
Abstract
Bovine leukemia virus (BLV) is the etiological agent of enzootic bovine leukosis, which is the most common neoplastic disease of cattle. BLV infects cattle worldwide, imposing a severe economic impact on the dairy cattle industry. However, there are no comprehensive studies on the distribution of BLV in the Philippines, and the genetic characteristics of Philippine BLV strains are unknown. Therefore, the aim of this study was to detect BLV infections in the Philippines and determined their genetic variability. Blood samples were obtained from 1116 cattle from different farms on five Philippine islands, and BLV provirus was detected by BLV-CoCoMo-qPCR-2 and nested PCR targeting BLV long terminal repeats. Out of 1116 samples, 108 (9.7 %) and 54 (4.8 %) were positive for BLV provirus, as determined by BLV-CoCoMo-qPCR-2 and nested PCR, respectively. Of the five islands, Luzon Island showed the highest prevalence of BLV infection (23.1 %). Partial env gp51 genes from 43 samples, which were positive for BLV provirus by both methods, were sequenced for phylogenetic analysis. Phylogenetic analysis based on a 423-bp fragment of the env gene revealed that Philippine BLV strains clustered into either genotype 1 or genotype 6. Substitutions were mainly found in antigenic determinants, such as the CD4+ T-cell epitope, the CD8+ T-cell epitope, the second neutralizing domain, B and E epitopes, and these substitutions varied according to genotype. This study provides comprehensive information regarding BLV infection levels in the Philippines and documents the presence of two BLV genotypes, genotypes 1 and 6, in this population.
Similar content being viewed by others

Introduction
Bovine leukemia virus (BLV) is an oncogenic member of the genus Deltaretrovirus of the family Retroviridae and is the etiological agent of enzootic bovine leukosis (EBL), which is the most common neoplastic disease of cattle [1]. BLV is closely related to human T-cell leukemia viruses type 1 and 2 (HTLV-1 and -2) [1]. In addition to cattle, BLV has also been found in human tissues, such as human breast tissue, suggesting a risk for the acquisition and proliferation of this virus in humans [2]. Cattle infected with BLV are usually asymptomatic; approximately 30 % of infected cattle progress to persistent lymphocytosis (PL), the polyclonal expression of non-neoplastic CD5+ B lymphocytes [1, 3]. By contrast, after 1–8 years of latency, 1–5 % of infected animals develop tumors, characterized as malignant CD5+ B-cell lymphoma [3, 4].
In addition to the structural and enzymatic Gag, Pol and Env proteins, the BLV genome contains regulatory genes including Tax, Rex, R3 and G4 [1]. BLV has two identical long terminal repeats (LTRs), which possess a U3 region, an unusually long R region and a U5 region; these LTRs only exert efficient transcriptional promoter activity in cells productively infected with BLV [1]. BLV integrates into the host genome as a provirus [5]. It appears that BLV-infected cattle retain at least one copy of the full-length proviral genome throughout the course of the disease [6]. Moreover, large or small deletions of the proviral genome are thought to be very rare events in BLV-infected cattle [6].
The above findings suggest that the BLV provirus remains integrated in cellular genomes [6, 7], even in the absence of detectable BLV antibodies. After infection, BLV expression in cattle is blocked at the transcriptional level during the latent period [8–10]. Therefore, in addition to the routine diagnosis of BLV infection using conventional serological techniques, such as agar gel immunodiffusion and enzyme-linked immunosorbent assay, diagnostic BLV PCR techniques that detect the integrated BLV proviral genome within the host genome are also commonly used [6]. BLV is endemic worldwide; however, there is no effective vaccine or therapy [11]. Detection and genetic variation analysis of BLV isolates are important for diagnostic purposes and vaccine development. Therefore, we recently developed a new quantitative real-time PCR method using coordination of common motifs (CoCoMo) primers to measure the proviral load of both known and novel BLV variants in BLV-infected animals [7, 12, 13]. The assay was highly effective in detecting BLV in cattle from a number of international locations. The BLV-CoCoMo-qPCR technique amplifies a single-copy host gene, the bovine leukocyte antigen (BoLA)-DRA gene, in parallel with viral genomic DNA, which effectively normalizes the level of viral genomic DNA. Recently, BLV-CoCoMo-qPCR-2 was improved from the original BLV-CoCoMo-qPCR by optimizing the primer degeneracy and PCR conditions and reconstructing the standard plasmid (Takeshima et al., unpublished work).
The Env glycoprotein plays an essential role in the viral life cycle. It is required for cell entry and is the target of neutralizing antibodies [14, 15]. The env gene also contains a variable region containing sequences coding for epitopes involved in antigen-antibody reactions and virus-host interactions [16, 17]. The BLV env gene encodes a polyprotein precursor (gPr72), which is cleaved to form the gp51 Env surface and gp30 transmembrane glycoprotein [1]. The N-terminal half of mature BLV gp51 plays an important role in viral infectivity and syncytium formation [18, 19] and contains the conformational epitopes F, G and H [15, 20], whilst the C-terminal half of BLV gp51 contains the linear epitopes A, B, D and E [15].
