
Abstract
Bovine leukemia virus (BLV), an oncogenic member of the Deltaretrovirus genus, is closely related to human T-cell leukemia virus (HTLV-I and II). BLV infects cattle worldwide and causes important economic losses. In this review, we provide a summary of available information about commonly used diagnostic approaches for the detection of BLV infection, including both serological and viral genome-based methods. We also outline genotyping methods used for the phylogenetic analysis of BLV, including PCR restriction length polymorphism and modern DNA sequencing-based methods. In addition, detailed epidemiological information on the prevalence of BLV in cattle worldwide is presented. Finally, we summarize the various BLV genotypes identified by the phylogenetic analyses of the whole genome and env gp51 sequences of BLV strains in different countries and discuss the distribution of BLV genotypes worldwide.
Similar content being viewed by others

Background
Bovine leukemia virus (BLV) is a retrovirus, an oncogenic member of the Deltaretrovirus genus, and the causative agent of enzootic bovine leukosis (EBL) [1, 2]. The Deltaretrovirus genus also includes human T-cell lymphotropic virus types I and II (HTLV-I and -II) and simian T-cell lymphotropic virus (STLV) [3, 4]. EBL is a contagious lymphoproliferative disease of cattle, characterized by B-cell lymphosarcoma, which occurs throughout the world [2, 5]. Although BLV can infect various immune cell populations, including CD5+ IgM+ and CD5− IgM+ B-cells; CD2+, CD3+, CD4+, CD8+, and γ/δ T-cells; monocytes; and granulocytes in peripheral blood and lymphoid tissues of cattle [6,7,8,9,10,11], BLV-induced tumors usually arise from the CD5+ IgM+ B-cell subpopulation [12].
BLV infection can result in a variety of clinical outcomes [2]. The majority of BLV-infected cattle are asymptomatic carriers of the virus, neither showing any clinical signs nor any changes in lymphocyte count; however, a recent study showed that although lymphocyte counts were not elevated in BLV-infected but clinically normal cattle, CD5+ IgM+ B-cells were increased [11], and there is substantial evidence suggesting that BLV-infected but clinically normal cattle may exhibit a degree of immunological dysregulation leading to economic losses for various reasons including reduced milk production [13], a high incidence of infectious disease [14], and reproductive inefficiency [15]. Approximately one-third of infected cattle develop a benign form of non-malignant proliferation of untransformed B-lymphocytes, termed persistent lymphocytosis (PL). PL is typically characterized by a permanent and stable increase in the number of CD5+ IgM+ B-cells circulating in the peripheral blood. Less than 5% of infected cattle develop malignant B-cell lymphoma originating from mono- or oligo-clonal accumulation of CD5+ IgM+ B-cells after a relatively long period of latency. This malignant form of B-cell lymphoma is predominantly detected in cattle over 4–5 years old [16]. Such malignancies induce disruption of the spleen and remarkable enlargement of the lymph nodes, which can be visible under the skin. BLV-induced neoplastic cells can penetrate into the abomasums, right auricle of the heart, intestine, kidney, lung, liver, and uterus. The clinical signs of BLV-induced tumors are varied and primarily involve digestive disturbance, weight loss, weakness, reduced milk production, loss of appetite, and enlarged lymph nodes [17].
BLV genome structure
The BLV genome consists of 8714 nucleotides (nt) [18] including essential structural protein and enzyme coding genes and a pX region, flanked by two identical long terminal repeats (LTRs) (Fig. 1a). The structural protein and enzyme coding genes, namely, gag, pro, pol, and env, have essential and indispensable roles in the viral lifecycle, viral infectivity, and the production of infectious virions [19,20,21,22,23,24]. The gag gene of BLV is translated as the precursor, Pr45 Gag, and processed to generate three mature proteins [19, 23]: the matrix protein, p15, which binds viral genomic RNA and interacts with the lipid bilayer of the viral membrane [25]; the capsid protein, p24, which is the major target of the host immune response, with high antibody titers against this molecule found in the serum of infected animals [26, 27]; and the nucleocapsid protein, p12, which binds to packaged genomic RNA [28] (Fig. 1b). The env gene encodes the mature extracellular protein, gp51, and a transmembrane protein, gp30 [19]. The pX region, which is located between env and the 3′ LTR [2], encodes the regulatory proteins Tax and Rex, and the accessory proteins R3 and G4 (Fig. 1a). The regulatory proteins are important for regulation of viral transcription, transformation of BLV-induced leukemogenesis, and nuclear export of viral RNA into the cytoplasm [29,30,31,32,33,34,35,36]. The R3 and G4 accessory proteins contribute to the maintenance of high viral loads [37, 38]. In addition to the genes described above, the BLV genome also contains RNA polymerase-III-encoded viral microRNAs (miRNAs) between the env and pX regions. Viral miRNAs are strongly expressed in preleukemic and malignant cells, and may have roles in tumor onset and progression [39, 40] through their effects on proviral load and consequently viral replication in the natural host [41]. Besides, Van Driessche et al. revealed the recruitment of positive epigenetic marks on BLV miRNA cluster, inducing strong antisense promoter activity [42]. They also identified cis-acting elements of an RNAPII-dependent promoter [42].

Schematic representations of the BLV genome structure (a) and viral particle (b). The structural and enzymatic genes, gag, pro, pol, and env; regulatory genes, tax and rex; accessory genes R3 and G4; and microRNA (miRNA) are indicated in (a). Proteins encoded by structural and enzymatic genes, including the Env glycoproteins (gp51 and gp30) encoded by the env gene, the Gag proteins (p12, p24, and p15) encoded by the gag gene, reverse transcriptase and integrase (RT-IN) encoded by the pol gene, and protease (Pro) encoded by the pro gene are indicated in (b)
BLV diagnosis
A variety of techniques have been developed for diagnosis of BLV and implemented worldwide. These diagnostic methods can be assigned into two main groups, consisting of antibody-based serological tests and detection of the proviral genome by nucleic acid-based polymerase chain reaction (PCR) assays (summarized in Table 1).
Serological tests
For indirect BLV diagnostic methods, particularly antibody-based tests, antibodies recognizing the p24 capsid protein encoded by the gag gene and the extracellular gp51 protein encoded by env-gp51 are targeted. This is because antibodies against these proteins are produced shortly after BLV infection, can be detected 2–3 weeks post-infection, and remain detectable for the life of the host animal [43]. In addition, the p24 capsid protein is a major target for host immune responses, inducing high antibody titers [44], and gp51 invokes the expression of massive amounts of specific antibodies in infected animals [24, 45, 46]. Therefore, antibodies against these proteins are targeted for BLV diagnostics using conventional serological techniques such as agar gel immunodiffusion (AGID) [43, 47,48,49,50], passive hemagglutination assay (PHA) [43, 51], enzyme-linked immunosorbent assay (ELISA) [43, 49, 50, 52, 53], and radio immunoassay (RIA) [54]. Most of these serological methods aim to detect antibodies in bovine serum and milk, and the supernatants of BLV-infected cell cultures. AGID is relatively inexpensive and can be used to screen many serum samples simultaneously; however, it is not sufficiently sensitive [55] and it is not suitable for analysis of milk samples. ELISA is a highly sensitive and easily implemented procedure, and can be used to analyze both serum and milk samples; however, it requires a number of controls and produces both false-negative result in serum samples from cattle in the early phase of infection [55] and false-positive results in calves that contain maternally-derived antibodies [56]. PHA aims to detect BLV glycoproteins, but, PHA test efficiency is sensitive to pH, temperature, and trypsin. RIA is suitable for diagnosing BLV soon after animals are exposed, but not suitable for the purpose of mass screening [57]. Overall, these antibody-based detection methods cannot be used to test calves less than 6 months old, due to the presence of maternal antibodies, which may trigger false-positive results [58].
