
Abstract
Background
Aspergillus niger is a filamentous fungus used for the majority of global citric acid production. Recent developments in genome editing now enable biotechnologists to engineer and optimize A. niger. Currently, however, genetic-leads for maximizing citric acid titers in industrial A. niger isolates is limited.
Results
In this study, we try to engineer two citric acid A. niger production isolates, WT-D and D353, to serve as platform strains for future high-throughput genome engineering. Consequently, we used genome editing to simultaneously disrupt genes encoding the orotidine-5′-decarboxylase (pyrG) and non-homologous end-joining component (kusA) to enable use of the pyrG selection/counter selection system, and to elevate homologous recombination rates, respectively. During routine screening of these pyrG mutant strains, we unexpectedly observed a 2.17-fold increase in citric acid production when compared to the progenitor controls, indicating that inhibition of uridine/pyrimidine synthesis may increase citric acid titers. In order to further test this hypothesis, the pyrG gene was placed under the control of a tetracycline titratable cassette, which confirmed that reduced expression of this gene elevated citric acid titers in both shake flask and bioreactor fermentation. Subsequently, we conducted intracellular metabolomics analysis, which demonstrated that pyrG disruption enhanced the glycolysis flux and significantly improved abundance of citrate and its precursors.
Conclusions
In this study, we deliver two citric acid producing isolates which are amenable to high throughput genetic manipulation due to pyrG/kusA deletion. Strikingly, we demonstrate for the first time that A. niger pyrG is a promising genetic lead for generating citric acid hyper-producing strains. Our data support the hypothesis that uridine/pyrimidine biosynthetic pathway offer future avenues for strain engineering efforts.

Similar content being viewed by others
Background
Citric acid is the most important bulk industrial organic acid with the worldwide market of nearly 2 million tons per year [1] and has been widely applied in various industries [1,2,3]. Aspergillus niger is the important industrial cell factory for the citric acid production, which contributes approximately 80% of world-wide citric acid [4]. Over the past 20 years, numerous technological advances have occurred in the A. niger field [5], including the development of recyclable selection markers, disruption of non-homologous end-joining systems for high throughput gene targeting, and, most recently, genome editing [6,7,8,9,10,11,12,13]. Most of these tools and techniques have been developed and optimized in fully sequenced strain backgrounds. This includes isolates CBS 513.88 or ATCC 1015, which are industrial isolates used for glucoamylase and citric acid fermentation respectively [14, 15]. As sequencing and genome editing technologies are increasingly cheap and simple to develop, it is now possible to expand the full A. niger toolkit to other industrially harnessed strains. This is an attractive strategy, as optimization efforts in specific isolates can rapidly be applied at a commercial scale. Currently, this toolkit remains a key objective for biotechnologists as genetic leads for generating A. niger citric acid hyper-producing isolates are limited [5, 16, 17]. For example, several studies have used gene knock-down of the chitin synthase chsC [18] or amino acid transporter Brsa-25 [19] to elevate citric acid titers about 42.6% [18] and 10% [19], respectively. Alternatively, the over-expression of the organic acid transporter cexA has been used for hyperproduction [20]. Despite these studies, the majority of genes with potential industrial applications to elevate citric acid production remain hypothetical and lack functional characterization in the laboratory [5, 16, 17].
Arguably, the first and most important technology for genetic manipulation of any given A. niger isolate is development of an efficient selection marker for transformation. The orotidine-5′-decarboxylase encoding gene pyrG has been widely applied as a recyclable transformation marker in many fungal species [8, 11, 20,21,22,23]. The use of this system requires that a strain of interest firstly has pyrG disrupted/deleted, generating a uridine auxotrophy as the orotidine-5′-monophosphate (OMP) decarboxylase. PyrG is an essential enzyme involved in uridine biosynthesis [21]. Subsequently, a functional pyrG encoded in a transformation cassette serves as an efficient selection marker by restoring prototrophy. Additionally, as PyrG enables catalysis of the non-toxic 5-fluoroorotic acid (5-FOA) to the toxic product as 5-fluorouracil, isolates containing pyrG marker can be counter selected. Several Aspergillus spp. transformation cassettes are designed to facilitate efficient excision of the pyrG selection marker from recipient genomes under 5-FOA counter selection, ultimately generating a fully recyclable marker system. More recently, an additional benefit of using the pyrG marker has been demonstrated, as pyrG resides in a highly expressed locus on chromosome III [24]. Consequently, the pyrG marker and genomic locus are highly convenient for gene over-expression studies.
An additional objective for enabling high throughput gene functional analysis in a strain of interest is disruption/deletion of non-homologous end-joining pathways, most commonly kusA in A. niger [25]. Deletion of this gene elevates homologous recombination rates, therefore increasing the targeting efficiency of exogenous DNA cassettes with the recipient genome. Recently, concerns regarding genome stability in kusA mutants have been disproven by sequencing NHEJ mutants and progenitor controls, confirming their application in biotechnology [26]. In this study, we simultaneously disrupted both pyrG and kusA by a highly efficient CRISPR/Cas9 system based on 5S rRNA. Surprisingly, during routine screening of the mutant strains and progenitor controls, it demonstrated that the pyrG gene is promising target to modulate citric acid production during submerged industrial fermentation.
