
Abstract
Background
Oat (Avena sativa L.), a hexaploid crop with unknown genome, has valuable nutritional, medicinal and pharmaceutical uses. However, no suitable RGs (reference genes) for qPCR (quantitative real-time PCR) has been documented for oat yet. Single-copy gene is often selected as RG, which is challengeable or impactable in unexplored polyploids.
Results
In this study, eleven candidate RGs, including four duplicated genes, were selected from oat transcriptome. The stability and the optimal combination of these candidate RGs were assessed in 18 oat samples by using four statistical algorithms including the ΔCt method, geNorm, NormFinder and BestKeeper. The most stable RGs for “all samples”, “shoots and roots of seedlings”, “developing seeds” and “developing endosperms” were EIF4A (Eukaryotic initiation factor 4A-3), UBC21 (Ubiquitin-Conjugating Enzyme 21), EP (Expressed protein) and EIF4A respectively. Among these RGs, UBC21 was a four-copy duplicated gene. The reliability was validated by the expression patterns of four various genes normalized to the most and the least stable RGs in different sample sets.
Conclusions
Results provide a proof of concept that the duplicated RG is feasible for qPCR in polyploids. To our knowledge, this study is the first systematic research on the optimal RGs for accurate qPCR normalization of gene expression in different organs and tissues of oat.
Similar content being viewed by others
Background
Oat (Avena sativa L.) is an allohexaploid (2n = 6 × = 42) cereal crop with estimated 13 Gb genome [1]. With an upswing in food and industrial utilization, oats are now cultivated worldwide and form an important dietary staple on a global scale [2, 3]. As a wholegrain product, rolled oats are a rich source of minerals, starch and lipids, and they are a predominant supply of soluble fiber β-glucan [4, 5]. Particularly, unlike other cereals, most of the lipids in oat seeds are deposited in cells of oat endosperms which also accumulate starch [6, 7]. Due to the rich constituents, oats also possess different pharmacological purposes like antioxidative, immunomodulatory, antidiabetic and anti-cholesterolaemic effects [8, 9]. Additionally, oat plants are more adapted to severe weather compared to other monocot crops, and they comparatively require fewer pesticide and fertilizers than other food cereals [10, 11]. These features boost oat as an eco-friendly crop with valuable nutrition and pharmaceutical applications. Many classic breeding approaches are already underway to explore and improve oats [12]. Moreover, with the combined advances in molecular biological research and omics technologies, there has been an increasing number of oat studies that focuses on specific genes in molecular breeding endeavors [13, 14].
Gene expression analysis is becoming increasingly important for exploring functions of candidate genes in biological research. Because gene expression is mainly regulated at the transcription level, studies of it are often carried out at the level of mRNA. Techniques for measuring gene expressions commonly include Northern blot, in situ hybridization, semiquantitative reverse transcription PCR, reverse transcription-PCR, microarray and RNA-seq. Among them, quantitative real-rime polymerase chain reaction (qPCR) is more commonly used for measuring mRNA levels of specific genes for its specificity, sensitivity, flexibility, scalability, and most importantly its potential for high throughput [15, 16]. The fluorescent reporter molecules are used in qPCR to monitor the amplification production during each cycle of the PCR reaction. The amounts of qPCR products are generally calculated by the relative quantification compared with stably expressed genes, which is the most robust and straightforward method of accurately quantifying subtle changes [17]. Reference gene (RG) is the prerequisite for gene expression normalization in relative quantification analysis. An unsuitable RG in gene expression assays usually leads to confounding results [18]. Therefore, the validity of an RG is critical for generating reliable and accurate qPCR results [19, 20].
Some housekeeping genes, such as glyceraldehyde-3-phosphate dehydrogenase, beta-actin, 18S ribosomal RNA, elongation factor-1 alpha and ubiquitin, are generally selected as RGs [21, 22]. Nevertheless, previous studies pointed out that the commonly used housekeeping genes might not be suitable for all materials under different experimental conditions [23, 24]. Accordingly, an increasing number of studies have been conducted to identify reliable RGs for various plant materials at different developmental stages. Meanwhile, several statistical algorithms such as geNorm [25], NormFinder [26] and BestKeeper [27], have been developed for the evaluation of RGs for qPCR analysis.
To our best knowledge, RG selection and evaluation in oat have not been reported. Especially, as an allohexaploid crop similar to wheat, oat may mainly contain duplicated genes, and each copy of these duplicated genes may not uniformly express in different samples, which makes it complicated to search proper RGs or design optimal primers [28, 29]. Polyploids such as tobacco, potato, rapeseed, camelina and wheat are widely cultivated and economically important. Single-copy genes are usually used as RGs, although they only account for a small proportion in the genomes of polyploids [30, 31]. In fact, it is worth noting that most researches on RG selection in polyploids neither display nor discuss the copy number of candidate RGs [32,33,34,35,36]. However, with the widespread of omics technology, some of these “single-copy” RGs were proven to be duplicated genes. Moreover, gene duplication cannot be simply determined in a polyploid without sequenced genome, such as oat. Therefore, the examination and validation of duplicated RGs are common concerns for researchers who are facing a polyploid with unknown genome. Taken together, it is indispensable to identify and verify appropriate RGs in oat, and it is also worth evaluating duplicated RGs in such genome unknown species.