Proviral DNA detection
BLV can integrate into dispersed sites within the host genome [59] and appears to be transcriptionally silent in vivo [60,61,62] and remain in cellular genomes, even in the absence of detectable BLV antibodies. Indeed, transcription of the BLV genome in fresh tumor or peripheral blood mononuclear cell samples from infected individuals is almost undetectable by conventional techniques [60, 63]. Interestingly, one copy of the full-length proviral genome can be detected in BLV-infected cattle throughout the course of the disease [64]. Another study also demonstrated that BLV-induced tumors and BLV-infected cells contain provirus, with approximately four copies of proviral DNA in each tumor [65]. Hence, in addition to the routine diagnosis of BLV infection using the conventional serological techniques described above, nucleic acid-based PCR methods can greatly accelerate the detection of BLV prevalence.
A variety of PCR methods, including standard PCR [49, 50], nested PCR [33, 52, 64], real-time quantitative PCR (qPCR) [43, 66,67,68,69,70,71], and direct blood-based PCR [72, 73], have been extensively applied worldwide for BLV detection (Table 1). A variety of genes in the BLV genome are targeted for detection of BLV infection prevalence by direct diagnostic PCR methods, including the LTR region [43, 70, 71, 73,74,75,76,77], and the gag [78], pol [69, 79, 80], env [55, 79], and tax [68, 79] genes.
Importantly, the BLV provirus copy number is generally very low compared with that of host genes therefore, the majority of PCR systems designed to detect BLV used a nested design [64, 74, 76]. These nested assays are extremely sensitive, but also obtain false-positive results due to DNA contamination. However, the method requires expensive real-time PCR machines and reagents and involves difficult sample preparation protocols. Recently, a novel blood-based PCR system that amplifies target DNA regions without a requirement for DNA isolation and purification was developed [72, 73]. The assay can detect BLV provirus with high specificity and at low cost, facilitating timely identification of BLV-infected cattle.
As discussed above, PCR-based genome screening methods for diagnosis of BLV broaden the range of samples that can be used, increase testing sensitivity, specificity, and efficiency, and are less time consuming. PCR also allows the detection of BLV infection in cattle several weeks before it is possible to detect antibodies [81]; however, PCR-based provirus screening involves complicated sample preparation processes, which can lead to false-positive results if cross contamination occurs. In addition, PCR-based BLV detection methods require specific laboratory facilities, including PCR machines, and the design of specific primers and probes is also necessary. The CoCoMo algorithm, is a method used to design degenerate primer sets that amplify all available sequences within a target region. Recently, the BLV-CoCoMo-qPCR assay was developed to measure the BLV proviral load with extremely high sensitivity and to amplify both known and novel BLV variants [43, 70, 71]. This assay enabled us to demonstrate that the proviral load correlates not only with BLV infection capacity but also with BLV disease progression [43, 82], and identification of risk factor associated with increased BLV proviral load in infected cattle [82, 83] and detection of BLV provirus in nasal secretion and saliva samples [84].
Other methods
In addition to the techniques described above, other BLV diagnostic approaches, including detection of viral proteins by western blotting [21, 31, 33, 85], a syncytium formation assay [85], and detection of BLV antigens by indirect immunofluorescent assay [47], have also been described.
BLV genotyping and identification of ten distinct genotypes
Studies of BLV genotypes for phylogenetic and epidemiological analyses have primarily focused on the env gene, the env gp51 gene in particular, because of its biological functions. The extracellular gp51 protein has key roles in the viral lifecycle and is indispensable for viral entry into host cells [20, 86]. In addition, because of the surface localization of the gp51 glycoprotein, it is also the target of neutralizing antibodies [87]. The conformational epitopes, F, G, and H, located in the N-terminal half of gp51, are important in syncytium formation and viral infectivity [87, 88]. Therefore, the env gp51 sequence region is frequently used for BLV phylogenetic analysis.
Over the years, a number of methods have been applied for BLV genotyping, as summarized in Table 2. In the early days of BLV genotyping, researchers clustered or genotyped BLV strains from different geographical regions based on restriction fragment length polymorphisms (RFLP) of PCR-products, generated using various restriction enzymes [86, 89,90,91,92,93,94,95,96]. BLV clusters and genotypes were named after the geographical region of sample isolation, such as “Argentine type” or “Australian type”, or with reference to phylogenetic clustering (e.g., “cluster one”). A total of seven BLV clusters/genotypes were determined by PCR-RFLP [91]; however, PCR-RFLP genotyping studies were not consistent or comprehensive.
In 2007, Rodriguez et al. reported sequencing of the env gene (all of gp51 and part of gp30) of 28 BLV field strains, performed phylogenetic analysis of these sequences in comparison with published sequence data representative of established genetic groups by neighbor-joining, maximum likelihood, and Bayesian inference methods, and assigned BLV sequences into seven genotypes [97]. Subsequently, a new genotype, genotype-8, was identified in BLV samples from Croatia by Balic et al. [98], who concluded that BLV may be more divergent than previously thought, speculating that additional genotypes might be discovered in the future. Indeed, the presence of eight BLV genotypes was later confirmed in different geographical locations [74, 77, 99,100,101]. Finally, in 2016, the novel BLV genotypes, genotype-9 and -10, were discovered in Bolivia [75], Thailand [102], and Myanmar [76], a totaling ten BLV genotype clusters (Fig. 2). Previously, almost all phylogenetic studies of BLV genotypes focused on the partial or entire env gene. However, for the first time in their study [75, 76], Polat et al. successfully concluded the existence of genotypes-1, −2, −4, −6, −9 and −10 among ten BLV genotypes (Fig. 3) by phylogenetic analysis using complete sequences of BLV strains newly determined by next generation sequencing and sequencing cloned, overlapping PCR products in their studies, and using complete BLV genome sequences available in the database (NCBI & DDBJ). These phylogenetic analysis of complete BLV genomes demonstrated that each BLV genotype encodes specific amino acid substitutions in both structural and non-structural gene regions.

Maximum likelihood phylogenetic tree constructed based on partial BLV env sequences identified in geographical locations around the world. A maximum likelihood (ML) phylogenetic tree was constructed based on sequences from known BLV strains, representing ten different BLV genotypes derived from viruses isolated worldwide. Nucleotide sequences were obtained from the GenBank nucleotide sequence database. Sequences are labeled with their accession numbers and countries of origin. Genotypes are indicated by numbers to the right of the figure. One thousand replications were performed to calculate bootstrap values (indicated on the tree). The bar at the bottom of the figure indicates evolutionary distance

Maximum likelihood (ML) phylogenetic tree constructed from complete BLV genomic sequences. The ML phylogenetic tree was constructed using complete BLV genomic sequences from the GenBank nucleotide sequence database. One thousand replications were performed to calculate bootstrap values (indicated on the tree). The strains identified in this study are indicated by the sample identification number and country name. Genotypes are indicated by numbers to the right of the figure. The bar at the bottom of the figure indicates evolutionary distance
BLV prevalence
BLV has spread to all continents via the trade in breeding animals, and is prevalent in cattle worldwide. BLV infection levels vary between and within countries, as shown in Table 3 (data obtained on March 17th, 2017; updated and detailed information is available at http://www.oie.int/wahis_2/public/wahid.php/Diseaseinformation/statuslist) [17, 103]. BLV eradication programs and control measures have been established in European Community member countries since the second half of the twentieth century, and eradication programs have been very successful in the majority of western Europe [104,105,106,107]; indeed, some countries, including Denmark, Finland, Switzerland, Estonia, The Netherlands and Poland, are completely free of BLV [104, 108,109,110]. Despite the majority of countries in Western Europe being free from disease, EBL still exists in eastern European nations, including Poland, Ukraine, and Croatia [98, 100, 111,112,113]. In addition, in Italy, Portugal, Belarus, Latvia, Greece, Romania, and Bulgaria, BLV is present, although disease is either absent or limited to specific areas [103].