Results and discussion
Gene disruption of pyrG and pyrG/kusA constructed by CRISPR/Cas9 system in A. niger
In yeast and filamentous fungi, the pyrG gene has been widely used as nutritional/auxotrophic marker for fungal genetic manipulation [21, 22, 27,28,29,30]. With the advantage of counter selection using 5-FOA, pyrG could is utilized as the bidirectional selection marker for recyclable genome editing.
To establish genetic manipulation of citric acid producing isolates WT-D and D353, we chose the traditional selection marker pyrG gene and key gene kusA involved in NHEJ system as targets [10]. To simultaneously disrupt these two targets, two double strand breaks were introduced at respective loci using Cas9 with corresponding guides and then were repaired by homologous recombination system (Fig. 1). For each strain, 24 primary transformants were randomly picked and subcultured onto either MM agar, MM agar supplemented with uridine, and MM supplemented with 5′-FOA, in order to screen the pyrG deficient mutants. As shown in Additional file 1: Figure. S1, pyrG deficient mutants were able to grow on the MM with uridine and MM with 5′-FOA, but can’t grow on the MM plate without uridine. For both WT-D and D353, 11 pyrG deficient mutants among 24 detected transformants were selected for further PCR verification. The genomes of these pyrG deficient transformants were extracted and verified with the corresponding primer pairs (Fig. 1). For A. niger WT-D, two mutants D10 and D20 were confirmed as the pyrG/kusA double mutants, while for A. niger D353, D353.8 was verified as the pyrG/kusA mutant.

Simultaneous inactivation of pyrG and kusA in A. niger D and D353. a Schematic diagram of simultaneous disrupted mutagenesis of both pyrG and kusA mediated by integrating the donor DNA with 40-bp micro-homology arms via CRISPR/Cas9 system. The donor DNAs of MHi-pyrG1-hph and MHi-kusA-hph were co-transformed with linear sgRNA constructs (sgRNA-pyrG1 and sgRNA-kusA) and Cas9 expression plasmid pCas9-hyh into the protoplasts of A. niger D and D353. Two DSBs were generated by the Cas9 under the guide of sgRNA, and then were repaired by HR with the integration of donor DNAs. b, c Diagnostic PCR analysis of the selected pyrG deficient transformants. The expected sizes of PCR products of the mutants were 3240-bp (pyrG-g-F/pyrG-g-R1) and 2702-bp (kusA-g-F/kusA-g-R), when the selection marker hph cassette were correctly inserted at the loci of pyrG or kusA. The expected sizes of PCR products of the hosts were 1345-bp (pyrG-g-F/pyrG-g-R1) and 804-bp (kusA-g-F/kusA-g-R), when the hph markers were not inserted at the loci of pyrG or kusA. For A. niger D353, the PCR products of some mutants using pyrG-g-F/pyrG-g-R1 were smaller than the expected size. After sequencing theses PCR products, it’s found that the smaller inserted fragments were the sgRNA-pyrG1 expression cassette
To test whether the pyrG and kusA deficient mutants can be used for further genome editing, the A. niger D10 and D353.8 were selected as progenitor strains for disruption of the polyketide synthase encoding gene albA, which is required for synthesis of DHN melanin and conidial pigmentation. With the selection for pyrG integration into the genomes, all the primary transformants were confirmed to be prototrophic, which can be grow normally on the MM agar without uridine. Due to the high efficiency of CRIPSR/Cas9 and high frequency of homologous recombination in the NHEJ system deficient strains, all the selected transformants were showed demonstrated to be genome edited (Additional file 1:Figure S2).
We simultaneously disrupted both pyrG and kusA in two citric acid producing strains (Fig. 1) and constructed the fungal chassis which the further recyclable genome editing (Additional file 1: Figure S2). The results here was consistent with the previous study reported in Aspergillus orzye, in which the pyrG gene was also disrupted with the other important gene involved in NHEJ, the ligD gene, to generate the mutant with highly efficient gene targeting [29]. Taken together, it demonstrated that the pyrG and kusA disrupted mutants could be as utilized as citric acid producing chassis to facilitate the recyclable genome editing.