In this study, eleven candidate RGs with one or more copies were selected from the transcriptome of hexaploid oat seeds. Because of the nutritious seeds [4, 5], the unique oily endosperms [6, 7], and the different roles of shoots and roots play in stress tolerance [10], seven stages of developing seeds and corresponding endosperms, as well as shoots and roots from two-leaf and three-leaf stages were collected as oat samples separately. The qPCR assays of these 18 samples were performed with specific primer pairs for the left ten candidate RGs after the evaluation of primers designed for them. And the expression stabilities were evaluated using four statistical algorithms including the ΔCt method, geNorm, Normfinder and BestKeeper. The comprehensive ranking of the optimal RG for each sample sets was generated by geometric means of four rankings. The expression levels of four various genes in different sample sets were normalized to the most and the least stable RGs for verifying the reliability of the evaluation results. The results of this study present a comprehensive screening of RGs in diverse samples of oat for the first time, and furthermore provide a foundation of accurate gene expression analysis for this crop. Moreover, this study also demonstrates the feasible use of duplicated RGs in hexaploid oat, and an effective system dealing with selection of duplicated RGs in polyploids was also discussed.
Results
Selection of oat candidate reference genes
Due to the absence of genomic sequencing for oat, the exclusive released transcriptome of oat seeds [1] was used as the BLAST database, and the sequences of up to 19 RGs published in previous articles [37,38,39,40] or collected online were used as query data in TBLASTN to search their homologs in oat seeds transcriptome. However, several query sequences were not found with any BLAST hits in that oat transcriptome. Among matched subject transcripts, some of them were too short for qPCR primer designing. Consequently, a total of 11 candidate RGs, namely Protein Phosphatase 2A Subunit A3 (PP2A), Polyubiquitin 10 (UBQ10), Ubiquitin-Conjugating Enzyme 21 (UBC21), Elongation factor 1-Alpha (EF1A), Glyceraldehyde-3-phosphate Dehydrogenase C Subunit 1 (GAPDH1), 18S ribosomal RNA (18S), Heterogeneous nuclear ribonucleoprotein 27C (HNR), Expressed protein (EP), TBC1 domain family member 22A (TBC), Tubulin alpha-6 chain (TUA6) and Eukaryotic initiation factor 4A-3 (EIF4A), were identified as candidates for qPCR primer designing and their sequences were listed in Additional file 1. In hexaploid oat, it was not surprising that duplicated genes in its transcriptome was identified. Among 11 candidate RGs, 18S and GAPDH1 were matched with two copies, while TUA6 and UBC21 had three and four transcripts respectively, and others only had one best BLAST hit (Table 1).
Considering the limited information from one set of oat transcriptome data, the genes with only one BLAST hit were insufficient to be considered as single-copy genes. Potential additional copies of them and their expression differentiation among tissues might affect their validity as RGs. Therefore, the genes with multiple hits were also included in the test of their feasibility as RGs. Clearly, it was impractical to design qPCR primers for duplicate genes which share relatively low similarities (i.e. less than 60%). Thus, the sequence similarities of these duplicated genes and their expression levels were characterized first. Sequence alignment showed that two 18S genes had up to 99.84% similarity in their coding regions, followed by 97% similarity among four UBC21 genes and 83.33% between two GAPDH1 genes, and three TUA6 genes merely displayed approximately 72% similarity. Additionally, the expression level indicated by the RPKM (Reads Per Kilobase per Million mapped reads) value of each transcript from the same homologs could be similar to each other or vary considerably (Table 1). In details, the expression levels of two 18S genes were almost the same at 1,067 and 1,053. Contrary to 18S genes, among four UBC21 genes, two of them showed two times more mRNA accumulation than other two, and the RPKM value of one TUV6 gene was even seven-fold higher than the other two. These results suggested that duplicated genes could be differentially expressed and one transcript of them might not represent them all at least at the expression level, even only in one specific organ. Therefore, primers for qPCR of these duplicated genes were designed in their identical regions.
Primer verification and PCR amplification efficiency
The primer specificity for candidate RGs was verified by both regular PCR and qPCR, and the cDNA of oat shoots from three-leaf stage seedlings was used as templates. Based on agarose gel electrophoresis, the amplification product sizes ranged from 87 bp of EF1A to 288 bp of GAPDH1 (Fig. 1, Table 1). Specific amplicon was amplified by most pairs of primers, apart from those of TUA6 (Fig. 1). Similar conclusion was drawn by the number of peaks in melting curve analysis. Only the melt curve of TUA6 amplicon contained an evident peak noise, which further confirmed the inevitable mispairing of this pair of primers. Meanwhile, other primer pairs produced specific amplificons based on the single peak in their melt curves (Fig. 2). In primers designed for qPCR, over two different mispairing nucleotides on primer can lead to a distinction of two similar sequences [41]. Because there is no other identical region on three TUA6 transcripts as an alternative priming position, TUA6 was excluded in following experiments.

Specificity of primers and amplificon lengths. Specific product lengths of each reference gene were indicated after 1.5% agarose gel electrophoresis. Marker represents Marker DL2000

Melt curves of qPCR amplification of 11 candidate reference genes
The amplification efficiencies of other ten RG primers varied from 92.7% for TBC to 112.4% for EP, all of which were in the reliable section from 90 to 115% [42]. The correlation coefficient values (R2) ranged from 0.993 for EIF4A and UBC21 to 1.000 for HNR, which indicated that these primer pairs were highly specific to their targeted region. Other information including the primer sequences and primer characteristics of candidate RGs were all summarized in Table 1.
Expression stabilities of candidate RGs
To evaluate whether the ten candidate RGs are suitable for qPCR analysis in various organs and tissues of oat, 18 samples were named as four experimental sets: all samples, shoot and root (seedlings), developing seed (seeds) and developing endosperm (endosperms).