Analysis of BLV gp51 env gene sequences from different geographic regions has revealed the presence of different genetic groups that correlate with the geographic origin of the isolates [21–29]. Recent phylogenetic studies on the BLV env gene of strains isolated worldwide demonstrate that the virus can be classified into eight genotypes [27, 30, 31]. However, there are few studies on the distribution of BLV infections on Philippine farms, and until now, no study has focused on the genetic variability of BLV in the Philippines. In this study, we investigate the spread of BLV infection in the Philippines by amplification of BLV LTRs using a combination of BLV-CoCoMo-qPCR-2 and nested PCR, and we investigate the genetic variability of Philippine BLV strains by DNA sequencing and phylogenetic analyses based on a 423-bp sequence of the BLV env gene. This is the first study to identity the BLV genotypes in the Philippines.
Materials and methods
Animals and extraction of genomic DNA
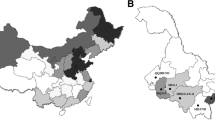
Blood samples were taken from 1116 cattle from different farms on five Philippine islands as shown in Fig. 1. All animals were handled by veterinarians from the RIKEN and the Philippine Carabao Center in strict accordance with good animal practice following the Philippine Carabao Center institutional guidelines. This experiment was approved by the Institutional Animal Care and Use Committee (IACUC) at the Philippine Carabao Center on the Ethics of Animals for Research. Genomic DNA was extracted from 40 μl of whole blood spotted onto Whatman FTA elute cards (GE Healthcare Japan Corp., Tokyo, Japan) according to the manufacturer’s instructions.

Map of the Philippine Islands showing the number of cattle from islands included in the study. The five islands where sampling was performed are indicated and colored in the figure
Detection of BLV provirus by BLV-CoCoMo-qPCR-2
BLV LTR regions were detected by BLV-CoCoMo-qPCR-2 as described previously (Takeshima et al., unpublished work) [12]. Briefly, a 120-bp region of the BLV-LTR was amplified using degenerate primer pairs as shown in Table 1. A single copy of the bovine leukocyte antigen (BoLA)-DRA gene was also amplified, which effectively normalizes the level of viral genomic DNA. Both viral and host genes were detected with a FAM probe. Sterilized water was used as a negative control for PCR.
Detection of BLV provirus by nested PCR
BLV LTR regions were detected by nested PCR, using the primers shown in Table 1. Briefly, the first PCR amplification was performed with primers BLTR256F and BLTR453R. As an internal control, the BoLA-DRA gene was amplified using primers BDRA488F and BDRA1145R as described above. Each primer at a final concentration of 5 pM, 2 μl of 10 × rTaq PCR Buffer (Toyobo, Osaka, Japan), 2 μl of 25 pM MgCl2, 2 μl of 2 mM dNTP, 0.1 μl of 5 U/μl rTaq and 10.4 μl of nuclease-free water were added to each sample, which was amplified in a final volume of 20 μl for 45 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s and then a final extension step of 4 min. Two microliters of Exo-SAP IT (USB Corp., Cleveland, OH) was applied to purify the first PCR products under conditions of 37 °C for 15 min and 80 °C for 15 min. The first PCR amplicons were subsequently applied to the second PCR with primers BLTR306F and BLTR408R as shown in Table 1. Each primer at a final concentration of 5 pM, 2 μl of 10 × rTaq PCR Buffer, 2 μl of 25 pM MgCl2, 2 μl of 2 mM dNTP, 0.1 μl of 5 U/μl rTaq and 11.9 μl of nuclease-free water were added to 1 μl of each purified PCR product. Sterilized water was used as a negative control for PCR.
PCR amplification of BLV env gene fragments and nucleotide sequences
The partial BLV env gene was amplified by nested PCR. PCR amplification was performed using PrimeSTAR GXL DNA Polymerase (Takara Bio Inc., Otsu, Japan) using primers described previously [28, 32], as shown in Table 1. The reaction mixture contained 13.5 μl (1st PCR) and 14.5 μl (2nd PCR) of distilled water, 5 μl of 5 × PS GXL Buffer, 2 μl of 2.5 mM dNTP mixture, 0.5 μl of PrimeSTAR GXL and 1 μl of each primer (10 μM). PCR amplification consisted of 30 cycles of 98 °C for 15 s, 60 °C for 20 s and 68 °C for 60 s. The external primers resulted in amplification of a 913-bp DNA fragment, and internal primers amplified a 597-bp fragment of the gp51 region of the env gene.