Gene disruption of pyrG significantly elevated citric acid titers in shake flaks culture
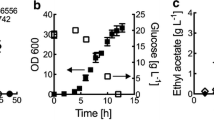
During routine quality control of transformant isolates and progenitor strains, we observed an unexpected increase of citric acid titers in the culture supernatants for mutants of both WT-D and D353. To confirm this observation, five pyrG deficient mutants derived from either A. niger WT-D and D353 were selected for citric acid fermentation in shake flasks. This assay demonstrated that significantly increased citric acid titers were observed for all the pyrG deficient mutants (Fig. 2). For pyrG mutants derived from A. niger WT-D, citric acid increased from 1.92-fold (D14, 30.94 ± 0.58 g/L) to 2.17-fold (D13, 33.59 ± 3.24 g/L), compare to the progenitor control strain (WT-D, 16.98 ± 1.91 g/L) (Fig. 2a). Additionally, mutants derived from A. niger D353 also demonstrated increased citric acid titers from 1.24-fold (D353.20, 38.92 ± 1.17 g/L) to 1.36-fold (D353.4, 42.69 ± 1.48 g/L), compared to control (31.28 ± 0.88 g/L) (Fig. 2b). Normalized citric acid titers to biomass of all isolates confirmed increased citric acid production in mutant strains (Fig. 2c, d). For the pyrG/kusA double mutants, D10, D20 and D353.8 showed the similar citric acid production as these pyrG single mutants did, and the kusA single mutants did not influence the citric acid titers (data not shown). While it can be noted that the increase in citric acid titers following pyrG deletion was greater in isolate WT-D when compared to D353 (Fig. 2), magnitude of increased citric acid titer was sufficiently high in both backgrounds to warrant further investigation. Thus, we hypothesized that pyrG deletion may increase citric acid production in A. niger.

Citric acid fermentation characteristics of the pyrG disrupted mutants. a, b citric acid titer and c, d normalized citric acid titer (g citric acid/g dry weight) were calculated for each strain. 1 × 105 spores/mL were inoculated in 20 mL citrate fermentation (CitFM) media and incubated at 34 °C for 96 h. The extracellular citric acid was determined by the method of HPLC. WT-D and D353, the parent strains used as the control; D10, D12, D13, D14, and D20, the pyrG deficient mutants derived from WT-D; D353.4, D353.8, D353.10, D353.18, D353.20, the pyrG deficient mutants derived from D353. Among them, D10, D20 and D353.8 were the pyrG and kusA double deficient mutants. Pairwise Student’s t-tests were conducted between conditional expression mutant relative to the parent strains. p values are indicated as (< 0.05, *; < 0.01, **; < 0.001, ***)
Generation of a pyrG conditional expression mutant in A. niger
The pyrG gene encoding orotidine-5′-monophosphate (OMP) decarboxylase plays an essential role in uridine biosynthesis [21], but there is no research to unveil its impact on the other cell metabolism. To further investigate the influence of pyrG expression on citric acid production, we constructed the conditional expressed mutant of pyrG by in situ integration of the Tet-on system at the upstream of pyrG encoding sequences (Fig. 3). Consistent with the previous studies [31,32,33,34], Tet-on system is a useful method to functionally analyze essential genes, enabling analyses of null, loss-of-function, and over-expression in a single strain by addition of the inducer Dox. After co-transformation, ten randomly selected primary transformants were subcultured on MM plates, MM supplemented with 20 µg/mL Dox and MM supplemented with uridine. Six transformants were able to grow on MM with uridine but were unable to grow on MM plate, suggesting that these transformants were pyrG deficient mutants. Among these mutants, only two transformants resembled to the pyrG+ phenotype when 20 µg/mL Dox was supplemented into the MM plate. After verifying these genotypes via genomic PCR diagnosis (Additional file 1: Figure S3), isolate XMD1.6 was used as the conditional expression mutant for the further experiments.

Titratable expression of pyrG mutagenesis constructed in A. niger. a Schematic diagram of pyrG titratalbe expression mutagenesis mediated by integrating the donor DNA with 40-bp micro-homology arms via CRISPR/Cas9 system based 5S rRNA. The donor DNA MHi-pyrG2-hyh:Tet-on, containing the Tet-on cassette, were co-transformed with linear sgRNA construct sgRNA-pyrG2 and Cas9 expression plasmid pCas9-hyh into the protoplasts of A. niger D353. DSBs at the locus of the upstream of pyrG encoding sequences, were generated by the Cas9 under the guide of sgRNA-pyrG2, and then were repaired by HR with the integration of donor DNA MHi-pyrG2-hyh:Tet-on, resulting in the replacement of pyrG native promoter. b Phenotypic screening of pyrG conditional expression mutants on solid plates. 1 × 103, 1 × 102, and 1 × 101 spores were inoculated in 2 µL volumes onto the MM supplemented with various concentrations of Dox and MM with uridine as control. Plates were incubated at 30 °C in the dark for 48 h. Representative images are shown for technically triplicated experiments. Control, A. niger D353 as the positive controls; XMD1.6, the pyrG conditional expression mutants
In order to confirm titratable pyrG expression in isolate XMD1.6, phenotypic screens of the conditional mutant were conducted on MM solid agar supplemented with 0, 0.2, 2, 20 and 50 µg/mL Dox, respectively. No growth of the conditional expression mutant was observed in the absence of Dox in growth media, and the mutant was indistinguishable from the control on the MM plate with uridine but without Dox (Fig. 3). However, the titration of Dox in MM agar without uridine enabled isolate XMD1.6 to prototrophy, and ultimately to generate colonies which resembled the parental strain. These data confirm Dox dependent titration of pyrG expression in the XMD1.6 mutant.