Nationwide BLV eradication and control programs were introduced in Australia and New Zealand in 1983 and 1996, respectively, and 99.7% of Australian dairy herds were declared free from EBL in December 2013, while those in New Zealand have been free from BLV-induced EBL since 2008 [113, 114].
In North America, an epidemiological study of BLV prevalence in US dairy cattle conducted by the Department of Agriculture’s National Animal Health Monitoring System demonstrated that 83.9% of dairy cattle were BLV-positive at herd level and 39% of beef herds had at least one BLV-infected animal [115]. In Canada, studies of BLV prevalence revealed that up to 37.2% of cows and 89% of herds were BLV-positive [116,117,118]. BLV is also present in both beef and dairy cattle in Mexico [119]; however, disease is either absent or limited to specific areas [17] (accessed on 22 Dec 2016).
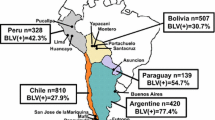
In South America, relatively high levels of BLV prevalence have been observed, and BLV-induced leukosis is present in the majority of countries. In Brazil, BLV prevalence varies among states, with infection rates ranging from 17.1% to 60.8% [120,121,122,123]. Individual and herd level BLV prevalence in Argentina are as high as 77.4% and 90.9%, respectively [75, 95, 124]. Moreover, individual infection rates between 19.8% and 54.7% have been reported in Chile, Bolivia, Peru, Venezuela, Uruguay, Paraguay, and Columbia [75, 94, 125,126,127,128,129,130,131].
BLV infection is widespread in Chinese dairy farms. Infection rates are up to 49.1% among individual dairy cattle, while 1.6% of beef cattle are BLV-positive [132]. Moreover, serological tests revealed that 20.1% of yaks in China were BLV-positive [133]. Epidemiological studies in Japan revealed varying levels of BLV prevalence throughout the country, based on different detection methods [83, 134,135,136], and BLV infection rates of 40.9% of dairy and 28.7% of beef cattle, with infection rates in animals over 2-years-old reaching 78% in dairy herds and 69% in beef cattle herds [136]. Less than 6% of cattle were infected with BLV in Mongolia (3.9%) [77], Cambodia (5.3%) [137], and Taiwan (5.8%) [48], while a serological survey in Iran revealed that the prevalence of BLV was between 22.1% and 25.4% in that country [138, 139]. Lee et al. [102] demonstrated an average prevalence of BLV of 58.7% in Thailand, reaching maxima of 87.8% and 100% of cattle when assayed using PCR and ELISA, respectively. In Korea, 54.2% of dairy cattle and 86.8% of dairy herds were BLV-positive, whereas only 0.14% of beef cattle were infected with BLV [101]. BLV infection levels in The Philippines ranged from 4.8% to 9.7% [74] while it was 9.1% in Myanmar [76]. BLV infections in Middle Eastern countries are relatively low. The prevalence of BLV infection is approximately 5% in Israel [140], while in Saudi Arabia, 20.2% of dairy cattle tested as BLV-positive [141]. Compared to these countries, BLV infection rates in Turkey are higher, with 48.3% of dairy herds including sero-positive animals [142].
Distribution of BLV genotypes worldwide
As mentioned above, phylogenetic analyses of whole genome (Fig. 3) and env gp51 sequences (Fig. 2) of BLV strain showed that BLV can be classified into ten genotypes. Three genotypes of BLV, namely genotype-1, genotype-4 and genotype-6, were mainly detected from across the world, as shown in Table 4. Genotype-1 is the most dominant genotype of BLV and is distributed across almost all continents, including Europe, America, Asia, and Australia. In particularly, genotype-1 spread to South and North America, and these continents still have a high prevalence of BLV infection. In addition, genotype-1 continues to spread worldwide, including Asian countries. The second most widely distributed genotype is genotype-4, which is primarily detected in Europe and some American countries. However, it is only found in Mongolia among Asian nations. Interestingly, although genotype-4 used to exist in Europe, it decreased because of BLV eradication in European countries. Genotype 6 may have come from South America and spread to South Asia by animal trading. Of the other genotypes, genotype-2 is restricted to South American countries and is only found in Japan among Asian nations, while genotype-8 is restricted to Europe. Genotypes-5 (in Brazil and Costa Rica) and −10 (in Thailand and Myanmar) are only observed in geographically proximal areas, where there may be an exchange of animals across national boundaries [76, 102]. By contrast, genotypes-7 is distributed across geographically dispersed regions [74, 77].
In detail, in Europe, a total of five different BLV genotypes have been detected (genotypes −1, −3, −4, −7, and −8): genotype-4 in Belarus [100] and Belgium [86, 143]; genotypes-4, −7, and −8 in Russia and Ukraine [100]; genotype-8 in Croatia [98]; genotypes −4 and −7 in Poland [100]; genotypes −3 and −4 in France [86]; genotypes −1 and −4 in Germany [91]; and genotype-7 in Italy [144]. In Australia, only genotype-1 was detected [90]. In North America, genotypes −1, −3, and −4 have been detected in the USA [86, 143, 145], and genotype-1 was reported in the Caribbean [146]. In Central America, genotypes −1 and −5 were detected in Costa Rica [143]. A variety of BLV genotypes (−1, −2, −4, −5, −7, and −9) were detected in South America: genotypes −1, −2, −4, and −6 in Argentina [93, 95, 97, 147, 148]; genotypes −1, −2, −5, −6, and −7 in Brazil [122, 123, 127]; genotypes −4 and −7 in Chile [94]; genotypes −1, −2, −6, and −9 in Bolivia [75]; genotypes −1, −2, and −6 in Peru and Paraguay [75]; and genotype-1 in Uruguay [126]. In Asia, a total of seven BLV genotypes have been confirmed (−1, −2, −3, −4, −6, −7, and −10): genotypes −1 and −3 in Korea [101, 149]; genotypes −1, −2, and −3 in Japan [93, 99, 143, 150]; genotypes −1 and −6 in The Philippines [74]; genotypes −1, −6, and −10 in Thailand [102]; genotypes −1, −4, and −7 in Mongolia [77]; genotype-10 in Myanmar [76]; and genotypes −1 and −6 in Jordan [151].
Based on the European Food Safety Authority panel on animal health and welfare, BLV-induced EBL may have originated and spread widely from an area of Memel in East Prussia (now Klaipeda in Lithuania) [113, 152]. The worldwide distribution of the disease occurred due to the introduction of cattle from European countries into herds in other countries free of the disease, and also through the international trade of bred animals [113]. Interestingly, genotype-4 existed primarily in East Prussia as shown in Table 4. Then, infected cattle were reintroduced into some European countries; for example, BLV was introduced into the UK via bred animals from Canada in 1968 and 1973 [113]. As detailed in some previous publications, the widespread distribution of BLV genotypes within and between distant geographical locations may be driven by the spread of virus through the movement of live animal populations, associated with human migration and animal domestication, and also with viral transmission during close contact between individual animals [97].
Future prospects
It appears that at least ten different BLV genotypes of BLV strains are circulating in various geographical locations worldwide. The completion of whole genome sequencing of these BLV strains has revealed that BLV genomes contain a number of unique genotype specific substitutions not only in the env region, but also in the LTR, Gag, Pro, Pol, Tax, Rex, R3, G4, and miRNA encoding regions, distinguishing each genotype [75]. However, the BLV genome sequences of strains from different geographic origins, especially the important sites on the regulation of viral replication of BLV, are relatively stable and highly conserved among BLV strains, assigned to different genotypes. By contrast, several groups recently reported that the expression or pathogenesis of BLV does not depend on strains, but rather, is related with the specific site of mutation in their BLV genome [153, 154]. These results clearly demonstrate that BLV strain should be determined by full genome sequencing. However, although BLV is present worldwide, BLV genotyping studies are limited to certain areas, as shown in Table 4. Therefore, the accumulation of the full genome sequencing of BLV strains, assigned to different genotypes worldwide may define the genotype-dependent pathogenesis and association between genetic variability in each genotype and its infectivity, and differences in its functions in the future.