Positive second-round PCR products were purified using 5 × Exo-SAP IT (USB Corp., Cleveland, OH) and were sequenced on an ABI3730xl DNA Analyzer using an ABI PRISM BigDye Terminator v 3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). Sequences included a 423-bp region of the env gene, corresponding to nucleotide positions 5126 to 5548 of the BLV genome. Editing, alignment and identification of nucleotide sequences were performed using MEGA 5.1 software [33].
Construction of a phylogenetic tree
The 43 partial BLV env sequences from the Philippines were aligned together with 72 BLV env sequences from GenBank, which are representative of eight known BLV genotypes, by MEGA 5.1 software [33]. Phylogenetic analyses of a 423-bp region of the env gene were conducted using MEGA 5.1 [33]. Phylogenetic trees were constructed using the neighbor-joining algorithm [34] with the Tamura-Nei model of nucleotide substitution [35] based on alignments of the partial env sequence.
Results
Detection of BLV infection by BLV-CoCoMo-qPCR-2 and nested PCR
A total of 1116 samples obtained from different farms on five Philippine islands were screened for BLV infection. A total of 174 cattle samples, including 72 cattle from Bohol Island and 102 cattle from Cebu Island, were negative for BLV provirus, as determined by BLV-CoCoMo-qPCR-2 and nested PCR, which detected BLV LTRs, present at two copies per provirus (Tables 2 and 3). Cattle were classified as Philippine native cattle, Philippine native × Brahman cattle, Holstein × Sahiwal cattle, Holstein × Jersey cattle and Holstein × Brahman × Sahiwal cattle. On Leyte Island, only 4 of 372 cattle samples were positive for BLV provirus, as determined by BLV-CoCoMo-qPCR-2, but they were negative for BLV, as determined by nested PCR (Table 4). All of these four cattle were Philippine native cattle. Of the 159 samples collected from six farms on Iloilo Island, only three were positive for BLV provirus by both BLV-CoCoMo-qPCR-2 and nested PCR, with only six samples BLV positive by BLV-CoCoMo-qPCR-2 but not determined by nested PCR (Table 5). Eight of these nine cattle were Philippine native cattle.
In contrast to cattle living in Bohol, Cebu, Iloilo and Leyte Islands, which are located in the southern Philippines, of 411 samples collected on Luzon Island, which is located in the northern Philippines, 95 samples (23.1 %) and 51 (12.4 %) were positive for BLV provirus, as determined by BLV-CoCoMo-qPCR-2 and by nested PCR, respectively (Table 6). Thus, Luzon Island showed the highest frequency of BLV infection. Interestingly, BLV-infected cattle found on Luzon Island were mainly Holstein × Sahiwal (64.2 %) and Brahman cattle (29.5 %), rather than Philippine native cattle (0.1 %). In particular, 52.6 % of BLV-infected cattle originated from “G” farm of “V” City, Luzon Island. This farm is a dairy farm raising Holstein × Sahiwal cattle. In addition, cattle from beef farms raising Brahman breeds were the next most frequently infected on Luzon Island.
These results suggest that the prevalence of BLV infection in Philippine cattle is different in two populations: southern (Bohol, Cebu, Iloilo, and Leyte Islands) and northern (Luzon Island) Philippines.
Phylogenetic analysis of a partial sequence of BLV env-gp51
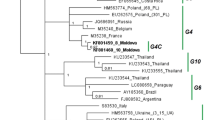
Of the 1116 samples screened, 54 showed positive results for BLV detection by both BLV-CoCoMo-qPCR-2 and nested PCR. Only samples that were positive by both methods were used for further analysis in terms of successful amplification of BLV env-gp51. Therefore, to gain insight into the degree of genetic variability of BLV strains in the Philippines, 43 field strains, which were representative of different breeds on the BLV-infected farms, were selected for phylogenetic characterization. After direct sequencing, 423-bp sequences corresponding to nucleotide positions 5126 to 5548 of the full-length BLV genome (BLV isolate LS3, GenBank accession number HE 967303) were aligned with 72 corresponding sequences from known BLV strains representing all BLV genotypes, including genotypes 1-8 [27, 29–31]. A neighbor-joining phylogenetic tree was then constructed using the Tamura-Nei model of nucleotide substitution [35]. The phylogenetic tree is shown in Fig. 2. The results were similar to those of previous studies where BLV strains were divided into eight genotypes [27, 30, 31] and an extra Iranian isolate cluster [36]. Interestingly, the 43 Philippine BLV strains clustered either with genotype 1 or genotype 6.