Reduced pyrG expression improves citric acid titers in A. niger submerged culture
In order to test the influence of pyrG expression levels on citric acid production, we conducted citric acid fermentation with various Dox concentrations using pyrG conditional expression mutant XMD1.6. As shown in Fig. 4, pyrG expression significantly influenced the citric acid production of A. niger, with titers and proportion of total acid gradually reduced with the increase of the Dox supplementation into fermentation media. These data further suggest that PyrG has a negative impact on in A. niger citric acid production. Without Dox or with 0.2 μg/mL Dox supplemented, the citric acid production of pyrG conditional expressed mutants XMD1.6 increased about 1.38-fold (37.8 ± 0.63 g/L) and 1.27-fold (35.02 ± 1.16 g/L) as that of the control D353 (27.43 ± 1.03 g/L) respectively, which is similar to the results of pyrG disrupted mutants (Fig. 2). With 2 μg/mL Dox supplemented, the citric acid production of pyrG conditional expressed mutants XMD1.6 slightly increased about 1.17-fold (33.5 ± 0.76 g/L) as that of the control strain (Fig. 4). It should be noted that pyrG null or low expression also reduced the cell growth. However, normalization of citric acid titers against biomass of XMD1.6 confirmed 1.83-fold, 1.66-fold, and 1.38-fold increased citric acid titers without or with 0.2 and 2 μg/mL Dox supplemented, compared to that of the D353 control. In contrast, when 20 or 50 µg/mL Dox was supplemented to growth media, the citric acid production of XMD1.6 showed no statistically significant difference compared to the parent strain D353 (Fig. 4). The similar results were also obtained when citric acid fermentations were carried out in 5 L bioreactor (Fig. 5). Without the Dox supplemented, the final citric acid production of XMD1.6 reached up to 50.30 ± 1.55 g/L, with the max citric acid productivity of 1.00 g/L/h, which increase about 1.57-fold as to the parent strain (32.08 ± 0.59 g/L). In contrast, when 20 µg/mL Dox supplemented, the citric acid titer of XMD1.6 (30.23 ± 0.52 g/L) resembled to the parent strain. As to by-products, only a small amount of oxalic acid and succinic acid were detected in the end-point samples, while acetic acid and fumaric acid were not detected (Additional file 1: Figure S4). For instance, without the Dox supplemented, the final titer of oxalic acid and succinic acid of XMD1.6 were 1.35 ± 0.07 g/L and 2.16 ± 0.05 g/L, respectively. These data therefore support the hypothesis that citric acid production improved due to the disruption or down-regulated expression of pyrG.

Citric acid fermentation characteristics of pyrG titratable expression mutant. a Citric acid titer and b normalized citric acid titer (g citric acid/g dry weight) was detected and calculated for each Dox concentration. 1 × 105 spores/mL were inoculated in 20 mL citrate fermentation (CitFM) media with different Dox concentration and incubated at 34 °C in the dark for 96 h. The extracellular citric acid was determined by the method of HPLC. Pairwise Student’s t-tests were conducted between conditional expression mutant relative to the parent strains. p values are indicated as (< 0.05, *; < 0.01, **; < 0.001, ***)

Citric acid production of pyrG titratable expression mutant in 5 L bioreactor. Citric acid titers of pyrG titratable expression mutant under 0 and 20 μg/mL Dox were compared with its parent strains in the 5 L bioreactor. 1 × 105 spores/mL were inoculated in 3 L citrate fermentation (CitFM) media at 34 °C in the dark for 124 h. The extracellular citric acid was determined by the method of HPLC. Control, the parent strains D353; XMD1.6, the pyrG conditional expression mutants
Metabolite profile analysis of A. niger D353 and pyrG disrupted mutant
In order to elucidate the impact of pyrG disruption on the A. niger intracellular metabolite profile, metabolomics analyses of parent strain D353 and pyrG deficient mutant D353.8 were conducted by our established LC–MS/MS pipeline [35]. Samples were taken at the end of citric acid fermentation in shake flasks (Fig. 6). Metabolite analysis demonstrated that most of the Embden–Meyerhof pathway (EMP) intermediates decreased in concentration, while citrate and its precursor Actyl-CoA and oxaloacetate significantly increased in the pyrG mutant when compared to the control. With regard of TCA cycle intermediates, 2-oxoglutarate decreased significantly in the mutant, but the malate, fumarate and succinate increased in different degrees (Fig. 6). The data suggest that pyrG disruption dramatically disturbed the intracellular central metabolism and improved the intracellular level of the citric acid and its precursor, which may lead to the extracellular citric acid accumulation. Considering the EMP intermediates decreased while the citric acid precursors acetyl-CoA and oxaloacetate were significantly increased, it suggested that this metabolic change may be caused by the accelerated glycolysis pathway and the insufficient activity of the citrate synthetase. In Aspergillus nidulans, it is reported that the genes involved in carbon metabolism, such as α-glucosidase B (agdB) and sugar transporter, were significantly up-regulated in a pyrG deleted mutant [36], which is consistent with the EMP metabolism changes in this study. Therefore, we speculated that higher citrate synthetase expression could further improve citric acid production when pyrG expression was disrupted or reduced.

