendritic spines (S). Astrocyte branches are indicated (G). Note that astrocytes have very high densities (Lehre et al. 1995; Ginsberg et al. 1995; Lehre and Danbolt 1998) of both EAAT2 (red dots) and EAAT1 (blue dots). The highest densities of EAAT1 and EAAT2 are in the astrocyte membranes facing neuropil, while the membranes facing the endothelium have low levels. Also note that glutamate transporters have not been detected in the endothelium. EAAT1 is selective for astrocytes (Lehre et al. 1995; Ginsberg et al. 1995), while EAAT2 is predominantly expressed in astrocytes (Danbolt et al. 1992), but there is also some (about 10 %) in hippocampal nerve terminals (Furness et al. 2008). EAAT3 (green dots) is selective for neurons, but is expressed at levels two orders of magnitude lower than EAAT2 and is targeted to dendrites and cell bodies (Holmseth et al. 2012b). Also note that the endfeet may actually overlap with no gaps in between them (Mathiisen et al. 2010) (Copyright: Neurotransporter AS; Reproduced with permission)
EAAT1 (GLAST; slc1a3) is selectively expressed in astrocytes throughout the CNS (Lehre et al. 1995). This conclusion is supported both by in situ hybridization and immunocytochemistry (e.g. Ginsberg et al. 1995; Rothstein et al. 1995 Schmitt et al. 1997; Berger and Hediger 1998, 2000) and appears to be valid for all parts of the central nervous system including the regions where EAAT1 is the predominant transporter (Lehre et al. 1995; Lehre and Danbolt 1998; Takatsuru et al. 2007; Takayasu et al. 2009): the retina (Rauen et al. 1996; Lehre et al. 1997; Rauen et al. 1998; Rauen 2000; Rauen and Wiessner 2000), the inner ear (Furness and Lehre 1997; Takumi et al. 1997), and the circumventricular organs (Berger and Hediger 2000). Thus, there is no disagreement here. Other statements can be found in the literature, but these have been corrected by the authors themselves.
After having determined the cell types expressing EAAT1 (Lehre et al. 1995), immunogold was performed to obtain additional information (Chaudhry et al. 1995). This revealed that EAAT1 is preferentially targeted to the plasma membranes, and that plasma membranes facing neuropil have higher densities than those facing cell bodies, pia mater and endothelium (Fig. 1).
Mice lacking EAAT1 (Watase et al. 1998) develop normally, but show symptoms of insufficient glutamate uptake in regions where EAAT1 is the major glutamate transporter (Watase et al. 1998; Hakuba et al. 2000; Harada et al. 1998). The EAAT1 knockout mice also display poor nesting behavior; abnormal sociability, reduced alcohol intake and reward (Watase et al. 1998; Stoffel et al. 2004; Karlsson et al. 2009, 2012). Lack of GLAST does not lead to spontaneous seizures like those seen in connection with EAAT2-deficiency (Tanaka et al. 1997), but GLAST deficiency increases seizure duration and severity (Watanabe et al. 1999). EAAT1 mutations in humans are linked to episodic ataxia (Bröer and Palacin 2011; Jen et al. 2005; de Vries et al. 2009).
EAAT2 (GLT-1; slc1a2) was the first glutamate transporter to be localized immunocytochemically. In the mature and normal brain it is predominantly expressed in astrocytes (Danbolt et al. 1992; Levy et al. 1993; Rothstein et al. 1994; Lehre et al. 1995). There is no disagreement here either, and this conclusion is supported both by later immunocytochemistry (e.g. Schmitt et al. 1996; Kugler and Schmitt 2003; Berger et al. 2005; Holmseth et al. 2009) and in situ hybridization (Torp et al. 1994, 1997; Berger and Hediger 2000, 2001) as well as by data obtained with EAAT2 eGFP BAC reporter mice (de Vivo et al. 2010a).
EAAT2 is the only one of the EAAT-type of glutamate transporters that is required for survival under non-challenging conditions (Tanaka et al. 1997; Danbolt 2001). This is in agreement with biochemical data showing that the EAAT2 protein represents about 1 % of the total forebrain protein and that it is about four times more abundant than EAAT1 in the hippocampus and six times less abundant than EAAT1 in the cerebellum (Lehre and Danbolt 1998). Based on immunoadsorption of transport activity EAAT2 was shown to account for 95 % of the total glutamate uptake activity in young adult forebrain tissue (Danbolt et al. 1992; Haugeto et al. 1996). This conclusion was confirmed by deletion of the EAAT2 gene in mice (Tanaka et al. 1997; Voutsinos-Porche et al. 2003; Matsugami et al. 2006; Kiryk et al. 2008; Holmseth et al. 2012b) as well as by electrophysiological recordings of glutamate transporter currents (Otis and Kavanaugh 2000).
The discussion about EAAT2 distribution concerns expression in neurons. Having said that, there is consensus that EAAT2 is expressed in cultured neurons from hippocampus and neocortex; in particular if these are cultured in the absence of astrocytes (Mennerick et al. 1998; Wang et al. 1998; Plachez et al. 2000) in agreement with observations that EAAT2 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression (Yamada et al. 1998). There is also consensus that EAAT2 is present in neurons in the normal and mature mammalian retina (Rauen et al. 1996, 1999; Rauen and Kanner 1994; Euler and Wassle 1995; Rauen 2000).
The controversy is related to expression of EAAT2 in neurons in the normal and mature brain (cerebrum and cerebellum). All studies, however, agree that there is EAAT2 mRNA in CA3 hippocampal neurons (Torp et al. 1994, 1997; Berger and Hediger 2000, 2001; de Vivo et al. 2010a) and that their axon-terminals express the protein, at least in the CA1 (Chen et al. 2004; Furness et al. 2008; Melone et al. 2009, 2011). Further, all of the glutamate uptake activity in glutamatergic terminals in CA1 is due to EAAT2 (Furness et al. 2008).
The remaining controversy concerns (a) the expression of EAAT2 in axon-terminals in other parts of the brain, and (b) the physiological importance of the uptake into terminals. Why was about half of all d-aspartate taken up by hippocampus slices found in axon-terminals when terminals only contain around 10 % of the EAAT2 protein (Furness et al. 2008)? This disproportionally large uptake cannot simply be disregarded as an in vitro artifact due to a higher rate of heteroexchange than net uptake (Zhou et al. 2013), but it might still be an artifact because the possibility has not been ruled out that astrocytes release glutamate via anion channels or similar. Preliminary data from selective deletion of EAAT2 in axon-terminals indicate disturbances in synaptic transmission (Sun et al. 2012), and thereby may suggest that EAAT2 in terminals is functionally relevant. However, further studies are required before definite conclusions can be made.
In contrast to EAAT1, there is very little EAAT2 in mice and rats at birth and in the first postnatal week (Ullensvang et al. 1997; Furuta et al. 1997). This explains why EAAT2-knockout mice are inconspicuous at birth. But at 3 weeks, when the EAAT2-levels in wild-type mice have increased to 50 % of adult levels, the EAAT2-deficient mice can readily be identified because they are hyperactive, epileptic and smaller than their wild-type littermates. They have increased extracellular glutamate levels (Mitani and Tanaka 2003; Takasaki et al. 2008), and about half of them die from spontaneous seizures before they reach 4 weeks of age (Tanaka et al. 1997). The heterozygote EAAT2 knockout mice (±) have only half the EAAT2-concentrations as wild-type mice, but do not show any apparent morphological brain abnormalities (Kiryk et al. 2008), but are more vulnerable to traumatic spinal cord injury (Lepore et al. 2011).
EAAT3 (EAAC1; slc1a1) has been particular hard to localize. Nevertheless, the first studies were basically correct (Kanai and Hediger 1992; Rothstein et al. 1994). EAAT3 is a neuronal transporter, and is not expressed in glial cells (Holmseth et al. 2012b; Shashidharan et al. 1997). It appears to be expressed in the majority if not all neurons throughout the CNS, but has a unique sorting motif (Cheng et al. 2002) selectively targeting it to somata and dendrites avoiding axon terminals (Holmseth et al. 2012b; Shashidharan et al. 1997).
The highest levels of EAAT3 in the brain are found in the hippocampus and neocortex, but the total tissue content in young adult rat brains is about 100 times lower than that of EAAT2 (Holmseth et al. 2012b). It is also expressed in the kidney and in the ileum. In agreement, mice lacking EAAT3 (Peghini et al. 1997) develop dicarboxylic aminoaciduria, but do not show signs of neurodegeneration at young age and do not have epilepsy (Peghini et al. 1997; Aoyama et al. 2006; Berman et al. 2011). Humans lacking EAAT3 develop dicarboxylic aminoaciduria (Bailey et al. 2011) and EAAT3 polymorphisms are associated with obsessive–compulsive disorders (Brandl et al. 2012; Walitza et al. 2010).
