
Abstract
Tumor microenvironment (TME) is a specialized ecosystem of host components, designed by tumor cells for successful development and metastasis of tumor. With the advent of 3D culture and advanced bioinformatic methodologies, it is now possible to study TME’s individual components and their interplay at higher resolution. Deeper understanding of the immune cell’s diversity, stromal constituents, repertoire profiling, neoantigen prediction of TMEs has provided the opportunity to explore the spatial and temporal regulation of immune therapeutic interventions. The variation of TME composition among patients plays an important role in determining responders and non-responders towards cancer immunotherapy. Therefore, there could be a possibility of reprogramming of TME components to overcome the widely prevailing issue of immunotherapeutic resistance. The focus of the present review is to understand the complexity of TME and comprehending future perspective of its components as potential therapeutic targets. The later part of the review describes the sophisticated 3D models emerging as valuable means to study TME components and an extensive account of advanced bioinformatic tools to profile TME components and predict neoantigens. Overall, this review provides a comprehensive account of the current knowledge available to target TME.
Similar content being viewed by others

Background
Significant developments made in the field of cancer therapy over the last decade have led to the improvement in the life expectancy of cancer patients [1]. However, these approaches are not equally effective across different tumor types and patients. Even the response to a specific line of therapy varies broadly depending on the type of tumor, i.e., benign, locally advanced or metastatic [2]. These therapeutic challenges have been exceedingly attributed to the ability of cancer cells to hijack the host machinery to create a niche, i.e., tumor microenvironment for themselves, and progressively modulate it from anti-tumoral to pro-tumoral responses [3]. Furthermore, a lack of unanimity in defining TME in tumor proximal and distal locations, and unavailability of model systems that accurately mimic the interaction of cancer cells with its microenvironment are the two prime reasons for our limited understanding of TME and exploring its therapeutic potentials [4, 5].
Tumor microenvironment is an ecosystem created by the cancer cells and comprised of components contributed by both tumor and host. Different factors in the TME unanimously ensure the development, progression and expansion of tumor through an uninterrupted supply of nutrients and oxygen, hampering the immune surveillance reach, and efficient drugs carting [5]. A dynamic interaction between cancer cells with these cellular and acellular components of TME is essential for generating heterogeneity, clonal evolution and enhancing multi drug resistance in tumor cells [6]. Broadly, six distinct specialized microenvironments within TME have been identified namely the hypoxic niche, acidic niche, innervated niche, metabolism microenvironment, immune microenvironment, and mechanical microenvironment [4].
Presently, most of the therapeutic efforts involving the TME either focusses on targeting TME components or the development of experimental model systems that accurately mimics complexities of TME. Moreover, TME-targeting strategies can be achieved by stimulating the intrinsic host immune system, which encompass either activation of anti-tumor immune cells or inhibiting pro-tumoral immune cells in the TME. Recent studies have shown that these specialized TME microenvironments and niches have potential to act as target of cancer therapy through reprogramming [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22].
Various modes of cancer treatment have been applied in clinic, which include surgery, chemotherapy, hormone therapy, radiation therapy, immunotherapy, targeted therapy, hyperthermia, photodynamic therapy and stem cell transplant [23]. More recently, immunotherapy has emerged as the preferred regimen because of its ability to induce durable responses, and low degrees of side-effects as compared to other types of treatment methods [24]. Cancer immunotherapy (CIT) strategies include adoptive T-cell transfer, immune checkpoint inhibitors, monoclonal antibodies, non-specific immune stimulation, oncolytic virus immunotherapy and vaccinations [25]. The major challenges posed by CIT approaches include lack of therapeutic responses and acquired resistance [26]. Therefore, lack of understanding of real-time TME dynamics and identification of cancer specific neoantigen is the major challenge in developing effective immunotherapy. Advanced 3D models provide the opportunity to reconstruct and comprehend the heterogeneous TME and its dynamic interactions more precisely by using human cells, and tightly regulated cellular compositions, physiological conditions and physical parameters [27,28,29,30,31]. Furthermore, bioinformatic tools are emerging as prospective means to profile and predict neoantigen. In this review, we described different components of TME as barrier and opportunity to target cancer, the 3D models available to understand TME, and the bioinformatics advancements towards profiling of tumor microenvironments for identification of novel immunotherapeutic targets.
Salient features of TME
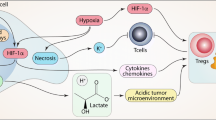
The main elements which define the variability in the TME includes but are not limited to genomic instability, tumor type, tumor location, presence of mutations such as KRAS, EGFR, PTEN etc., presence of lymph nodes, adipose tissue in the vicinity, and therapeutic interventions [32]. However, there are certain characteristics which are constant and encompass the hallmarks of the TME. These hallmarks include the presence of stromal cells, endothelial cells, components of both innate and adaptive immune cells and extracellular matrix [33]. Altogether, the intricate interactions among these TME elements led to the development of localized and specified microenvironments within TME which in turn defines the resilience and immunogenicity of the tumor as summarized in Fig. 1.
Specialized microenvironments of the TME
Hypoxic niche
Uncontrolled proliferation of cancer cells and limited vascularization from host cells give rise to oxygen crisis in different areas of a tumor [34]. These oxygen-restricted areas of the TME are known as the hypoxic niche as depicted in Fig. 1. The adaptation of cancer cells to a hypoxic environment is largely mediated by hypoxia inducible factor-1 (HIF-1) [35]. HIF-1 initiates the angiogenic process through activation of multiple factors including the most prominent angiogenic ligand, vascular endothelial growth factors (VEGF), and its receptors including VEGFR2 [36]. HIF-1 regulates expression of cancer stem cell markers like KLF4, MYC, OCT4, SOX2, and NANOG, and thus help cancer cells to survive through hypoxic crisis [37,38,39]. In addition to pluripotent factors, HIF-1 also regulates other epigenetic modifiers involved in regulating stemness, including BMI1 and SIRT1 [40]. Hypoxia signaling can promote EMT mainly through HIF-1-mediated transactivation of EMT inducing factors (EIFs) such as TWIST, SNAIL, and ZEB1 [41]. In addition, HIF-1 can also activate transforming growth factor beta (TGF-β), WNT, and NOTCH signaling and inhibit the Hippo signaling pathway to promote survival of cancer stem cells [42,43,44,45]. Several compounds capable of inhibiting hypoxia inducible factor-1 (HIF-1) or its targets have shown competence in inhibiting tumor progression and are in clinical trials Fig. 2B. For instance, topotecan, a topoisomerase 1 inhibitor, has been approved by the US Food and Drug administration (FDA) to be used as a second line of treatment for small cell lung and ovarian cancers [46]. Another drug targeting the hypoxic niche of TME, metformin, is presently in a clinical trial for head and neck squamous cell carcinoma (NCT03510390) [47]. Moreover, several hypoxia-reactive prodrugs, which become activated in the TME’s hypoxic niche have been developed [48]. TME also uses hypoxic response to rewire the metabolic mechanisms and transcriptomic profiles, which can be used as a therapeutic target in combinatorial therapy [49, 50]. Wigerup et al. has described various strategies to target hypoxia, and the drugs under exploration in a comprehensive review [51].
Acidic microenvironment
Tumor cells prefer glycolysis as the major mode of glucose metabolism, even in the presence of oxygen [52, 53]. Hypoxia and reduced vascularization further triggers glycolysis and suppress oxidative phosphorylation in tumor tissue. The elevated level of glycolysis causes increased lactate accumulation in the TME, which results in the extracellular low pH [54]. Acidic microenvironment at its initial stage of formation acts as a hostile niche, and triggers apoptosis in cancer cells. However, persistent TME acidification prompts cancer-cell adaptation, and results in aggressive tumors and hence act as barrier for effective therapy [55]. Furthermore, low pH facilitates activation of pro-tumorigenic macrophages, neutrophils, dendritic cells (DCs), and inhibition of tumor-infiltrating lymphocytes (TILs) cytotoxic activity, hence impairing immunosurveillance [4]. Targeting dysregulated pH zones of TME is therefore a potential opportunity for effective therapeutic intervention (Fig. 1). Several small-molecule inhibitors to target the acidic niche of TME are under exploration. In addition, pH-responsive drug release systems have also been recently developed to deliver cytotoxic drugs to the acidic TME. Recently, several small molecular inhibitors have been developed to target acidic tumor microenvironment, and Zhong et al. provides a detailed description of such class of inhibitors [56].
Inflammatory microenvironment
Inflammation plays critical role in initiation, progression and metastasis of cancer [57, 58]. The chemo-attractants produced by cancer cells stimulates the infiltration of immune cells like neutrophils, macrophages, dendritic cells, eosinophils and mast cells in TME. These inflammatory cells secrete pro-inflammatory cytokines like IL-1, IL-6, IL-15, IL-17, IL-23, tumor necrosis factor-α (TNF-α), and other molecules like IFN-γ, reactive oxygen species (ROS), serine and cysteine proteases, matrix metalloproteinases and membrane-perforating agents which are cytotoxic to tumor cells. Inflammation also potentiates the proliferation of residential myeloid cells and enhance the secretion of inflammatory factors such as histamines, cytokines etc., within TME. Additionally, it activates adaptive immune cells, and results in recruitment of anti-tumorigenic cytotoxic T lymphocytes (CTLs) [59]. Acute inflammation therefore creates a hostile condition for the tumor growth and progression [60,61,62]. However, persistent inflammation, hypoxic condition and a nutrient-restricted microenvironment within tumor results in the formation of an immunosuppressive microenvironment through accumulation of large number of immunosuppressive cells like M2 macrophages, MDSCs, Treg cells, Breg cells etc., These cells secrete pro-tumor cytokines such as IL-6, IL-1β, IL-17, IL-11, and growth factors like TNF-α which promotes tumor growth, proliferation,metastasis [63, 64] and therapy resistance which act as a barrier for effective therapeutic intervention (Fig. 1). Furthermore, multiple signaling pathways, such as NF-kB, JAK-STAT, TLR pathways, cGAS/STING, and MAPK pathways are known to play important role in regulating pro-tumor inflammation [59]. Targeting inflammatory microenvironment is therefore a potential opportunity, and at present, many anti-inflammatory drugs are undergoing clinical trials (Fig. 2A). One such drug, Statins have shown promising anti-tumor effect in different cancer types including CRC, breast cancer etc. [65, 66]. Aspirin, a non-steroid anti-inflammatory drug (NSAID) have shown beneficial effect in many kinds of cancer [67, 68]. Sulindac, another NSAIDs and selective COX-2 inhibitors are given to patients who are at high risk of getting colorectal cancer [68]. Targeting IL-6 is also emerging as an attractive strategy for cancer prevention and Tocilizumab, an IL-6R-specific antibody is under clinical trials. Chimeric monoclonal antibody siltuximab that binds IL6 is currently investigated for the treatment of several tumor types including prostate cancer and metastatic renal cell cancer. Several natural anti-inflammatory agents like curcumin, resveratrol, ursolic acid, capsaicin, silibinin, silymarin, guggulsterone, and plumbagin have also been enormously explored in cancer prevention. The recent review by Zhao et al. has comprehensively described the tumor-associated inflammation, cytokines/chemokines involved, and the targeting drugs under clinical trials [59].

Tumor microenvironment (TME): TME is a complex ecosystem of cellular. niche, acidic niche, inflammation etc. Extracellular matrix (ECM), the major non-cellular and acellular components, and several specialized microenvironments such as hypoxic component, provides architectural support, and act as a store house for factors such as chemokines, cytokines, growth factors etc., required for continuous tumor transformation process. Cellular components consist of non-immune and immune cell populations. Non- immune cell types include tumor cell, cancer associated fibroblast (CAF), neuron, and endothelial cell (blood vessel) that helps in tumor invasion, progression, and metastasis. Immune cells within TME comprise of tumor-associated macrophages (TAMs), tumor- associated neutrophils (TANs), dendritic cell (DCs), regulatory T cell (Treg), B cell, Natural killer (NK) cell, and cytotoxic T lymphocytes (CTLs). In immuno-competent conditions, CTL identifies and bring about tumor cell killing by releasing cytotoxic molecules such as granzyme-B, interferon-γ (IFN-γ), perforins etc. The figure is prepared by using BioRender software and publication license is obtained
Innervated microenvironment
Neuronal involvement in promoting tumor progression and metastasis is another level of complexity within TME. Intra-tumoral nerves are either newly formed or recruited fibers that infiltrate the TME from nearby tissues [19, 69]. Moreover, neuronal progenitor cells migrate from the brain to home in the developing tumor [70]. These progenitor cells facilitate tumoral neurogenesis through the formation and recruitment of functional neurons within the tumor [20, 71]. Additionally, TME also secretes chemokines that stimulates the nearby nerves to sprout and grow in the tumor [72, 73]. Collectively, this phenomenon has been termed as tumor innervation, and is found to be associated with an aggressive tumor phenotype, cancer-related pain, and poor prognosis in clinical studies [19, 74, 75]. Innervation within TME relies on the release of neurotransmitters or neuropeptides, such as dopamine, catecholamine, and acetylcholine. Crosstalk between the nerves and other TME components such as ECM, immune cells, stromal cells, endothelial cells and tumor cells has also been coupled with the poor prognosis and acquired resistance to current targeted therapies [76]. Therefore, assessment of tumor innervation might serve as a potential predictive marker of disease severity. Some studies have shown that surgically severing of nerves entering the tumor can inhibit tumor growth and metastasis [19, 77, 78]. Anti-neurotrophic therapies have also shown the promising outcomes in targeting densely innervated therapy-resistant tumors (Fig. 2). However, targeting TME innervation is relatively new field, and further studies are required to clearly understand the role of nerves in tumor progression. In a recent review, Li et al. describes the present status, future perspectives and the associated challenges associated with targeting of tumor innervation [79].