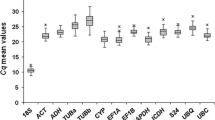
The qPCR results were firstly displayed using Ct values in boxplot analysis (Fig. 3) and then evaluated by algorithms including the ΔCt method, geNorm, NormFinder and BestKeeper (Table 2). Corresponding index values for determining gene expression stability were listed in the brackets of each RG (Table 2). The lower these index values are, the higher the gene expression stabilities are. In all sample set, the Ct values ranged from 7.05 of 18S to 29.23 of TBC (Fig. 3a). The 18S displayed the highest expression and the least variation, whereas GADPH1 showed the least stability (Table 2). The geNorm and NormFinder analyses both exhibited that HNR and EIF4A were the top two stable RGs while 18S was the least. But similar to the results of the ΔCt method, 18S ranked the first in the BestKeeper analysis.

Boxplot analysis of Ct values of ten candidate reference genes in all samples (a), seedlings including shoots and roots (b), developing seeds (c) and developing endosperms (d). The boxes indicates the 25th and 75th percentiles. The line across the box represents the median. The circle in the middle of the box shows the mean value. The whisker shows the maximum and minimum values, respectively. × represents the extremum values
In seedling set, the STDEV values of all candidate RGs were obviously lower than any other sample sets (Fig. 3b; Table 2). The Ct values varied from 7.01 of 18S to 27.76 of TBC. Based on the ΔCt method and the BestKeeper analysis, UBC21 had the most stable expression, followed by GAPDH1 and EP. However, GAPDH1 and 18S were the least stably expressed RGs calculated by geNorm and NormFinder. Furthermore, HNR performed relatively better than most of RGs in all four algorithms.
In seed set, candidate RGs displayed the most variation and the least stability among four experimental sets (Fig. 3c; Table 2), which indicated the complicated regulation network of gene expressions during developing oat seeds. 18S still had the minimum Ct value of 7.13, which indicated its highest expression level among different stages of oat seeds and accorded with the highest RPKM values in oat seed transcriptome (Table 1). GAPDH1 ranked last in the ΔCt method and BestKeeper analysis, and ranked the second last in geNorm and NormFinder, respectively. Additionally, the rankings of candidate RGs in geNorm and NormFinder were exactly the same, among which EF1A and EP were the most two stable RGs.
In endosperm set, the distribution of Ct values was similar to that of oat seeds, but the variations and stabilities were distinctly different between them (Fig. 3d; Table 2). Similar conclusions were drawn by the ΔCt method and the BestKeeper analyses, that 18S and EIF4A were the most reliable RGs while GAPDH1 was the least. In the geNorm analysis, though HNR was the greatest RG, the M value of it was very close to that of PP2A, UBC21 and EIF4A.
Previous research had proven that the conventional use of a single RG for normalization led to relatively large errors in qPCR results [25]. The pairwise variation analysis provided an accurate standard to select the minimum number of RGs by comparing the Vn/Vn + 1 values with 0.15. Once the Vn/Vn + 1 value was lower than 0.15, the top n RGs should be combined as the internal standard gene set. As shown in Fig. 4, the V2/V3 values of four experimental sets were all less than 0.15, which indicated that the two RG combinations were reliable enough for result normalization in them. According to this conclusion, the geometric means of all candidate RGs’ ranking values given by four algorithms were calculated, and the best two RGs for each sample set were listed in Table 3. Meanwhile, the least stable RGs of four sample sets were also displayed. To be precise, EIF4A + HNR was the most stable RG set across all samples and for developing endosperms. The best RG set for seedling samples was UBC21 + HNR, and the optimal RG set for developing seeds was EP + EF1A. However, GAPDH1 was the least recommended RG for both seeds and endosperms, and 18S was particularly unstable for shoots and roots of seedling.

Pairwise variation (Vn/Vn + 1) analysis of the optimal number among ten candidate reference genes in different experimental sets
Validation of candidate reference genes
PKP1 (Plastidial Pyruvate Kinase 1) and AGPL2 (ADP-glucose pyrophosphorylase large subunit 2) play important roles in glycolysis [43] and starch biosynthesis [44], respectively. Homologs of SGT1 [suppressor of the G2 allele of SKP1 (S-phase kinase-associated protein 1)] and SCL14 (Scarecrow-Like 14) in wheat were reported in wheat seedling growth studies [45, 46]. To confirm the reliability of the selected best RG sets after the comprehensive analysis above, the expression levels of AsPKP1 and AsAGPL2 were detected in developing seeds and developing endosperms, while those of AsSGT1 and AsSCL14 were detected in shoots and roots of seedlings, respectively. In the meantime, the RG with the comprehensively lowest stability in each sample set was used for gene expression normalization as a negative control (Table 3; Fig. 5).