Intracellular metabolite profiling of the A. niger D353 and pyrG mutant D353.8. Intracellular metabolite profiling of the intermediates involved in central metabolism (a) and pyrimidine biosynthesis pathway (b). The relative level of each metabolite in A. niger D353.8 was calculated by dividing its normalized peak area by that of the control D353. Red arrows represent the relative quantitative change of the metabolite increase in A. niger D353.8, while green arrows represent the relative quantitative change of the metabolite decrease. The number next to the arrow represented the fold change of each intracellular metabolite in A. niger D353.8 against the ones in D353. OMP orotidine 5′-phosphate, UMP uridine monophosphate, UDP uridine biphosphate, UTP uridine triphosphate
What’s more, the intermediates in the uridine pathway were also observed. PyrG is essential for the de novo pyrimidine biosynthesis, which catalyzes the carboxylation of OMP into uridine monophosphate (UMP) [21, 36]. When the pyrG gene is deleted, the UMP is synthesized by the salvage pathway from uridine or uracil [36]. As shown in Fig. 6, compared to the control, the intracellular uracil and uridine increased 7.56-fold and 3.48-fold in the pyrG mutant, respectively, while the intracellular OMP were not detected. Sun et al. also observed the similar phenomenon that the pyrG mutation leads to uracil and uridine accumulation in the cells of A. nidulans [36]. These metabolite profiling data indicated that the pyrG deletion may influence the global metabolic network. Future studies, such as dynamic metabolomic and transcriptomic experiments, will be carried out to fully elucidate the molecular basis about the impact of the pyrG gene on citric acid production.
Conclusions
In this study, we demonstrate that pyrG impacts organic acid metabolism in A. niger leading to potential industrial benefits. Both gene disruption and down-regulation of pyrG significantly increases of citric acid production in A. niger. We suggest that pyrG is a promising target for metabolic engineering of filamentous fungal cell factories.
Methods
Strains and cultivation conditions
The strains used in this study are listed in Table 1. The citric acid producing strains A. niger WT-D and D353 were purchased from Shanghai Industrial Microbiology Institute Tech. Co. (Shanghai, China), Ltd. The wild-type A. niger WT-D was isolated on acid PDA media from soil samples, and the D353 was derived from WT-D after Co60 γ- ray radiation mutagenesis. Escherichia coli DH5α (Transgene, Beijing, China) was used for plasmid construction and cultured at 37 °C in Luria–Bertani broth containing ampicillin (100 μg/mL). A. niger strains were cultivated on defined minimal medium (MM) as reported previously [22], or on complete medium (CM) consisting of MM supplemented with 0.5% yeast extract and 0.1% casamino acids. 1.5% agar was supplemented for plates. When necessary, 150 μg/mL of hygromycin was added for the hph selection marker; 0.75% 5-fluoroorotic acid (5-FOA) and 10 mM uracil were used in the MM for the pyrG mutants.
Genetic manipulation
The plasmids used in this study were listed in Table 1. The protospacers and primers were listed in Additional file 1: Tables S1, S2, respectively. For recycling genome editing using the selection marker pyrG, the pyrG coding sequence was amplified with the A. nidulans genome as template and primers AnpyrG-F and AnpyrG-R, then cloned into the backbone of reverse amplified using pSilent-1 as temple and primers Ptrp-Rrev and TtrpC-Frev using the ClonExpress™ one step cloning kit (Vazyme, C113), resulting in pSM-AnpyrG. To construct the Cas9 expression plasmid hygromycin resistance, the selection marker hph cassette was amplified with the template pSilent-1 [37] and the primer pair of PtrpC-Fm and TtrpC-Rm. The plasmid backbone was amplified using pCas9 as the template with the primer pair of pGm-Frev and pGm-Rrev. The hph expression cassette and the AnpyrG expression cassette were then cloned into the plasmid backbond via the ClonExpress™ one step cloning kit (Vazyme, C113), resulting in pCas9-hph and pCas9-AnpyrG, respectively (Table 1).
To construct the sgRNA targeting different genes, protospacers were predicted sgRNA by the sgRNAcas9 software [38], and designed with the minimal off-target possibility. The targeting sgRNA constructs were built by digestion of sgRNA expression plasmids psgRNA6.0 [10] with BbsI, and ligation with synthetic double stranded oligonucleotides of the desired sequences. The linear sgRNA targeting expression cassettes with 5S rRNA gene as promoter for A. niger transformation were obtained by amplification using sequence verified corresponding plasmids as template and primers M13F and M13R as previously described [10]. The DNA sequences of sgRNA constructs are given in Additional file 1: Table S3.