EAAT4 (slc1a6) is predominantly found in the cerebellar Purkinje cells (Fairman et al. 1995; Dehnes et al. 1998) where it is targeted to the dendrites, the spines in particular (Dehnes et al. 1998), but there is also some EAAT4 in a subset of forebrain neurons (Dehnes et al. 1998; Massie et al. 2008; de Vivo et al. 2010b) and in vestibular hair cells and calyx endings (Dalet et al. 2012). EAAT4 knockout mice are viable and appear normal (Huang et al. 2004) albeit with some alteration of receptor activation (Nikkuni et al. 2007).
EAAT5 (slc1a7) is preferentially expressed in the retina, while the levels in the brain are low (Arriza et al. 1997; Eliasof et al. 1998). EAAT5 is also expressed in vestibular hair cells and calyx endings (Dalet et al. 2012). There is more than one isoform in the retina due to variable splicing (Eliasof et al. 1998). As explained above, EAAT4 and EAAT5 are not very efficient as transporters, but are efficient chloride channels suggesting that they may be more important as inhibitory glutamate receptors than as transporters. Some investigators have tried to determine the exact cellular and subcellular localization of EAAT5, but the validity of these studies is hard to judge at present because nobody has as yet made an EAAT5 knockout mouse that could serve as negative control for validation of the immunolabeling. We have previously shown how important this control is and also how inadequate the so called pre-adsorption test is (Holmseth et al. 2012a). So, validated information on EAAT5 distribution remains to be provided.
Comments on the glutamine-glutamate cycle
Glutamate taken up by astroglial cells can be metabolized via the tricarboxylic acid cycle and be used in protein synthesis or converted to glutamine. Glutamine can be released to the extracellular fluid by a sodium neutral amino transporter in the astrocytic membrane by SNAT3 (Boulland et al. 2002, 2003; Mackenzie and Erickson 2004; Nissen-Meyer et al. 2011) and SNAT5 (SN2; slc38a5) (Hamdani et al. 2012) because it is inactive in the sense that it cannot activate glutamate receptors (for review: Erecinska and Silver 1990; Danbolt 2001; Hertz 2013). The conversion of glutamate to glutamine is catalyzed by the enzyme glutamine synthetase (GLUL) in an ATP-dependent manner (Erecinska and Silver 1990; Marcaggi and Coles 2001). Glutamine synthetase plays important roles in the brain and in other organs from implantation to high age. This is evident from studies of glutamine deficiency in man and mice (He et al. 2007, 2010a, b; Haberle et al. 2011, 2012). Further, reduced glutamine synthetase levels are associated with some forms of epilepsy (Eid et al. 2004).
The prevailing view has been that glutamine from astrocytes is the predominant source of glutamate in glutamatergic terminals (Sibson et al. 2001; Hertz 2013), but this hypothesis implies that the supply of glutamine to terminals keeps up with glutamate release. And although there are many observations in cultured cells suggesting the existence of glutamine transporters in glutamatergic terminals, it is important to keep in mind that cultured astrocytes are different from mature astrocytes (e.g. Plachez et al. 2000; Cahoy et al. 2008). Further, it is important to note that glutamine transporters have so far not been positively identified in terminals in brain tissue (Mackenzie and Erickson 2004; Chaudhry et al. 2002; Conti and Melone 2006). The only positive identifications of SNAT2 (SAT2; slc38a2) and SNAT1 (SAT1; GlnT; slc38a1) are in dendrites and cell bodies of neurons (e.g. Jenstad et al. 2009; Solbu et al. 2010; Conti and Melone 2006). One possibility is that they have evaded detection in glutamatergic terminals due to methodological challenges. Another possibility is that they have not been detected simply because they are not there. This would be in line with studies suggesting that SNAT1 and SNAT2 play no role in delivering glutamine for glutamatergic transmission (Grewal et al. 2009). There could be other glutamine transporters, however. For instance, ASCT2 (slc1a5) has ability to transport glutamine (Bröer et al. 1999), but is expressed at low levels in the mature brain (Utsunomiya-Tate et al. 1996; Bröer and Brookes 2001). There are also other potential candidates within the slc38-family. On the other hand, lack of significant glutamine uptake activities in terminals would be is in line with some old reports (e.g. Hertz et al. 1980; Yu and Hertz 1982; McMahon and Nicholls 1990). Another possibility is whether glutamate may be formed in a glutamine-independent manner (Hassel and Bråthe 2000; McKenna et al. 2000), but this is also debated. A third source is direct uptake by glutamate transporters in terminals themselves (Gundersen et al. 1993). As explained above, there is EAAT2 in terminals and this uptake is highly active (Furness et al. 2008). Another complicating factor is that nerve terminals in different brain regions may differ. While terminals in several forebrain regions (e.g. neocortex, hippocampus and striatum) have been shown to posses glutamate uptake activity (e.g. Gundersen et al. 1993), this is more uncertain in the cerebellar cortex (e.g. Wilkin et al. 1982). In conclusion, the glutamine-glutamate cycle has been studied and debated for about 50 years and we still do not have the final answer!
Glutamate transporters at the blood brain barrier
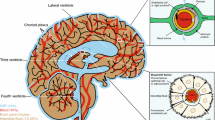
The nervous system isolates itself from blood by means of barriers (e.g. Abbott 2005; Alvarez et al. 2013). This is important for a number of reasons. One of them is the fact that serum glutamate is typically in the range 50–200 μm (Zlotnik et al. 2011a, b, c) which is orders of magnitude higher than the concentrations that are toxic to neurons (Danbolt 2001).
The blood–brain barrier is between blood and the interstitial fluid of the brain. It is in mammals formed by the endothelial cells after influence from brain cells. Another barrier is in the choroid plexus epithelium which secretes cerebrospinal fluid (CSF). These barriers are important both from a physiological point of view because they are essential for brain homeostasis, and from a pharmacological point of view because they prevent drugs from entering brain tissue (Deboer and Gaillard 2007; Teichberg 2007). The literature is extensive and full of conflicting reports. A full account is beyond the scope of this review. Here we only want to point out (Fig. 1) that brain barrier endothelial cells do not express significant levels of EAAT1-3 (Lehre et al. 1995; Berger and Hediger 2000; Holmseth et al. 2009, 2012b). There are, however, huge amounts of glutamate transporters in the astrocytic endfeet surrounding the blood vessels (Fig. 1). When isolating brain microvessels, the preparations are likely to be contaminated by endfeet and this may explain some of the data. Thus, it seems that no significant transport of glutamate can occur through a normal and intact blood–brain barrier. In agreement, injection of radiolabeled glutamate and aspartate does not result in accumulation of radioactivity in the brain (Klin et al. 2010). On the other hand, there is an efflux mechanism for glutamate as blood-mediated scavenging is reported to reduce glutamate in the cerebrospinal fluid (Gottlieb et al. 2003). There is some evidence that this may offer some protection (Zlotnik et al. 2008; Teichberg et al. 2009; Zlotnik et al. 2010; Nagy et al. 2010). The mechanism, however, of release from the brain remains to be identified. This illustrates that brain water homeostasis and transport mechanisms between the blood and the extracellular fluid in brain are incompletely understood. Recent work from Nedergaard and co-workers may represent a leap in our understanding. They introduce the term “glymphatics” (Iliff et al. 2012; Nedergaard 2013) to describe flow of fluid from the arachnoid space along blood vessels into brain tissue. This may reconcile a number of apparently conflicting reports. Perhaps this also will explain why the betaine-GABA transporter (BGT1; slc6a12; Zhou et al. 2012b) and the taurine transporting GABA transporter 2 (GAT2; slc6a13; Zhou et al. 2012a) are expressed in the leptomeninges.
Concluding remarks
As outlined above, substantial progress has been made over the last decades. But there are major gaps in our understanding of key processes. One example is transport of metabolites across the blood brain barrier. Another unknown is the uptake in glutamatergic nerve endings and the relevance of the glutamate-glutamine cycle for transmitter glutamate. A third topic is why the body needs several different glutamate transporters, and how they can be pharmacologically modulated.