Neighbor-joining phylogenetic tree based on 423-bp nucleotide sequences of env genes from 43 BLV strains in the Philippines and BLV strains isolated elsewhere. Philippine BLV strains are indicated by the sample ID together with the collection time and country name. The remaining isolates in the tree are indicated by accession number and country of origin. Philippine BLV strains are indicated by open (○) and filled (●) circles. Typical BLV isolates from identical samples were aligned as shown in Figure 3A and are indicated by filled circles (●). Genotypes are indicated by the numbers on the right of the figure. The bar at the bottom of the figure denotes distance
The nucleotide sequence similarity of the 423-bp BLV env gene sequence ranged from 97.2 % to 100 % for the 43 BLV strains studied here. These sequences were 94.8 % to 99.8 % similar to those corresponding to all known genotypes deposited in GenBank. Thirty-two of the 43 BLV strains showed the highest nucleotide sequence similarity to the reference sequence of genotype 1 (98.6 to 99.8 %). By contrast, the other 11 BLV strains were 98.8 to 99.1 % similar to genotype 6. These former strains were identified in close proximity to each other and were assigned to genotype 1, while the latter sequences clustered with genotype 6 and formed three subgroups, named G-6a, G-6b and G-6c. Of the 32 strains clustering to genotype 1, 31 were isolated in “V” City, and one was isolated in “II” City, Luzon Island. By contrast, 11 strains clustering to genotype 6 were isolated in several different cities, including “II” City, “IV” City, “V” City and “VI” City, on Luzon Island.
Nucleotide and amino acid substitutions of BLV env-gp51 from strains isolated in the Philippines
One typical sequence among those that showed 100 % identity was submitted to GenBank (accession numbers KJ668809-KJ668819). Nucleotide sequences for the 11 selected sequences from 43 strains were aligned with that of the Japanese K02120 strain as a reference sequence (Fig. 3A). All of these are indicated by filled circles in the neighbor-joining phylogenetic tree (Fig. 2). All of the Philippine BLV strains clustered in genotype 1 show one unique silent substitution in the third base of residue 129 (nucleotide 387), and all BLV strains clustered in genotype 6 show seven unique silent substitutions in the third base of residues 135 (nucleotide 405), 161 (nucleotide 483), 175 (nucleotide 525), 184 (nucleotide 552), 185 (nucleotide 555), 194 (nucleotide 582) and 205 (nucleotide 615) (Fig. 3). By contrast, all Philippine BLV strains belonging to genotypes 1 and 6 show two common substitutions in the third base of residues 121 (nucleotide 363) and 133 (nucleotide 399). Interestingly, of the ten silent substitutions, seven were located within epitopes, including the viral G epitope (nucleotide 363), the second neutralizing domain (ND) (nucleotides 399 and 405), the CD8+ T-cell epitope (nucleotides 483 and 525) and the E epitope (nucleotides 552 and 555), as shown in Fig. 3B. In addition to these, seven nucleotide substitutions caused amino acid variations.

Alignment of a partial nucleotide sequence and deduced amino acid sequence of the BLV env gene from strains in the Philippines. One typical BLV isolate from each identical sample was submitted to GenBank (accession numbers: KJ668809 (PCC130), KJ668810 (PCC67), KJ668811 (PCC99), KJ668812 (PCC63), KJ6688113 (PCC44), KJ668814 (PCC74), KJ668815 (PCC122), KJ668816 (CAM69), KJ668817 (PCC4), KJ668818 (PCC158), KJ668819 (PCC141)). Alignments were performed for 11 typical nucleotide sequences (A) and eight unique deduced amino acid sequences (B) of the env gene from 43 Philippine BLV strains. Philippine BLV strains are shown by the sample ID without the collection time and country name. Silent nucleotide substitutions are indicated by numbers and a filled triangle (▾). The first, second and third neutralizing domains (ND) and other epitopes are shown at the top of the alignment in B. Numbers above the sequences are amino acid residue numbers that indicate the start and end of each domain. Genotypes (G-1 or G-6) are indicated by the black bars at the far left of the figure. Dots indicate identity with K02120 (accession number: AY151262), which was used as a reference in this study
Eight deduced amino acid sequences for the 11 different Philippine BLV strains were aligned with the predicted amino acid sequence of K02120. Figure 3B shows the distribution of amino acid changes within the middle region of gp51, encompassing amino acid positions 101 to 241. This env gp51 region includes a portion of the first ND (residues 101–105), second ND (residues 131–149) and third ND (residues 210–225), a portion of the CD4+ T-cell epitope (residues 101–113) and CD8+ T-cell epitope (residues 154–182), and the viral G (residues 121), E (residues 175–194) and B (residues 228–238) epitopes [27]. A comparison of the predicted amino acid sequences of the partial env genes from Philippine BLV strains revealed highly conserved regions. Although most of the deduced amino acid sequences showed high homology to that of the Japanese K02120 strain (accession number: AY151262), seven different amino acid substitutions were found among the Philippine BLV strains. Three amino acid substitutions were specifically found in the genotype 1 strains, namely, substitutions of tyrosine to cysteine at residue 108 within the CD4+ T-cell epitope, alanine to proline at residue 119, and leucine to phenylalanine at residue 202. By contrast, four other amino acid substitutions were specifically found in the genotype 6 strains. Interestingly, all of the Philippine genotype 6 strains showed isoleucine-to-threonine substitutions at residue 144 within the second ND, which is a typical characteristic of the genotype 6 strains. Deduced amino acid sequences of BLV isolates that formed subgroup G-6b had one unique isoleucine-to-serine substitution at residue 231 within the viral B epitope, while subgroup G-6c had a unique serine-to-asparagine substitution at residue 234 in the B epitope. A lysine-to-glutamic acid substitution was found at residue 175 within a portion of the CD8+ T-cell epitope and the E epitope. However, no substitutions were found within the first ND. Thus, Philippine BLV isolates showed different amino acid substitutions according to genotype.