Conclusion
BLV is the etiologic agent of EBL, which is the most common neoplastic disease in cattle. It infects cattle worldwide, thereby imposing a severe economic burden on the dairy cattle industry. In this review, we summarized currently available detailed information on BLV infection worldwide, and indicated that BLV has spread to most countries except for some countries which are completely free of BLV by successful BLV eradication. We also outlined at least ten different BLV genotypes circulating in various geographical locations worldwide and the distribution of these BLV genotypes worldwide. This should be useful information to those investigating BLV for the potential development of diagnostic methods and vaccines, and for reducing the incidence of BLV in herds.
Abbreviations
- AGID:
-
Agar gel immunodiffusion
- BI:
-
Bayesian inference method
- BLV:
-
Bovine leukemia virus
- EBL:
-
Enzootic bovine leukosis
- ELISA:
-
Enzyme-linked immunosorbent assay
- EPAHW:
-
European Food Safety Authority panel on animal health and welfare
- HTLV-I &-II:
-
Human T-cell lymphotropic virus types I and II
- LTR:
-
Long terminal repeats
- miRNA:
-
microRNA
- ML:
-
Maximum likelihood method
- NJ:
-
Neighbor-joining method
- PCR:
-
Polymerase chain reaction
- PHA:
-
Passive hemagglutination assay
- PL:
-
Persistent lymphocytosis
- qPCR:
-
Quantitative PCR
- RFLP:
-
Restriction fragment length polymorphism
- RIA:
-
Radio immunoassay
- STLV:
-
Simian T-cell lymphotropic virus
References
Kettmann R, Portetelle D, Mammerickx M, Cleuter Y, Dekegel D, Galoux M, Ghysdael J, Burny A, Chantrenne H. Bovine leukemia virus: an exogenous RNA oncogenic virus. Proc Natl Acad Sci U S A. 1976;73:1014–8.
Aida Y, Murakami H, Takahashi M, Takeshima SN. Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front Microbiol. 2013;4:328.
Tozser J. Comparative studies on retroviral proteases: substrate specificity. Viruses. 2010;2:147–65.
Hajj HE, Nasr R, Kfoury Y, Dassouki Z, Nasser R, Kchour G, Hermine O, De The H, Bazarbachi A. Animal models on HTLV-1 and related viruses: what did we learn? Front Microbiol. 2012;3:333.
Kirkland PD, Rodwell BJ. Enzootic Bovine Leukosis. Australia and New Zealand Standard Diagnostic Procedures. 2005:1–14.
Williams DL, Barta O, Amborski GF. Molecular studies of T-lymphocytes from cattle infected with bovine leukemia virus. Vet Immunol Immunopathol. 1988;19:307–23.
Stott ML, Thurmond MC, Dunn SJ, Osburn BI, Stott JL. Integrated bovine leukosis proviral DNA in T helper and T cytotoxic/suppressor lymphocytes. J Gen Virol. 1991;72(Pt 2):307–15.
Schwartz I, Bensaid A, Polack B, Perrin B, Berthelemy M, Levy D. In vivo leukocyte tropism of bovine leukemia virus in sheep and cattle. J Virol. 1994;68:4589–96.
Mirsky ML, Olmstead CA, Da Y, Lewin HA. The prevalence of proviral bovine leukemia virus in peripheral blood mononuclear cells at two subclinical stages of infection. J Virol. 1996;70:2178–83.
Wu D, Takahashi K, Murakami K, Tani K, Koguchi A, Asahina M, Goryo M, Aida Y, Okada K. B-1a, B-1b and conventional B cell lymphoma from enzootic bovine leukosis. Vet Immunol Immunopathol. 1996;55:63–72.
Panei CJ, Takeshima SN, Omori T, Nunoya T, Davis WC, Ishizaki H, Matoba K, Aida Y. Estimation of bovine leukemia virus (BLV) proviral load harbored by lymphocyte subpopulations in BLV-infected cattle at the subclinical stage of enzootic bovine leucosis using BLV-CoCoMo-qPCR. BMC Vet Res. 2013;9:95.
Aida Y, Okada K, Amanuma H. Phenotype and ontogeny of cells carrying a tumor-associated antigen that is expressed on bovine leukemia virus-induced lymphosarcoma. Cancer Res. 1993;53:429–37.
Nekouei O, VanLeeuwen J, Stryhn H, Kelton D, Keefe G. Lifetime effects of infection with bovine leukemia virus on longevity and milk production of dairy cows. Prev Vet Med. 2016;133:1–9.
Sandev N, Koleva M, Binev R, Ilieva D. Influence of enzootic bovine leukosis virus upon the incidence of subclinical mastitis in cows at a different stage of infection. Veterinarski Archiv. 2004;76:411–6.
Bartlett PC, Norby B, Byrem TM, Parmelee A, Ledergerber JT, Erskine RJ. Bovine leukemia virus and cow longevity in Michigan dairy herds. J Dairy Sci. 2013;96:1591–7.
Ferrer JF, Marshak RR, Abt DA, Kenyon SJ. Persistent lymphocytosis in cattle: its cause, nature and relation to lymphosarcoma. Ann Rech Vet. 1978;9:851–7.
OIE. Manual of diagnostic tests and vaccines for terrestrial animals: chapter 2.4.11. Enzootic Bovine Leukosis. Seventh Edition edn. France: World organization for animal health; 2012.
Sagata N, Yasunaga T, Tsuzuku-Kawamura J, Ohishi K, Ogawa Y, Ikawa Y. Complete nucleotide sequence of the genome of bovine leukemia virus: its evolutionary relationship to other retroviruses. Proc Natl Acad Sci U S A. 1985;82:677–81.
Sagata N, Yasunaga T, Ohishi K, Tsuzuku-Kawamura J, Onuma M, Ikawa Y. Comparison of the entire genomes of bovine leukemia virus and human T-cell leukemia virus and characterization of their unidentified open reading frames. EMBO J. 1984;3:3231–7.
Callebaut I, Voneche V, Mager A, Fumiere O, Krchnak V, Merza M, Zavada J, Mammerickx M, Burny A, Portetelle D. Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. J Virol. 1993;67:5321–7.
Inabe K, Nishizawa M, Tajima S, Ikuta K, Aida Y. The YXXL sequences of a transmembrane protein of bovine leukemia virus are required for viral entry and incorporation of viral envelope protein into virions. J Virol. 1999;73:1293–301.
Jewell NA, Mansky LM. The beginning: genome recognition, RNA encapsidation and the initiation of complex retrovirus assembly. J Gen Virol. 2000;81:1889–99.
Hamard-Peron E, Muriaux D. Retroviral matrix and lipids, the intimate interaction. Retrovirology. 2011;8:15.
Bai L, Otsuki H, Sato H, Kohara J, Isogai E, Takeshima SN, Aida Y. Identification and characterization of common B cell epitope in bovine leukemia virus via high-throughput peptide screening system in infected cattle. Retrovirology. 2015;12:106.
Copeland TD, Morgan MA, Oroszlan S. Complete amino acid sequence of the nucleic acid-binding protein of bovine leukemia virus. FEBS Lett. 1983;156:37–40.
Mager A, Masengo R, Mammerickx M, Letesson JJ. T cell proliferative response to bovine leukaemia virus (BLV): identification of T cell epitopes on the major core protein (p24) in BLV-infected cattle with normal haematological values. J Gen Virol. 1994;75(Pt 9):2223–31.
Willems L, Kerkhofs P, Attenelle L, Burny A, Portetelle D, Kettmann R. The major homology region of bovine leukaemia virus p24gag is required for virus infectivity in vivo. J Gen Virol. 1997;78(Pt 3):637–40.
Katoh I, Yasunaga T, Yoshinaka Y. Bovine leukemia virus RNA sequences involved in dimerization and specific gag protein binding: close relation to the packaging sites of avian, murine, and human retroviruses. J Virol. 1993;67:1830–9.