Relative expression level of various genes in different sample sets using the most and the least stable reference genes for normalization. a, b Relative expression levels of AsPKP1 (a) and AsAGPL2 (b) in developing seeds. EP+EF1A represents the most stable RG set and GAPDH1 is the least stable RG. Error bars indicate SD of three replicates. The B, C, D, E, G, I and J refer to developing stages of oat seeds [7]. c, d Relative expression levels of AsPKP1 (c) and AsAGPL2 (d) in developing endosperms. EIF4A+HNR represents the most stable RG set and GAPDH1 is the least stable RG. Error bars indicate SD of three replicates. The B, C, D, E, G, I and J refer to developing stages of oat seeds [7]. e, f Relative expression levels of AsSGT1 (e) and AsSCL14 (f) in shoots and roots of seedlings at two-leaf and three-leaf stage. UBC21+HNR represents the most stable RG set and 18S is the least stable RG. Error bars indicate SD of three replicates
In developing oat seeds, it was evident that the expression patterns of AsPKP1 and AsAGPL2 normalized by EP + EF1A, EP and EF1A were mainly similar, and were obviously different from that by GAPDH1 (Fig. 5a, b). When the EP + EF1A set was used for normalization, the relative expression of AsPKP1 was 2.2 times higher at stage C than that of stage B, and increase slightly at stage D subsequently, followed by continuous decrease till stage J. However, almost no difference was shown in the expression trend of GAPDH1 normalization results from stage B to D (Fig. 5a). The expression level of AsGPL2 normalized by EP, EF1A and their combination showed an evident increase from stage B to C, while that normalized by GAPDH1 kept decreasing in same developing period (Fig. 5b). Similar differences were also found in developing endosperms (Fig. 5c, d). When the most stable RG set “EIF4A + HNR” was used for normalization together and separately, the transcript abundance of AsPKP1 dropped steadily during the whole developmental stages of endosperms. Conversely, the relative expression of AsPKP1 showed a sharp increase at stage C compared to that of stage B when GAPDH1 was used for normalization (Fig. 5c). The expression level of AsGPL2 varied much more when normalized by GAPDH1 compared with that normalized by EIF4A and HNR together or separately (Fig. 5d).
In two-leaf and three-leaf stages of oat seedlings, the relative expressions of AsSGT1 and AsSCL14 normalized by optimal UBC21, HNR and their combination showed almost the same levels, which were evidently different from those normalized by the least stable RG 18S (Fig. 5e, f). When UBC21 and HNR were used for normalization, AsSGT1 in seedling roots always showed tiny decreases compared with that in shoots at two different stages. However, the relative expression level of AsSGT1 even displayed twofold more change between shoots and roots at three-leaf stage, when 18S was used as RG (Fig. 5e). Thus, these results confirmed the validation and the reliability of the identified RG sets.
Discussion
qPCR is a powerful tool for analyzing gene expression, with good specificity, high accuracy, great efficiency, and excellent reproducibility. However, numerous studies have shown that the reference genes, such as GAPDH and Actin, which are used to normalize the data in qPCR studies, may not remain stable across all kinds of samples [47, 48]. In this study, RGs for various organs and tissues of oat were selected and evaluated systematically for the first time.
All candidate RGs in this study were frequently used housekeeping genes. Practically, other functional genes with relatively stable expression levels among different samples were also reported as candidate RGs, for example, a Dual Specificity Protein Phosphatase in Setaria viridis [24] and a s-Adenosyl methionine decarboxylase in Eriobotrya japonica [49]. However, the only released oat transcriptome provided data derived from pooled samples of four developmental stages [1], which also limits an exploration for new RGs in silicon.
Qualified RGs are suggested to have a low and consistent copy number in different varieties of the same species [50]. Polyploid crops are commonly cultured in agriculture and they normally have amount of multi-copy genes. In Brassica napus, a typical tetraploid cash crop, only 9.0% of genes own less than 2 copies (one copy: 3.98%; two copies: 5.02%) and up to 71.53% genes even have over six copies according to the genome-wide analysis. To search proper candidate RGs in Brassica napus, only genes with one or two copies were taken into consideration and corresponding qPCR primers were designed in the consistent part of multi-copy genes [31]. In wheat, another hexaploid crop closely related to oat, nearly 90% are present in at least three complete copies generated from the duplication in allopolyploidization [51]. But the criteria for choosing RGs in different wheat varieties or under different conditions remain similar. Basically, any candidate genes with more than two isoforms were excluded and the coincident regions among genes with less than two homologs were used for primer designing [30, 52]. In this study, due to the high estimated percentage of duplicated genes in oat, one to four copy numbers of RGs were all considered as candidates, among which 18S, GAPDH1, TUV6 and UBC21 are duplicated genes (Table 1). The two-copy 18S performed well in three sample sets when evaluated by the ΔCt method (Table 2), and the four-copy UBC21 even ranked as the most stable RG for oat seedlings (Table 3). These results provide a proof of concept that duplicated RG is feasible and valid in polyploid oat. Additionally, though the 72% similarity among three TUV6 copies was not quite low, it was still difficult to target continuous identical regions for qPCR primer designing. Thus, this study also put up that the primer designing for multi-copy RGs should also be based on the sequence similarity and continuity among all transcripts.
Polyploids are universally widespread and have important functionalities. However, the large proportion of duplicated genes in their genome leads to great difficulty of the unigene assembly and a high cost of the genome sequencing [51, 53]. As a typical example of polyploids without any reference genome, oat only has one released transcriptome of its developing seeds [13]. Theoretically, the copy number of assembled transcripts might not accord with that of duplicated genes in genome. In this study, candidate RGs with one to four copies were evaluated together, but the copy numbers for them require validation once the whole genome of oat gets completed. Besides, different copies of duplicated genes usually display diverse tissue specificity or display various expression levels even in the same organ [54, 55]. As shown in Table 1, different transcripts of TUV6 and UBC21 also showed two to seven-fold variation among their RPKM values in oat seed transcriptome. Further considering that these so-called “single-copy” genes were deduced from limited transcriptome data and have not been supported by genome sequences, they might not represent the total expression level of possible duplicated genes. Therefore, selecting duplicated genes and priming in their identical region may help to eliminate the effect of their potential differentiation on expression in different tissues. Meanwhile, to represent the integral expression level of an RG, the primers for qPCR should be designed in identical regions between homologs. In addition, to be clear, the stabilities evaluated in present study specifically referred to primers generated from existing sequences. The applicability of certain RG demands other assessment with more omics data.