Discussion
Our study has three major conclusions. First, the present study revealed the distribution of BLV in the Philippines, as determined by BLV-CoCoMo-qPCR-2 and nested PCR. A total of 1116 samples collected from different farms located on five Philippine islands showed relatively low levels of BLV infection. Among the five islands investigated, Bohol and Cebu were negative for BLV provirus. This result is in accordance with previous studies of BLV in the Philippines [37]. Likewise, BLV was detected at extremely low levels, 1.1 % (4/372) and 5.6 % (9/159), in Leyte and Iloilo, respectively. By contrast, BLV infection was remarkably high (23.1 %) in Luzon compared to the other four islands. This observation is similar to that of a previous study [38], where BLV proviral DNA was detected in samples obtained from water buffalos present in different parts of Luzon. Thus, our results suggest that the prevalence of BLV infection in Philippine cattle is different between the southern Philippines, such as Bohol, Cebu, Iloilo and Leyte Islands, and the northern Philippines, such as Luzon Island. Second, the present results clearly indicate that the sensitivity of BLV-CoCoMo-qPCR-2 was higher than that of nested PCR because we found a number of cattle on Leyte, Iloilo and Luzon Islands that were BLV-positive as determined by the BLV-CoCoMo-qPCR-2 but were negative for BLV provirus as determined by nested PCR. This result showed the same tendency as discussed in our previous publications [7, 12], showing that our original BLV-CoCoMo-qPCR is highly specific and sensitive and able to detect BLV in samples that are negative by the nested PCR assay. In addition, our previous paper [7] demonstrated that the positive rate for the nested PCR in cattle correlated with the proviral load determined by the original BLV-CoCoMo-qPCR as follows: 1) positive rates for the nested PCR ranged from 62.9 % to 98.5 % among animals with proviral copy numbers ranging from 100 to 104 copies per 105 cells, and 2) the positive rate for nested PCR was 100 % in cattle with high proviral loads (>104 copies per 105 cells). Therefore, cattle that were BLV-positive as determined by the BLV-CoCoMo-qPCR-2 but were negative for BLV provirus as determined by nested PCR may have a very low copy number of BLV, which cannot be detected by nested PCR. Indeed, only samples that were positive by both methods resulted in successful amplification of BLV env-gp51, and therefore these samples were used for further phylogenetic analysis. Interestingly, our previous study showed that the original BLV-CoCoMo-qPCR using highly degenerate primers to detect known and novel BLV variants in clinical cattle was able to detect various BLV strains from a broad geographical origin, including Japan, Peru, Bolivia, Chile and the U.S.A. [12]. Here, we analyzed whether BLV-CoCoMo-qPCR-2, which uses optimized degenerate primers, allows the highly sensitive detection of BLV by using two methods, BLV-CoCoMo-qPCR-2 and nested PCR. Our results showed that BLV-CoCoMo-qPCR-2 is highly effective in detecting BLV in cattle from the Philippines. Third, phylogenetic analysis based on a 423-bp fragment of the env gene demonstrated that Philippine BLV strains were generally of genotypes 1 and 6, out of the eight distinct BLV genotypes worldwide [27, 29, 30]. Interestingly, a number of substitutions were found in epitope regions, such as the CD4+ T-cell epitope, the CD8+ T-cell epitope, the second ND, B and E epitopes, and the substitutions varied according to genotype.
The majority of BLV-positive samples detected in this study were collected from relatively large dairy farms where milking is the most common activity. A tendency towards high rates of BLV infection on dairy farms was also found in a Japanese study [39]. Thompson and Miller [40] demonstrated that pathogens can be transmitted during the milking process. Therefore, it is possible that the high level of BLV infection on dairy farms in Luzon is a consequence of transmission of infection from residual infected milk present in a milking machine to a healthy receptive cow during the milking process. One interesting finding in this study is that the BLV-infected farms in “V” City on Luzon Island, where the highest rate of BLV infection was observed, belong to a totally confined setting. By contrast, cattle that were negative for the BLV provirus were raised on small farms. Previous studies focusing on risk factors for BLV transmission revealed that loose housing systems, the presence of hematophagous insects, blood-contaminated dehorning devices [41], physical contact [42], common use of needles and/or the introduction of infected animals [39] are the predominant risk factors. Therefore, we believe that the extremely high level of BLV present in farms in “V” City may be the result of cattle management procedures that involve transfer of infected blood, such as common use of needles, ear tattooing, physical contact and/or cattle exchange from one farm to another, as well as infection through milking.