Abbreviations
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- l-AP4:
-
l-2-Amino-4-phosphonobutanoate
- ATP:
-
Adenosine triphosphate
- CNS:
-
Central nervous system
- EAAC1:
-
Glutamate transporter number 3 (EAAT3; slc1a1; Kanai and Hediger 1992)
- EAAT:
-
Excitatory amino acid transporter (synonymous to glutamate transporter)
- GABA:
-
γ-Aminobutyric acid
- GLAST:
-
Glutamate transporter number 1 (EAAT1; slc1a3; Storck et al. 1992; Tanaka 1993a)
- GLT-1:
-
Glutamate transporter number 2 (EAAT2; slc1a2; Pines et al. 1992)
- GLUL:
-
Glutamine synthetase
- NMDA:
-
N-Methyl-d-aspartate
- TBOA:
-
dl-threo-β-benzyloxyaspartate
References
Abbott NJ (2005) Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol 25:5–23
Aihara Y, Mashima H, Onda H, Hisano S, Kasuya H, Hori T, Yamada S, Tomura H, Yamada Y, Inoue I, Kojima I, Takeda J (2000) Molecular cloning of a novel brain-type Na+-dependent inorganic phosphate cotransporter. J Neurochem 74:2622–2625
Akbar MT, Torp R, Danbolt NC, Levy LM, Meldrum BS, Ottersen OP (1997) Expression of glial glutamate transporters GLT-1 and GLAST is unchanged in the hippocampus in fully kindled rats. Neuroscience 78:351–359
Alvarez JI, Katayama T, Prat A (2013) Glial influence on the blood brain barrier. Glia 61:1939–1958. doi:10.1002/glia.22575
Amiry-Moghaddam M, Ottersen OP (2013) Immunogold cytochemistry in neuroscience. Nat Neurosci 16:798–804. doi:10.1038/nn.3418
Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA (2006) Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9:119–126. doi:10.1038/nn1609
Arriza JL, Kavanaugh MP, Fairman WA, Wu YN, Murdoch GH, North RA, Amara SG (1993) Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem 268:15329–15332
Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14:5559–5569
Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA 94:4155–4160
Bailey CG, Ryan RM, Thoeng AD, Ng C, King K, Vanslambrouck JM, Auray-Blais C, Vandenberg RJ, Bröer S, Rasko JE, Weinstein N, Hodgins HS, Ryan RM (2011) Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J Clin Invest 121:446–453. doi:10.1172/JCI44474
Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22:9134–9141
Balcar VJ, Johnston GA (1972) The structural specificity of the high affinity uptake of l-glutamate and l-aspartate by rat brain slices. J Neurochem 19:2657–2666
Balcar VJ, Johnston GA (1975) High affinity uptake of l-glutamine in rat brain slices. J Neurochem 24:875–879
Balcar VJ, Li Y (1992) Heterogeneity of high affinity uptake of l-glutamate and l-aspartate in the mammalian central nervous system. Life Sci 51:1467–1478
Bannai S (1986) Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 261:2256–2263
Barbour B, Szatkowski M, Ingledew N, Attwell D (1989) Arachidonic acid induces a prolonged inhibition of glutamate uptake into glial cells. Nature 342:918–920. doi:10.1038/342918a0
Beart PM (1976) The autoradiographic localization of l-[3H] glutamate in synaptosomal preparations. Brain Res 103:350–355
Beckstrøm H, Julsrud L, Haugeto Ø, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC (1999) Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer’s disease. J Neurosci Res 55:218–229
Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH (1998) The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J Neurosci 18:8648–8659
Bellocchio EE, Reimer RJ, Fremeau RT Jr, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957–960
Berger UV, Hediger MA (1998) Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol (Berl) 198:13–30
Berger UV, Hediger MA (2000) Distribution of the glutamate transporters GLAST and GLT-1 in rat circumventricular organs, meninges and dorsal root ganglia. J Comp Neurol 421:385–399
Berger UV, Hediger MA (2001) Differential distribution of the glutamate transporters GLT-1 and GLAST in tanycytes of the third ventricle. J Comp Neurol 433:101–114
Berger UV, Desilva TM, Chen WZ, Rosenberg PA (2005) Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol 492:78–89. doi:10.1002/cne.20737
Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19:1297–1308
Bergles DE, Diamond JS, Jahr CE (1999) Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol 9:293–298
Bergles DE, Roberts JD, Somogyi P, Jahr CE (2000) Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405:187–191. doi:10.1038/35012083
Bergles DE, Tzingounis AV, Jahr CE (2002) Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J Neurosci 22:10153–10162
Berl S, Lajtha A, Waelsch H (1961) Amino acid and protein metabolism. VI. Cerebral compartments of glutamic acid metabolism. J Neurochem 7:186–197
Berl S, Takagaki G, Clarke DD, Waelsch H (1962) Metabolic compartments in vivo: ammonia and glutamic acid metabolism in brain and liver. J Biol Chem 237:2562–2569
Berman AE, Chan WY, Brennan AM, Reyes RC, Adler BL, Suh SW, Kauppinen TM, Edling Y, Swanson RA (2011) N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1(−/−) mouse. Ann Neurol 69:509–520. doi:10.1002/ana.22162
Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A et al (2013) Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One 8:e61177. doi:10.1371/journal.pone.0061177
Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A (2004) Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci 7:613–620
Bjørås M, Gjesdal O, Erickson JD, Torp R, Levy LM, Ottersen OP, Degree M, Storm-Mathisen J, Seeberg E, Danbolt NC (1996) Cloning and expression of a neuronal rat brain glutamate transporter. Brain Res Mol Brain Res 36:163–168
Bjørnsen LP, Eid T, Holmseth S, Danbolt NC, Spencer DD, de Lanerolle NC (2007) Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis 25:319–330
Bonaccorso C, Micale N, Ettari R, Grasso S, Zappala M (2011) Glutamate binding-site ligands of NMDA receptors. Curr Med Chem 18:5483–5506
Boulland JL, Osen KK, Levy LM, Danbolt NC, Edwards RH, Storm-Mathisen J, Chaudhry FA (2002) Cell-specific expression of the glutamine transporter SN1 suggests differences in dependence on the glutamine cycle. Eur J Neurosci 15:1615–1631
Boulland JL, Rafiki A, Levy LM, Storm-Mathisen J, Chaudhry FA (2003) Highly differential expression of SN1, a bidirectional glutamine transporter, in astroglia and endothelium in the developing rat brain. Glia 41:260–275
Brandl EJ, Muller DJ, Richter MA (2012) Pharmacogenetics of obsessive-compulsive disorders. Pharmacogenomics 13:71–81. doi:10.2217/pgs.11.133
Bräuner-Osborne H, Nielsen B, Stensbol TB, Johansen TN, Skjaerbaek N, Krogsgaard-Larsen P (1997) Molecular pharmacology of 4-substituted glutamic acid analogues at ionotropic and metabotropic excitatory amino acid receptors. Eur J Pharmacol 335:R1–R3
Bridges RJ, Esslinger CS (2005) The excitatory amino acid transporters: pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacol Ther 107:271–285
Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR (1991) Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem 34:717–725
Bridges RJ, Kavanaugh MP, Chamberlin AR (1999) A pharmacological review of competitive inhibitors and substrates of high-affinity, sodium-dependent glutamate transport in the central nervous system. Curr Pharm Des 5:363–379
Bridges R, Lutgen V, Lobner D, Baker DA (2012a) Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev 64:780–802. doi:10.1124/pr.110.003889
Bridges RJ, Natale NR, Patel SA (2012b) System xc− cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 165:20–34
Bröer S (2008) Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88:249–286. doi:10.1152/physrev.0 0018.2006
Bröer S, Brookes N (2001) Transfer of glutamine between astrocytes and neurons. J Neurochem 77:705–719
Bröer S, Palacin M (2011) The role of amino acid transporters in inherited and acquired diseases. Biochem J 436:193–211. doi:10.1042/BJ20101912
Bröer A, Brookes N, Ganapathy V, Dimmer KS, Wagner CA, Lang F, Bröer S (1999) The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J Neurochem 73:2184–2194
Broman J, Hassel B, Rinvik E, Ottersen OP (2000) Biochemistry and anatomy of transmitter glutamate. In: Handbook of chemical neuroanatomy, vol 18. Elsevier, Amsterdam, pp 1–44
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. doi:10.1523/JNEUROSCI.4178-07.2008
Casado M, Bendahan A, Zafra F, Danbolt NC, Aragón C, Giménez C, Kanner BI (1993) Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem 268:27313–27317
Chaudhry FA, Lehre KP, Campagne MV, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720
Chaudhry FA, Reimer RJ, Edwards RH (2002) The glutamine commute: take the N line and transfer to the A. J Cell Biol 157:349–355
Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA (2004) The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136–1148. doi:10.1523/JNEUROSCI.1586-03.2004
Cheng C, Glover G, Banker G, Amara SG (2002) A novel sorting motif in the glutamate transporter excitatory amino acid transporter 3 directs its targeting in Madin-Darby canine kidney cells and hippocampal neurons. J Neurosci 22:10643–10652