Tumor vascularization
Tumor-associated dysfunctional and leaky blood vessels regulate tumor perfusion and maintains the immunosuppressive microenvironment necessary for tumor survival and progression. The abnormal morphology and behavior of these tumor blood vessels is because of the disorganized network of the tortuous endothelial cells and lack of the normal hierarchical artery-arteriole-capillary arrangement [80, 81]. However, irrespective of its tissue of origin, several critical functions such as regulation of transport of oxygen, nutrients and other solutes from bloodstream to tissues, maintaining blood flow by providing non-thrombogenic surface, and controlling the infiltration of leukocytes between tissues has been attributed to these endothelial cell populations [82]. Hence, under malignancies, inhibiting the formation of such dysfunctional and leaky blood vessels, interrupting the supply of oxygen and nutrients, and increasing the leukocytes infiltration could be a promising therapeutic intervention (Fig. 1). Therefore, tumor-associated endothelial cells can act as an attractive target for therapeutic purposes [83]. Several anti-angiogenic compounds have been developed and tested in clinical trials (Fig. 2D); the results are promising with an increase in the overall survival. Bevacizumab (Avastin), the first anti-angiogenic antibody approved by the FDA is already in use [84]. Furthermore, sunitinib (Sutent), a multi-tyrosine kinase inhibitor of VEGF, platelet-derived growth factor receptor (PDGFR), and receptor tyrosine kinase—oncogene c-KIT (KIT) is a potent anti-angiogenic drug approved for the treatment of various tumors [85]. Recent review by Zheng et al. has comprehensively described various approaches employed in targeting tumor vascularization [86].
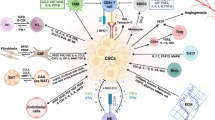
Major components of TME and targeting strategies
Stromal cells
Cancer cells and other TME cells secretes growth factors like transforming growth factor beta (TGF-β), fibroblast growth factor 2 (FGF2), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) which are key regulators of fibroblast recruitment and activation [87, 88]. Cancer associated fibroblasts (CAFs) provide the physical support to the cancer cells by secreting ECM (Fig. 1). CAFs also secrete MMPs, which are ECM-degrading proteases; hence regulates ECM turnover and plays crucial role in tumor invasion [89]. Additionally, CAFs also play a critical role in the angiogenesis, progression and metastasis of tumors by producing many growth factors and proinflammatory cytokines like vascular endothelial growth factor (VEGF), IL-6, CXC-chemokine ligand (CXCL12), TGFβ, NF-κB, TNF-α, IFN-γ, SDF-1α, EGF, galectin-1 [90,91,92,93,94,95]. CAFs therefore are the key determinant in the tumor development, and are emerging as a potential target for cancer therapies [96] as described in Fig. 2. One of the most successful approaches is targeting fibroblast activating protein (FAP) expressed by stromal cells. FAB is known to be involved in the proliferation of stromal cells as well as ECM secretion [97, 98]. Anti-FAB antibody conjugated with cytotoxic drug like maytansine (DM1) has shown promising results in xenograft mice models studies [99]. Besides, all-trans retinoic acid (ATRA) has also been observed to inhibit FAP, TGFβR and αSMA expression in CAFs, and therefore, currently being used for the treatment of multiple cancer types in conjunction with other drugs [100, 101]. Furthermore, drugs-like paricalcitol, a vitamin D receptor agonist, has been used to reprogram pro-tumor CAFs to a quiescent-like state [102]. In a comprehensive review, Chen et al. describes various strategies available to target CAFs, and different drugs under evaluation presently [103].
Extracellular matrix (ECM)
Extracellular matrix (ECM) is the major non-cellular component of TME and mainly comprise of collagen. In addition to providing structural framework, ECM also acts as a store house of several chemokines, cytokines and growth factors which plays important role in creating immune-suppressive tumor microenvironment [104]. Approximately 60% of the total mass of the solid tumors are comprised of ECM deposits that provides structural stiffness to them. Such ECM rigidity enables the cancer cells to proliferate aggressively and undergo directed cell migration [105, 106]. High matrix stiffness has been observed in aggressive tumors such as triple negative breast cancer and associated with poor prognosis [107] (Fig. 1). TGF-β, which regulates collagen synthesis, has been shown to inhibit tumor growth in several in vitro studies [108,109,110]. Fresolimumab, a monoclonal antibody that targets TGF-β mediated collagen synthesis, underwent several clinical trials (clinicaltrials.gov identifier: NCT01401062 and NCT02581787) [111,112,113]. Similarly, ronespartat (SST0001), showed strong anti-tumor effect in multiple myeloma in a Phase 1 clinical trial (Clinical Trial NCT01764880) [114]. Since the high expression of MMPs in the TME regulates ECM homeostasis and facilitates tumor invasion and metastasis, MMPs inhibitors have also been evaluated as potential therapeutic target and have shown promising results [115]. Drugs targeting MMPs like incyclinide, have been through several clinical trials for AIDS-related sarcomas (Clinical trials NCT00004147, NCT00003721, NCT00001683, and NCT00020683) [116]. Other MMPs-targeting agents include JNJ0966, which is highly selective towards MMP-9, and the antibody Fab 3369, which targets MMP-14 [117, 118]. In a recent review, Huang et al. has described various ECM targeting strategies in detail [119].
Major immune components—innate and adaptive
Immune cells are essential components of TME. Depending on the stage and complexity of the TME, immune cells can either be anti-tumorigenic or promote tumor growth. Chronic inflammation at the site of tumor growth activates the residential and circulatory immune cells to accumulate in the vicinity of tumor. Furthermore, components of tumor microenvironment especially stromal cells secrete cytokines and chemokines which attract infiltration of immune cells from both innate and adaptive immune system. Various review articles discussing diverse immune components of TME in detail are widely available in literature, hence, the description of immune components of TME is kept concise in the current review [33, 120,121,122,123,124,125,126].
Macrophages
Macrophages are specialized cells of the innate immune system that are derived from monocytes. Several soluble factors secreted by the cancer or stromal cells are responsible for recruitment of macrophages within TME. These soluble molecules include IL-3, colony stimulating factor 1 (CSF-1), and CCL2 [61]. CSF-1 induce monocyte transformation into highly plastic non-polarized (M0) macrophages. M0 macrophages can be stimulated by interferon-γ (IFN-γ) into activated anti-tumor M1 macrophages which exerts cytotoxic effect on tumor by secreting cytokines like IL-2 and TNFα. Conversely IL-4, IL-10 and IL-13 secreted by tumor or stromal cells can stimulate conversion of M0 to pro-tumor M2 macrophages. These tumor-associated macrophages (TAMs) secrete anti-tumor cytokines IL-6, IL-8 and IL-10 and matrix metalloproteinases (MMPs) which regulates neo-vasculogenesis in TME [127]. The cytokines and chemokines secreted by macrophages and tumor cells within TME further impede CD8 + T-cell infiltration, thus creating an immunosuppressive microenvironment which support tumor growth and metastasis [61]. High infiltration of TAM within TME has been shown to be associated with poor prognosis in many tumor types [128,129,130,131]. Targeting macrophages therefore is a promising strategy verified in many clinical trials; however complete removal of macrophages has shown severe liver toxicity. Various indirect approaches to target TAMs are presently under evaluation in several studies Fig. 2F, G. For example, targeting colony stimulating factor by CSF1R inhibitors alone or in combination with other agents have shown promising anti-tumor efficacy in various tumor types. Several studies targeting pro-inflammatory CCR2/CCL2 axis have shown initial promising outcomes. PF-04136309 (CCR2 inhibitor) is in a phase 1 clinical trial and has been tested in combination with FOLFIRINOX chemotherapy in PDAC patients [132]. Similarly, changing the polarization of pro-inflammatory M2 to anti-inflammatory M1 state within TME can enhance the anti-tumor effect of TAMs, and such strategy is under evaluation in several clinical trials. Recent developments regarding therapeutic targeting of macrophages within TME have been extensively described in the review by Pathriaet et al. [133].
Neutrophils
Tumor-associated inflammation drives accumulation of neutrophils within TME. The chemokine/cytokine composition of the TME determines the anti-tumor (N1) or pro-tumor (N2) phenotype of tumor-associated neutrophils (TANs). Anti-tumor N1 TANs possess high levels of TNFα, CCL3, ICAM-1, and are also involved in the production of ROS which is cytotoxic to tumor cells. In the later stages of tumor development, there is high infiltration of N2 neutrophils that support tumor growth and progression [134, 135]. IL-8 secreted by tumor cells in the TME stimulates neutrophils to release arginase enzyme which is responsible for degradation of arginine. Arginine is essential for the activation and proliferation of T-cell. Hence, TANs play important role in suppression of T-cell immune response. Additionally, TANs also suppress NK cells activation by regulating the secretion of IL-18. Tumor-associated neutrophils (TANs) therefore contribute substantially to tumor progression, invasion, and angiogenesis [136, 137]. A comprehensive account of targeting TAN in cancer immunotherapy has been recently described by Rahmy et al. [138].
Dendritic cells
Tumor infiltrating dendritic cells (DCs) are the chief antigen-presenting cells (APCs) which scan and phagocytose the tumor associated antigens, process the antigen peptide to present with MHC class II molecule and prime CD8 + T cells. The pro-inflammatory cytokines and other soluble factors present in TME such as, IL-15, IL-2, IL-21, IFN-α, and GM-CSF further enhance the anti-tumor characteristic of DC.
Tumor cells and other TME components like stromal cell, endothelial cells, tumor infiltrating immune cells also secrete cytokines, chemokines, prostaglandins and growth factors which can modulate the DCs to behave in a pro-tumor fashion. These soluble factors like IL-6, IL-10, IDO, M-CSF, TGF-β1, PGE2, VEGF present in TME reprogram DCs to possess inefficient antigen-presenting capabilities, and an immunosuppressive regulatory phenotype that supports tumor progression [139,140,141,142,143]. The balance between the levels of different cytokines and growth factors in TME determines the pro-tumor or anti-tumor nature of DC. Targeting these cytokines therefore can be promising strategy to potentiate the immunotherapies like immune checkpoint blockade and CAR-T therapy. Personalized vaccines comprise of patient derived DCs which are engineered and amplified and injected back to host circulation have shown promising tumor suppressing effect [144] Fig. 2J. Similarly, delivery of ligand for toll-like receptor 3 (TLR3) or STING agonist to activate DCs at the site of tumor are showing promising result in enhancing DC based immune response [145]. Furthermore, combination therapy of DC vaccines with anti-inflammatory drug like aspirin have shown promising outcome in pre-clinical models [146]. Wculek et al. has described the role of dendritic cells in TME and DC-targeting strategies in a comprehensive review [147].
Natural killer cells
Natural killer (NK) cells are key components of the innate immune system and are highly efficient in identifying and killing undifferentiated or poorly differentiated tumor cells in the tumor or in circulation [148, 149]. The major mode of action of NK cells is by releasing perforins and granzyme B to induce necrotic or apoptotic cell death. NK cells secrete a wide variety of anti-tumor cytokines such as IL-10, IL-5, IL-13, GM-CSF, IFN-γ, TNF-α [150, 151]. IFN-γ is one of the major anti-tumor cytokines secreted by NK cells. The balance between the cytokines secreted by tumor infiltrating immune cells, tumor cells or stromal cells in the tumor microenvironment determines the pro-tumor or anti-tumor characteristic of NK cells [152]. Immunosuppressive factors secreted by tumor cells include TGF-β, VEGF, indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), and adenosine which inhibit antitumor immune functions. Tregs, MDSC, and M2-macrophages secrete immunosuppressive cytokines such as IL-10 and TGF-β which inhibits NK cells activation and function. Therefore, enhancing the NK cells activity within the TME is a promising direction in cancer therapy [153, 154] Fig. 2I. Wu et al. has provided a detailed description of NK cell-based targeting strategies for cancer therapy [153].
T cells
T-cells forms the major line of anti-tumor adaptive immune response in TME. Tumor-infiltrating lymphocytes (TILs) present in TME include CD4 + helper cells, immunosuppressive CD4 + FOXP3 + regulatory T-cells (Tregs) and CD8 + cytotoxic T-cells (CTLs). Higher CD8 + T cells infiltration is mostly associated with better prognosis and therapy response. Naive CD8 + T cells gets activated and differentiate into cytotoxic effector T cells on encountering tumor-associated antigens (Fig. 1-Active T cell mediated tumor killing). Under normal circumstances, once the antigen is eliminated, most effector T cells undergo apoptosis while a small fraction differentiate into memory T cells. However, in cancer, persistent stimulation of CD8 + T cells in tumor microenvironment results in a hyporesponsive state of T cell known as T cell exhaustion. T cell exhaustion shows progressive loss of effector function like loss of IL-2, TNF-α, and IFN-γ production and sustained expression of inhibitory receptors. High expression of inhibitory receptors such as PD-1, CTLA-4, Tim-3, LAG-3, B- and T-lymphocyte attenuator (BTLA), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), NK cell receptor 2B4, and the glycoprotein CD160 are associated with the severity of the dysfunctional T cell phenotype. Reactivation of CD8 + T cells therefore presents a huge opportunity to target advanced tumors. Indeed, immune checkpoint blockers (ICBs) emerged as most promising therapeutic intervention in many types of cancer. At present, all the cancer immunotherapeutic approaches are based on the objectives of sustaining the activation of T-cells, and stimulating T-cell infiltration within the tumor [155] Fig. 2K. In the recent review, Waldman et al. has defined various T cell-based targeting approaches in cancer therapy [156]. The major immunotherapeutic strategies employed currently to attain these objectives include immune checkpoint blockers (ICBs), chimeric antigen-receptor (CAR)-based therapies, and cancer vaccines. In the following sections, the above-mentioned major cancer immunotherapeutic approaches have been described in detail.