As a traditional cereal crop, researches of oat mainly focus on the food science [4]. Scarce molecular biological studies of oat, especially about gene functions, limit the validation of chosen RGs. Among four genes used for validation in this study, only AsPKP1 was analyzed in oat previously [14]. Alternatively, three more genes well-studied in wheat were selected to provide more evidences. The expression patterns of these four genes normalized by the optimal RGs were highly consistent, and showed obvious difference compared to those normalized by the worst RG (Fig. 5). Besides, the expression pattern of AsPKP1 was quite similar to that of PKP1 homologs in other plant seeds, such as Arabidopsis [56]. As a homolog of AsSGT1 in wheat, the expression level of TaSGT1 in seedling shoots is also slightly higher than that in roots [45]. It would be more convincing to compare these expression results with transcriptome data. However, the only transcriptome of mixed developing oat seeds [1] restricts this attempt.
Previous studies on RG selection mainly focus on different organs and tissues from the same species or focus on various treatments and conditions for a certain organ or tissue [17, 21, 37]. One organ and its subordinate tissues were rarely studied together. As the two reproductive organs in the plant, the developing capsules and their seeds of Euscaphis konishii [57], and the developing fruits and their seeds of Eriobotrya japonica [49] were both collected as sample sets. The rankings of all chosen RGs were thoroughly different between its fruits and corresponding seeds, which illustrated that the RGs for two closely related organs still need to be verified experimentally. Oat endosperm is the largest component of mature oat seeds, accounting for about 90% of size [58]. The proportions of storage materials such as oil and carbohydrates are quite similar between oat seed and endosperm [6, 7]. In this study, the least stable RG of them both was GAPDH1 (Table 3). However, the optimal RG in developing oat seeds, namely EP, ranked in the middle or lower under four algorithms in endosperms (Tables 2, 3). The ranking of other RGs were not similar at all when evaluated by each algorithm (Table 2). And the Ct values of all ten RGs varied in larger range in developing seeds than in developing endosperms (Fig. 4c, d). Our results strongly suggest that organs and their appurtenant tissues should be treated independently as different sample sets when they are used for RG selection.
Conclusions
In this study, eleven candidate RGs were screened for qPCR in 18 samples of hexaploid oat, four of which were duplicated genes. With the analysis by the ΔCt method, geNorm, NormFinder and BestKeeper, our results provide a proof of concept that duplicated genes are feasible as RGs for qPCR assays in polyploid crops. The results suggested that EIF4A + HNR showed the highest stability than any other candidate RG sets across all tested samples and in developing endosperms. The combination of EP and EF1A was the best RG set for developing seeds. UBC21, an example of a four-copy duplicated RG, along with HNR was identified as the most stable RG set in shoots and roots of oat seedlings. Conversely, GAPDH1 was regarded as the least stable in both developing seeds and endosperms. The expression pattern analysis of four various genes verified the accuracy and the reliability of optimal RG sets in developing oat seeds, endosperms and seedlings. This work is the first report for RG validation in oat and will provide useful references for future studies of gene expression based on qPCR in oat. These findings will also facilitate similar research on other closely related crops and polyploid species.
Methods
Plant materials
Seeds of oat cultivar Baiyan No.9 were germinated in the field at the experimental station of Northwest A&F University, Shaanxi, China (34°09′ N, 108°08′ E). Shoots and roots were segmented from two-leaf stage and three-leaf stage of seedlings, respectively. Developing oat seeds were collected at seven stages designated as stage B, C, D, E, G, I and J, based on the definitions described by Ekman et al. [7]. Corresponding endosperms of seeds from the seven stages mentioned above were separated out carefully by removing hulls, brans and embryos. Eighteen samples, including seeds, endosperms, shoots and roots of seedlings, were acquired and frozen using liquid nitrogen, then stored at -80℃ for RNA extraction.
RNA extraction and cDNA synthesis
Total RNA of plant materials was extracted following the manufacturer’s instruction of the E.Z.N.A.® Plant RNA Kit (OMEGA) in biological triplicate. RNA quality was detected in 1% agarose gel and quantified with Nanodrop ND-2000 spectrophotometer (Thermo). cDNA synthesis was performed from 1 μg of total RNA via reverse transcription using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara).
Selection of candidate reference genes and primer design
Candidate RGs were selected from collected RGs in sequenced Gramineae species including wheat (Triticum aestivum), barley (Hordeum vulgare), sorghum (Sorghum bicolor), rice (Oryza sativa), maize (Zea mays) and Brachypodium distachyon in the internal control genes (ICG) database (https://icg.big.ac.cn/index.php/Species:Plant), and from other reported RGs in relevant articles. Corresponding sequences were retrieved by the accession numbers in NCBI (https://www.ncbi.nlm.nih.gov). A total number of nineteen candidate RGs gathered from above sources were used as query sequences to find homolog genes in oat seed transcriptome [1] by TBLASTN of NCBI local BLAST tool (blast-2.7.1 +), and subject sequences with E value no more than 1e−30 were chosen for BLASTX on Phytozome (https://phytozome.jgi.doe.gov) to verify their accuracy. In BLAST results, eight query RGs had no matched hits in oat seed transcriptome. Consequently, there were 11 oat RGs left and sequences of them were shown in Additional file 1.
Specific primers for qPCR were designed using Primer Premier 5.0 according to following parameters: primer length of 20–24 bp, melting temperature (Tm) of 55–65 °C, GC content of 45–60% and product size of 80–200 bp. Detailed information is listed in Table 1. As for those candidate RGs with more than one transcript, the highly conserved part of sequences was used for primer design. Multiple alignments of duplicated genes were conducted using DNAMAN 6. All primers were synthesized by TsingKe Biotech Co., Ltd (Xi’an, China) and their products of regular PCR were verified through 1.5% agarose gels.