Willems L, Heremans H, Chen G, Portetelle D, Billiau A, Burny A, Kettmann R. Cooperation between bovine leukaemia virus transactivator protein and ha-ras oncogene product in cellular transformation. EMBO J. 1990;9:1577–81.
Willems L, Grimonpont C, Heremans H, Rebeyrotte N, Chen G, Portetelle D, Burny A, Kettmann R. Mutations in the bovine leukemia-virus tax protein can abrogate the long terminal repeat-directed Transactivating activity without concomitant loss of transforming potential. Proc Natl Acad Sci U S A. 1992;89:3957–61.
Tajima S, Aida Y. The region between amino acids 245 and 265 of the bovine leukemia virus (BLV) tax protein restricts transactivation not only via the BLV enhancer but also via other retrovirus enhancers. J Virol. 2000;74:10939–49.
Felber BK, Derse D, Athanassopoulos A, Campbell M, Pavlakis GN. Cross-activation of the Rex proteins of HTLV-I and BLV and of the rev protein of HIV-1 and nonreciprocal interactions with their RNA responsive elements. New Biol. 1989;1:318–28.
Tajima S, Takahashi M, Takeshima SN, Konnai S, Yin SA, Watarai S, Tanaka Y, Onuma M, Okada K, Aida Y. A mutant form of the tax protein of bovine leukemia virus (BLV), with enhanced transactivation activity, increases expression and propagation of BLV in vitro but not in vivo. J Virol. 2003;77:1894–903.
Takahashi M, Tajima S, Takeshima SN, Konnai S, Yin SA, Okada K, Davis WC, Aida Y. Ex vivo survival of peripheral blood mononuclear cells in sheep induced by bovine leukemia virus (BLV) mainly occurs in CD5- B cells that express BLV. Microbes Infect. 2004;6:584–95.
Takahashi M, Tajima S, Okada K, Davis WC, Aida Y. Involvement of bovine leukemia virus in induction and inhibition of apoptosis. Microbes Infect. 2005;7:19–28.
Tajima S, Aida Y. Mutant tax protein from bovine leukemia virus with enhanced ability to activate the expression of c-fos. J Virol. 2002;76:2557–62.
Willems L, Kerkhofs P, Dequiedt F, Portetelle D, Mammerickx M, Burny A, Kettmann R. Attenuation of bovine leukemia virus by deletion of R3 and G4 open reading frames. Proc Natl Acad Sci U S A. 1994;91:11532–6.
Florins A, Gillet N, Boxus M, Kerkhofs P, Kettmann R, Willems L. Even attenuated bovine leukemia virus proviruses can be pathogenic in sheep. J Virol. 2007;81:10195–200.
Kincaid RP, Burke JM, Sullivan CS. RNA virus microRNA that mimics a B-cell oncomiR. Proc Natl Acad Sci U S A. 2012;109:3077–82.
Rosewick N, Momont M, Durkin K, Takeda H, Caiment F, Cleuter Y, Vernin C, Mortreux F, Wattel E, Burny A, et al. Deep sequencing reveals abundant noncanonical retroviral microRNAs in B-cell leukemia/lymphoma. Proc Natl Acad Sci U S A. 2013;110:2306–11.
Gillet NA, Hamaidia M, de Brogniez A, Gutierrez G, Renotte N, Reichert M, Trono K, Willems L. Bovine leukemia virus small noncoding RNAs are functional elements that regulate replication and contribute to Oncogenesis in vivo. PLoS Pathog. 2016;12:e1005588.
Van Driessche B, Rodari A, Delacourt N, Fauquenoy S, Vanhulle C, Burny A, Rohr O, Van Lint C. Characterization of new RNA polymerase III and RNA polymerase II transcriptional promoters in the bovine leukemia virus genome. Sci Rep. 2016;6:31125.
Jimba M, Takeshima SN, Murakami H, Kohara J, Kobayashi N, Matsuhashi T, Ohmori T, Nunoya T, Aida Y. BLV-CoCoMo-qPCR: a useful tool for evaluating bovine leukemia virus infection status. BMC Vet Res. 2012;8:167.
Walker PJ, Molloy JB, Rodwell BJ. A protein immunoblot test for detection of bovine leukemia virus p24 antibody in cattle and experimentally infected sheep. J Virol Methods. 1987;15:201–11.
Portetelle D, Bruck C, Mammerickx M, Burny A. In animals infected by bovine leukemia virus (BLV) antibodies to envelope glycoprotein gp51 are directed against the carbohydrate moiety. Virology. 1980;105:223–33.
Bai L, Takeshima SN, Isogai E, Kohara J, Aida Y. Novel CD8(+) cytotoxic T cell epitopes in bovine leukemia virus with cattle. Vaccine. 2015;33:7194–202.
Aida Y, Miyasaka M, Okada K, Onuma M, Kogure S, Suzuki M, Minoprio P, Levy D, Ikawa Y. Further phenotypic characterization of target cells for bovine leukemia virus experimental infection in sheep. Am J Vet Res. 1989;50:1946–51.
Wang CT. Bovine leukemia virus infection in Taiwan: epidemiological study. J Vet Med Sci. 1991;53:395–8.
Monti GE, Frankena K, Engel B, Buist W, Tarabla HD, de Jong MC. Evaluation of a new antibody-based enzyme-linked immunosorbent assay for the detection of bovine leukemia virus infection in dairy cattle. J Vet Diagn Investig. 2005;17:451–7.
Kurdi A, Blankenstein P, Marquardt O, Ebner D. Serologic and virologic investigations on the presence of BLV infection in a dairy herd in Syria. Berl Munch Tierarztl Wochenschr. 1999;112:18–23.
Fukai K, Sato M, Kawara M, Hoshi Z, Ueno S, Chyou N, Akashi H. A case of an embryo transfer calf infected with bovine leukemia virus from the recipient cow. Zentralbl Veterinarmed B. 1999;46:511–5.
Zaghawa A, Beier D, Abd El-Rahim IH, Karim I, El-ballal S, Conraths FJ, Marquardt O. An outbreak of enzootic bovine leukosis in upper Egypt: clinical, laboratory and molecular-epidemiological studies. J Vet Med B Infect Dis Vet Public Health. 2002;49:123–9.
Schoepf KC, Kapaga AM, Msami HM, Hyera JM. Serological evidence of the occurrence of enzootic bovine leukosis (EBL) virus infection in cattle in Tanzania. Trop Anim Health Prod. 1997;29:15–9.
Levy D, Deshayes L, Parodi AL, Levy JP, Stephenson JR, Devare SG, Gilden RV. Bovine leukemia virus specific antibodies among French cattle. II. Radioimmunoassay with the major structural protein (BLV p24). Int J Cancer. 1977;20:543–50.
Naif HM, Brandon RB, Daniel RCW, Lavin MF. Bovine leukemia Proviral DNA detection in cattle using the polymerase chain-reaction. Vet Microbiol. 1990;25:117–29.
Burridge MJ, Thurmond MC, Miller JM, Schmerr MJ, Van Der Maaten MJ. Fall in antibody titer to bovine leukemia virus in the periparturient period. Can J Comp Med. 1982;46:270–1.
Nguyen VK, Maes RF. Evaluation of an enzyme-linked immunosorbent assay for detection of antibodies to bovine leukemia virus in serum and milk. J Clin Microbiol. 1993;31:979–81.
Ohshima K, Morimoto N, Kagawa Y, Numakunai S, Hirano T, Kayano HA. Survey for maternal antibodies to bovine leukemia virus (BLV) in calves born to cows infected with BLV. Nihon Juigaku Zasshi. 1984;46:583–6.
Kettmann R, Meunier-Rotival M, Cortadas J, Cuny G, Ghysdael J, Mammerickx M, Burny A, Bernardi G. Integration of bovine leukemia virus DNA in the bovine genome. Proc Natl Acad Sci U S A. 1979;76:4822–6.