Colton CK, Kong Q, Lai L, Zhu MX, Seyb KI, Cuny GD, Xian J, Glicksman MA, Lin C-LG (2010) Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen 15:653–662
Conrad M, Sato H (2012) The oxidative stress-inducible cystine/glutamate antiporter, system xc−: cystine supplier and beyond. Amino Acids 42:231–246
Conti F, Melone M (2006) The glutamine commute: lost in the tube? Neurochem Int 48:459–464. doi:10.1016/j.neuint.2005.11.016
Conti F, Barbaresi P, Melone M, Ducati A (1999) Neuronal and glial localization of NR1 and NR2a/b subunits of the NMDA receptor in the human cerebral cortex. Cereb Cortex 9:110–120
Curtis DR, Phillis JW, Watkins JC (1959) Chemical excitation of spinal neurons. Nature 183:611
Curtis DR, Phillis JW, Watkins JC (1960) The chemical excitation of spinal neurons by certain acidic amino acids. J Physiol 150:656–682
Dalet A, Bonsacquet J, Gaboyard-Niay S, Calin-Jageman I, Chidavaenzi RL, Venteo S, Desmadryl G, Goldberg JM, Lysakowski A, Chabbert C (2012) Glutamate transporters EAAT4 and EAAT5 are expressed in vestibular hair cells and calyx endings. PLoS One 7:e46261. doi:10.1371/journal.pone.0046261
Danbolt NC (1994) The high affinity uptake system for excitatory amino acids in the brain. Prog Neurobiol 44:377–396
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Danbolt NC, Storm-Mathisen J (1986a) Na+-dependent “binding” of D-aspartate in brain membranes is largely due to uptake into membrane-bounded saccules. J Neurochem 47:819–824
Danbolt NC, Storm-Mathisen J (1986b) Inhibition by K+ of Na+-dependent D-aspartate uptake into brain membrane saccules. J Neurochem 47:825–830
Danbolt NC, Pines G, Kanner BI (1990) Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry 29:6734–6740
Danbolt NC, Storm-Mathisen J, Kanner BI (1992) An [Na+ + K+]coupled l-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 51:295–310
Danbolt C, Hansen TWR, Øyasaeter S, Storm-Mathisen J, Bratlid D (1993) In vitro binding of [3H]bilirubin to neurons in rat brain sections. Biol Neonate 63:35–39
Danbolt NC, Lehre KP, Dehnes Y, Chaudhry FA, Levy LM (1998) Localization of transporters using transporter-specific antibodies. Methods Enzymol 296:388–407
Davies LP, Johnston GA (1976) Uptake and release of d- and l-aspartate by rat brain slices. J Neurochem 26:1007–1014
De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, Vermoesen K, Bannai S, Sato H, Michotte Y, Smolders I, Massie A (2011) Loss of system x(c)− formula does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci 31:5792–5803
de Vivo L, Melone M, Bucci G, Rothstein JD, Conti F (2010a) Quantitative analysis of EAAT4 promoter activity in neurons and astrocytes of mouse somatic sensory cortex. Neurosci Lett 474:42–45. doi:10.1016/j.neulet.2010.03.003
de Vivo L, Melone M, Rothstein JD, Conti F (2010b) GLT-1 Promoter Activity in Astrocytes and Neurons of Mouse Hippocampus and Somatic Sensory Cortex. Front Neuroanat 3:31. doi:10.3389/neuro.05.031.2009
de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, Haan J, Terwindt GM, Boon EM, Howard BD, Frants RR, Baloh RW, Ferrari MD, Jen JC, van den Maagdenberg AM (2009) Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol 66:97–101. doi:10.1001/archneurol.2008.535
Deboer AG, Gaillard PJ (2007) Drug targeting to the brain. Annu Rev Pharmacol Toxicol 47:323–355
Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate- gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619
del Arco A, Satrustegui J (1998) Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem 273:23327–23334
Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62:649–671
Eid T, Thomas MJ, Spencer DD, Rundenpran E, Lai JCK, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, Delanerolle NC (2004) Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363:28–37
El Mestikawy S, Wallen-Mackenzie A, Fortin GM, Descarries L, Trudeau L-E (2011) From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci 12:204–216
Eliasof S, Arriza JL, Leighton BH, Amara SG, Kavanaugh MP (1998) Localization and function of five glutamate transporters cloned from the salamander retina. Vis Res 38:1443–1454
Erecinska M, Silver IA (1990) Metabolism and role of glutamate in mammalian brain. Prog Neurobiol 35:245–296
Euler T, Wassle H (1995) Immunocytochemical identification of cone bipolar cells in the rat retina. J Comp Neurol 361:461–478. doi:10.1002/cne.903610310
Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375:599–603. doi:10.1038/375599a0
Fairman WA, Sonders MS, Murdoch GH, Amara SG (1998) Arachidonic acid elicits a substrate-gated proton current associated with the glutamate transporter EAAT4. Nat Neurosci 1:105–113. doi:10.1038/355
Ferkany J, Coyle JT (1986) Heterogeneity of sodium-dependent excitatory amino acid uptake mechanisms in rat brain. J Neurosci Res 16:491–503. doi:10.1002/jnr.490160305
Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE (2002) Identification of the mitochondrial glutamate transporter: bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem 277:19289–19294
Fletcher EJ, Johnston GA (1991) Regional heterogeneity of l-glutamate and l-aspartate high-affinity uptake systems in the rat CNS. J Neurochem 57:911–914
Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42:1–11
Fontana AC, Beleboni RO, Wojewodzic MW, Dos SWF, Coutinho-Netto J, Grutle NJ, Watts SD, Danbolt NC, Amara SG (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom. Mol Pharmacol 72:1228–1237. doi:10.1124/mol.107.037127
Forsythe ID, Barnes-Davies M (1997) Synaptic transmission: well-placed modulators. Curr Biol 7:R362–R365
Furness DN, Lehre KP (1997) Immunocytochemical localization of a high-affinity glutamate-aspartate transporter, GLAST, in the rat and guinea-pig cochlea. Eur J Neurosci 9:1961–1969
Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC (2008) A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157:80–94. doi:10.1016/j.neuroscience.2008.08.043
Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17:8363–8375
Gameiro A, Braams S, Rauen T, Grewer C (2011) The discovery of slowness: low-capacity transport and slow anion channel gating by the glutamate transporter EAAT5. Biophys J 100:2623–2632. doi:10.1016/j.bpj.2011.04.034
Gegelashvili G, Civenni G, Racagni G, Danbolt NC, Schousboe I, Schousboe A (1996) Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. Neuroreport 8:261–265
Gegelashvili G, Danbolt NC, Schousboe A (1997) Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem 69:2612–2615
Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A (2000) The high-affinity glutamate transporters GLT1, GLAST and EAAT4 are regulated via different signalling mechanisms. Neurochem Int 37:163–170
Gegelashvili G, Robinson MB, Trotti D, Rauen T (2001) Regulation of glutamate transporters in health and disease. Prog Brain Res 132:267–286. doi:10.1016/S0079-6123(01)32082-4
Ginsberg SD, Martin LJ, Rothstein JD (1995) Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J Neurochem 65:2800–2803
Gonda X (2012) Basic pharmacology of NMDA receptors. Curr Pharm Des 18:1558–1567
Gonzales AL, Lee W, Spencer SR, Oropeza RA, Chapman JV, Ku JY, Eskandari S (2007) Turnover rate of the gamma-aminobutyric acid transporter GAT1. J Membr Biol 220:33–51
Gonzalez MI, Robinson MB (2004) Neurotransmitter transporters: why dance with so many partners? Curr Opin Pharmacol 4:30–35
Gottlieb M, Wang Y, Teichberg VI (2003) Blood-mediated scavenging of cerebrospinal fluid glutamate. J Neurochem 87:119–126
Gouaux E (2009) Review. The molecular logic of sodium-coupled neurotransmitter transporters. Philos Trans R Soc Lond B Biol Sci 364:149–154. doi:10.1098/rstb 2008.0181
Gregory KJ, Noetzel MJ, Niswender CM (2013) Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Prog Mol Biol Transl Sci 115:61–121. doi:10.1016/B978-0-12-394587-7.00002-6
Grewal S, Defamie N, Zhang X, De Gois S, Shawki A, Mackenzie B, Chen C, Varoqui H, Erickson JD (2009) SNAT2 amino acid transporter is regulated by amino acids of the SLC6 gamma-aminobutyric acid transporter subfamily in neocortical neurons and may play no role in delivering glutamine for glutamatergic transmission. J Biol Chem 284:11224–11236
Grewer C, Rauen T (2005) Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203:1–20. doi:10.1007/s00232-004-0731-6
Gundersen V, Danbolt NC, Ottersen OP, Storm-Mathisen J (1993) Demonstration of glutamate/aspartate uptake activity in nerve endings by use of antibodies recognizing exogenous D-aspartate. Neuroscience 57:97–111
Haberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T (2011) Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab 103:89–91
Haberle J, Shahbeck N, Ibrahim K, Schmitt B, Scheer I, O’Gorman R, Chaudhry FA, Ben-Omran T (2012) Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet J Rare Dis 7:48. doi:10.1186/1750-1172-7-48