The donor DNAs with micro-homology (40-bp) flanks were generated as previously described [10]. These micro-homology sequences were designed adjacent to the 5′ and 3′ regions of the target sequence without the PAM site. The linear donor DNA constructs MHi-kusA-hph and MHi-pyrG1-hyh for homologous recombination of selection marker hph into the kusA and pyrG were generated by PCR using pSilent-1 [37] as template and primers MHi-kusA-Fm/MHi-kusA-Rm and MHi-pyrG-Fm/MHi-pyrG-Rm, respectively. Similarly, the donor DNA MHi-albA-AnpyrG was amplified with pSM-AnpyrG as template and the primers MHi-albA-Fm/MHi-albA-Rm. The linear donor DNA MHi-pyrG2-hyh:Tet-on was generated by PCR with the template pTC1.13 and the primer pair MHi-pyrG2-Fm/MHi-pyrG2-Rm. After purification by PCR products purification kit, PCR products were used for A. niger transformation. DNA sequences of donor DNA are given in Additional file 1: Table S4.
DNA transformation
The standard protocol of A. niger genome editing using the CRISPR/Cas9 system based 5S rRNA was performed as previously described [10]. For simultaneous gene insertion of kusA and pyrG, 2 μg the donor DNA constructs MHi-kusA-hph and MHi-pyrG1-hyh as HR template were co-transformed into the protoplasts of A. niger D and D353 together with their corresponding sgRNA targeting constructs sgRNA-kusA and sgRNA-pyrG1 and the pCas9-hyh. For gene insertion of albA, 2 μg sgRNA-albA constructs and 2 μg donor DNA constructs MHi-albA-ANpyrG into the protoplasts of kusA/pyrG double mutants A. niger D.10 and D353.8, respectively. For the pyrG titratible expression mutants, the donor DNA MHi-pyrG2-hyh:Tet-on, containing the Tet-on cassette, were co-transformed with linear sgRNA construct sgRNA-pyrG2 and Cas9 expression plasmid pCas9-hyh into the protoplasts of A. niger D353. After twice subculture and purification, genomic DNA of random selected transformants was extracted and verified via diagnostic PCR and sequencing analysis with the corresponding primers.
Phenotypic analysis on solid media
To analyse the phenotype of pyrG mutants, A. niger conidia were harvested in 0.9% NaCl solution from 5-day cultivated CM agar plates. Spores of A. niger isolates were spotted with 2 µl at the contraction of 105/mL on MM agar plates without uridine, with uridine and with uridine and 5-FOA, which were incubated for 4 days at 30 °C. For the pyrG titratable expression mutants, 1 × 103, 1 × 102, and 1 × 101 spores were inoculated in 2 µl volumes onto the MM supplemented with various concentrations of Dox and MM with uridine as control. Plates were incubated at 30 °C in the dark for 48 h. Phenotypic detections were conducted in technical triplicate.
Citric acid fermentation
Citrate fermentation was carried out using the liquefied corn media [35]. The final concentration of 1 × 105 spores/mL was inoculated in 20 mL liquefied corn media with different concentration of Dox in 100 mL shake flasks, which were cultivated at 34 °C and 220 rpm for 96 h. The weight of the shake flasks were measured before and after the citric acid fermentation to eliminate measurement errors caused by evaporation. For the citric acid production in the 5 L bioreactor with a stirring paddle device, most identical fermentation parameters were utilized for 124 h, but the aeration rate was coupled to dissolved oxygen concentration (> 60%).
Supernatants were filtered from cultures using filter paper. Total acids were first titrated using 0.1429 M NaOH with 20 μL 0.1% phenolphthalein as pH indicator. Next, the supernatants were diluted in sterile distilled water depending on the estimated total acid. Samples were boiled for 15 min at 100 °C, after which supernatants were centrifuged at 12,000 rpm for 5 min and filtered through a 0.22 μm sterile filter membrane. Extracellular organic acids were detected by Prominence UFLC equipped with a UV detector (Shimadzu, Kyoto, Japan) and a Bio-Rad Aminex HPX-87H column (300 × 7.8 mm) according to the procedure described previously.
Intracellular metabolite analysis by LC–MS/MS
The samples for intracellular metabolite profiling were prepared and detected based on the standardized and improved LC–MS/MS metabolomics methodology [35]. Briefly, 5 mL (± 0.5) of A. niger mycelial culture was fast filtered by a -20 °C pre-cooled vacuum filter with six layers Miracloth (CalBiochem, Merck, Darmstadt, Germany). After washing with 25 mL pro-cooled PBS buffer, the mycelial samples were flash frozen into liquid nitrogen. After pulverization by pestle and mortar in liquid nitrogen, about 0.1 g (± 0.01) of sample (equal to about 10 mg dry weight) was suspended in 1 mL pre-heated 75% ethanol and incubated at 100 °C for 15 min. Then, the mixture was centrifuged at 12,000 rpm for 5 min at 0 °C and the supernatant was collected into a precooled tube. The cell pellets were re-suspended with another 1 mL of extraction solution and processed by boiling-water bath for 15 min. The second mixture was centrifuged again and the supernatant extraction were mixed with the first supernatant and centrifuged at 12,000 rpm for 30 min at 0 °C. The supernatant was then collected, freeze dried and detected by the LC–MS/MS platform containing Ultra-performance liquid chromatography (UPLC) 30A (Shimadzu, Kyoto, Japan) and TripleTOF™ 6600 mass spectrometer (Applied Biosystem Sciex, USA) as described previously [35]. The LC–MS/MS data were normalized by sample weight, and then metabolites involved in central metabolism were identified according to the protocol described [35].