Kettmann R, Deschamps J, Cleuter Y, Couez D, Burny A, Marbaix G. Leukemogenesis by bovine leukemia virus: proviral DNA integration and lack of RNA expression of viral long terminal repeat and 3′ proximate cellular sequences. Proc Natl Acad Sci U S A. 1982;79:2465–9.
Tajima S, Tsukamoto M, Aida Y. Latency of viral expression in vivo is not related to CpG methylation in the U3 region and part of the R region of the long terminal repeat of bovine leukemia virus. J Virol. 2003;77:4423–30.
Tajima S, Aida Y. Induction of expression of bovine leukemia virus (BLV) in blood taken from BLV-infected cows without removal of plasma. Microbes Infect. 2005;7:1211–6.
Kettmann R, Cleuter Y, Mammerickx M, Meunier-Rotival M, Bernardi G, Burny A, Chantrenne H. Genomic integration of bovine leukemia provirus: comparison of persistent lymphocytosis with lymph node tumor form of enzootic. Proc Natl Acad Sci U S A. 1980;77:2577–81.
Tajima S, Ikawa Y, Aida Y. Complete bovine leukemia virus (BLV) provirus is conserved in BLV-infected cattle throughout the course of B-cell lymphosarcoma development. J Virol. 1998;72:7569–76.
Burny A, Cleuter Y, Kettmann R, Mammerickx M, Marbaix G, Portetelle D, Vandenbroeke A, Willems L, Thomas R. Bovine leukemia - facts and hypotheses derived from the study of an infectious cancer. Vet Microbiol. 1988;17:197–218.
Brym P, Rusc A, Kaminski S. Evaluation of reference genes for qRT-PCR gene expression studies in whole blood samples from healthy and leukemia-virus infected cattle. Vet Immunol Immunopathol. 2013;153:302–7.
Tawfeeq MM, Horiuchi N, Kobayashi Y, Furuoka H, Inokuma H. Evaluation of gene expression in peripheral blood cells as a potential biomarker for enzootic bovine Leukosis. J Vet Med Sci. 2013;75:1213–7.
Somura Y, Sugiyama E, Fujikawa H, Murakami K. Comparison of the copy numbers of bovine leukemia virus in the lymph nodes of cattle with enzootic bovine leukosis and cattle with latent infection. Arch Virol. 2014;159:2693–7.
Lew AE, Bock RE, Miles J, Cuttell LB, Steer P, Nadin-Davis SA. Sensitive and specific detection of bovine immunodeficiency virus and bovine syncytial virus by 5’Taq nuclease assays with fluorescent 3’minor groove binder-DNA probes. J Virol Methods. 2004;116:1–9.
Jimba M, Takeshima SN, Matoba K, Endoh D, Aida Y. BLV-CoCoMo-qPCR: Quantitation of bovine leukemia virus proviral load using the CoCoMo algorithm. Retrovirology. 2010;7:91.
Takeshima SN, Kitamura-Muramatsu Y, Yuan Y, Polat M, Saito S, Aida Y. BLV-CoCoMo-qPCR-2: improvements to the BLV-CoCoMo-qPCR assay for bovine leukemia virus by reducing primer degeneracy and constructing an optimal standard curve. Arch Virol. 2015;160:1325–32.
Nishimori A, Konnai S, Ikebuchi R, Okagawa T, Nakahara A, Murata S, Ohashi K. Direct polymerase chain reaction from blood and tissue samples for rapid diagnosis of bovine leukemia virus infection. J Vet Med Sci. 2016;78:791–6.
Takeshima SN, Watanuki S, Ishizaki H, Matoba K, Aida Y. Development of a direct blood-based PCR system to detect BLV provirus using CoCoMo primers. Arch Virol. 2016;161:1539–46.
Polat M, Ohno A, Takeshima SN, Kim J, Kikuya M, Matsumoto Y, Mingala CN, Onuma M, Aida Y. Detection and molecular characterization of bovine leukemia virus in Philippine cattle. Arch Virol. 2015;160:285–96.
Polat M, Takeshima SN, Hosomichi K, Kim J, Miyasaka T, Yamada K, Arainga M, Murakami T, Matsumoto Y, de la Barra Diaz V, et al. A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology. 2016;13:4.
Polat M, Moe HH, Shimogiri T, Moe KK, Takeshima SN, Aida Y. The molecular epidemiological study of bovine leukemia virus infection in Myanmar cattle. Arch Virol. 2016.
Ochirkhuu N, Konnai S, Odbileg R, Nishimori A, Okagawa T, Murata S, Ohashi K. Detection of bovine leukemia virus and identification of its genotype in Mongolian cattle. Arch Virol. 2016;161:985–91.
Dus Santos MJ, Trono K, Lager I, Wigdorovitz A. Development of a PCR to diagnose BLV genome in frozen semen samples. Vet Microbiol. 2007;119:10–8.
Martin D, Arjona A, Soto I, Barquero N, Viana M, Gomez-Lucia E. Comparative study of PCR as a direct assay and ELISA and AGID as indirect assays for the detection of bovine leukaemia virus. J Vet Med B Infect Dis Vet Public Health. 2001;48:97–106.
Heenemann K, Lapp S, Teifke JP, Fichtner D, Mettenleiter TC, Vahlenkamp TW. Development of a bovine leukemia virus polymerase gene-based real-time polymerase chain reaction and comparison with an envelope gene-based assay. J Vet Diagn Investig. 2012;24:649–55.
Kelly EJ, Jackson MK, Marsolais G, Morrey JD, Callan RJ. Early detection of bovine leukemia virus in cattle by use of the polymerase chain reaction. Am J Vet Res. 1993;54:205–9.
Miyasaka T, Takeshima SN, Jimba M, Matsumoto Y, Kobayashi N, Matsuhashi T, Sentsui H, Aida Y. Identification of bovine leukocyte antigen class II haplotypes associated with variations in bovine leukemia virus proviral load in Japanese black cattle. Tissue Antigens. 2013;81:72–82.
Ohno A, Takeshima SN, Matsumoto Y, Aida Y. Risk factors associated with increased bovine leukemia virus proviral load in infected cattle in Japan from 2012 to 2014. Virus Res. 2015;210:283–90.
Yuan Y, Kitamura-Muramatsu Y, Saito S, Ishizaki H, Nakano M, Haga S, Matoba K, Ohno A, Murakami H, Takeshima SN, Aida Y. Detection of the BLV provirus from nasal secretion and saliva samples using BLV-CoCoMo-qPCR-2: comparison with blood samples from the same cattle. Virus Res. 2015;210:248–54.
Inabe K, Ikuta K, Aida Y. Transmission and propagation in cell culture of virus produced by cells transfected with an infectious molecular clone of bovine leukemia virus. Virology. 1998;245:53–64.
Mamoun RZ, Morisson M, Rebeyrotte N, Busetta B, Couez D, Kettmann R, Hospital M, Guillemain B. Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J Virol. 1990;64:4180–8.
Portetelle D, Couez D, Bruck C, Kettmann R, Mammerickx M, Van der Maaten M, Brasseur R, Burny A. Antigenic variants of bovine leukemia virus (BLV) are defined by amino acid substitutions in the NH2 part of the envelope glycoprotein gp51. Virology 1989; 169:27-33.
Bruck C, Mathot S, Portetelle D, Berte C, Franssen JD, Herion P, Burny A. Monoclonal antibodies define eight independent antigenic regions on the bovine leukemia virus (BLV) envelope glycoprotein gp51. Virology. 1982;122:342–52.
Kettmann R, Couez D, Burny A. Restriction endonuclease mapping of linear unintegrated proviral DNA of bovine leukemia virus. J Virol. 1981;38:27–33.
Coulston J, Naif H, Brandon R, Kumar S, Khan S, Daniel RC, Lavin MF. Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: comparison with other isolates. J Gen Virol. 1990;71:1737–46.