Cancer immunotherapeutic approaches
Immune checkpoint blockers
Drugs targeting immune checkpoints, or their associated ligands have emerged as one of the most successful cancer immunotherapeutic approaches in several cancer types such as melanoma, non-small cell lung cancer (NSCLC), microsatellite instability high colorectal cancer, gastric cancer etc. Immune cell populations like T-cells, B-cells and cancer cells express repressor proteins, which when activated marks the termination of the immune response. These checkpoint proteins help in maintaining homeostasis of the immune response by controlling hyper immune activity to prevent autoimmunity. The major checkpoint proteins expressed by immune cells are programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte-associated protein-4 (CTLA4). Recent studies have reported a few other checkpoint proteins, which include VISTA, TIM3, TIGIT, and LAG3. T- cell exhaustion is associated with high expression of checkpoint protein which inhibits T-cells clonal expansion and inactivates T cell immune response (Fig. 1—Immune evasion). Additionally, cancer cells also express high levels of checkpoint protein as well as its ligands (Fig. 1). TME containing high levels of exhausted T cells expressing checkpoint proteins benefits most from CPBs. Mostly, a three-factor metric system is used to predict the efficacy of immune checkpoint inhibitors. These factors include 1) expression levels of checkpoint proteins and their ligands, 2) tumor mutation burden, and 3) presence of CD8 + T cells within the tumor. However, these factors cannot be considered as the gold standard to ensure the therapeutic response, because primary resistance can strongly limit the efficacy of immune checkpoint therapy in a majority of cancer patients.
T-cell based therapeutic strategies
Adoptive T-cell therapy is based on the principle of expanding the patient’s T-cell pool to enhance the immune system’s ability to identify and destroy tumor cells. Adoptive cell therapy involves isolation of autologous or allogenic T-cells from the patient or donor respectively, followed by ex vivo expansion and injection into the patient. Adoptive cell therapy has become a potential therapeutic option in many types of cancer [157, 158]. Infusing tumor-infiltrating lymphocytes (TIL) along with interleukins like IL-2 and lympho-conditioning showed promising results in a subset of patients with metastatic melanoma [159]. However, major disadvantages associated with these therapies include difficulty in predicting which patients will respond, and the cost and time involved in ex vivo production of T lymphocytes [160]. Recently, Morotti et al. has described in detail the promises and challenges of adoptive T cell therapy [161].
CAR-T cell therapy involves production of genetically engineered T cells expressing synthetic T-cell receptors (TCRs) specific to the tumor antigens. CAR-T cells have been proved to be very impactful cancer therapy in most of the relapsed or refractory hematological cancers. Several clinical trials involving CAR-T cell therapies have shown promising results [157]. The astonishing outcomes of these trials has resulted in the approval of several drugs by the European Commission (EC) and U. S. Food and Drug Association (FDA) between 2017 and early 2021 [162]. The major drawback associated with CAR-T cell therapy is the development of systemic inflammatory toxicity in the host body [163, 164]. Further studies are ongoing, which aim to reduce the bottlenecks associated with this line of cancer therapy. Fesnak et al. has provided a detailed review on the promises and challenges associated with CAR-T cell therapy [165].
Alternatively, CAR-NK cellular immunotherapy has been developed to overcome the CAR T-cells therapy limitations like graft-versus-host disease (GVHD), cytokine release syndrome (CRS), neurotoxicity etc., [166, 167]. Abundant availability from various sources and no HLA-matching restriction are the two major advantages associated with CAR engineered NK cells [168]. In contrast to CAR T-cells, CAR-NK cells are considered as safer because they may rarely cause GVHD, CRS and neurotoxicity. Additionally, CAR-NK cells secrete a totally different set of cytokines (e.g., GM-CSF and INF-γ) as compared to those pro-inflammatory released by CAR T-cells (e.g., TNF-α, IL-2, IL-6) [169]. Moreover, CAR-NK cells can identify and neutralize tumor cells both in CAR-dependent as well as CAR-independent manner [167]. CAR-NK cells pro-tumor effects are contributed by their ability to release of immunosuppressive cytokines (e.g., TGF-β, adenosine and indoleamine 2,3-dioxygenase) within immuno-suppressive TME, and also to express inhibitory receptors (e.g., TIGIT, PD-1, CTLA-1, NKG2A, CISH) [170, 171]. Therefore, the future efforts should aim at increasing the efficacy of the CAR-NK cell immunotherapy. Recently, a clinical trial (NCT03056339) was conducted on 11 patients to target CD19-expressing B-cell malignancies through CAR-NK cells derived from umbilical cord blood (UCB) [59]. 8/11 patients in this trial showed clinical response, and seven of those had rapid response and complete remission [172]. Altogether, the results from this trial and other ongoing clinical trials suggest that the CAR-NK cell therapy may represent the future opportunity in cancer immunotherapy. In the review by Albinger et al., an extensive comparative study describing the potential, limitations and ongoing clinical trials of CAR-based therapies i.e., the CAR-T and CAR-NK, has been described extensively.
Cancer vaccines
Therapeutic cancer vaccines are personalized to patients and are based on the principle of activating host T cells by exposing them to tumor-specific neo-antigens (i.e., mutated proteins on cancer cells). The major hurdle to this therapeutic strategy is to identify and obtain the tumor-specific neoantigens. In several studies, cancer cells obtained from biopsies or whole cell lysate were used as vaccines, but in most of the cases it had failed to activate the host immunity-possibly due to an insufficient amount of tumor-specific neo-antigens. Hence, other approaches using dendritic cells are employed, in which host DCs are activated by tumor-specific neo-antigens, primed, expanded and injected back in the host circulation [173]. One such vaccine sipuleucel-T showed some initial success and was ultimately approved by the FDA in 2010 [174, 175]. mRNA vaccines are recently emerging as a mode to express neo-antigen peptides. However, these strategies are far from getting into the treatment regimens due to the complications associated with the production and administration [23]. Recently, Hu et al. in their review has provided a detailed account of the status of cancer vaccines in cancer therapy [176].
Despite the promising response observed with cancer immunotherapies, a large section of patients does not respond to the initial treatment or develop resistance later during the treatment. Several possible intrinsic and extrinsic factors contribute to such primary or acquired resistance. Aberrant intracellular signaling pathways in tumor cells is one of the key factor which inhibits T cell function and infiltration in the TME. One of the major pathways reported to be overactive in many cancer types, is the MAPK/ERK pathway which results in over-production of VEGF and IL-8, and establishment of an immunosuppressive environment for the T-cells [177]. Genetic mutations in EGFR and loss of function mutation in PTEN can activate the MAPK or PI3K pathways, which are reported to cause resistance to immune checkpoint therapy [178]. Genetic alterations in interferon-γ (IFN-γ), a prominent anti-tumor factor secreted by T effector cells, can also lead to immunotherapy resistance [179]. Aberrant β/Wnt signaling is also associated with primary or acquired resistance in many cancer types [180]. Furthermore, tumor cells are proficient in hiding from cytotoxic T-cells either by 1) preventing exposure of tumor-specific antigens on major antigen presenting cells by altering the MHC, or 2) by extensive trimming or modification of the antigen to be recognized by the T cells which lead to primary or acquired resistance. Furthermore, overexpression of immune checkpoint proteins and their corresponding ligands in the TME is a characteristic feature of many cancer types and is associated with T-cell exhaustion. This is one of the prominent factors involved in acquiring resistance against immunotherapy.
The extrinsic factors that contribute to primary or acquired resistance to immunotherapy include the components of the TME other than tumor cells. Myeloid lineage cells in the TME including tumor-associated macrophages, neutrophils, monocytes etc., inhibit T cells trafficking in the tumor, and sustain tolerance towards immunotherapy. Tregs, professional checkpoint cells, control T cells activity by secreting inhibitory cytokines or direct killing. High recruitment of Tregs in TME is associated with immunotherapy resistance [181]. TME’s acellular components such as hypoxia, ECM, low pH etc., also creates a highly hostile nutrient depleted, low oxygenated, acidic niche which favors T cell depletion or inactivation, thus playing a crucial role in acquiring resistance to various immunotherapies. Sharma et al. in their latest review provides a comprehensive account of the mechanism involved in immunotherapy resistance [2].
Highly efficient preclinical models are therefore urgently warranted to understand the complexity of the TME and it’s possible reprogramming for therapeutic interventions and the mechanism associated with therapy resistance.

Targeting different TME components for cancer therapy: Current strategies available for targeting major TME components for effective cancer therapy are shown. A Targeting inflammation, B targeting hypoxic TME, C targeting TME nerve supply, D targeting TME vascularization and cellular components like E targeting cancer associated fibroblasts (CAFs), targeting innate immune components by F Inducing M1Polarization, G Inhibiting M2 Polarization, H targeting Neutrophils, I targeting Natural Killer cells, J targeting Dendritic cells, and targeting adaptive immune components by K Activation of CTLs and L Targeting B cells are promising targets. Various drugs/inhibitors/antibodies targeting these components are in preclinical studies, under clinical trial or FDA approved for cancer treatment. The figure is prepared by using BioRender software and publication license is obtained
Models to study interactions of the TME
Several attempts have been made towards in vivo and in vitro modelling of the TME interactions, and most of these studies are based on 2D co-cultures, xenografts, humanized mice, and syngeneic mouse models [5, 182, 183]. Undoubtedly, 2D models offers a cost-effective model system with high degree of reproducibility of the conditions. However, the cells in 2D co-cultures form a monolayer, and thus the models are unable to accurately mimic the TME’s complex cellular interactions and signaling pathways [30, 184, 185]. Furthermore, the 2D models are unable to maintain the original morphology and polarization of the TME components. On the other hand, animal model systems are very expensive. Also, very often these in vivo model systems lack true representations of human-specific immune response [186, 187]. This partially explains why only 27% of the drugs with high efficacy in animal models can successfully reach to phase II clinical trials [188, 189]. Bioengineered 3D in vitro model systems can overcome several of the abovementioned challenges posed by 2D and animal models [182]. In the following sections, various 3D in vitro models and their utility in studying complex interactions within the TME will be described.
Cell/extracellular matrix-based 3D models
In concordance with the fact that ECM is a major component of the TME with tumor-inducing capabilities, several cell/ECM-based 3D models have been devised to study interactions of the TME components [182].
Tumor cells are cultured within decellularized native tissues or natural/synthetic biomaterial scaffolds that provide proper cell adhesion, differentiation, and migration properties, and closely mimic the cell-ECM interactions [190,191,192,193,194,195,196,197,198,199]. While synthetic polymer provides higher degree of control and modulation of scaffold’s properties, the natural or decellularized ECM (dECM) recreate biochemical and structural environments similar to that of in vivo conditions by maintaining ECM and tissue-specific architecture [198, 199]. dECM have an additional advantage over other scaffolds as it considers ECM environment as a whole, whereas scaffolds focus on specific ECM components. The major concern with dECM-based 3D models is the intactness of the tissues, as decellularization procedure involves treatment with enzymes and detergents [200]. Along with selection of scaffolds biomaterials, cell types and physical/chemical conditions are other critical factors that determine the experimental outcomes of tumor modeling. For instance, seeding porous scaffolds with different cell populations of the TME, and allowing them to proliferate and rearrange, result in a 3D matured scaffold culture condition. Whereas cells cultured in a hydrogel-based scaffolds, after proliferation and undergoing rearrangement generate a matured cell-laden hydrogel. Further, depending on the hydrogel and cell type used for seeding, such matured cell-laden hydrogels can form cell clusters/organoids. While maintaining the original hydrogel network, matured and rearranged cell populations can also produce ECM in such in vitro 3D models. The hydrogel-based model’s mechanical properties can be modulated in order to closely mimic tumor ECM [182].
Based on these considerations, Rijal et al. developed a native ECM based tissue matrix scaffold (TMS) [201]. This in vitro model consists of multilayered tissue culture platform derived from mouse mammary tissue. Culturing of the stromal and cancer cells in compartmentalized manner, induces the expression of extracellular and intracellular biomarkers of breast cancer, thus confirming the correct proliferation and cancer growth. Therefore, this model mimics mammary tissue, and can be used for specific tumor biomarkers screening [201]. Similarly, Hume et al. created a 3D tumor model by culturing tumor cells and adipocytes in collagen scaffolds [197]. Presence of adipocytes was observed to promote cancer cell invasion by increasing tumor cells migration, while decreasing the total number of drifting cells. Thus, this 3D tumor model clearly demonstrates the heterogeneous cell behavior within TME [197].