Our analysis of partial BLV env sequences resulted in the identification of 17 nucleotide substitutions, ten of which were silent substitutions, and seven were amino acid substitutions. Nucleotide substitutions at residues 231 and 234 were located in the B epitope. The substitution at residue 108 was located in the CD4+ T-cell epitope, and the substitution at residues 144 was located between the second ND. Another substitution at residue 175 was located between the CD8+ T-cell epitope and the E epitope. Thus, most of the amino acid changes occurred within epitope regions. Of all the amino acid substitutions, substitutions at codons 108, 119 and 202 were a unique characteristic of the genotype 1 Philippine BLV strains, while those at residues 144, 175, 231 and 234 were unique to genotype 6 BLV strains. This result is in accordance with a previous finding that most substitutions in gp51 are found within epitopes rather than at random locations [19].
In this study, two genotypes (genotype 1 and genotype 6) were identified in BLV strains from the Philippines. The classification was based on the percentage of nucleotide sequence similarity, silent nucleotide substitution patterns, the phylogenetic tree, and amino acid substitutions within a partial env gene sequence, together with phylogenetic analysis. Most of the Philippine BLV strains clustered into genotype 1 together with two BLV strains from Japan, and these Philippine BLV strains were identified in samples located in close proximity. All of the Philippine BLV strains clustering into genotype 1 were isolated from dairy cattle, Holstein × Sahiwal breeds, and were collected from “II” City and “V” City in Luzon Island. The rest of the Philippine BLV strains clustered into genotype 6, together with isolates from Brazil and Argentina. The Philippine BLV strains clustering into genotype 6 consisted of Brahman cattle from “VI” City and Holstein × Sahiwal cattle from “II” City, “IV” City and “V” City in Luzon Island. Notably, the Philippine National Dairy Authority imported Holstein × Sahiwal cattle from New Zealand and Australia to Luzon Island (http://www.nda.da.gov.ph/na123.htm; http://pcic.gov.ph/index.php/news/press-release/pcic-covers-13th-batch-of-nda-cattle-importation/#). These two countries are known to have BLV-infected cattle [43–45] (www.daff.gov.au/animal-planthealth/pests-diseases-weeds/animal/ebl). This observation was also mentioned in a previous study [37]. In addition to the above, farmers in the Philippines are also encouraged to cross Holstein cattle with Sahiwal cattle to increase milk productivity (http://www.nda.da.gov.ph/2013/PROGRAMS/programs2013.html). Collectively, our study is the first report to show that BLV strains isolated from Luzon Island were the most similar to BLV strains from Japan, Brazil and Argentina.
Another interesting finding was that both genotype 1 and genotype 6 exist only on farms in “II” City and “V” City on Luzon Island, which belong to a totally confined setting, but they were not present on other farms, especially on small farms. The presence of more than one genotype in the same herd has previously been noted in open herds [46] and closed herds. The presence of two genotypes in “II” City and “V” City may be the consequence of separate infections of different viral origins in two cities and further transmission of the virus through exchange of infected cattle.
In conclusion, two different BLV genotypes (1 and 6) are present in the Philippines. These results provide important information on BLV infection levels and will enable the implementation of appropriate cattle-management policies in addition to providing supplementary information for the development of more-effective methods of BLV eradication in the Philippines.