Fechner H, Blankenstein P, Looman AC, Elwert J, Geue L, Albrecht C, Kurg A, Beier D, Marquardt O, Ebner D. Provirus variants of the bovine leukemia virus and their relation to the serological status of naturally infected cattle. Virology. 1997;237:261–9.
Licursi M, Inoshima Y, Wu D, Yokoyama T, Gonzalez ET, Sentsui H. Genetic heterogeneity among bovine leukemia virus genotypes and its relation to humoral responses in hosts. Virus Res. 2002;86:101–10.
Licursi M, Inoshima Y, Wu D, Yokoyama T, Gonzalez ET, Sentsui H. Provirus variants of bovine leukemia virus in naturally infected cattle from Argentina and Japan. Vet Microbiol. 2003;96:17–23.
Felmer R, Munoz G, Zuniga J, Recabal M. Molecular analysis of a 444 bp fragment of the bovine leukaemia virus gp51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet Microbiol. 2005;108:39–47.
Monti G, Schrijver R, Beier D. Genetic diversity and spread of bovine leukaemia virus isolates in argentine dairy cattle. Arch Virol. 2005;150:443–58.
Asfaw Y, Tsuduku S, Konishi M, Murakami K, Tsuboi T, Wu D, Sentsui H. Distribution and superinfection of bovine leukemia virus genotypes in Japan. Arch Virol. 2005;150:493–505.
Rodriguez SM, Golemba MD, Campos RH, Trono K, Jones LR. Bovine leukemia virus can be classified into seven genotypes: evidence for the existence of two novel clades. J Gen Virol. 2009;90:2788–97.
Balic D, Lojkic I, Periskic M, Bedekovic T, Jungic A, Lemo N, Roic B, Cac Z, Barbic L, Madic J. Identification of a new genotype of bovine leukemia virus. Arch Virol. 2012;157:1281–90.
Matsumura K, Inoue E, Osawa Y, Okazaki K. Molecular epidemiology of bovine leukemia virus associated with enzootic bovine leukosis in Japan. Virus Res. 2011;155:343–8.
Rola-Luszczak M, Pluta A, Olech M, Donnik I, Petropavlovskiy M, Gerilovych A, Vinogradova I, Choudhury B, Kuzmak J. The molecular characterization of bovine leukaemia virus isolates from Eastern Europe and Siberia and its impact on phylogeny. PLoS One. 2013;8:e58705.
Lee E, Kim EJ, Joung HK, Kim BH, Song JY, Cho IS, Lee KK, Shin YK. Sequencing and phylogenetic analysis of the gp51 gene from Korean bovine leukemia virus isolates. Virol J. 2015;12:64.
Lee E, Kim EJ, Ratthanophart J, Vitoonpong R, Kim BH, Cho IS, Song JY, Lee KK, Shin YK. Molecular epidemiological and serological studies of bovine leukemia virus (BLV) infection in Thailand cattle. Infect Genet Evol. 2016;41:245–54.
OIE. World animal health infromation database-version: 1.4. Paris France: World organisation for animal Health; 2009.
Nuotio L, Rusanen H, Sihvonen L, Neuvonen E. Eradication of enzootic bovine leukosis from Finland. Prev Vet Med. 2003;59:43–9.
Acaite J, Tamosiunas V, Lukauskas K, Milius J, Pieskus J. The eradication experience of enzootic bovine leukosis from Lithuania. Prev Vet Med. 2007;82:83–9.
Kautzsch S, Schluter H. Prognosis and economic-aspects relating to control of enzootic bovine Leukosis. Monatsh Veterinarmed 1990; 45:41-45.
Maresca C, Costarelli S, Dettori A, Felici A, Iscaro C, Feliziani F. Enzootic bovine leukosis: report of eradication and surveillance measures in Italy over an 8-year period (2005-2012). Prev Vet Med. 2015;119:222–6.
Gottschau A, Willeberg P, Franti CE, Flensburg JC. The effect of a control program for enzootic bovine leukosis. Changes in herd prevalence in Denmark, 1969-1978. Am J Epidemiol. 1990;131:356–64.
Stark KD. Animal health monitoring and surveillance in Switzerland. Aust Vet J. 1996;73:96–7.
EFSA Panel on Animal Health and Welfare, More SJ, Bøtner A, Butterworth A, Calistri P, Depner K, Bicout D. Assessment of listing and categorisation of animal diseases within the framework of the Animal Health Law (Regulation (EU) No 2016/429): enzootic bovine leukosis (EBL). EFSA J. 2017;15(8):1–28.
Zhao XR, Jimenez C, Sentsui H, Buehring GC. Sequence polymorphisms in the long terminal repeat of bovine leukemia virus: evidence for selection pressures in regulatory sequences. Virus Res 2007; 124:113–124.
Sandev N, Illieva D, Rusenova N, Marasheva V. Prevalence of enzootic Bobivne Leukosis in Bulgaria. Bulletin UASVM Veterinary Medicine. 2015;72:43–6.
European Panal on Animal Health and Welfare (EPAHW). Scientific opinion on enzootic bovine leukosis. EFSA J. 2015;13:63.
Chethanond U-S. The epidemiology of enzootic bovine leukosis in diary cattle in New Zealand. Massey University; 1999.
APHIS. Bovine Leukosis Virus (BLV) on U.S.Dairy Operations, 2007. United states department of agriculture; 2008.
VanLeeuwen JA, Keefe GP, Tremblay R, Power C, Wichtel JJ. Seroprevalence of infection with Mycobacterium Avium subspecies paratuberculosis, bovine leukemia virus, and bovine viral diarrhea virus in maritime Canada dairy cattle. Can Vet J. 2001;42:193–8.
VanLeeuwen JA, Forsythe L, Tiwari A, Chartier R. Seroprevalence of antibodies against bovine leukemia virus, bovine viral diarrhea virus, Mycobacterium Avium subspecies paratuberculosis, and Neospora caninum in dairy cattle in Saskatchewan. Can Vet J. 2005;46:56–8.
Nekouei OA. Study of prevalence, risk factors, and lifetime impacts of infection with bovine keukemia virus in the Canadian dairy industry. University of Prince Edward Island, Atlanti veterinary college, department of. Health Management. 2015;
Suzan VM, Onuma M, Aguilar RE, Murakami Y. Prevalence of bovine Herpesvirus-1, Para-Influenza-3, bovine rotavirus, bovine viral diarrhea, bovine Adenovirus-7, bovine leukemia-virus and bluetongue virus-antibodies in cattle in Mexico. Jap J Vet Res. 1983;31:125–32.
Samara SI, Lima EG, Nascimento AA. Monitoring of enzootic bovine leukosis in dairy cattle from the Pitangueiras region in São Paulo, Brazil. Braz J Vet Res Anim Sci. 1997;34:349–51.
D'Angelino JL, Garcia M, Birgel EH. Epidemiological study of enzootic bovine leukosis in Brazil. Trop Anim Health Prod. 1998;30:13–5.
Camargos MF, Stancek D, Rocha MA, Lessa LM, Reis JK, Leite RC. Partial sequencing of env gene of bovine leukaemia virus from Brazilian samples and phylogenetic analysis. J Vet Med B Infect Dis Vet Public Health. 2002;49:325–31.
Camargos MF, Pereda A, Stancek D, Rocha MA, dos Reis JK, Greiser-Wilke I, Leite RC. Molecular characterization of the env gene from Brazilian field isolates of bovine leukemia virus. Virus Genes. 2007;34:343–50.
Trono KG, Perez-Filgueira DM, Duffy S, Borca MV, Carrillo C. Seroprevalence of bovine leukemia virus in dairy cattle in Argentina: comparison of sensitivity and specificity of different detection methods. Vet Microbiol. 2001;83:235–48.
Bovine CAH. Leukaemia virus infection in Peru. Trop Anim Health Prod. 1983;15:61.
Marín C, de López NM, Alvarez L, Lozano O, España W, Castaños H, León A. Epidemiology of bovine leukemia in Venezuela. Ann Rech Vet. 1978;9:4.