Scaffolds can also be used to develop more recent in vitro 3D bioprinted models, and investigate spatio-temporal patterning of cells in tumor [187]. In 3D bioprinting technique, bioinks (sorted individual cell types and/or scaffold/hydrogel) are deposited in a predefined manner using a 3D bioprinter and then crosslinked to the carrier material (scaffold/hydrogel) to generate a stable culture. Depending on the bioinks and the specific aim of the study, different types of mature 3D bioprinted models can be obtained after cells proliferation and rearrangement, i.e., scaffold-free culture, semi-scaffold free culture and scaffold-based cultures. Such 3D in vitro model’s composition and architecture is well-defined with higher degree of reproducibility. These model systems have enabled us to introduce tumor cells into the complex TME, and to study ECM deposition, interaction of different TME cell types, and self-organization of the tissue itself [202]. Recently, Langer et al. has bioengineered a more desmoplastic TME by sequentially incorporating breast cancer cell core surrounded by human mammary fibroblasts, umbilical vein endothelial cells, primary human preadipocytes and mesenchymal stem cells [202]. Using this 3D tumor model, self-organization of tissue, interaction of tumor and stromal cells, and ECM deposition in complex tumor microenvironment could be learned [202].
Bioprinted 3D in vitro models can also be used to investigate time-dependent studies such as the kinetics or dynamics of administered drugs, growth factors and metastatic dissemination of cancer cells over time [203]. For instance, Meng et al. bioprinted 3D model to recreate TME that primes metastatic spread of lung cancer [203]. Taken together, with the advent of 3D bioprinting technique, creation of spatially defined 3D in vitro models has been improved. Well-defined composition and architecture, provision to use variety of materials with higher precision while fabrication, and commercial availability has further enhanced the utility of these 3D model systems. The common limitations associated with development and utilization of such in vitro 3D models include toxicity of bioinks, slow printing speeds and lack of reproducibility to devise state-of-the-art model system [204, 205].
Cell-based 3D models
In cell-based 3D models, the cells are present as aggregates, and usually they formulate their own intrinsic ECM. Thus, these models closely mimic tumorigenesis, the physiological framework of tumor organization, and tumor-stromal interactions within the TME [182]. Depending upon the differences in the initial stages of constitution, cell-based 3D models can be broadly classified as 3D (hetero)spheroids and organoids.
3D (hetero)spheroids model system
3D (hetero)spheroids are the most extensively used 3D model system in cancer biology and are specifically used for anticancer drugs testing. Such spheroid models are formed as a result of forced aggregation of selected cells that manifest high deposition of ECM or high cell–cell contact [182]. The main advantage of using cell-based 3D (hetero)spheroids model systems is its ability to precisely recreate important in vivo tumor microenvironment features such as morphology constituted by multiple cell layers, cell–cell/cell-ECM interactions, cellular heterogeneity, gene expression patterns, and cell signaling pathways [206]. Moreover, these 3D (hetero)spheroids provide flexibility to maintain different types of physical/chemical gradients and integrate multiple cell types [182, 206]. In conjunction with 3D bioprinting technologies, 3D (hetero)spheroids are cost-effective 3D culture method with an ability to create more physiologically representative tumor model. Although the cell-based 3D (hetero)spheroids model systems have immensely advanced our cancer biology knowledge, there are few constraints associated to it such as slow fabrication rate and more costly as compared to 2D cultures [182]. Furthermore, lack of homogeneity in the size/morphology of cell aggregates in spheroids compromise the reproducibility, and thus impedes the development efforts of the cell-based 3D standard models [207].
Organoids- 3D tumor model system
Organoids are 3D tumor models generated as a result of proliferation and self-organization of a single progenitor cell. Therefore, these models can closely mimic the architecture and the complexity of the tissue of origin [208]. Unlike spheroids, organoids are developed based on the genetic programming of the progenitor cell, thus mimicking the actual tumor development more closely. The major advantage of organoid 3D models is their distinct capability to follow different stages of native tumor progression trajectory, and therefore its capability of retaining the cellular heterogeneity and maintaining the pathophysiology of the tumors in vitro [209]. Moreover, as organoids can retain salient tumor features in three-dimensional space, these models are suitable for studying tumor-stroma interactions. However, organoids has some limitations such as its development is time-consuming with high degree of variability between experiments, some of the mature organoid models does not truly represent in vivo conditions, and lack stroma and vasculature system [210]. Furthermore, the tumor organoid models lack immune-competent microenvironment and stromal components because of the epithelial origin of the progenitor cells. This drawback has been overcome by co-culturing organoids with stromal and patient’s immune cells [211, 212]. More advanced organoid culture systems with well-defined architecture, cellular composition and signaling profiles can be developed by combining 3D bioprinting technique with organoids [211, 213]. Such combined strategy has the potential to ensure both precise spatial arrangement of cells in 3D models and maintaining the hierarchical-like architecture of the TME [182].
3D organoid models developed using patient-derived tissues are known as patient-derived organoids (PDOs). These tumor model systems have removed the bottleneck of extrapolation of results from animal and patient-derived xenografts (PDX) models [210, 211]. PDOs can be generated from various types of adult stem cells (ASCs) and pluripotent stem cells (PSCs) from distinct tissues through a procedure like human organogenesis [214, 215]. PDOs derived from ASCs contain epithelial cells and are suitable model system to study tissue regeneration and homeostasis. Contrastingly, PSCs derived PDOs can contain cells of both epithelial and non-epithelial origins, and are appropriate to study organ development [216]. Stability of gene expression profiles and reproducibility of model system are the major advantages associated with the usage of PDOs [217]. PDOs recreates basic characteristics of primary tumors while maintaining genomic and transcriptomic profiles of primary tumors [209, 218,219,220,221,222,223,224,225,226]. The heterogeneities specific to patients and cancer subtypes can be captured using individual PDOs, which has been reported to be in more consent with the actual patients response to drugs [227]. Additionally, PDOs can also be very useful in developing patient-specific treatment strategies. By combining organoids derived from healthy and tumor tissues from a patient, the efficacy of large number of drugs can be tested at the same time. The best drug for a patient responds by selectively killing tumor cells without damaging the healthy ones [227]. Moreover, the ease of manipulation of PDOs through CRISPER/Cas9 approach has encouraged its implementation for tumor modelling and identifying significant driver mutations involved in the tumor development and progression [228]. Recently, circulating tumor cells (CTCs) isolated through non-invasive liquid biopsies of cancer patients has also been used to develop PDOs. These CTC-derived organoids could be pivotal in gaining genetic and epigenetics insights about cancers in patient-specific manner [229]. Collectively, the development of PDOs has enormously transformed the drug and target discovery research arena, providing new avenues for drug testing, and designing personalized therapeutic interventions in a pre-clinical setting [230].
While PDOs models have many advantages, there are several limitations associated to it. These cell-based 3D models have been reported mostly to be deficient of essential components like blood vessels, stromal cells, immune cells, surrounding mesenchyme and neurons, thus lacking the typical TME [208]. Deficiency of immune cells within PDOs TME appears to limit its value in studies evolving tumor immunotherapy approaches [227]. For instance, at present, the efficacy determination of the inhibitors of immune checkpoints programmed death-1/programmed death ligand-1 (PD-1/PD-L1) using PDOs model cannot be conclusive and need further research attention [231]. Recently, there have been efforts to create a native immune microenvironment within PDOs by co-culturing of more complex immune and stromal components in a more compartmentalized manner [232, 233].
In summary, PDOs are good substitute model for understanding tumor biology, drug screening, and development of therapeutic approaches because of the aforesaid advantages. Currently, research involving the development of PDOs and its clinical applications are still in its early stage. In future, PDOs-oriented research should focus on the improvement of TME by overcoming present barriers and conducting more clinical trials on PDOs model systems with precisely defined composition and architecture.
Microfluidics models
Development of microfluidic systems, and their application in building organ-on-chip devices (platforms that can model physiological functions of tissues and organs) have revolutionized the field of tumor biology [182]. The main advantage of these in vitro 3D culture models is their flexibility to modulate various parameters independently. In these models, optimized cell survival conditions can be achieved by maintaining desired cellular heterogeneity and localization, chemical gradients, tissue interfaces orientations and mechanical forces [234]. Such tumor biology-specific microfluidic cancer-on-a-chip (CoC) models are now becoming preferred systems due to their microscale volume requirement which makes it cost-effective as compared to other 3D culture protocols and bioreactors [234,235,236,237,238]. Moreover, heterotypic cancer-on-a-chip 3D model systems generated by culturing multiple cell types in a dynamic microenvironment of the microfluidic chip can be used to understand distinct interactions/communications between tumor cells and various TME cellular and acellular components [239]. For instance, T-cell infiltration rates within TME has been studied on a heterotypic 3D microfluidic platform developed from breast cancer cell, human umbilical vein epithelial cells (HUVECs) and monocytes confined spatially in a gelatin hydrogel, and T cells dispersed in the medium [239]. The results showed higher rates of T-cells infiltration in presence of monocytes in the medium as well as extreme hypoxic conditions stimulated by using tumor spheroids instead of diffused cancer cells [239]. Furthermore, the effects of different growth factors (GFs) or drugs in a biomimetic microenvironment and stroma-driven ECM remodeling can also be studied using these microfluidic chip-based in vitro 3D models with a high degree of monitoring [240, 241]. Carvalho et al. have developed a microfluidic chip-based in vitro 3D model to mimic human colorectal cancer TME’s microvascular tissue functions. This model system allowed radial drug access into solid tumors and was used to assess the dynamic interaction between endothelial cells and colorectal tumor cells in a time-dependent manner [240]. Additionally, Gioiella et al. made a tumor-on-a-chip model with a stromal compartment to investigate tumor-stroma activation-dependent ECM remodeling [241]. Recently, innovative 3D in vitro models have been developed by combining the features of tumor spheroids/organoids and microfluidic chip systems. These models can be used to study tumor-stroma interactions and their systemic effects simultaneously [27, 242, 243]. For example, a 3D microvascular network of endothelial cells generated in two distinct microenvironments i.e., bone and muscle with osteo-differentiated cells and smooth muscle mesenchymal stromal cells (MSCs) respectively, showed different tumor cell extravasation rates, and thus confirming the TME’s role in cancer progression [244]. Moreover, in contrast to inadequately aligned endothelial cells grown in 2D culture system, cells in this model manifested phenotypes similar to in vivo conditions.
Although these microfluidic models have enabled us to define the different aspects of TME, there are certain limitations associated to its usage. Microfluidic model system needs to be more robust and reliable such that they should not be affected by any external or internal impairments like air bubbles, hindered laminar flow etc., [245]. Moreover, development of a new chip fabrication material is urgently needed, as polydimethylsiloxane (PDMS), the most commonly used material, can retain small molecules non-specifically [27, 246]. Taken together, these microfluidic systems in combination with other 3D in vitro tumor models and emerging integration/fabrication technologies have potential to reveal the tumor-stromal interactions at higher resolutions.
In conclusion, 3D culture model systems are successful up to a certain extent in reconstituting the complex TME in vitro. However, they are still in their early stage of development, and therefore, limitations must be addressed appropriately before deriving any conclusions. One of the most critical features to consider in clinical tumor samples is the patient heterogeneity contributed by patient-specific tumor burden, immune cell types within TME, and the tumor stroma content. The incorporation of all these variables in in vitro models is paramount, however, it makes the model complex, and may compromise the reproducibility. Moreover, at present, the development of novel culture methods/protocols that allows long term in vitro maintenance of different cell sub-populations, and integration of multiple cell types in a single model is urgently required. Additionally, research should also be focused on the development of strategies required for the inclusion and continuous renewal of the diverse immune cell populations within 3D models. Along with overcoming challenges at experimentation front, the advanced in silico approaches can partially complement the pace of understanding different underlying features of the TME and defining its therapeutic potential. In the following sections, different aspects of TME profiling that can be achieved using state-of-the-art computational approaches have been described.
In silico approaches of TME profiling
In the backdrop of biological significance, the extent of the success of cancer immunotherapy approaches highly relies on the intricate interplay between tumor cells and TME’s immune and stromal components. Detailed molecular- and cellular-level characterization of this dynamic ecosystem can delineate strategies for designing more effective therapeutic interventions and identify novel biomarkers capable of classifying therapy responders and non-responders [247]. With the advent of high-throughput technologies, it is now possible to study TME complexities experimentally at the genomics, epigenomics, transcriptomics and proteomics levels at resolutions ranging from whole organisms to single cells. However, most of these assays require dissociation of the tumor tissues, which in turn can modify cells phenotypic and population representations [248, 249]. In order to overcome the above-mentioned challenges, computational methods have been devised. These in silico methods have helped us to understand the complexities of the TME and derive inferences from bulk tissue gene expression profiles. In the following sections, we will discuss various in silico analysis tools developed to determine tumor purity, immune repertoire profiling, and neoantigen predictions. We will also describe the computational methodology and models developed to screen prognostic genes in the TME.
Tumor purity and TME immune cell types profiling
Computational approaches to determine tumor tissue composition can be broadly classified into two categories, namely enrichment methods and deconvolution methods [250]. The success of both these classes of approaches depends on the prior acquaintance of the marker genes with the cell types of interest. The enrichment strategies aim at identifying tissue-specific differentially expressed gene sets or pathways that depict distinct cell populations [251]. However, such methods are unable to compute proportions of discrete cell types and cannot differentiate between cell subtypes with common gene markers [250]. In contrast, deconvolution methods can perform in silico evaluation of the proportions of distinct cell types along with closely related cell subpopulations. Deconvolution strategies can also overlay gene expression data from bulk tissue transcriptomes to specific cell types [249, 252,253,254]. Additionally, ATAC-seq and DNA methylation profiles can be used to evaluate tumor tissue compositions [255, 256]. In the recent years, the combination of automated tissue dissection protocols and scRNA-seq data has emerged as the preferred methodology to explore novel cell sates in bulk tissues [247].