References
Aida Y, Murakami H, Takahashi M, Takeshima SN (2013) Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front Microbio 4:328
Buehring GC, Shen HM, Jensen HM, Choi KY, Sun DJ, Nuovo G (2014) Bovine leukemia virus DNA in human breast tissue. Emerg Infect Dis 20:772–782
Mirsky ML, Olmstead CA, Da Y, Lewin HA (1996) The prevalence of proviral bovine leukemia virus in peripheral blood mononuclear cells at two subclinical stages of infection. J Virol 70:2178–2183
Aida Y, Okada K, Amanuma H (1993) Phenotype and ontogeny of cells carrying a tumor-associated antigen that is expressed on bovine leukemia virus-induced lymphosarcoma. Cancer Res 53:429–437
Kettmann R, Meunier-Rotival M, Cortadas J, Cuny G, Ghysdael J, Mammerickx M, Burny A, Bernardi G (1979) Integration of bovine leukemia virus DNA in the bovine genome. Proc Natl Acad Sci USA 76:4822–4826
Tajima S, Ikawa Y, Aida Y (1998) Complete bovine leukemia virus (BLV) provirus is conserved in BLV-infected cattle throughout the course of B-cell lymphosarcoma development. J Virol 72:7569–7576
Jimba M, Takeshima SN, Murakami H, Kohara J, Kobayashi N, Matsuhashi T, Ohmori T, Nunoya T, Aida Y (2012) BLV-CoCoMo-qPCR: a useful tool for evaluating bovine leukemia virus infection status. BMC Vet Res 8:167
Kettmann R, Deschamps J, Cleuter Y, Couez D, Burny A, Marbaix G (1982) Leukemogenesis by bovine leukemia virus: proviral DNA integration and lack of RNA expression of viral long terminal repeat and 3’ proximate cellular sequences. Proc Natl Acad Sci USA 79:2465–2469
Lagarias DM, Radke K (1989) Transcriptional activation of bovine leukemia virus in blood cells from experimentally infected, asymptomatic sheep with latent infections. J Virol 63:2099–2107
Tajima S, Aida Y (2005) Induction of expression of bovine leukemia virus (BLV) in blood taken from BLV-infected cows without removal of plasma. Microbes infect 7:1211–1216
Gillet N, Florins A, Boxus M, Burteau C, Nigro A, Vandermeers F, Balon H, Bouzar AB, Defoiche J, Burny A, Reichert M, Kettmann R, Willems L (2007) Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology 4:18
Jimba M, Takeshima SN, Matoba K, Endoh D, Aida Y (2010) BLV-CoCoMo-qPCR: quantitation of bovine leukemia virus proviral load using the CoCoMo algorithm. Retrovirology 7:91
Panei CJ, Takeshima SN, Omori T, Nunoya T, Davis WC, Ishizaki H, Matoba K, Aida Y (2013) Estimation of bovine leukemia virus (BLV) proviral load harbored by lymphocyte subpopulations in BLV-infected cattle at the subclinical stage of enzootic bovine leucosis using BLV-CoCoMo-qPCR. BMC Vet Res 9:95
Johnston ER, Radke K (2000) The SU and TM envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions. J Virol 74:2930–2935
Mamoun RZ, Morisson M, Rebeyrotte N, Busetta B, Couez D, Kettmann R, Hospital M, Guillemain B (1990) Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J Virol 64:4180–4188
Ban J, Czene S, Altaner C, Callebaut I, Krchnak V, Merza M, Burny A, Kettmann R, Portetelle D (1992) Mapping of sequential epitopes recognized by monoclonal antibodies on the bovine leukaemia virus external glycoproteins expressed in Escherichia coli by means of antipeptide antibodies. J Gen Virol 73(Pt 9):2457–2461
Johnston ER, Albritton LM, Radke K (2002) Envelope proteins containing single amino acid substitutions support a structural model of the receptor-binding domain of bovine leukemia virus surface protein. J Virol 76:10861–10872
Callebaut I, Voneche V, Mager A, Fumiere O, Krchnak V, Merza M, Zavada J, Mammerickx M, Burny A, Portetelle D (1993) Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. J Virol 67:5321–5327
Portetelle D, Couez D, Bruck C, Kettmann R, Mammerickx M, Van der Maaten M, Brasseur R, Burny A (1989) Antigenic variants of bovine leukemia virus (BLV) are defined by amino acid substitutions in the NH2 part of the envelope glycoprotein gp51. Virology 169:27–33
Bruck C, Mathot S, Portetelle D, Berte C, Franssen JD, Herion P, Burny A (1982) Monoclonal antibodies define eight independent antigenic regions on the bovine leukemia virus (BLV) envelope glycoprotein gp51. Virology 122:342–352
Beier D, Blankenstein P, Marquardt O, Kuzmak J (2001) Identification of different BLV provirus isolates by PCR, RFLPA and DNA sequencing. Berl Munch Tierarztl Wochenschr 114:252–256
Camargos MF, Stancek D, Rocha MA, Lessa LM, Reis JK, Leite RC (2002) Partial sequencing of env gene of bovine leukaemia virus from Brazilian samples and phylogenetic analysis. J Vet Med B Infect Dis Vet Public Health 49:325–331
Camargos MF, Pereda A, Stancek D, Rocha MA, dos Reis JK, Greiser-Wilke I, Leite RC (2007) Molecular characterization of the env gene from Brazilian field isolates of Bovine leukemia virus. Virus Genes 34:343–350