Quantitative real-time PCR validation of candidate reference genes
A standard curve was generated using a series of five diluted cDNAs to calculate the amplification efficiency (E) and correlation coefficients (R2) of each candidate RG. The calculation of E values is as follows: E (%) = (10 − 1/slope − 1) × 100. The detailed information for all primer pairs of eleven candidate RGs is listed in Table 1.
Diluted aliquots of the reverse-transcribed cDNAs were used as templates in qPCR assays. qPCR was performed in three biological replicates with three technical replicates on QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems, US) using ChamQ SYBR Color qPCR Master Mix (High ROX Premixed; Vazyme, Nanjing, China). Each 20 μL reaction mixture contained 10 μL 2 × qPCR Mix, 0.4 μL forward primer (10 μM), 0.4 μL reverse primer (10 μM), 1 μL cDNA (200 μg/μL) and 8.2 μL ddH2O. The qPCR program was as follows: 50 °C for 2 min and 94 °C for 30 s, 42 cycles of 94 °C for 5 s and 60 °C for 30 s. Melting curves were generated to analyze the primer specificity. To verify the stabilities of the screened RG sets, the expression patterns of AsPKP1 (Forward primer: 5′-TCAAGAACCACATGAGCGAAAT-3′, Reverse primer: 5′-CAGACGGGCGGTAATGACTAA-3′), AsAGPL2 (Forward primer: 5′-ATCGTCACATTCACCGCACCT-3′, Reverse primer: 5′-ATCGCCCGACAAGATCAAAATG-3′), AsSGT1 (Forward primer: 5′-GCTGTCTTGAGGTTGGTTCTT-3′, Reverse primer: 5′-CCTGTATTTGGGCTTGCTTGG-3′) and AsSCL14 (Forward primer: 5′-TCTGTTCTTCTATTCTGCCCTGT-3′, Reverse primer: 5′-GCTCCACCCTATCTGTACCCTCA-3′) were detected using the most and the least stable RGs, then the qPCR results were calculated by 2−ΔΔCt method.
Data analysis
The Ct values of each RG in qPCR were used to evaluate the stability using the ΔCt method, geNorm [25], NormFinder [26] and BestKeeper [27]. In geNorm analysis, the stability value (M) of each RG was generated based on the average pairwise variation (V) between all tested genes. Candidate RGs with lower M values have more stable expression. In NormFinder analysis, the stability value was evaluated by determining inter- and intra-group variations through an ANOVA-based model. The lower stability value and inter- and intra-group variation represent a more stable candidate RG. In BestKeeper analysis, the expression stability of candidate RGs was determined by the calculation of the standard deviation (SD) and coefficient of variance (CV). The lowest CV value indicates the highest stability. The geometric mean was computed to rank the stability of candidate RGs. The lower geometric mean shows the higher stability. All assays were carried out in triplicates, and the data represent the mean ± SD.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Abbreviations
- 18S:
-
18S Ribosomal RNA
- EF1A:
-
Elongation factor 1-Alpha
- EIF4A:
-
Eukaryotic initiation factor 4A-3
- EP:
-
Expressed protein
- GAPDH1:
-
Glyceraldehyde-3-phosphate Dehydrogenase C Subunit 1
- HNR:
-
Heterogeneous nuclear ribonucleoprotein 27C
- PP2A:
-
Protein Phosphatase 2A Subunit A3
- RG:
-
Reference gene
- qPCR:
-
Quantitative real-time polymerase chain reaction
- TBC:
-
TBC1 domain family member 22A
- TUA6:
-
Tubulin alpha-6 chain
- UBC21:
-
Ubiquitin-Conjugating Enzyme 21
- UBQ10:
-
Polyubiquitin 10
References
Gutierrez-Gonzalez JJ, Tu ZJ, Garvin DF. Analysis and annotation of the hexaploid oat seed transcriptome. BMC Genom. 2013;14:471.
Singh R, De S, Belkheir A. Avena sativa (Oat), a potential neutraceutical and therapeutic agent: an overview. Crit Rev Food Sci Nutr. 2013;53:126–44.
Butt MS, Tahir-Nadeem M, Khan MKI, Shabir R, Butt MS. Oat: unique among the cereals. Eur J Nutr. 2008;47:68–79.
Ben Halima N, Ben Saad R, Khemakhem B, Fendri I, Abdelkafi S. Oat (Avena sativa L.): oil and nutriment compounds valorization for potential use in industrial applications. J Oleo Sci. 2015;64:915–32.
Sang S, Chu Y. Whole grain oats, more than just a fiber: role of unique phytochemicals. Mol Nutr Food Res. 2017;61:1600715.
Banaś A, Debski H, Banas W, Heneen WK, Dahlqvist A, Bafor M, Gummeson PO, Marttila S, Ekman Å, Carlsson AS, et al. Lipids in grain tissues of oat (Avena sativa): differences in content, time of deposition, and fatty acid composition. J Exp Bot. 2007;58:2463–70.
Ekman Å, Hayden DM, Dehesh K, Bülow L, Stymne S. Carbon partitioning between oil and carbohydrates in developing oat (Avena sativa L.) seeds. J Exp Bot. 2008;59:4247–57.
Gilissen L, van der Meer IM, Smulders MJM. Why oats are safe and healthy for celiac disease patients. Med Sci. 2016;4:21.