Determination of fungal biomass
To determine fungal biomass after citric acid fermentation, cultures were vacuum filtered through filter paper, washed in 5-fold sterile water, and added to pre-weighed falcon tubes. Biomass was incubated at 50 °C until dry (minimum of 24 h), after which dry weight was determined.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
References
Tong Z, Zheng X, Tong Y, Shi YC, Sun J. Systems metabolic engineering for citric acid production by Aspergillus niger in the post-genomic era. Microb Cell Fact. 2019;18:28.
Karaffa L, Kubicek CP. Aspergillus niger citric acid accumulation: do we understand this well working black box? Appl Microbiol Biotechnol. 2003;61:189–96.
Legisa M, Mattey M. Changes in primary metabolism leading to citric acid overflow in Aspergillus niger. Biotechnol Lett. 2007;29:181–90.
Dhillon GS, Brar SK, Verma M, Tyagi RD. Recent advances in citric acid bio-production and recovery. Food Bioprocess Technol. 2011;4:505–29.
Cairns TC, Nai C, Meyer V. How a fungus shapes biotechnology: 100 years of Aspergillus niger research. Fungal Biol Biotechnol. 2018;5:13.
Nodvig CS, Nielsen JB, Kogle ME, Mortensen UH. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 2015;10:e0133085.
Zheng X, Zheng P, Sun J, Kun Z, Ma Y. Heterologous and endogenous U6 snRNA promoters enable CRISPR/Cas9 mediated genome editing in Aspergillus niger. Fungal Biol Biotechnol. 2018;5:2.
Dong H, Zheng J, Yu D, Wang B, Pan L. Efficient genome editing in Aspergillus niger with an improved recyclable CRISPR-HDR toolbox and its application in introducing multiple copies of heterologous genes. J Microbiol Methods. 2019;163:105655.
Kwon MJ, Schutze T, Spohner S, Haefner S, Meyer V. Practical guidance for the implementation of the CRISPR genome editing tool in filamentous fungi. Fungal Biol Biotechnol. 2019;6:15.
Zheng X, Zheng P, Zhang K, Cairns TC, Meyer V, Sun J, Ma Y. 5S rRNA promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger. ACS Synth Biol. 2019;8:1568–74.
Sarkari P, Marx H, Blumhoff ML, Mattanovich D, Sauer M, Steiger MG. An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger. Bioresour Technol. 2017;245:1327–33.
Kuivanen J, Wang YMJ, Richard P. Engineering Aspergillus niger for galactaric acid production: elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9. Microb Cell Fact. 2016;15:210.
Kuivanen J, Arvas M, Richard P. Clustered genes encoding 2-keto-l-gulonate reductase and l-idonate 5-dehydrogenase in the novel fungal d-glucuronic acid pathway. Front Microbiol. 2017;8:225.
Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 2011;21:885–97.
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–31.
Meyer V, Andersen MR, Brakhage AA, Braus GH, Caddick MX, Cairns TC, de Vries RP, Haarmann T, Hansen K, Hertz-Fowler C, et al. Current challenges of research on filamentous fungi in relation to human welfare and a sustainable bio-economy: a white paper. Fungal Biol Biotechnol. 2016;3:6.
Cairns TC, Zheng X, Zheng P, Sun J, Meyer V. Moulding the mould: understanding and reprogramming filamentous fungal growth and morphogenesis for next generation cell factories. Biotechnol Biofuels. 2019;12:77.
Sun X, Wu H, Zhao G, Li Z, Wu X, Liu H, Zheng Z. Morphological regulation of Aspergillus niger to improve citric acid production by chsC gene silencing. Bioprocess Biosyst Eng. 2018;41:1029–38.
Dai Z, Mao X, Magnuson JK, Lasure LL. Identification of genes associated with morphology in Aspergillus niger by using suppression subtractive hybridization. Appl Environ Microbiol. 2004;70:2474–85.
Meyer V, Arentshorst M, Flitter SJ, Nitsche BM, Kwon MJ, Reynaga-Pena CG, Bartnicki-Garcia S, van den Hondel CA, Ram AF. Reconstruction of signaling networks regulating fungal morphogenesis by transcriptomics. Eukaryot Cell. 2009;8:1677–91.
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA. Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet. 1987;206:71–5.
Carvalho ND, Arentshorst M, Jin Kwon M, Meyer V, Ram AF. Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol. 2010;87:1463–73.
Xu Y, Shan L, Zhou Y, Xie Z, Ball AS, Cao W, Liu H. Development of a Cre-loxP-based genetic system in Aspergillus niger ATCC1015 and its application to construction of efficient organic acid-producing cell factories. Appl Microbiol Biotechnol. 2019;103:8105–14.