Identification of diverse immune cell types in tumor based on precise signatures is an urgent task. The presence of hierarchical sub-clonal populations, and differences in the TME background composition with that of normal cells pose additional intrinsic complexities [257]. However, a deeper understanding of the immune cells repertoire in TME both qualitative and quantitative is essential for designing successful therapeutic interventions. Similar to TME composition’s delineating computational efforts, several tools have been developed specifically for estimating relative proportions of different immune cell types and subtypes within a sample using their specific gene expression profiles [254, 258]. Details of the bioinformatics tools commonly used to assess tumor purity, estimate stromal or immune fractions from bulk tumor transcriptomes, and identify immune cell types are tabulated in Table 1.
Few of the tools listed in Table 1 have already been used to discover potential prognostic and therapeutic biomarkers [259,260,261]. ESTIMATE in particular, has emerged as one of the widely used method, and is currently employed in several standard analysis pipelines of The Cancer Genome Atlas (TCGA). It determines the general fractions of stromal and immune components of the tumor based on stromal and immune scores derived from the gene set enrichment analysis (GSEA) of the stromal and tumor signature genes. Integration of immune and stromal scores resulted in an estimate score that defines the tumor purity in a sample. Recently, four distinct consensus molecular subtypes (CMSs) of colorectal cancer has been identified based on the immune subtype signatures characterization using ESTIMATE [262]. These subtypes have distinguishing features: CMS1 (14%) highly mutated, strong immune activation, microsatellite unstable; CMS2 (37%) epithelial origin, marked activation of MYC and WNT signaling pathways; CMS3 (13%) epithelial origin, highly dysregulated metabolic pathways; CMS4 (23%) mesenchymal origin, marked TGF-β activation, highly aggressive stromal invasion and angiogenesis. Approximately 13% of colorectal cancer samples in the study cohort showed features of multiple consensus molecular subtypes, which can be probably linked to intra-tumoral heterogeneity or transition phenotypes. DeMix, a linear model-based tool, computes the proportion of stromal and tumor cells in samples by considering the transcripts contributed by the epithelial and stromal component of a tumor sample [263]. As an input, this tool requires gene expression profile of at least one gene of each cell type. Recently, the characterization of heterogeneity among 333 primary prostate carcinomas samples has been performed using DeMix, and has led to the identification molecular targets with therapeutic potential [264]. Moreover, the Bayesian statistics-based PurBayes tool evaluates the tumor purity and sub-clonality of tumor samples by utilizing the expression data of tumor, stroma and matched normal signature genes [265].
Initial methods devised to estimate the relative fractions of distinct immune cell types within the tumor samples require some degree of prior information regarding defined cell types, their relative proportion, and specific gene signatures with or without expression profiles [257]. However, the recent developments utilize data mining approaches, and aims at minimizing this dependency of prior knowledge by defining distinct immune cell types based on marker genes [263, 266, 267]. csSAM and Dsection are two popular bioinformatics approaches that capitalizes on these recent developments [254, 268]. csSAM, a linear regression model, uses information of known cell proportions in the sample to determine the cell-specific expression profiles. Using csSAM, the comparative gene expression profiling of whole-blood from patients with stable kidney transplant and those experiencing acute rejection has revealed several hundreds of differentially expressed genes that has remained undetectable previously [254]. Dsection, an implementation of a probabilistic approach, determines cell type proportions in heterogeneous tissue samples, and differential cell-specific expression patterns under various experimental conditions by using previously estimated cell proportions and their reference expression profiles [268]. Nanodissection is a supervised machine learning-based iterative framework that identifies cell/tissue specific transcripts within the sample. For model training purpose, nanodissection method require a small set of marker genes and the reference expression profiles [269]. Quigley et al. has used this algorithm to identify cell type-specific transcripts, and used them successfully to examine the presence of cytotoxic T-lymphocytes, T-helper 1, T-helper 2 and B cells in the breast tissue. This study also shows higher cytotoxic T-lymphocytes infiltration in integrative cluster 10 (IC10)/basal-like breast cancers with wild type TP53 mutation, thus suggesting association between inactivation of TP53 and tumor immunosurveillance failure [270]. CIBERSORT is another popular supervised machine learning algorithm used to quantify immune cell types in a highly heterogeneous transcriptome samples. As an input, this tool requires precisely defined signature genes specific to various immune cell types populations and their proportions [271]. In clinical setting, CIBERSORT has been recently used to identify leukocyte diversity and prognostic genes within and across 25 tumor types from TCGA database [270]. In addition, Gentles et al. applied CIBERSORT on bulk transcriptomics data from 40,000 tumors to investigate immune and tumor heterogeneity, and identified intricate correlation between 22 leukocyte cell types and survival outcomes [272]. In another study, Thorsson et al. integrated tumor-infiltrating lymphocytes genomics, hematoxylin & eosin imaging data, CIBERSORT deconvoluted transcriptomic data of immune cell fractions, TCR & BCR repertoire, neoantigen prediction, expression of immune gene, somatic DNA alterations, and viral RNA expression. By implementing this multi-omics approach, six immune cell types shared across multiple tumor categories were identified [11]. An implementation of CIBERSORT, i.e. MethylCIBERSORT uses DNA methylation data from bulk tumors, deconvolution estimates and hot/cold tumors data from TCGA to define composition of tumor tissues. Singh et al. has identified distinct tumor clusters based on immune cell proportions in glioblastomas. In contrast to isocitrate dehydrogenase (IDH) wild type glioblastoma cases, where five optimum clusters were recognized based on immune cell types proportions, the IDH mutant glioblastoma samples have only two optimal consensus clusters [273]. Also, in IDH wild-type glioblastomas, tumor clusters were found to be associated with oncogenic alterations like CDKN2A/B deletion and EGFR amplification [273]. Similarly, chromatin accessibility profiles (estimated using ATAC-seq protocol) of tumors has also been used to define 16 major cell types of normal hematopoietic and leukemic hierarchies in human blood from 12 acute myeloid leukemia (AML) and 9 healthy humans [255]. Taken together, the advancements to determine tumor purity and immune cell heterogeneity within TME has contributed immensely to our present understanding of both tumor and immune biology.
Screening of TME-related prognostic genes
The identification of prognostic TME-related genes for predicting outcomes has enormous potential [274, 275]. The first step towards building a prognostic model is the acquisition of gene expression and clinical data for the given cancer type. Single-sample gene set enrichment analysis for each sample in the cohort generates stromal and immune scores. Patients are then grouped as high/low stromal and high/low immune subgroups based on the median values of the respective scores. For each of the stromal and immune score groups, differentially expressed genes are identified. Functional enrichment analysis of the intersecting differentially expressed genes across different score subgroups helps to elucidate the potential and significant biologic functions [275]. Furthermore, survival analysis is performed to analyze intersecting differentially expressed genes and their prognostic association with patient’s overall survival (OS). Protein–protein interaction (PPI) networks are constructed for differentially expressed genes with prognostic values, and central genes with higher degrees of connection are identified. Further univariate and multivariate regression analyses are performed to obtain the most significant prognostic value genetic signature, which in turn is used to establish a risk score formula for predictive purposes.
Recently, Chen et al. has systematically investigated pancreatic tumor microenvironment, and established biomarkers associated with tumor/stromal cell populations. Overall survival analysis showed that the high immune/stromal group of pancreatic patients are closely related with poor prognosis [274]. In this study, four signature genes COL2A1, CXCL10, TRPC7 and CUX2 emerged as independent prognostic factors. The prognostic model created using these signature genes assigned high risk scores to KRAS and TP53 mutations. Additionally, at single cell resolution, CXCR3 was found to be highly expressed on T cells, whereas its ligand CXCL10 is abundant on tumor associated macrophage population [274]. Similarly, Ye et al. performed thorough examination of breast cancer (BC) microenvironment, and identified three signature genes namely SIT1, KLRB1 and GZMM as prognostic factors [275]. All these three genes were found to be negatively correlated with tumor purity, and positively associated with the intrusion of immune cells like B cells, neutrophils, CD4 T cells, CD8 T cells, macrophages and dendritic cells in BC microenvironment [275]. In conjunction with the necessary experimental validation, these identified prognostic genes could be promising candidates for therapeutic interventions.
Immune cell receptor profiling and neoantigen prediction
Immune cell repertoires i.e., T-cell receptors (TCRs) and B-cell receptors (BCRs), recognize and neutralize a highly diverse range of antigens [247]. Quantitative studies of these immune cells repertoires in various cellular compartments can be performed using high-throughput sequencing. Lymphocyte-specific TCR and BCR sequencing approaches have enabled the analysis and tracking of diverse lymphoid cell populations, which in turn has further increased our understanding of intra-tumoral, inter-tumoral and clinical outcomes heterogeneity. The ability to profile only a subset of cellular heterogeneity, and inability to discriminate between cellular states, i.e., naive vs. activated, are the two major shortcomings of these immune cell receptors profiling methods [247].
Immune repertoire sequencing has provided opportunity to study immune cell heterogeneity in the TME of various cancer types [276, 277]. For instance, TCR sequencing has been used routinely to understand the T cells repertoires after immune checkpoint blockade therapy. In melanoma patients, anti-CTLA-4 and anti-PD-1 therapies have been shown to increase the diversity of TCRs, and T cell clonotypes [278, 279]. The major challenge for tools analyzing immune cells repertoires include making a clear distinction between rare clones in bulk data, and identifying sequencing and/or PCR errors. Furthermore, recent technological advances have enabled simultaneous transcriptomics analysis and BCR/TCR profiling at single-cell resolution. Various bulk and single-cell repertoire analysis tools are listed in Table 2.
Early methods of immune cell repertoire profiling include IgBLAST, IHMMUNE-ALIGN and IMGT/V-QUEST [280,281,282]. MiXCR is a more recent and widely used sequencing-based approach of bulk BCR and TCR profiling. This method is capable of correcting PCR errors, and identifying germline hypermutations by applying multilayer clustering algorithm [283]. Ma et al. analyzed the impact of functional DNA damage repair (DDR) gene polymerase epsilon (POLE) mutations on tumor immune microenvironment post immune checkpoint blockade (ICB) therapy [284]. The authors observed upregulation of immune-related pathways in post-ICB PoleP286R tumors. Evaluation of the TCR-beta CDR3 clonotypes isolated through MiXCR showed a higher rate of clonal expansion, richness and decreased evenness in post-ICB PoleP286R tumors [284]. Another method GLIPH, clusters TCRs based on their global similarity between CDR3 sequences, and the conserved motifs that provide common specificity to the receptors [285]. Subudhi et al. have shown that the autoreactive T cell clonal expansion and diversification of T cells repertoire occurs in prostate cancer patients treated with CTLA-4 blocking antibody or those experiencing immune-related adverse events (irAEs). This supports the hypothesis that newly established responsiveness to shared antigens may led to inflammatory response in cancer patients undergoing blockade therapy [286].
With the advent of single cell technologies, it is now possible to perform single-cell TCR analysis. Such analysis has enabled the pairing TCR of α and β chains sequences. TRACER tool generate all possible pair of TCR α and β chains by aligning all the possible V and J segments, then assembling the reads in contiguous sequences [287]. Zheng et al. implemented TRACER tool for TCR profiling of > 5 k single T cells isolated from blood, tumor and adjacent normal tissues of hepatocellular cancer patients. The coupled TCR and transcriptional profiles facilitated the identification of functionally 11 T cell subsets, and their developmental path [288]. Moreover, to understand the activity of cells and their correlation with antigen receptor sequences, new methods of RNA-seq and repertoire sequencing from the same cell has been developed.
In addition to TCR sequencing of T cells in the TME, the prediction of neoantigens from patient’s DNA or RNA represents a major step towards personalized therapeutic approaches [289]. Neoantigens are present only on tumor cells but not on normal cells; therefore, neoantigens can elicit tumor-specific immune responses. These mutation-associated cancer antigens are cleaved, and short peptides are presented to TCRs on MHC molecules. In view of the unprecedented possibilities of neoantigens, TCR rearrangements, and large variations in MHC molecules, there is an urgent need of neoantigens prediction tools [290]. Neoantigen prediction is a three-step process: identifying the mutation-associated cancer proteins, HLA typing, and determining neoantigen affinity towards MHC binding [290]. List of in silco tools and pipelines developed to analyze different steps of neoantigen prediction independently or combined are listed in Table 3.