Hakuba N, Koga K, Gyo K, Usami SI, Tanaka K (2000) Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. J Neurosci 20:8750–8753
Hamdani EH, Gudbrandsen M, Bjorkmo M, Chaudhry FA (2012) The system N transporter SN2 doubles as a transmitter precursor furnisher and a potential regulator of NMDA receptors. Glia 60:1671–1683
Hamilton NB, Attwell D (2010) Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11:227–238. doi:10.1038/nrn2803
Harada T, Harada C, Watanabe M, Inoue Y, Sakagawa T, Nakayama N, Sasaki S, Okuyama S, Watase K, Wada K, Tanaka K (1998) Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc Natl Acad Sci USA 95:4663–4666
Hassel B, Bråthe A (2000) Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci 20:1342–1347
Haugeto Ø, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC (1996) Brain glutamate transporter proteins form homomultimers. J Biol Chem 271:27715–27722
Hayashi T (1954) Effects of sodium glutamate on the nervous system. Keio J Med 3:183–192
He Y, Hakvoort TB, Vermeulen JL, Lamers WH, Van Roon MA (2007) Glutamine synthetase is essential in early mouse embryogenesis. Dev Dyn 236:1865–1875
He Y, Hakvoort TB, Kohler SE, Vermeulen JL, de Waart DR, de Theije C, ten Have GA, van Eijk HM, Kunne C, Labruyere WT, Houten SM, Sokolovic M, Ruijter JM, Deutz NE, Lamers WH (2010a) Glutamine synthetase in muscle is required for glutamine production during fasting and extrahepatic ammonia detoxification. J Biol Chem 285:9516–9524
He Y, Hakvoort TB, Vermeulen JL, Labruyere WT, De Waart DR, Van Der Hel WS, Ruijter JM, Uylings HB, Lamers WH (2010b) Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia 58:741–754
Hediger MA (1999) Glutamate transporters in kidney and brain. Am J Physiol 277:F487–F492
Hediger MA, Clemencon B, Burrier RE, Bruford EA (2013) The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol Aspects Med 34:95–107. doi:10.1016/j.mam.2012.12.009
Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741. doi:10.1523/JNEUROSCI.3009-07.2007
Hertz L (2013) The glutamate-glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (Lausanne) 4:59. doi:10.3389/fendo.2013.00059
Hertz L, Yu A, Svenneby G, Kvamme E, Fosmark H, Schousboe A (1980) Absence of preferential glutamine uptake into neurons–an indication of a net transfer of TCA constituents from nerve endings to astrocytes? Neurosci Lett 16:103–109
Hofmann K, Duker M, Fink T, Lichter P, Stoffel W (1994) Human neutral amino acid transporter ASCT1: structure of the gene (SLC1a4) and localization to chromosome 2p13–p15. Genomics 24:20–26
Holmseth S, Dehnes Y, Bjørnsen LP, Boulland JL, Furness DN, Bergles D, Danbolt NC (2005) Specificity of antibodies: unexpected cross reactivity of antibodies directed against the EAAT3 (EAAC) glutamate transporter. Neuroscience 136:649–660. doi:10.1016/j.neuroscience.2005.07.022
Holmseth S, Lehre KP, Danbolt NC (2006) Specificity controls for immunocytochemistry. Anat Embryol (Berl) 211:257–266. doi:10.1007/s00429-005-0077-6
Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC (2009) The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 162:1055–1071. doi:10.1016/j.neuroscience.2009.03.048
Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC (2012a) The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 32:6000–6013. doi:10.1523/JNEUROSCI.5347-11.2012
Holmseth S, Zhou Y, Follin-Arbelet VV, Lehre KP, Bergles DE, Danbolt NC (2012b) Specificity controls for immunocytochemistry: the antigen pre-adsorption test can lead to inaccurate assessment of antibody specificity. J Histochem Cytochem 60:174–187. doi:10.1369/0022155411434828
Huang YH, Dykeshoberg M, Tanaka K, Rothstein JD, Bergles DE (2004) Climbing fiber activation of EAAT4 transporters and kainate receptors in cerebellar Purkinje cells. J Neurosci 24:103–111
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. doi:10.1126/scitranslmed.3003748
Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, Gerber U (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA 96:8733–8738
Jabaudon D, Scanziani M, Ghwiler BH, Gerber U (2000) Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci USA 97:5610–5615
Jen JC, Wan J, Palos TP, Howard BD, Baloh RW (2005) Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65:529–534. doi:10.1212/01.wnl.0000172638.58172.5a
Jensen AA, Bräuner-Osborne H (2004) Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence- based membrane potential assay. Biochem Pharmacol 67:2115–2127
Jensen JB, Pickering DS, Schousboe A (2000) Depolarization-induced release of [3H]D-aspartate from GABAergic neurons caused by reversal of glutamate transporters. Int J Dev Neurosci 18:309–315
Jenstad M, Quazi AZ, Zilberter M, Haglerod C, Berghuis P, Saddique N, Goiny M, Buntup D, Davanger S, SHaug F-M, Barnes CA, McNaughton BL, Ottersen OP, Storm-Mathisen J, Harkany T, Chaudhry FA (2009) System A transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cereb Cortex 19:1092–1106
Kanai Y, Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360:467–471. doi:10.1038/360467a0
Kanai Y, Smith CP, Hediger MA (1993) The elusive transporters with a high affinity for glutamate. Trends Neurosci 16:365–370
Kanner BI (2007) Gate movements in glutamate transporters. ACS Chem Biol 2:163–166. doi:10.1021/cb700040e
Kanner BI (2013) Substrate-induced rearrangements in glutamate-transporter homologs. Nat Struct Mol Biol 20:1142–1144. doi:10.1038/nsmb.2685
Karakossian MH, Spencer SR, Gomez AQ, Padilla OR, Sacher A, Loo DD, Nelson N, Eskandari S (2005) Novel properties of a mouse gamma- aminobutyric acid transporter (GAT4). J Membr Biol 203:65–82
Karlsson R-M, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A (2009) Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34:1578–1589. doi:10.1038/npp.2008.215
Karlsson R-M, Adermark L, Molander A, Perreau-Lenz S, Singley E, Solomon M, Holmes A, Tanaka K, Lovinger DM, Spanagel R, Heilig M (2012) Reduced alcohol intake and reward associated with impaired endocannabinoid signaling in mice with a deletion of the glutamate transporter GLAST. Neuropharmacology 63:181–189. doi:10.1016/j.neuropharm.2012.01.027
Kimelberg HK, Mongin AA (1998) Swelling-activated release of excitatory amino acids in the brain: relevance for pathophysiology. In: Lang F (ed) Contrib Nephrol. Karger, CH-4009 Basel, Switzerland, pp 240–257
Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA (1990) Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10:1583–1591
Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, Knapska E, Filipkowski RK, Kaczmarek L, Danysz W (2008) Behavioral characterization of GLT1 (±) mice as a model of mild glutamatergic hyperfunction. Neurotox Res 13:19–30
Klin Y, Zlotnik A, Boyko M, Ohayon S, Shapira Y, Teichberg VI (2010) Distribution of radiolabeled l-glutamate and d-aspartate from blood into peripheral tissues in naive rats: significance for brain neuroprotection. Biochem Biophys Res Commun 399:694–698. doi:10.1016/j.bbrc.2010.07.144
Klöckner U, Storck T, Conradt M, Stoffel W (1993) Electrogenic l-glutamate uptake in Xenopus laevisoocytes expressing a cloned rat brain l-glutamate/l-aspartate transporter (GLAST-1). J Biol Chem 268:14594–14596
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T (1999) The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet 22:159–163
Krebs HA (1935) Metabolism of amino acids. IV. Synthesis of glutamine from glutamic acid and ammonia, and the enzymatic hydrolysis of glutamine in animal tissue. Biochem J 29:1951–1969
Kugler P, Schmitt A (2003) Complementary neuronal and glial expression of two high-affinity glutamate transporter GLT1/EAAT2 forms in rat cerebral cortex. Histochem Cell Biol 119:425–435
Kullmann DM (1999) Synaptic and extrasynaptic roles of glutamate in the mammalian hippocampus. Acta Physiol Scand 166:79–83. doi:10.1046/j.1365-201x.1999.00546.x
Lauriat TL, Mcinnes LA (2007) EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry 12:1065–1078. doi:10.1038/sj.mp.4002065
Leary GP, Holley DC, Stone EF, Lyda BR, Kalachev LV, Kavanaugh MP (2011) The central cavity in trimeric glutamate transporters restricts ligand diffusion. Proc Natl Acad Sci USA 108:14980–14985
Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757
Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853
Lehre KP, Davanger S, Danbolt NC (1997) Localization of the glutamate transporter protein GLAST in rat retina. Brain Res 744:129–137
Lehre AC, Rowley NM, Zhou Y, Holmseth S, Guo C, Holen T, Hua R, Laake P, Olofsson AM, Poblete-Naredo I, Rusakov DA, Madsen KK, Clausen RP, Schousboe A, White HS, Danbolt NC (2011) Deletion of the betaine-GABA transporter (BGT1; slc6a12) gene does not affect seizure thresholds of adult mice. Epilepsy Res 95:70–81. doi:10.1016/j.eplepsyres.2011.02.014
Lepore A, O’donnell J, Kim A, Yang E, Tuteja A, Haidet-Phillips A, O’Banion C, Maragakis N (2011) Reduction in expression of the astrocyte glutamate transporter, GLT1, worsens functional and histological outcomes following traumatic spinal cord injury. Glia 59:1996–2005. doi:10.1002/glia.21241