Grundy MM, Fardet A, Tosh SM, Rich GT, Wilde PJ. Processing of oat: the impact on oat’s cholesterol lowering effect. Food Funct. 2018;9:1328–43.
Zhao G, Ma B, Ren C. Growth, gas exchange, chlorophyll fluorescence, and ion content of naked oat in response to salinity. Crop Sci. 2007;47:123–31.
Givens D, Davies T, Laverick R. Effect of variety, nitrogen fertiliser and various agronomic factors on the nutritive value of husked and naked oats grain. Anim Feed Sci Technol. 2004;113:169–81.
Stewart D, McDougall G. Oat agriculture, cultivation and breeding targets: implications for human nutrition and health. Br J Nutr. 2014;112(Suppl 2):S50–S5757.
Grimberg Å, Carlsson AS, Marttila S, Bhalerao R, Hofvander P. Transcriptional transitions in Nicotiana benthamiana leaves upon induction of oil synthesis by WRINKLED1 homologs from diverse species and tissues. BMC Plant Biol. 2015;15:192.
Yang Z, Liu X, Li N, Du C, Wang K, Zhao C, Wang Z, Hu Y, Zhang M. WRINKLED1 homologs highly and functionally express in oil-rich endosperms of oat and castor. Plant Sci. 2019;287:110193.
Bustin SA, Benes V, Nolan T, Pfaffl MW. Quantitative real-time RT-PCR–a perspective. J Mol Endocrinol. 2005;34:597–601.
Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005;6:279–84.
Huis R, Hawkins S, Neutelings G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol. 2010;10:71.
Udvardi MK, Czechowski T, Scheible WR. Eleven golden rules of quantitative RT-PCR. Plant Cell. 2008;20:1736–7.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE Guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2019;55:611–22.
Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat Protoc. 2006;1:1559–822.
Hao X, Horvath DP, Chao WS, Yang Y, Wang X, Xiao B. Identification and evaluation of reliable reference genes for quantitative real-time PCR analysis in tea plant (Camellia sinensis (L.) O. Kuntze). Int J Mol Sci. 2014;15:22155–72.
Zhong H, Simons JW. Direct comparison of GAPDH, beta-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochem Biophys Res Commun. 1999;259:523–6.
Suzuki T, Higgins PJ, Crawford DR. Control selection for RNA quantitation. Biotechniques. 2000;29:332–7.
Nguyen DQ, Eamens AL, Grof CPL. Reference gene identification for reliable normalisation of quantitative RT–PCR data in Setaria viridis. Plant Methods. 2018;14:24.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real–time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:1–11.
Andersen CL, Jensen JL, Ørntoft TF. Normalization of real–time quantitative reverse transcription-PCR data: a model–based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–50.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–15.
Chaffin AS, Huang YF, Smith S, Bekele WA, Babiker E, Gnanesh BN, Foresman BJ, Blanchard SG, Jay JJ, Reid RW, et al. A consensus map in cultivated hexaploid oat reveals conserved grass synteny with substantial subgenome rearrangement. Plant Genome. 2016;9:1–21.
Zeng L, Deng R, Guo Z, Yang S, Deng X. Genome-wide identification and characterization of Glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genomics. 2016;17:240.
Mu J, Chen L, Gu Y, Duan L, Han S, Li Y, Yan Y, Li X. Genome-wide identification of internal reference genes for normalization of gene expression values during endosperm development in wheat. J Appl Genet. 2019;60:233–41.
Yang H, Liu J, Huang S, Guo T, Deng L, Hua W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene. 2014;538:113–22.
Chao WS, Wang H, Horvath DP, Anderson JV. Selection of endogenous reference genes for qRT-PCR analysis in Camelina sativa and identification of FLOWERING LOCUS C allele-specific markers to differentiate summer- and winter-biotypes. Ind Crops Prod. 2019;129:495–502.
Fuentes A, Ortiz J, Saavedra N, Salazar LA, Meneses C, Arriagada C. Reference gene selection for quantitative real-time PCR in Solanum lycopersicum L. inoculated with the mycorrhizal fungus Rhizophagus irregularis. Plant Physiol Biochem. 2016;101:124–31.
Sabeh M, Duceppe MO, St-Arnaud M, Mimee B. Transcriptome-wide selection of a reliable set of reference genes for gene expression studies in potato cyst nematodes (Globodera spp.). PLoS ONE. 2018;13:e0193840.
Schmidt GW, Delaney SK. Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol Genet Genomics. 2010;283:233–41.
Yang L, Quan S, Zhang D. Endogenous reference genes and their quantitative real-time PCR assays for genetically modified bread wheat (Triticum aestivum L.) detection. In: Bhalla PL, Singh MB, editors. Wheat biotechnology. New York: Humana Press; 2017. p. 259–268.
Chen C, Wu J, Hua Q, Tel-Zur N, Xie F, Zhang Z, Chen J, Zhang R, Hu G, Zhao J, et al. Identification of reliable reference genes for quantitative real-time PCR normalization in pitaya. Plant Methods. 2019;15:70.
Galli V, da Silva MR, dos Anjos e Silva SD, Rombaldi CV. Selection of reliable reference genes for quantitative real-time polymerase chain reaction studies in maize grains. Plant Cell Rep. 2013;32:1869–77.
Gimenez MJ, Piston F, Atienza SG. Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta. 2011;233:163–73.
Lambret-Frotte J, de Almeida LC, de Moura SM, Souza FL, Linhares FS, Alves-Ferreira M. Validating internal control genes for the accurate normalization of qPCR expression analysis of the novel model plant Setaria viridis. PLoS ONE. 2015;10:e0135006.