MUTECT and GENOME ANALYSIS TOOLKIT (GATK) are two well-known variant analysis tools that implement Bayesian classifier principle to detect single nucleotide polymorphism [291, 292]. A limitation associated with such variant analysis tools is to determine the functional implications of a variant on different transcripts, so the choice of tools and database is critical. In the second step of neoantigen prediction, i.e. HLA typing, a sensitive assembly or mapping strategy is involved, which in turn relies on well-annotated reference genome. OptiType and PolySolver are examples of some of the popular HLA typing tools [293, 294]. Park et al. used the HLA-I genotype information of > 1500 patients suffering from 11 different cancer types from two independent studies using OptiType and PolySolver tools for prediction. The results revealed that HLA-I heterozygosity is positively correlated with early onset of tumor [295]. Finally, at the MHC-binding affinity estimation step the non-linear, machine-learning based methods such as NETMHC, NETMHCII have shown improved prediction accuracy as compared to those early sequence-based methods like SYFPEITHI and BIMAS [296,297,298,299,300]. EDGE, a neural network-based computational model for epitope prediction has been developed using HLA mass spectrometry neoantigen peptides and genomic data of 74 cancer patients [301]. In comparison to tumor sets binding-affinity features, EDGE have a nine-fold higher positive predictive value.
Bioinformatics analysis pipelines integrating the abovementioned multiple steps of neoantigen prediction are tabulated in Table 3. For instance, FRED 2 provides a unified immunoinformatics framework for T-cell epitope prediction, selection, assembly and HLA typing [302]. Loffler et al. implemented FRED2 pipeline for defining different aspects of tumor neoantigens in hepatocellular carcinoma, and concluded that the mutated HLA ligands derived from exome represent the limited targets for personalized immunotherapy approaches [303]. Therefore, an increase in the neoantigen search space is needed to identify potential targets and enhance the efficacy of tumor immunotherapy approaches, especially in case of malignancies with lower or similar mutational burden. NetTepi is another integrated approach of T cell epitope discovery that combine predictions of peptide-MHC (pMHC) binding affinity, stability and T cell propensity [304]. Recently, Buckley et al. identified NetTepi as one of the best peptide immunogenicity prediction tools in an unbiased comparative evaluation of existing models [305].
In a nutshell, the development of bioinformatics tools and methodologies has created new opportunities for the researchers to study tumor characteristics and its microenvironment complexities in silico. In addition to providing deeper understanding of the intrinsic heterogeneity associated with tumor development, progression and immune evasion, these tools have guided us toward therapeutic and diagnostic discoveries. By applying the current computational tools, it is now possible to predict the distinct immune and stromal components diversity within TME. Also, the available methods can create high resolution TME cell type-specific interaction network, which may eventually help in improving our understanding of cancer therapy responders and non-responders patients as well as to the development of immune-modulatory drugs.
Conclusion
In this review, various aspects of TME, including its therapeutic potential has been described. TME heterogeneity is contributed by its highly diverse and non-uniformly distributed cellular and acellular components (Fig. 1). Variability of these components within the TME gives rise to several physiologically different specialized tumor microenvironments such as the acidic niche, hypoxic, innervated and inflammatory microenvironments. In the recent years, these specialized tumor microenvironments have emerged as the hotspots for therapeutic interventions. TME-specific therapeutic strategies can be broadly categorized based on the TME components targeted such as ECM, non-immune cells and immune cells. Major cancer immunotherapy approaches involving targeting of the immune components of TME include adoptive-T lymphocytes and CAR-based therapies, cancer vaccines, and employing immune checkpoint inhibitors. Thus, various TME components provides an opportunity to target tumor progression and metastasis through therapeutic interventions as summarized in Fig. 2. However, one of the major limitations associated to these cancer immunotherapy approaches targeting TME include primary or acquired resistance due to various extrinsic and intrinsic factors. A deeper understanding of the dynamic TME components and their real-time interaction could help in overcoming these limitations. Towards this end, several in vitro 3D experimental model systems have been developed to precisely mimic the TME conditions. Also, bioinformatics tools can be used to estimate tumor purity, immune repertoire profiling, predict neoantigens and prognostic genes in the TME. Altogether, this article gives an overview of TME components, and their promising future potential as therapeutic targets in the light of knowledge gained through experimental 3D model systems and bioinformatics predictions.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ASCs:
-
Adult stem cells
- ATRA:
-
All-trans retinoic acid
- ALI:
-
Air–liquid interface
- APCs:
-
Antigen-presenting cells
- BTLA:
-
B- and T-lymphocyte attenuator
- BC:
-
Breast cancer
- BCRs:
-
B-cell receptors
- CAR:
-
Chimeric antigen-receptor
- CIT:
-
Cancer immunotherapy
- CTLs:
-
Cytotoxic T lymphocytes
- CAFs:
-
Cancer-associated fibroblasts
- CSSs:
-
Cancer stem cells
- CXCL12:
-
CXC-chemokine ligand 12
- CSF-1:
-
Colony stimulating factor 1
- CTLA4:
-
Cytotoxic T-lymphocyte associated protein 4
- CRS:
-
Cytokine release syndrome
- CTCs:
-
Circulating tumor cells
- CMSs:
-
Consensus molecular subtypes
- dECM:
-
Decellularized ECM
- DCs:
-
Dendritic cells
- DDR:
-
DNA damage repair
- EC:
-
European Commission
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-to-mesenchymal transformation
- EIFs:
-
EMT inducing factors
- EndMT:
-
Endothelial-to-mesenchymal transformation
- EGF:
-
Epidermal growth factor
- FAP:
-
Fibroblast activating protein
- FGF2:
-
Fibroblast growth factor 2
- FDA:
-
US Food and Drug Administration
- GVHD:
-
Graft-versus-host disease
- GFs:
-
Growth factors
- GATK:
-
Genome Analysis Toolkit
- GSEA:
-
Gene set enrichment analysis
- HUVECs:
-
Human umbilical vein epithelial cells
- HIF-1:
-
Hypoxia inducible factor-1
- IFN-γ:
-
Interferon-γ
- IC10:
-
Integrative cluster 10
- IDH:
-
Isocitrate dehydrogenase
- irAEs:
-
Immune-related adverse events
- IDO:
-
Indoleamine 2,3-dioxygenase
- ICBs:
-
Immune checkpoint blockers
- MSCs:
-
Mesenchymal stromal cells
- MMPs:
-
Matrix metalloproteinases
- MHC:
-
Major Histocompatibility complex
- NK:
-
Natural Killer cells
- NSAID:
-
Non-steroid anti-inflammatory drug
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed death ligand-1
- PGE2:
-
Prostaglandin E2
- PDAC:
-
Pancreatic ductal carcinoma
- PSC:
-
Pancreatic stellate cells
- PDOs:
-
Patient-derived organoids
- PDX:
-
Patient-derived xenografts
- PSCs:
-
Pluripotent stem cells
- POLE:
-
Polymerase epsilon
- PDMS:
-
Polydimethylsiloxane
- PPI:
-
Protein–protein interaction
- pMHC:
-
Peptide-MHC
- ROS:
-
Reactive oxygen species
- TGF-β:
-
Transforming growth factor beta
- TILs:
-
Tumor-infiltrating lymphocytes
- TNF-α:
-
Tumor necrosis factor-α
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- TLR3:
-
Toll-like receptor 3
- Tregs:
-
Regulatory T-cells
- TIGIT:
-
T-cell immunoreceptor with Ig and ITIM domains
- TCRs:
-
T-cell receptors
- TME:
-
Tumor microenvironment
- TMS:
-
Tissue matrix scaffold
- TCGA:
-
The Cancer Genome Atlas
- VEGF:
-
Vascular endothelial growth factor
- UCB:
-
Umbilical cord blood
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19–20):1267–84.
Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):166.
Shelton SE, Nguyen HT, Barbie DA, Kamm RD. Engineering approaches for studying immune-tumor cell interactions and immunotherapy. iScience. 2021;24(1):101985.
Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18(1):59.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Abou Khouzam R, Goutham HV, Zaarour RF, Chamseddine AN, Francis A, Buart S, et al. Integrating tumor hypoxic stress in novel and more adaptable strategies for cancer immunotherapy. Semin Cancer Biol. 2020;65:140–54.
Qiu GZ, Jin MZ, Dai JX, Sun W, Feng JH, Jin WL. Reprogramming of the tumor in the hypoxic niche: the emerging concept and associated therapeutic strategies. Trends Pharmacol Sci. 2017;38(8):669–86.
Ros XR, Vermeulen L. Turning cold tumors hot by blocking TGF-beta. Trends Cancer. 2018;4(5):335–7.
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–30.
Toor SM, Sasidharan Nair V, Decock J, Elkord E. Immune checkpoints in the tumor microenvironment. Semin Cancer Biol. 2020;65:1–12.
Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol. 2014;24(8):472–8.
Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. 2016;16(10):663–73.
Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52.
Garcia-Canaveras JC, Chen L, Rabinowitz JD. The tumor metabolic microenvironment: lessons from lactate. Cancer Res. 2019;79(13):3155–62.
Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol. 2020;82:103–26.
Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer. 2017;17(10):577–93.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341(6142):1236361.
Saloman JL, Albers KM, Rhim AD, Davis BM. Can stopping nerves, stop cancer? Trends Neurosci. 2016;39(12):880–9.
Zahalka AH, Frenette PS. Nerves in cancer. Nat Rev Cancer. 2020;20(3):143–57.
Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009;69(10):4167–74.
Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30(6):507–19.
Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35.
Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of cancer resistance to immunotherapy. Front Oncol. 2020;10:1290.
Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 2019;19(2):65–81.
Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21(12):745–54.
Langhans SA. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front Pharmacol. 2018;9:6.
Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, et al. Modeling physiological events in 2D vs. 3D cell culture. Physiology (Bethesda). 2017;32(4):266–77.
Jensen C, Teng Y. Is it time to start transitioning from 2D to 3D cell culture? Front Mol Biosci. 2020;7:33.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921–5.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309.
Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer. 2016;2(12):758–70.
Otrock ZK, Hatoum HA, Awada AH, Ishak RS, Shamseddine AI. Hypoxia-inducible factor in cancer angiogenesis: structure, regulation and clinical perspectives. Crit Rev Oncol Hematol. 2009;70(2):93–102.
Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102(5):789–95.
Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71(13):4640–52.
Zhang Q, Han Z, Zhu Y, Chen J, Li W. Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol Med Rep. 2021;23(1):1.
Qin J, Liu Y, Lu Y, Liu M, Li M, Li J, et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep. 2017;7(1):10592.
Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, et al. HIF-1α promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS ONE. 2015;10(6): e0129603.
Suzuki A, Kusakai G, Shimojo Y, Chen J, Ogura T, Kobayashi M, et al. Involvement of transforming growth factor-beta 1 signaling in hypoxia-induced tolerance to glucose starvation. J Biol Chem. 2005;280(36):31557–63.
Mori H, Yao Y, Learman BS, Kurozumi K, Ishida J, Ramakrishnan SK, et al. Induction of WNT11 by hypoxia and hypoxia-inducible factor-1α regulates cell proliferation, migration and invasion. Sci Rep. 2016;6:21520.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105(17):6392–7.
Ma B, Chen Y, Chen L, Cheng H, Mu C, Li J, et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat Cell Biol. 2015;17(1):95–103.
Tsunetoh S, Terai Y, Sasaki H, Tanabe A, Tanaka Y, Sekijima T, et al. Topotecan as a molecular targeting agent which blocks the Akt and VEGF cascade in platinum-resistant ovarian cancers. Cancer Biol Ther. 2010;10(11):1137–46.
Curry JM, Johnson J, Mollaee M, Tassone P, Amin D, Knops A, et al. Metformin clinical trial in HPV+ and HPV- head and neck squamous cell carcinoma: impact on cancer cell apoptosis and immune infiltrate. Front Oncol. 2018;8:436.
Li Y, Zhao L, Li XF. Targeting hypoxia: hypoxia-activated prodrugs in cancer therapy. Front Oncol. 2021;11: 700407.
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–6.
Matschke J, Riffkin H, Klein D, Handrick R, Lüdemann L, Metzen E, et al. Targeted inhibition of glutamine-dependent glutathione metabolism overcomes death resistance induced by chronic cycling hypoxia. Antioxid Redox Signal. 2016;25(2):89–107.
Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–69.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Jiang B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017;4(1):25–7.
Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol. 2010;94(1):102–9.
Zhong S, Jeong JH, Chen Z, Chen Z, Luo JL. Targeting tumor microenvironment by small-molecule inhibitors. Transl Oncol. 2020;13(1):57–69.
Jang JH, Kim DH, Surh YJ. Dynamic roles of inflammasomes in inflammatory tumor microenvironment. NPJ Precis Oncol. 2021;5(1):18.
Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41.
Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):263.
Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13:118–37.
De Simone V, Franzè E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34(27):3493–503.
Chen XW, Zhou SF. Inflammation, cytokines, the IL-17/IL-6/STAT3/NF-κB axis, and tumorigenesis. Drug Des Devel Ther. 2015;9:2941–6.
Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J Clin Oncol. 2005;23(34):8606–12.
Maisonneuve P, Lowenfels AB. Statins and the risk of colorectal cancer. N Engl J Med. 2005;353(9):952–4.
Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377(9759):31–41.
Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10(5):501–7.
Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res. 2008;14(23):7593–603.
Mauffrey P, Tchitchek N, Barroca V, Bemelmans AP, Firlej V, Allory Y, et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature. 2019;569(7758):672–8.
Cervantes-Villagrana RD, Albores-García D, Cervantes-Villagrana AR, García-Acevez SJ. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther. 2020;5(1):99.
Nijs J, Meeus M, Versijpt J, Moens M, Bos I, Knaepen K, et al. Brain-derived neurotrophic factor as a driving force behind neuroplasticity in neuropathic and central sensitization pain: a new therapeutic target? Expert Opin Ther Targets. 2015;19(4):565–76.