Lerma J, Marques JM (2013) Kainate receptors in health and disease. Neuron 80:292–311. doi:10.1016/j.neuron.2013.09.045
Levi G, Raiteri M (1993) Carrier-mediated release of neurotransmitters. Trends Neurosci 16:415–419
Levy LM, Lehre KP, Rolstad B, Danbolt NC (1993) A monoclonal antibody raised against an [Na+ − K+]coupled l-glutamate transporter purified from rat brain confirms glial cell localization. FEBS Lett 317:79–84
Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC (1995) Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci 7:2036–2041
Levy LM, Warr O, Attwell D (1998) Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci 18:9620–9628
Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P (2013) The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18:522–555. doi:10.1089/ars 2011.4391
Li Y, Zhou Y, Danbolt NC (2012) The rates of postmortem proteolysis of glutamate transporters differ dramatically between cells and between transporter subtypes. J Histochem Cytochem 60:811–821. doi:10.1369/0022155412458589
Li D, Herault K, Silm K, Evrard A, Wojcik S, Oheim M, Herzog E, Ropert N (2013) Lack of evidence for vesicular glutamate transporter expression in mouse astrocytes. J Neurosci 33:4434–4455. doi:10.1523/JNEUROSCI.3667-12.2013
Logan WJ, Snyder SH (1971) Unique high affinity uptake systems for glycine, glutamic and aspartic acids in cetral nervous tissue of the rat. Nature 234:297–299
Logan WJ, Snyder SH (1972) High affinity uptake systems for glycine, glutamic and aspartic acids in synaptosomes of rat central nervous tissues. Brain Res 42:413–431
Longuemare MC, Swanson RA (1995) Excitatory amino acid release from astrocytes during energy failure by reversal of sodium-dependent uptake. J Neurosci Res 40:379–386
MacAulay N, Gether U, Klaerke DA, Zeuthen T (2001) Water transport by the human Na+-coupled glutamate cotransporter expressed in Xenopuso ocytes. J Physiol 530:367–378
MacAulay N, Hamann S, Zeuthen T (2004) Water Transport in the Brain: role of Cotransporters. Neuroscience 129:1031–1044. doi:10.1016/j.neuroscience.2004.06.045
Mackenzie B, Erickson JD (2004) Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch 447:784–795
Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA (1993) Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopuso ocytes. Neuron 10:177–188
Malarkey EB, Parpura V (2008) Mechanisms of glutamate release from astrocytes. Neurochem Int 52:142–154
Marcaggi P, Coles JA (2001) Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol 64:157–183
Massie A, Cnops L, Smolders I, McCullumsmith R, Kooijman R, Kwak S, Arckens L, Michotte Y (2008) High-affinity Na(+)/K(+)-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J Comp Neurol 511:155–172. doi:10.1002/cne.21823
Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP (2010) The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58:1094–1103
Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T, Ogawa M, Obata K, Watanabe M, Hashikawa T, Tanaka K (2006) Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci USA 103:12161–12166. doi:10.1073/pnas.0509144103
McKenna MC (2007) The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 85:3347–3358
McKenna MC, Stevenson JH, Huang X, Hopkins IB (2000) Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem Int 37:229–241
McLennan H (1976) The autoradiographic localization of l-[3H]glutamate in rat brain tissue. Brain Res 115:139–144
McMahon HT, Nicholls DG (1990) Glutamine and aspartate loading of synaptosomes: a reevaluation of effects on calcium-dependent excitatory amino acid release. J Neurochem 54:373–380
Melone M, Bellesi M, Conti F (2009) Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia 57:108–117. doi:10.1002/glia.20744
Melone M, Bellesi M, Ducati A, Iacoangeli M, Conti F (2011) Cellular and synaptic localization of EAAT2a in human cerebral cortex. Front Neuroanat 4:151. doi:10.3389/fnana.2010.00151
Mennerick S, Dhond RP, Benz A, Xu WY, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF (1998) Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci 18:4490–4499
Mim C, Balani P, Rauen T, Grewer C (2005) The glutamate transporter subtypes EAAT4 and EAATs 1-3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J Gen Physiol 126:571–589
Minchin MC, Beart PM (1975) Compartmentation of amino acid metabolism in the rat dorsal root ganglion; a metabolic and autoradiographic study. Brain Res 83:437–449
Mitani A, Tanaka K (2003) Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci 23:7176–7182
Mitrovic AD, Plesko F, Vandenberg RJ (2001) Zn2+inhibits the anion conductance of the glutamate transporter EAAT4. J Biol Chem 276:26071–26076
Nagy D, Knapp L, Marosi M, Farkas T, Kis Z, Vecsei L, Teichberg VI, Toldi J (2010) Effects of blood glutamate scavenging on cortical evoked potentials. Cell Mol Neurobiol 30:1101–1106. doi:10.1007/s10571-010-9542-8
Nedergaard M (2013) Neuroscience. Garbage truck of the brain. Science 340:1529–1530. doi:10.1126/science.1240514
Nedergaard M, Verkhratsky A (2012) Artifact versus reality–how astrocytes contribute to synaptic events. Glia 60:1013–1023. doi:10.1002/glia.22288
Ni B, Rosteck PR Jr, Nadi NS, Paul SM (1994) Cloning and expression of a cDNA encoding a brain-specific Na+-dependent inorganic phosphate cotransporter. Proc Natl Acad Sci USA 91:5607–5611
Nicholls DG (1993) The glutamatergic nerve terminal. Eur J Biochem 212:613–631
Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP (2011) Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60:1017–1041. doi:10.1016/j.neuropharm.2010.10.022
Nikkuni O, Takayasu Y, Iino M, Tanaka K, Ozawa S (2007) Facilitated activation of metabotropic glutamate receptors in cerebellar Purkinje cells in glutamate transporter EAAT4-deficient mice. Neurosci Res 59:296–303
Nissen-Meyer LS, Popescu MC, Hamdani EH, Chaudhry FA (2011) Protein kinase C-mediated phosphorylation of a single serine residue on the rat glial glutamine transporter SN1 governs its membrane trafficking. J Neurosci 31:6565–6575
Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322
Omote H, Miyaji T, Juge N, Moriyama Y (2011) Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport. Biochemistry 50:5558–5565. doi:10.1021/bi200567k
Otis TS, Jahr CE (1998) Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci 18:7099–7110
Otis TS, Kavanaugh MP (2000) Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci 20:2749–2757
Owe SG, Marcaggi P, Attwell D (2006) The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J Physiol 577:591–599. doi:10.1113/jphysiol.2006.116830
Palmieri F (2013) The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med 34:465–484
Peghini P, Janzen J, Stoffel W (1997) Glutamate transporter EAAC-1- deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J 16:3822–3832. doi:10.1093/emboj/16.13.3822
Penmatsa A, Gouaux E (2013) How LeuT shapes our understanding of the mechanisms of sodium-coupled neurotransmitter transporters. J Physiol. doi:10.1113/jphysiol.2013.259051
Petralia RS, Rubio ME, Wenthold RJ (1999) Cellular and subcellular distribution of glutamate receptors. In: Jonas P, Monyer H (eds) Ionotropic glutamate receptors in the CNS. Springer, Berlin, pp 143–171
Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI (1992) Cloning and expression of a rat brain l-glutamate transporter. Nature 360:464–467. doi:10.1038/360464a0
Plachez C, Danbolt NC, Recasens M (2000) Transient expression of the glial glutamate transporters GLAST and GLT in hippocampal neurons in primary culture. J Neurosci Res 59:587–593
Rauen T (2000) Diversity of glutamate transporter expression and function in the mammalian retina. Amino Acids 19:53–62
Rauen T, Kanner BI (1994) Localization of the glutamate transporter GLT-1 in rat and macaque monkey retinae. Neurosci Lett 169:137–140
Rauen T, Wiessner M (2000) Fine tuning of glutamate uptake and degradation in glial cells: common transcriptional regulation of GLAST1 and GS. Neurochem Int 37:179–189
Rauen T, Jeserich G, Danbolt NC, Kanner BI (1992) Comparative analysis of sodium-dependent l-glutamate transport of synaptosomal and astroglial membrane vesicles from mouse cortex. FEBS Lett 312:15–20
Rauen T, Rothstein JD, Wassle H (1996) Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res 286:325–336
Rauen T, Taylor WR, Kuhlbrodt K, Wiessner M (1998) High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res 291:19–31
Rauen T, Fischer F, Wiessner M (1999) Glia-neuron interaction by high-affinity glutamate transporters in neurotransmission. Adv Exp Med Biol 468:81–95
Ritzen A, Mathiesen JM, Thomsen C (2005) Molecular pharmacology and therapeutic prospects of metabotropic glutamate receptor allosteric modulators. Basic Clin Pharmacol Toxicol 97:202–213