Nagaraj S, Ramlal S, Venkataswamachari BP, Paul S, Kingston J, Batra HV. Differentiation of entC1 from entC2/entC3 with a single primer pair using simple and rapid SYBR Green-based RT-PCR melt curve analysis. Appl Microbiol Biotechnol. 2016;100:8495–506.
Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45.
Maeo K, Tokuda T, Ayame A, Mitsui N, Kawai T, Tsukagoshi H, Ishiguro S, Nakamura K. An AP2-type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW-box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 2009;60:476–87.
Bahaji A, Li J, Sánchez-López ÁM, Baroja-Fernández E, Muñoz FJ, Ovecka M, et al. Starch biosynthesis, its regulation and biotechnological approaches to improve crop yields. Biotechnol Adv. 2014;32:87–106.
Wang G, Fan R, Wang X, Wang D, Zhang X. TaRAR1 and TaSGT1 associate with TaHsp90 to function in bread wheat (Triticum aestivum L.) seedling growth and stripe rust resistance. Plant Mol Biol. 2015;87:577–89.
Chen K, Li H, Chen Y, Zheng Q, Li B, Li Z. TaSCL14, a novel wheat (Triticum aestivum L.) GRAS gene, regulates plant growth, photosynthesis, tolerance to photooxidative stress, and senescence. J Genet Genom. 2015;42:21–322.
Ambroise V, Legay S, Guerriero G, Hausman JF, Cuypers A, Sergeant K. Selection of appropriate reference genes for gene expression analysis under abiotic stresses in Salix viminalis. Int J Mol Sci. 2019;20:4210.
Wang E, Wang K, Chen D, Wang J, He Y, Long B, Yang L, Yang Q, Geng Y, Huang X, et al. Evaluation and selection of appropriate reference genes for real-time quantitative PCR analysis of gene expression in nile tilapia (Oreochromis niloticus) during vaccination and infection. Int J Mol Sci. 2015;16:9998–10015.
Su W, Yuan Y, Zhang L, Jiang Y, Gan X, Bai Y, Peng J, Wu J, Liu Y, Lin S. Selection of the optimal reference genes for expression analyses in different materials of Eriobotrya japonica. Plant Methods. 2019;15:7.
Chaouachi M, El Malki R, Berard A, Romaniuk M, Laval V, Brunel D, Bertheau Y. Development of a real-time PCR method for the differential detection and quantification of four Solanaceae in GMO analysis: potato (Solanum tuberosum), tomato (Solanum lycopersicum), eggplant (Solanum melongena), and pepper (Capsicum annuum). J Agric Food Chem. 2008;56:1818–28.
Appels R, Eversole K, Stein N, Feuillet C, Keller B, Rogers J, Pozniak CJ, Choulet F, Distelfeld A, Poland J, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361:eaar7191.
Kiarash JG, Dayton Wilde H, Amirmahani F, Mehdi Moemeni M, Zaboli M, Nazari M, Saeed Moosavi S, Jamalvandi M. Selection and validation of reference genes for normalization of qRT-PCR gene expression in wheat (Triticum durum L.) under drought and salt stresses. J Genet. 2018;97:1433–44.
Hoang NV, Furtado A, Mason PJ, Marquardt A, Kasirajan L, Thirugnanasambandam PP, Botha FC, Henry RJ. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017;18:395.
Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA. The fate of duplicated genes in a polyploid plant genome. Plant J. 2013;73:143–53.
Zhao C, Li H, Zhang W, Wang H, Xu A, Tian J, Zou J, Taylor DC, Zhang M. BnDGAT1s function similarly in oil deposition and are expressed with uniform patterns in tissues of Brassica napus. Front Plant Sci. 2017;8:2205.
Baud S, Wuilleme S, To A, Rochat C, Lepiniec L. Role of WRINKLED1 in the transcriptional regulation of glycolytic and fatty acid biosynthetic genes in Arabidopsis. Plant J. 2009;60:933–47.
Liang W, Zou X, Carballar-Lejarazu R, Wu L, Sun W, Yuan X, Wu S, Li P, Ding H, Ni L, et al. Selection and evaluation of reference genes for qRT-PCR analysis in Euscaphis konishii Hayata based on transcriptome data. Plant Methods. 2018;14:42.
Fulcher R. Morphological and chemical organization of the oat kernel. In: Webster FH, editor. Oats: chemistry and technology. Saint Paul: American Association of Cereal Chemists; 1986.
Acknowledgements
We thank Prof. Yingang Hu from Northwest A&F University for providing oat seeds and Mr. Lifu Zhang from University of Alberta for polishing our manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (31972964), the Key International Cooperation Project of Shaanxi province (2020KWZ-012) and Programme of Introducing Talents of Innovative Discipline to Universities (Project 111) from the State Administration of Foreign Experts Affairs (#B18042) “Crop breeding for disease resistance and genetic improvement”. The funding bodies have no role in the design of the study and collection, analysis and interpretation of data, and in writing the manuscript, but just provide the financial supports.
Author information
Authors and Affiliations
Contributions
ZY and MZ conceived the project and wrote the manuscript. ZY designed the experiments. ZY, KW and UA performed the most of the experiments and analyzed the data. CZZ carried on the field experiments and edited the manuscript. MZ supervised the experiments. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Sequences of 11 candidate RGs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, Z., Wang, K., Aziz, U. et al. Evaluation of duplicated reference genes for quantitative real-time PCR analysis in genome unknown hexaploid oat (Avena sativa L.). Plant Methods 16, 138 (2020). https://doi.org/10.1186/s13007-020-00679-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-020-00679-1