Moratorio G, Obal G, Dubra A, Correa A, Bianchi S, Buschiazzo A, Cristina J, Pritsch O. Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch Virol. 2010;155:481–9.
Rama G, Moratorio G, Greif G, Obal G, Bianchi S, Tomé L, Carrion F, Meikle A, Pritsch O. Development of a real time PCR assay using SYBR Green chemistry for bovine leukemia virus detection. Retrovirology. 2011: 8.
Alfonso R, Almansa JE, Barrera JD. Serological prevalence and evaluation of risk factors of enzootic bovine leukosis in the Sabana de Bogota region and the Ubate and Chiquinquira valleys of Colombia. Revue Scientifique Et Technique De L Office International Des Epizooties. 1998;17:723–32.
Hernández-Herrera DY, Posso-Terranova A, Benavides J, Muñoz-Flórez J, Giovambattista G, Álvarez-Franco L. Bovine leukosis virus detection in Creole Colombian breeds using nested-PCR. ACTA AGRONÓMICA. 2011;60:311–7.
Benavides B, Quevedo D, Cruz M. Epidemiological study of bovine leukemia virus in dairy cows in six herds in the municipality of Pasto Nariño. Revista Lasallista de Investigación. 2013;10:18–26.
Yang Y, Fan W, Mao Y, Yang Z, Lu G, Zhang R, Zhang H, Szeto C, Wang C. Bovine leukemia virus infection in cattle of China: association with reduced milk production and increased somatic cell score. J Dairy Sci. 2016;99:3688–97.
Ma JG, Zheng WB, Zhou DH, Qin SY, Yin MY, Zhu XQ, Hu GX. First Report of Bovine Leukemia Virus Infection in Yaks (Bos mutus) in China. Biomed Res Int. 2016:9170167.
Kobayashi S, Tsutsui T, Yamamoto T, Hayama Y, Kameyama K, Konishi M, Murakami K. Risk factors associated with within-herd transmission of bovine leukemia virus on dairy farms in Japan. BMC Vet Res. 2010;6:1.
Murakami K, Kobayashi S, Konishi M, Kameyama K, Yamamoto T, Tsutsui T. The recent prevalence of bovine leukemia virus (BLV) infection among Japanese cattle. Vet Microbiol. 2011;148:84–8.
Murakami K, Kobayashi S, Konishi M, Kameyama K, Tsutsui T. Nationwide survey of bovine leukemia virus infection among dairy and beef breeding cattle in Japan from 2009-2011. J Vet Med Sci. 2013;75:1123–6.
Meas S, Ohashi K, Tum S, Chhin M, Te K, Miura K, Sugimoto C, Onuma M. Seroprevalence of bovine immunodeficiency virus and bovine leukemia virus in draught animals in Cambodia. J Vet Med Sci. 2000;62:779–81.
Nekoei S, Hafshejani TT, Doosti A, Khamesipour F. Molecular detection of bovine leukemia virus in peripheral blood of Iranian cattle, camel and sheep. Pol J Vet Sci. 2015;18:703–7.
Mousavi S, Haghparast A, Mohammadi G, Tabatabaeizadeh SE. Prevalence of bovine leukemia virus (BLV) infection in the northeast of Iran. Vet Res Forum. 2014;5:135–9.
Trainin Z, Brenner J. The direct and indirect economic impacts of bovine leukemia virus infection on dairy cattle. Israel Journal of Veterinary Medicine. 2005;60:90–105.
Hafez SM, Sharif M, Al-Sukayran A, Dela-Cruz D. Preliminary studies on enzootic bovine leukosis in Saudi dairy farms. Dtsch Tierarztl Wochenschr. 1990;97:61–3.
Burgu I, Alkan F, Karaoglu T, Bilge-Dagalp S, Can-Sahna K, Gungor B, Demir B. Control and eradication programme of enzootic bovine leucosis (EBL) from selected dairy herds in Turkey. Dtsch Tierarztl Wochenschr. 2005;112:271–4.
Zhao X, Buehring GC. Natural genetic variations in bovine leukemia virus envelope gene: possible effects of selection and escape. Virology. 2007;366:150–65.
Molteni E, Agresti A, Meneveri R, Marozzi A, Malcovati M, Bonizzi L, Poli G, Ginelli E. Molecular characterization of a variant of proviral bovine leukaemia virus (BLV). Zentralbl Veterinarmed B. 1996;43:201–11.
Derse D, Diniak AJ, Casey JW, Deininger PL. Nucleotide sequence and structure of integrated bovine leukemia virus long terminal repeats. Virology. 1985;141:162–6.
Yang Y, Kelly PJ, Bai J, Zhang R, Wang C. First molecular characterization of bovine leukemia virus infections in the Caribbean. PLoS One. 2016;11:e0168379.
Dube S, Dolcini G, Abbott L, Mehta S, Dube D, Gutierrez S, Ceriani C, Esteban E, Ferrer J, Poiesz B. The complete genomic sequence of a BLV strain from a Holstein cow from Argentina. Virology. 2000;277:379–86.
Dube S, Abbott L, Dube DK, Dolcini G, Gutierrez S, Ceriani C, Juliarena M, Ferrer J, Perzova R, Poiesz BJ. The complete genomic sequence of an in vivo low replicating BLV strain. Virol J. 2009;6:120.
Lim SI, Jeong W, Tark DS, Yang DK, Kweon CH. Agar gel immunodiffusion analysis using baculovirus-expressed recombinant bovine leukemia virus envelope glycoprotein (gp51/gp30(T-)). J Vet Sci. 2009;10:331–6.
Inoue E, Matsumura K, Maekawa K, Nagatsuka K, Nobuta M, Hirata M, Minagawa A, Osawa Y, Okazaki K. Genetic heterogeneity among bovine leukemia viruses in Japan and their relationship to leukemogenicity. Arch Virol. 2011;156:1137–41.
Ababneh MM, Al-Rukibat RK, Hananeh WM, Nasar AT, Al-Zghoul MB. Detection and molecular characterization of bovine leukemia viruses from Jordan. Arch Virol. 2012;157:2343–8.
Rodriguez SM, Florins A, Gillet N, de Brogniez A, Sanchez-Alcaraz MT, Boxus M, Boulanger F, Gutierrez G, Trono K, Alvarez I, et al. Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Viruses. 2011;3:1210–48.
Inoue E, Matsumura K, Soma N, Hirasawa S, Wakimoto M, Arakaki Y, Yoshida T, Osawa Y, Okazaki K. L233P mutation of the tax protein strongly correlated with leukemogenicity of bovine leukemia virus. Vet Microbiol. 2013;167:364–71.
Murakami H, Asano S, Uchiyama J, Sato R, Sakaguchi M, Tsukamoto K. Bovine leukemia virus G4 enhances virus production. Virus Res. 2017;238:213–7.
Acknowledgments
We thank our collaborators for kindly assisting with the large-scale sampling from many farms in the Philippine, Myanmar, South America (Argentina, Peru, Paraguay, Chile and Bolivia) and Japan.
Funding
The studies on BLV were supported by Grants-in-Aid for Scientific Research [A (08021470), A (16H02590), B (10004294), and C (25450405)] from the Japan Society for the Promotion of Science (JSPS), by a grant from Integration Research for Agriculture and Interdisciplinary Fields in Japan (14538311), and by a grant from the Project of the NARO Bio-oriented Technology Research Advancement Institution (the special scheme project on regional developing strategy) (Grant No. 16817983)].
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
Yoko Aida designed the concept of the review article, edited and revised the manuscript. Meripet Polat wrote the manuscript, constructed Tables and Figures, and edited and revised the manuscript. Shin-nosuke Takeshima wrote some part and helped with the revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Polat, M., Takeshima, Sn. & Aida, Y. Epidemiology and genetic diversity of bovine leukemia virus. Virol J 14, 209 (2017). https://doi.org/10.1186/s12985-017-0876-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0876-4