Robinson MB (2006) Acute regulation of sodium-dependent glutamate transporters: a focus on constitutive and regulated trafficking. Handb Exp Pharmacol 175:251–275
Robinson MB, Hunter-Ensor M, Sinor J (1991) Pharmacologically distinct sodium-dependent l-[3H]glutamate transport processes in rat brain. Brain Res 544:196–202
Robinson MB, Sinor JD, Dowd LA, Kerwin JF (1993) Subtypes of sodium- dependent high-affinity l-[3H]glutamate transport activity - pharmacologic specificity and regulation by sodium and potassium. J Neurochem 60:167–179
Roettger V, Lipton P (1996) Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience 75:677–685
Rogawski MA (2013) AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol Scand Suppl 197:9–18. doi:10.1111/ane.12099
Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403:316–321. doi:10.1038/35002090
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. doi:10.1002/ana.410380114
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77. doi:10.1038/nature03180
Ryan RM, Mindell JA (2007) The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat Struct Mol Biol 14:365–371. doi:10.1038/nsmb1230
Sacher A, Nelson N, Ogi JT, Wright EM, Loo DD, Eskandari S (2002) Presteady-state and steady-state kinetics and turnover rate of the mouse gamma-aminobutyric acid transporter (MGAT3). J Membr Biol 190:57–73
Sagot E, Jensen AA, Pickering DS, Pu X, Umberti M, Stensbol TB, Nielsen B, Assaf Z, Aboab B, Bolte J, Gefflaut T, Bunch L (2008) Chemo-enzymatic synthesis of (2S,4R)-2-amino-4-(3-(2,2-diphenylethylamino)-3-oxopropyl)pentanedioic acid: a novel selective inhibitor of human excitatory amino acid transporter subtype 2. J Med Chem 51:4085–4092
Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC (2012) Novel NMDA receptor modulators: an update. Expert Opin Ther Pat 22:1337–1352. doi:10.1517/13543776.2012.728587
Sato H, Tamba M, Ishii T, Bannai S (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274:11455–11458
Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang HY, Ishii T, Mann GE, Bannai S (2001) Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem 276:10407–10412
Sato H, Tamba M, Okuno S, Sato K, Keinomasu K, Masu M, Bannai S (2002) Distribution of cystine/glutamate exchange transporter, system x(C)(-), in the mouse brain. J Neurosci 22:8028–8033
Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S (2004) Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 325:109–116
Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T, Takahashi S, Bannai S (2005) Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 280:37423–37429
Sattler R, Rothstein JD (2006) Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol 175:277–303
Schmitt A, Asan E, Puschel B, Jons T, Kugler P (1996) Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 71:989–1004
Schmitt A, Asan E, Puschel B, Kugler P (1997) Cellular and regional distribution of the glutamate transporter GLAST in the CNS of rats: nonradioactive in situ hybridization and comparative immunocytochemistry. J Neurosci 17:1–10
Schneider N, Cordeiro S, Machtens J-P, Braams S, Rauen T, Fahlke C (2014) Functional Properties of the Retinal Glutamate Transporters GLT-1c and EAAT5. J Biol Chem 289:1815–1824. doi:10.1074/jbc.M113.517177
Schousboe A (1981) Transport and metabolism of glutamate and GABA in neurons and glial cells. Int Rev Neurobiol 22:1–45
Seal RP, Amara SG (1999) Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol 39:431–456. doi:10.1146/annurev.pharmtox.39.1.431
Shafqat S, Tamarappoo BK, Kilberg MS, Puranam RS, McNamara JO, Guadanoferraz A, Fremeau RT (1993) Cloning and expression of a novel Na+-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J Biol Chem 268:15351–15355
Shashidharan P, Huntley GW, Murray JM, Buku A, Moran T, Walsh MJ, Morrison JH, Plaitakis A (1997) Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res 773:139–148
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333–355. doi:10.1016/j.neuint.2007.03.012
Shelton MK, McCarthy KD (1999) Mature hippocampal astrocytes exhibit functional metabotropic and ionotropic glutamate receptors in situ. Glia 26:1–11
Shigeri Y, Seal RP, Shimamoto K (2004) Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev 45:250–265
Shimamoto K (2008) Glutamate transporter blockers for elucidation of the function of excitatory neurotransmission systems. Chem Rec 8:182–199. doi:10.1002/tcr.20145
Shimamoto K, Shigeri Y (2006) Elucidation of glutamate transporter functions using selective inhibitors. Cent Nerv Syst Agents Med Chem 6:59–71
Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG (2001) In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during. J Neurochem 76:975–989
Sims KD, Robinson MB (1999) Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol 13:169–197
Solbu TT, Bjorkmo M, Berghuis P, Harkany T, Chaudhry FA (2010) SAT1, A Glutamine Transporter, is Preferentially Expressed in GABAergic Neurons. Front Neuroanat 4:1
Sontheimer H (2004) Ion channels and amino acid transporters support the growth and invasion of primary brain tumors. Mol Neurobiol 29:61–71
Sontheimer H (2008) A role for glutamate in growth and invasion of primary brain tumors. J Neurochem 105:287–295
Steinhauser C, Gallo V (1996) News on glutamate receptors in glial cells. Trends Neurosci 19:339–345
Stern JR, Eggleston LV, Hems R, Krebs HA (1949) Accumulation of glutamic acid in isolated brain tissue. Biochem J 44:410–418
Stoffel W, Korner R, Wachtmann D, Keller BU (2004) Functional analysis of glutamate transmission of GLAST1 and transporters in excitatory synaptic GLAST1/EAAC1 deficient mice. Brain Res Mol Brain Res 128:170–181. doi:10.1016/j.molbrainres.2004.06.026
Storck T, Schulte S, Hofmann K, Stoffel W (1992) Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA 89:10955–10959
Storm-Mathisen J (1981) Autoradiographic and microchemical localization of high affinity glutamate uptake. In: Roberts PJ, Storm-Mathisen J, Johnston GA (eds) Glutamate: transmitter in the central nervous system. Wiley, Chichester, New York, Brisbane, Toronto, pp 89–115
Storm-Mathisen J, Wold JE (1981) In vivo high-affinity uptake and axonal transport of D-[2,3-3H]aspartate in excitatory neurons. Brain Res 230:427–433
Sun Y, Petr GT, Frederick NM, Aoki CJ, Rotenberg A, Dhamne SC, Hameed MQ, Goodrich GS, Armsen W, Rosenberg PA (2012) Cell-type specific expression and function of the glutamate transporter GLT-1 at excitatory synapses probed with conditional deletion. Soc Neurosci Abstr 2012(332):16
Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407:189–194
Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3:798–803
Takasaki C, Okada R, Mitani A, Fukaya M, Yamasaki M, Fujihara Y, Shirakawa T, Tanaka K, Watanabe M (2008) Glutamate transporters regulate lesion-induced plasticity in the developing somatosensory cortex. J Neurosci 28:4995–5006. doi:10.1523/JNEUROSCI.0861-08.2008
Takatsuru Y, Iino M, Tanaka K, Ozawa S (2007) Contribution of glutamate transporter GLT-1 to removal of synaptically released glutamate at climbing fiber-Purkinje cell synapses. Neurosci Lett 420:85–89. doi:10.1016/j.neulet.2007.04.062
Takayasu Y, Iino M, Takatsuru Y, Tanaka K, Ozawa S (2009) Functions of glutamate transporters in cerebellar Purkinje cell synapses. Acta Physiol (Oxf) 197:1–12
Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, Nagatani K, Otani N, Osada H, Uozumi Y, Matsuo H, Nawashiro H (2013) Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 72:33–41 discussion 41
Takumi Y, Matsubara A, Danbolt NC, Laake JH, Storm-Mathisen J, Usami S, Shinkawa H, Ottersen OP (1997) Discrete cellular and subcellular localization of glutamine synthetase and the glutamate transporter GLAST in the rat vestibular end organ. Neuroscience 79:1137–1144
Tanaka K (1993a) Expression cloning of a rat glutamate transporter. Neurosci Res 16:149–153
Tanaka K (1993b) Cloning and expression of a glutamate transporter from mouse brain. Neurosci Lett 159:183–186
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT- 1. Science 276:1699–1702
Teichberg VI (2007) From the liver to the brain across the blood-brain barrier. Proc Natl Acad Sci USA 104:7315–7316. doi:10.1073/pnas.0702450104
Teichberg VI, Cohen-Kashi-Malina K, Cooper I, Zlotnik A (2009) Homeostasis of glutamate in brain fluids: an accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience 158:301–308
Tessler S, Danbolt NC, Faull RLM, Storm-Mathisen J, Emson PC (1999) Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 88:1083–1091
Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM (2005) Inhibitors of the glutamate vesicular transporter (VGLUT). Curr Med Chem 12:2041–2056
Torp R, Danbolt NC, Babaie E, Bjørås M, Seeberg E, Storm-Mathisen J, Ottersen OP (1994) Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci 6:936–942