Schape P, Kwon MJ, Baumann B, Gutschmann B, Jung S, Lenz S, Nitsche B, Paege N, Schutze T, Cairns TC, Meyer V. Updating genome annotation for the microbial cell factory Aspergillus niger using gene co-expression networks. Nucleic Acids Res. 2019;47:559–69.
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CA, Ram AF. Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol. 2007;128:770–5.
Alvarez-Escribano I, Sasse C, Bok JW, Na H, Amirebrahimi M, Lipzen A, Schackwitz W, Martin J, Barry K, Gutierrez G, et al. Genome sequencing of evolved aspergilli populations reveals robust genomes, transversions in A. flavus, and sexual aberrancy in non-homologous end-joining mutants. BMC Biol. 2019;17:88.
d’Enfert C. Selection of multiple disruption events in Aspergillus fumigatus using the orotidine-5′-decarboxylase gene, pyrG, as a unique transformation marker. Curr Genet. 1996;30:76–82.
Takahashi T, Hatamoto O, Koyama Y, Abe K. Efficient gene disruption in the koji-mold Aspergillus sojae using a novel variation of the positive-negative method. Mol Genet Genomics. 2004;272:344–52.
Zhu L, Maruyama J, Kitamoto K. Further enhanced production of heterologous proteins by double-gene disruption (DeltaAosedD DeltaAovps10) in a hyper-producing mutant of Aspergillus oryzae. Appl Microbiol Biotechnol. 2013;97:6347–57.
Nguyen KT, Ho QN, Do L, Mai LTD, Pham DN, Tran HTT, Le DH, Nguyen HQ, Tran VT. A new and efficient approach for construction of uridine/uracil auxotrophic mutants in the filamentous fungus Aspergillus oryzae using Agrobacterium tumefaciens-mediated transformation. World J Microbiol Biotechnol. 2017;33:107.
Cairns TC, Feurstein C, Zheng X, Zhang LH, Zheng P, Sun J, Meyer V. Functional exploration of co-expression networks identifies a nexus for modulating protein and citric acid titres in Aspergillus niger submerged culture. Fungal Biol Biotechnol. 2019;6:18.
Cairns TC, Feurstein C, Zheng X, Zheng P, Sun J, Meyer V. A quantitative image analysis pipeline for the characterization of filamentous fungal morphologies as a tool to uncover targets for morphology engineering: a case study using aplD in Aspergillus niger. Biotechnol Biofuels. 2019;12:149.
Fiedler MRM, Cairns TC, Koch O, Kubisch C, Meyer V. Conditional expression of the small GTPase ArfA impacts secretion, morphology, growth, and actin ring position in Aspergillus niger. Front Microbiol. 2018;9:878.
Amanullah A, Christensen LH, Hansen K, Nienow AW, Thomas CR. Dependence of morphology on agitation intensity in fed-batch cultures of Aspergillus oryzae and its implications for recombinant protein production. Biotechnol Bioeng. 2002;77:815–26.
Zheng X, Yu J, Cairns TC, Zhang L, Zhang Z, Zhang Q, Zheng P, Sun J, Ma Y. Comprehensive improvement of sample preparation methodologies facilitates dynamic metabolomics of Aspergillus niger. Biotechnol J. 2019;14:e1800315.
Sun X, Zhu J, Bao L, Hu C, Jin C, Harris SD, Liu H, Li S. PyrG is required for maintaining stable cellular uracil level and normal sporulation pattern under excess uracil stress in Aspergillus nidulans. Sci China Life Sci. 2013;56:467–75.
Nakayashiki H, Hanada S, Quoc NB, Kadotani N, Tosa Y, Mayama S. RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet Biol. 2005;42:275–83.
Xie S, Shen B, Zhang C, Huang X, Zhang Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE. 2014;9:e100448.
Acknowledgements
We thank Prof. Vera Meyer for providing the Tet-on inducible system.
Funding
This study was supported by the National Key R&D Program of China (2018YFA0900500), National Natural Sciences Foundation of China (31961133021 and 31700085).
Author information
Authors and Affiliations
Contributions
PZ and DW coordinated the project. XZ designed all the experiments. LZ and XZ carried out the experiments, interpreted the results and wrote the manuscript. ZZ took part in the intracellular metabolite profiling. TC revised the manuscript and give valuable suggestion for the final text. PZ, DW and JS revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Phenotypic comparison of pyrG and kusA mutants derived by A. niger D and D353. Figure S2. Insertion inactivation of the albA gene in A. niger D10 and D353.8. Figure S3. Titratable expression of pyrG mutagenesis constructed in A. niger. Figure S4. Extracellular organic acids of pyrG titratable expression mutant in 5 L bioreactor. Table S1. Protospacers used in this study, Table S2. Primers used in this study. Table S3. DNA sequences of sgRNA constructs used in this study. Table S4. DNA sequences of donor DNAs used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Zheng, X., Cairns, T.C. et al. Disruption or reduced expression of the orotidine-5′-decarboxylase gene pyrG increases citric acid production: a new discovery during recyclable genome editing in Aspergillus niger. Microb Cell Fact 19, 76 (2020). https://doi.org/10.1186/s12934-020-01334-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01334-z