Demir IE, Friess H, Ceyhan GO. Neural plasticity in pancreatitis and pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2015;12(11):649–59.
Reavis HD, Chen HI, Drapkin R. Tumor innervation: cancer has some nerve. Trends Cancer. 2020;6(12):1059–67.
Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep. 2013;4(2):287–301.
Jobling P, Pundavela J, Oliveira SM, Roselli S, Walker MM, Hondermarck H. Nerve-cancer cell cross-talk: a novel promoter of tumor progression. Cancer Res. 2015;75(9):1777–81.
Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6(250):250–115.
Coarfa C, Florentin D, Putluri N, Ding Y, Au J, He D, et al. Influence of the neural microenvironment on prostate cancer. Prostate. 2018;78(2):128–39.
Li X, Peng X, Yang S, Wei S, Fan Q, Liu J, et al. Targeting tumor innervation: premises, promises, and challenges. Cell Death Discov. 2022;8(1):131.
Warren BA, Shubik P, Wilson R, Garcia H, Feldman R. The microcirculation in two transplantable melanomas of the hamster. I. In vivo observations in transparent chambers. Cancer Lett. 1978;4(2):109–16.
Konerding MA, Malkusch W, Klapthor B, van Ackern C, Fait E, Hill SA, et al. Evidence for characteristic vascular patterns in solid tumours: quantitative studies using corrosion casts. Br J Cancer. 1999;80(5–6):724–32.
Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2(3): a006536.
Zhang H, Shen YW, Zhang LJ, Chen JJ, Bian HT, Gu WJ, et al. Targeting endothelial cell-specific molecule 1 protein in cancer: a promising therapeutic approach. Front Oncol. 2021;11: 687120.
Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86: 102017.
Leone Roberti Maggiore U, Valenzano Menada M, Venturini PL, Ferrero S. The potential of sunitinib as a therapy in ovarian cancer. Expert Opin Investig Drugs. 2013;22(12):1671–86.
Zheng R, Li F, Li F, Gong A. Targeting tumor vascularization: promising strategies for vascular normalization. J Cancer Res Clin Oncol. 2021;147(9):2489–505.
Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther. 2021;6(1):218.
Chen SY, Lin JS, Lin HC, Shan YS, Cheng YJ, Yang BC. Dependence of fibroblast infiltration in tumor stroma on type IV collagen-initiated integrin signal through induction of platelet-derived growth factor. Biochim Biophys Acta. 2015;1853(5):929–39.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303(5659):848–51.
Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130(10):2337–48.
Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94(6):715–25.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
LeBedis C, Chen K, Fallavollita L, Boutros T, Brodt P. Peripheral lymph node stromal cells can promote growth and tumorigenicity of breast carcinoma cells through the release of IGF-I and EGF. Int J Cancer. 2002;100(1):2–8.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86.
Kakarla S, Song XT, Gottschalk S. Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy. 2012;4(11):1129–38.
Cheng JD, Dunbrack RL Jr, Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002;62(16):4767–72.
Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2(3):187–93.
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66(8):4426–33.
Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014;345(1):132–9.
Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat Commun. 2020;11(1):4841.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115.
Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2019;6:160.
Barker HE, Chang J, Cox TR, Lang G, Bird D, Nicolau M, et al. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011;71(5):1561–72.
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406.
Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb). 2015;7(10):1120–34.
Murata H, Zhou L, Ochoa S, Hasan A, Badiavas E, Falanga V. TGF-beta3 stimulates and regulates collagen synthesis through TGF-beta1-dependent and independent mechanisms. J Invest Dermatol. 1997;108(3):258–62.
Liu S, Ren J, Ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6(1):8.
Huang CY, Chung CL, Hu TH, Chen JJ, Liu PF, Chen CL. Recent progress in TGF-β inhibitors for cancer therapy. Biomed Pharmacother. 2021;134: 111046.
Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811.
Bedinger D, Lao L, Khan S, Lee S, Takeuchi T, Mirza AM. Development and characterization of human monoclonal antibodies that neutralize multiple TGFβ isoforms. MAbs. 2016;8(2):389–404.
Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. 2014;9(3): e90353.
Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17(6):1382–93.
Fields GB. The rebirth of matrix metalloproteinase inhibitors: moving beyond the dogma. Cells. 2019;8(9):984.
Dezube BJ, Krown SE, Lee JY, Bauer KS, Aboulafia DM. Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi’s sarcoma: an AIDS Malignancy Consortium Study. J Clin Oncol. 2006;24(9):1389–94.
Scannevin RH, Alexander R, Haarlander TM, Burke SL, Singer M, Huo C, et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J Biol Chem. 2017;292(43):17963–74.
Ling B, Watt K, Banerjee S, Newsted D, Truesdell P, Adams J, et al. A novel immunotherapy targeting MMP-14 limits hypoxia, immune suppression and metastasis in triple-negative breast cancer models. Oncotarget. 2017;8(35):58372–85.
Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6(1):153.
Arneth B. Tumor microenvironment. Medicina (Kaunas). 2019;56(1):15.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50.
Guillerey C. NK Cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1273:69–90.
Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1224:117–40.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. 2020;11: 583084.
Kim J, Bae JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016;2016:6058147.
Wu L, Saxena S, Singh RK. Neutrophils in the tumor microenvironment. Adv Exp Med Biol. 2020;1224:1–20.
Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. 2020;10:188.
Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–72.
Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105(1):1–8.
Qiu SQ, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schröder CP. Tumor-associated macrophages in breast cancer: innocent bystander or important player? Cancer Treat Rev. 2018;70:178–89.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62.
Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–27.
McFarlane AJ, Fercoq F, Coffelt SB, Carlin LM. Neutrophil dynamics in the tumor microenvironment. J Clin Invest. 2021;131(6).
Di Carlo E, Forni G, Musiani P. Neutrophils in the antitumoral immune response. Chem Immunol Allergy. 2003;83:182–203.
Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179(3):1455–70.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8.
Rahmy S, Lu X. Targeting tumor-associated neutrophils in immunotherapy. Systemic Drug Delivery Strategies2022. p. 147–61.
Boissonnas A, Licata F, Poupel L, Jacquelin S, Fetler L, Krumeich S, et al. CD8+ tumor-infiltrating T cells are trapped in the tumor-dendritic cell network. Neoplasia. 2013;15(1):85–94.
Perrot I, Blanchard D, Freymond N, Isaac S, Guibert B, Pachéco Y, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol. 2007;178(5):2763–9.
Khazaie K, Blatner NR, Khan MW, Gounari F, Gounaris E, Dennis K, et al. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011;30(1):45–60.
Kim-Schulze S, Kim HS, Fan Q, Kim DW, Kaufman HL. Local IL-21 promotes the therapeutic activity of effector T cells by decreasing regulatory T cells within the tumor microenvironment. Mol Ther. 2009;17(2):380–8.
Petrella TM, Tozer R, Belanger K, Savage KJ, Wong R, Smylie M, et al. Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J Clin Oncol. 2012;30(27):3396–401.
Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun. 2019;10(1):5408.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564(7736):439–43.
Pelly VS, Moeini A, Roelofsen LM, Bonavita E, Bell CR, Hutton C, et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021;11(10):2602–19.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24.
Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. J Immunol. 2015;195(8):4010–9.
Brittenden J, Heys SD, Ross J, Eremin O. Natural killer cells and cancer. Cancer. 1996;77(7):1226–43.
Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–76.
Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66(1):517–26.
Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8:760.
Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer. 2020;19(1):120.
Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14(1):7.
Stampouloglou E, Cheng N, Federico A, Slaby E, Monti S, Szeto GL, et al. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020;18(1): e3000591.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68.
Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71.
Gilham DE, Anderson J, Bridgeman JS, Hawkins RE, Exley MA, Stauss H, et al. Adoptive T-cell therapy for cancer in the United kingdom: a review of activity for the British Society of Gene and Cell Therapy annual meeting 2015. Hum Gene Ther. 2015;26(5):276–85.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–7.
Qian X, Wang X, Jin H. Cell transfer therapy for cancer: past, present, and future. J Immunol Res. 2014;2014: 525913.
Morotti M, Albukhari A, Alsaadi A, Artibani M, Brenton JD, Curbishley SM, et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br J Cancer. 2021;124(11):1759–76.
Detela G, Lodge A. EU regulatory pathways for ATMPs: standard, accelerated and adaptive pathways to marketing authorisation. Mol Ther Methods Clin Dev. 2019;13:205–32.
Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–31.
Rivera AM, May S, Lei M, Qualls S, Bushey K, Rubin DB, et al. CAR T-cell-associated neurotoxicity: current management and emerging treatment strategies. Crit Care Nurs Q. 2020;43(2):191–204.
Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–81.
Hay KA. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br J Haematol. 2018;183(3):364–74.
Lu H, Zhao X, Li Z, Hu Y, Wang H. From CAR-T cells to CAR-NK cells: a developing immunotherapy method for hematological malignancies. Front Oncol. 2021;11: 720501.
Zhang L, Chu J, Yu J, Wei W. Cellular and molecular mechanisms in graft-versus-host disease. J Leukoc Biol. 2016;99(2):279–87.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3: e28147.
Murray S, Lundqvist A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: On being in the right place at the right time, with resilience. Hum Vaccin Immunother. 2016;12(3):607–11.
Bi J, Tian Z. NK cell dysfunction and checkpoint immunotherapy. Front Immunol. 2019;10:1999.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Sabado RL, Bhardwaj N. Dendritic cell immunotherapy. Ann N Y Acad Sci. 2013;1284:31–45.
Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115(16):3670–9.
Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29(36):4828–36.
Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18(3):168–82.
Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19(2):393–403.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–16.
Possick JD. Pulmonary toxicities from checkpoint immunotherapy for malignancy. Clin Chest Med. 2017;38(2):223–32.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5.
Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110(7):2080–9.
Rodrigues J, Heinrich MA, Teixeira LM, Prakash J. 3D In vitro model (R)evolution: unveiling tumor-stroma interactions. Trends Cancer. 2021;7(3):249–64.
Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018;8(11):1358–65.
Mabry KM, Payne SZ, Anseth KS. Microarray analyses to quantify advantages of 2D and 3D hydrogel culture systems in maintaining the native valvular interstitial cell phenotype. Biomaterials. 2016;74:31–41.
Melissaridou S, Wiechec E, Magan M, Jain MV, Chung MK, Farnebo L, et al. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019;19:16.
Sung KE, Beebe DJ. Microfluidic 3D models of cancer. Adv Drug Deliv Rev. 2014;79–80:68–78.
Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater Today (Kidlington). 2015;18(10):539–53.
Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9(11):4227–39.
Day CP, Carter J, Bonomi C, Hollingshead M, Merlino G. Preclinical therapeutic response of residual metastatic disease is distinct from its primary tumor of origin. Int J Cancer. 2012;130(1):190–9.
Rodenhizer D, Dean T, D’Arcangelo E, McGuigan AP. The current landscape of 3D in vitro tumor models: what cancer hallmarks are accessible for drug discovery? Adv Healthc Mater. 2018;7(8): e1701174.
Feng S, Duan X, Lo PK, Liu S, Liu X, Chen H, et al. Expansion of breast cancer stem cells with fibrous scaffolds. Integr Biol (Camb). 2013;5(5):768–77.
Long TJ, Sprenger CC, Plymate SR, Ratner BD. Prostate cancer xenografts engineered from 3D precision-porous poly(2-hydroxyethyl methacrylate) hydrogels as models for tumorigenesis and dormancy escape. Biomaterials. 2014;35(28):8164–74.
Rijal G, Li W. 3D scaffolds in breast cancer research. Biomaterials. 2016;81:135–56.
Taubenberger AV, Bray LJ, Haller B, Shaposhnykov A, Binner M, Freudenberg U, et al. 3D extracellular matrix interactions modulate tumour cell growth, invasion and angiogenesis in engineered tumour microenvironments. Acta Biomater. 2016;36:73–85.
Grolman JM, Zhang D, Smith AM, Moore JS, Kilian KA. Rapid 3D extrusion of synthetic tumor microenvironments. Adv Mater. 2015;27(37):5512–7.
Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza-Alvarez P, et al. 3D-3-culture: a tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials. 2018;163:185–97.
Hume RD, Pensa S, Brown EJ, Kreuzaler PA, Hitchcock J, Husmann A, et al. Tumour cell invasiveness and response to chemotherapeutics in adipocyte invested 3D engineered anisotropic collagen scaffolds. Sci Rep. 2018;8(1):12658.
Ferreira LP, Gaspar VM, Mano JF. Decellularized extracellular matrix for bioengineering physiomimetic 3D in vitro tumor models. Trends Biotechnol. 2020;38(12):1397–414.
Lü WD, Zhang L, Wu CL, Liu ZG, Lei GY, Liu J, et al. Development of an acellular tumor extracellular matrix as a three-dimensional scaffold for tumor engineering. PLoS ONE. 2014;9(7): e103672.
Gill BJ, West JL. Modeling the tumor extracellular matrix: Tissue engineering tools repurposed towards new frontiers in cancer biology. J Biomech. 2014;47(9):1969–78.
Rijal G, Li W. A versatile 3D tissue matrix scaffold system for tumor modeling and drug screening. Sci Adv. 2017;3(9): e1700764.
Langer EM, Allen-Petersen BL, King SM, Kendsersky ND, Turnidge MA, Kuziel GM, et al. Modeling tumor phenotypes in vitro with three-dimensional bioprinting. Cell Rep. 2019;26(3):608-23.e6.