Felmer R, Munoz G, Zuniga J, Recabal M (2005) Molecular analysis of a 444 bp fragment of the bovine leukaemia virus gp51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet Microbiol 108:39–47
Molteni E, Agresti A, Meneveri R, Marozzi A, Malcovati M, Bonizzi L, Poli G, Ginelli E (1996) Molecular characterization of a variant of proviral bovine leukaemia virus (BLV). Zentralbl Veterinarmed B 43:201–211
Monti G, Schrijver R, Beier D (2005) Genetic diversity and spread of Bovine leukaemia virus isolates in Argentine dairy cattle. Arch Virol 150:443–458
Balic D, Lojkic I, Periskic M, Bedekovic T, Jungic A, Lemo N, Roic B, Cac Z, Barbic L, Madic J (2012) Identification of a new genotype of bovine leukemia virus. Arch Virol 157:1281–1290
Moratorio G, Obal G, Dubra A, Correa A, Bianchi S, Buschiazzo A, Cristina J, Pritsch O (2010) Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch Virol 155:481–489
Rodriguez SM, Golemba MD, Campos RH, Trono K, Jones LR (2009) Bovine leukemia virus can be classified into seven genotypes: evidence for the existence of two novel clades. J Gen Virol 90:2788–2797
Matsumura K, Inoue E, Osawa Y, Okazaki K (2011) Molecular epidemiology of bovine leukemia virus associated with enzootic bovine leukosis in Japan. Virus Res 155:343–348
Rola-Luszczak M, Pluta A, Olech M, Donnik I, Petropavlovskiy M, Gerilovych A, Vinogradova I, Choudhury B, Kuzmak J (2013) The molecular characterization of bovine leukaemia virus isolates from Eastern Europe and Siberia and its impact on phylogeny. PloS one 8:e58705
Asfaw Y, Tsuduku S, Konishi M, Murakami K, Tsuboi T, Wu D, Sentsui H (2005) Distribution and superinfection of bovine leukemia virus genotypes in Japan. Arch Virol 150:493–505
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Hemmatzadeh F (2007) Sequencing and phylogenetic analysis of gp51 gene of bovine leukaemia virus in Iranian isolates. Vet Res Commun 31:783–789
Uera JA, Lazaro JV, Mingala CN (2012) Detection of enzootic bovine leukosis in cattle using nested polymerase chain reaction assay. Thai J Vet Med 42:319–324
Mingala CN, Konnai S, Cruz LC, Onuma M, Ohashi K (2009) Comparative moleculo-immunological analysis of swamp- and riverine-type water buffaloes responses. Cytokine 46:273–282
Kobayashi S, Hidano A, Tsutsui T, Yamamoto T, Hayama Y, Nishida T, Muroga N, Konishi M, Kameyama K, Murakami K (2014) Analysis of risk factors associated with bovine leukemia virus seropositivity within dairy and beef breeding farms in Japan: a nationwide survey. Res Vet Sci 96:47–53
Thompson PD, Miller RH (1974) Retrograde flow of milk within machine-milked teats. J Dairy 57:1489–1496
Lassauzet ML, Thurmond MC, Johnson WO, Stevens F, Picanso JP (1990) Effect of brucellosis vaccination and dehorning on transmission of bovine leukemia virus in heifers on a California dairy. Can J Vet Res 54:184–189
Kono Y, Sentsui H, Arai K, Ishida H, Irishio W (1983) Contact transmission of bovine leukemia virus under insect-free conditions. Nihon Juigaku Zasshi 45:799–802
Thompson KG, Johnstone AC, Hilbink F (1993) Enzootic bovine leukosis in New Zealand: a case report and update. N Z Vet J 41:190–194
Lew AE, Bock RE, Molloy JB, Minchin CM, Robinson SJ, Steer P (2004) Sensitive and specific detection of proviral bovine leukemia virus by 5′ Taq nuclease PCR using a 3′ minor groove binder fluorogenic probe. J Virol Methods 115:167–175
Coulston J, Naif H, Brandon R, Kumar S, Khan S, Daniel RC, Lavin MF (1990) Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: comparison with other isolates. J Gen Virol 71(Pt 8):1737–1746
Licursi M, Inoshima Y, Wu D, Yokoyama T, Gonzalez ET, Sentsui H (2002) Genetic heterogeneity among bovine leukemia virus genotypes and its relation to humoral responses in hosts. Virus Res 86:101–110
Acknowledgments
We thank Drs. T. Miyasaka, M.A. Villanueva, A.J. Salces, Libertado C. Cruz and Mr. M. Ohashi, and the management and staff of the Philippine Carabao Center for kindly assisting with the large-scale sampling from many farms in the Philippines. We are grateful to the Support Unit at the Bio-material Analysis, RIKEN BSI Research Resources Center, for help with sequence analysis. This work was supported by Grants-in-Aid for Scientific Research (B and C) from the Japan Society for the Promotion of Science (JSPS), by A-STEP (Adaptable & Seamless Technology Transfer Program through Target-driven R&D) and by a grant from the Program for the Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry in Japan.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Polat, M., Ohno, A., Takeshima, Sn. et al. Detection and molecular characterization of bovine leukemia virus in Philippine cattle. Arch Virol 160, 285–296 (2015). https://doi.org/10.1007/s00705-014-2280-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2280-3