Torp R, Hoover F, Danbolt NC, Storm-Mathisen J, Ottersen OP (1997) Differential distribution of the glutamate transporters GLT1 and rEAAC1 in rat cerebral cortex and thalamus: an in situ hybridization analysis. Anat Embryol (Berl) 195:317–326
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. doi:10.1124/pr.109.002451
Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, Racagni G, Danbolt NC (1995) Arachidonic acid inhibits a purified and reconstituted glutamate transporter directly from the water phase and not via the phospholipid membrane. J Biol Chem 270:9890–9895
Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A (1996) Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem 271:5976–5979
Trotti D, Danbolt NC, Volterra A (1998) Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci 19:328–334
Tzingounis AV, Wadiche JI (2007) Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8:935–947. doi:10.1038/nrn2274
Tzingounis AV, Lin CL, Rothstein JD, Kavanaugh MP (1998) Arachidonic acid activates a proton current in the rat glutamate transporter EAAT4. J Biol Chem 273:17315–17317
Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1997) Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci 9:1646–1655
Utsunomiya-Tate N, Endou H, Kanai Y (1996) Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem 271:14883–14890
Van den Berg CJ, Garfinkel D (1971) A simulation study of brain compartments—metabolism of glutamate and related substances in mouse brain. Biochem J 123:211–218
Vandenberg RJ, Ryan RM (2013) Mechanisms of glutamate transport. Physiol Rev 93:1621–1657. doi:10.1152/physrev.00007.2013 0007.2013
Vandenberg RJ, Mitrovic AD, Johnston GA (1998) Molecular basis for differential inhibition of glutamate transporter subtypes by zinc ions. Mol Pharmacol 54:189–196
Vernadakis A (1996) Glia-neuron intercommunications and synaptic plasticity. Prog Neurobiol 49:185–214
Veruki ML, Morkve SH, Hartveit E (2006) Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat Neurosci 9:1388–1396. doi:10.1038/nn1793
Volterra A, Bezzi P, Rizzini BL, Trotti D, Ullensvang K, Danbolt NC, Racagni G (1996) The competitive transport inhibitor l-trans-pyrrolidine-2,4- dicarboxylate triggers excitotoxicity in rat cortical neuron- astrocyte co-cultures via glutamate release rather than uptake inhibition. Eur J Neurosci 8:2019–2028
Voutsinos-Porche B, Bonvento G, Tanaka K, Steiner P, Welker E, Chatton JY, Magistretti PJ, Pellerin L (2003) Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 37:275–286
Wadiche JI, Amara SG, Kavanaugh MP (1995a) Ion fluxes associated with excitatory amino acid transport. Neuron 15:721–728
Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP (1995b) Kinetics of a human glutamate transporter. Neuron 14:1019–1027
Walitza S, Wendland JR, Gruenblatt E, Warnke A, Sontag TA, Tucha O, Lange KW (2010) Genetics of early-onset obsessive-compulsive disorder. Eur Child Adolesc Psychiatry 19:227–235. doi:10.1007/s00787-010-0087-7
Wang GJ, Chung HJ, Schnuer J, Pratt K, Zable AC, Kavanaugh MP, Rosenberg PA (1998) High affinity glutamate transport in rat cortical neurons in culture. Mol Pharmacol 53:88–96
Wang F, Smith NA, Xu Q, Goldman S, Peng W, Huang JH, Takano T, Nedergaard M (2013) Photolysis of caged Ca2 + but not receptor-mediated Ca2 + signaling triggers astrocytic glutamate release. J Neurosci 33:17404–17412. doi:10.1523/JNEUROSCI.2178-13.2013
Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K (1999) Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST- deficient mice. Brain Res 845:92–96
Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K (1998) Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10:976–988
Wenthold RJ, Roche KW (1998) The organization and regulation of non- NMDA receptors in neurons. Prog Brain Res 116:133–152
Wilkin GP, Garthwaite J, Balazs R (1982) Putative acidic amino acid transmitters in the cerebellum. II. Electron microscopic localization of transport sites. Brain Res 244:69–80
Wofsey AR, Kuhar MJ, Snyder SH (1971) A unique synaptosomal fraction, which accumulates glutamic and aspartic acids, in brain tissue. Proc Natl Acad Sci USA 68:1102–1106
Xing X, Chang L-C, Kong Q, Colton CK, Lai L, Glicksman MA, Lin C-LG, Cuny GD (2011) Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett 21:5774–5777
Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, Inoue Y (1998) Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J Neurosci 18:5706–5713
Yasuda T, Yamaguchi N, Kobayashi K, Nishi I, Horinouchi H, Jalil MA, Li MX, Ushikai M, Iijima M, Kondo I, Saheki T (2000) Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum Genet 107:537–545
Ye ZC, Rothstein JD, Sontheimer H (1999) Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci 19:10767–10777
Yernool D, Boudker O, Folta-Stogniew E, Gouaux E (2003) Trimeric subunit stoichiometry of the glutamate transporters from Bacillus Caldotenaxand Bacillus stearothermophilus. Biochemistry 42:12981–12988. doi:10.1021/bi030161q
Yernool D, Boudker O, Jin Y, Gouaux E (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431:811–818. doi:10.1038/nature03018
Young WS, Kuhar MJ (1979) A new method for receptor autoradiography: [3H]opioid receptors in rat brain. Brain Res 179:255–270
Yu AC, Hertz L (1982) Uptake of glutamate, GABA, and glutamine into a predominantly GABA-ergic and a predominantly glutamatergic nerve cell population in culture. J Neurosci Res 7:23–35. doi:10.1002/jnr.490070104
Zerangue N, Kavanaugh MP (1996a) Interaction of l-cysteine with a human excitatory amino acid transporter. J Physiol 493:419–423
Zerangue N, Kavanaugh MP (1996b) Flux coupling in a neuronal glutamate transporter. Nature 383:634–637. doi:10.1038/383634a0
Zerangue N, Arriza JL, Amara SG, Kavanaugh MP (1995) Differential modulation of human glutamate transporter subtypes by arachidonic acid. J Biol Chem 270:6433–6435
Zhou Y, Danbolt NC (2013) GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 4:165. doi:10.3389/fendo.2013.00165
Zhou Y, Holmseth S, Guo C, Hassel B, Hofner G, Huitfeldt HS, Wanner KT, Danbolt NC (2012a) Deletion of the gamma-aminobutyric acid transporter 2 (GAT2 and SLC6A13) gene in mice leads to changes in liver and brain taurine contents. J Biol Chem 287:35733–35746. doi:10.1074/jbc.M112.368175
Zhou Y, Holmseth S, Hua R, Lehre AC, Olofsson AM, Poblete-Naredo I, Kempson SA, Danbolt NC (2012b) The betaine-GABA transporter (BGT1, slc6a12) is predominantly expressed in the liver and at lower levels in the kidneys and at the brain surface. Am J Physiol Renal Physiol 302:F316–F328. doi:10.1152/ajprenal.00464.2011
Zhou Y, Wang XY, Tzingounis AV, Danbolt NC, Larsson HP (2013) Modeling of glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. Soc Neurosci Abstr 2013(703):05
Zhou Y, Waanders LF, Holmseth S, Guo C, Berger UV, Li Y, Lehre A-C, Lehre KP, Danbolt NC (2014) Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J Biol Chem 289:1329–1344. doi:10.1074/jbc.M113.529065
Zlotnik A, Gurevich B, Cherniavsky E, Tkachov S, Matuzani-Ruban A, Leon A, Shapira Y, Teichberg VI (2008) The contribution of the blood glutamate scavenging activity of pyruvate to its neuroprotective properties in a rat model of closed head injury. Neurochem Res 33:1044–1050. doi:10.1007/s11064-007-9548-x
Zlotnik A, Klin Y, Kotz R, Dubilet M, Boyko M, Ohayon S, Shapira Y, Teichberg VI (2010) Regulation of blood l-glutamate levels by stress as a possible brain defense mechanism. Exp Neurol 224:465–471. doi:10.1016/j.expneurol.2010.05.009
Zlotnik A, Gruenbaum BF, Mohar B, Kuts R, Gruenbaum SE, Ohayon S, Boyko M, Klin Y, Sheiner E, Shaked G, Shapira Y, Teichberg VI (2011a) The effects of estrogen and progesterone on blood glutamate levels: evidence from changes of blood glutamate levels during the menstrual cycle in women. Biol Reprod 84:581–586. doi:10.1095/biolreprod.110.088120
Zlotnik A, Klin Y, Gruenbaum BF, Gruenbaum SE, Ohayon S, Boyko M, Sheiner E, Aricha-Tamir B, Shapira Y, Teichberg VI (2011b) The activation of beta2-adrenergic receptors in naive rats causes a reduction of blood glutamate levels: relevance to stress and neuroprotection. Neurochem Res 36:732–738. doi:10.1007/s11064-010-0388-8
Zlotnik A, Ohayon S, Gruenbaum BF, Gruenbaum SE, Mohar B, Boyko M, Klin Y, Sheiner E, Shaked G, Shapira Y, Teichberg VI (2011c) Determination of factors affecting glutamate concentrations in the whole blood of healthy human volunteers. J Neurosurg Anesthesiol 23:45–49. doi:10.1097/ANA.0b013e3181f82a8f
Acknowledgments
The authors thank Gunnar Lothe for help with Fig. 1. This work was supported by stimulation funds from the University of Oslo, by the Norwegian Research Council (FUGE II-183727-S10) and by private funds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhou, Y., Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm 121, 799–817 (2014). https://doi.org/10.1007/s00702-014-1180-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-014-1180-8