Meng F, Meyer CM, Joung D, Vallera DA, McAlpine MC, Panoskaltsis-Mortari A. 3D bioprinted in vitro metastatic models via reconstruction of tumor microenvironments. Adv Mater. 2019;31(10): e1806899.
Moroni L, Burdick JA, Highley C, Lee SJ, Morimoto Y, Takeuchi S, et al. Biofabrication strategies for 3D in vitro models and regenerative medicine. Nat Rev Mater. 2018;3(5):21–37.
Heinrich MA, Liu W, Jimenez A, Yang J, Akpek A, Liu X, et al. 3D bioprinting: from benches to translational applications. Small. 2019;15(23): e1805510.
Costa EC, Moreira AF, de Melo-Diogo D, Gaspar VM, Carvalho MP, Correia IJ. 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol Adv. 2016;34(8):1427–41.
Rodrigues T, Kundu B, Silva-Correia J, Kundu SC, Oliveira JM, Reis RL, et al. Emerging tumor spheroids technologies for 3D in vitro cancer modeling. Pharmacol Ther. 2018;184:201–11.
Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18(7):407–18.
Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172(1–2):373-86.e10.
Dutta D, Heo I, Clevers H. Disease modeling in stem cell-derived 3D organoid systems. Trends Mol Med. 2017;23(5):393–410.
Fan H, Demirci U, Chen P. Emerging organoid models: leaping forward in cancer research. J Hematol Oncol. 2019;12(1):142.
Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. 2018;18(1):335.
Mollica PA, Booth-Creech EN, Reid JA, Zamponi M, Sullivan SM, Palmer XL, et al. 3D bioprinted mammary organoids and tumoroids in human mammary derived ECM hydrogels. Acta Biomater. 2019;95:201–13.
Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125.
Huch M, Koo BK. Modeling mouse and human development using organoid cultures. Development. 2015;142(18):3113–25.
Kretzschmar K, Clevers H. Organoids: modeling development and the stem cell niche in a dish. Dev Cell. 2016;38(6):590–600.
Liu L, Yu L, Li Z, Li W, Huang W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J Transl Med. 2021;19(1):40.
Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359(6378):920–6.
Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–38.
van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–45.
Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424–35.
Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–72.
Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods. 2014;11(1):106–12.
Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–87.
Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 2018;173(2):515-28.e17.
Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76(8):2465–77.
Yang H, Sun L, Liu M, Mao Y. Patient-derived organoids: a promising model for personalized cancer treatment. Gastroenterol Rep (Oxf). 2018;6(4):243–5.
Lannagan TRM, Lee YK, Wang T, Roper J, Bettington ML, Fennell L, et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut. 2019;68(4):684–92.
Praharaj PP, Bhutia SK, Nagrath S, Bitting RL, Deep G. Circulating tumor cell-derived organoids: current challenges and promises in medical research and precision medicine. Biochim Biophys Acta Rev Cancer. 2018;1869(2):117–27.
Driehuis E, Kretzschmar K, Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc. 2020;15(10):3380–409.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
Nozaki K, Mochizuki W, Matsumoto Y, Matsumoto T, Fukuda M, Mizutani T, et al. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J Gastroenterol. 2016;51(3):206–13.
Dao V, Yuki K, Lo YH, Nakano M, Kuo CJ. Immune organoids: from tumor modeling to precision oncology. Trends Cancer. 2022;8:870.
Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32(8):760–72.
Avnet S, Lemma S, Cortini M, Di Pompo G, Perut F, Baldini N. Pre-clinical models for studying the interaction between mesenchymal stromal cells and cancer cells and the induction of stemness. Front Oncol. 2019;9:305.
Jia W, Gungor-Ozkerim PS, Zhang YS, Yue K, Zhu K, Liu W, et al. Direct 3D bioprinting of perfusable vascular constructs using a blend bioink. Biomaterials. 2016;106:58–68.
Homan KA, Kolesky DB, Skylar-Scott MA, Herrmann J, Obuobi H, Moisan A, et al. Bioprinting of 3D Convoluted Renal Proximal Tubules on Perfusable Chips. Sci Rep. 2016;6:34845.
Datta P, Ayan B, Ozbolat IT. Bioprinting for vascular and vascularized tissue biofabrication. Acta Biomater. 2017;51:1–20.
Aung A, Kumar V, Theprungsirikul J, Davey SK, Varghese S. An engineered tumor-on-a-chip device with breast cancer-immune cell interactions for assessing T-cell recruitment. Cancer Res. 2020;80(2):263–75.
Carvalho MR, Barata D, Teixeira LM, Giselbrecht S, Reis RL, Oliveira JM, et al. Colorectal tumor-on-a-chip system: a 3D tool for precision onco-nanomedicine. Sci Adv. 2019;5(5):eaaw1317.
Gioiella F, Urciuolo F, Imparato G, Brancato V, Netti PA. An engineered breast cancer model on a chip to replicate ECM-activation in vitro during tumor progression. Adv Healthc Mater. 2016;5(23):3074–84.
Shang M, Soon RH, Lim CT, Khoo BL, Han J. Microfluidic modelling of the tumor microenvironment for anti-cancer drug development. Lab Chip. 2019;19(3):369–86.
Rothbauer M, Zirath H, Ertl P. Recent advances in microfluidic technologies for cell-to-cell interaction studies. Lab Chip. 2018;18(2):249–70.
Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, et al. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc Natl Acad Sci U S A. 2015;112(1):214–9.
Shemesh J, Jalilian I, Shi A, Heng Yeoh G, Knothe Tate ML, Ebrahimi WM. Flow-induced stress on adherent cells in microfluidic devices. Lab Chip. 2015;15(21):4114–27.
Halldorsson S, Lucumi E, Gómez-Sjöberg R, Fleming RMT. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens Bioelectron. 2015;63:218–31.
Liu CC, Steen CB, Newman AM. Computational approaches for characterizing the tumor immune microenvironment. Immunology. 2019;158(2):70–84.
van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods. 2017;14(10):935–6.
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–82.
Newman AM, Alizadeh AA. High-throughput genomic profiling of tumor-infiltrating leukocytes. Curr Opin Immunol. 2016;41:77–84.
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018;37:38.
Wang Z, Cao S, Morris JS, Ahn J, Liu R, Tyekucheva S, et al. Transcriptome deconvolution of heterogeneous tumor samples with immune infiltration. iScience. 2018;9:451–60.
Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3(4):346-60.e4.
Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, et al. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7(4):287–9.
Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–203.
Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol. 2014;15(3):R50.
Clancy T, Dannenfelser R, Troyanskaya O, Malmberg KJ, Hovig E, Kristensen V. Bioinformatics approaches to profile the tumor microenvironment for immunotherapeutic discovery. Curr Pharm Des. 2017;23(32):4716–25.
Yadav VK, De S. An assessment of computational methods for estimating purity and clonality using genomic data derived from heterogeneous tumor tissue samples. Brief Bioinform. 2015;16(2):232–41.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Shen Q, Hu J, Jiang N, Hu X, Luo Z, Zhang H. contamDE: differential expression analysis of RNA-seq data for contaminated tumor samples. Bioinformatics. 2016;32(5):705–12.
Anghel CV, Quon G, Haider S, Nguyen F, Deshwar AG, Morris QD, et al. ISOpureR: an R implementation of a computational purification algorithm of mixed tumour profiles. BMC Bioinformatics. 2015;16:156.
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–6.
Ahn J, Yuan Y, Parmigiani G, Suraokar MB, Diao L, Wistuba II, et al. DeMix: deconvolution for mixed cancer transcriptomes using raw measured data. Bioinformatics. 2013;29(15):1865–71.
The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–25.
Larson NB, Fridley BL. PurBayes: estimating tumor cellularity and subclonality in next-generation sequencing data. Bioinformatics. 2013;29(15):1888–9.
Clancy T, Hovig E. Profiling networks of distinct immune-cells in tumors. BMC Bioinformatics. 2016;17(1):263.
Clancy T, Pedicini M, Castiglione F, Santoni D, Nygaard V, Lavelle TJ, et al. Immunological network signatures of cancer progression and survival. BMC Med Genomics. 2011;4:28.
Erkkilä T, Lehmusvaara S, Ruusuvuori P, Visakorpi T, Shmulevich I, Lähdesmäki H. Probabilistic analysis of gene expression measurements from heterogeneous tissues. Bioinformatics. 2010;26(20):2571–7.
Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. 2013;23(11):1862–73.
Quigley D, Silwal-Pandit L, Dannenfelser R, Langerød A, Vollan HK, Vaske C, et al. Lymphocyte invasion in IC10/Basal-like breast tumors is associated with wild-type TP53. Mol Cancer Res. 2015;13(3):493–501.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45.
Singh O, Pratt D, Aldape K. Immune cell deconvolution of bulk DNA methylation data reveals an association with methylation class, key somatic alterations, and cell state in glial/glioneuronal tumors. Acta Neuropathol Commun. 2021;9(1):148.
Chen S, Huang F, Chen S, Chen Y, Li J, Li Y, et al. Bioinformatics-based identification of tumor microenvironment-related prognostic genes in pancreatic cancer. Front Genet. 2021;12: 632803.
Ye Q, Han X, Wu Z. Bioinformatics analysis to screen key prognostic genes in the breast cancer tumor microenvironment. Bioengineered. 2020;11(1):1280–300.
Wang T, Wang C, Wu J, He C, Zhang W, Liu J, et al. The different T-cell receptor repertoires in breast cancer tumors, draining lymph nodes, and adjacent tissues. Cancer Immunol Res. 2017;5(2):148–56.
Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, et al. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol. 2013;231(4):433–40.
Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238–70.
Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934-49.e16.
Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34-40.
Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT(®) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604.
Gaëta BA, Malming HR, Jackson KJ, Bain ME, Wilson P, Collins AM. iHMMune-align: hidden Markov model-based alignment and identification of germline genes in rearranged immunoglobulin gene sequences. Bioinformatics. 2007;23(13):1580–7.
Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380–1.
Ma X, Riaz N, Samstein RM, Lee M, Makarov V, Valero C, et al. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity. Nat Genet. 2022;54(7):996–1012.
Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547(7661):94–8.
Subudhi SK, Aparicio A, Gao J, Zurita AJ, Araujo JC, Logothetis CJ, et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci U S A. 2016;113(42):11919–24.
Stubbington MJT, Lönnberg T, Proserpio V, Clare S, Speak AO, Dougan G, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods. 2016;13(4):329–32.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342-56.e16.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9.
Hackl H, Charoentong P, Finotello F, Trajanoski Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet. 2016;17(8):441–58.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30(23):3310–6.
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152–8.
Park B, Heo SJ, Lee YJ, Seo MK, Hong J, Shin EC, et al. HLA-I-restricted CD8(+) T cell immunity may accelerate tumorigenesis in conjunction with VHL inactivation. iScience. 2022;25(6):104467.
Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–75.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–9.
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–12.
Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296.
Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8(1):33.
Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol. 2018;37:55.
Schubert B, Walzer M, Brachvogel HP, Szolek A, Mohr C, Kohlbacher O. FRED 2: an immunoinformatics framework for Python. Bioinformatics. 2016;32(13):2044–6.
Löffler MW, Mohr C, Bichmann L, Freudenmann LK, Walzer M, Schroeder CM, et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019;11(1):28.
Trolle T, Nielsen M. NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics. 2014;66(7–8):449–56.
Buckley PR, Lee CH, Ma R, Woodhouse I, Woo J, Tsvetkov VO, et al. Evaluating performance of existing computational models in predicting CD8+ T cell pathogenic epitopes and cancer neoantigens. Brief Bioinform. 2022;23(3).
Wang N, Gong T, Clarke R, Chen L, Shih Ie M, Zhang Z, et al. UNDO: a Bioconductor R package for unsupervised deconvolution of mixed gene expressions in tumor samples. Bioinformatics. 2015;31(1):137–9.
Quon G, Morris Q. ISOLATE: a computational strategy for identifying the primary origin of cancers using high-throughput sequencing. Bioinformatics. 2009;25(21):2882–9.
Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics. 2013;29(8):1083–5.
Kuhn A, Thu D, Waldvogel HJ, Faull RL, Luthi-Carter R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods. 2011;8(11):945–7.
Zuckerman NS, Noam Y, Goldsmith AJ, Lee PP. A self-directed method for cell-type identification and separation of gene expression microarrays. PLoS Comput Biol. 2013;9(8):e1003189.
Li Y, Xie X. A mixture model for expression deconvolution from RNA-seq in heterogeneous tissues. BMC Bioinformatics. 2013;14(Suppl 5):S11.
Liebner DA, Huang K, Parvin JD. MMAD: microarray microdissection with analysis of differences is a computational tool for deconvoluting cell type-specific contributions from tissue samples. Bioinformatics. 2014;30(5):682–9.
Qiao W, Quon G, Csaszar E, Yu M, Morris Q, Zandstra PW. PERT: a method for expression deconvolution of human blood samples from varied microenvironmental and developmental conditions. PLoS Comput Biol. 2012;8(12): e1002838.
Abbas AR, Wolslegel K, Seshasayee D, Modrusan Z, Clark HF. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS ONE. 2009;4(7): e6098.
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6.
Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220.
Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174.
Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 2018;9(1):3220.
Wang X, Park J, Susztak K, Zhang NR, Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun. 2019;10(1):380.
Frishberg A, Peshes-Yaloz N, Cohn O, Rosentul D, Steuerman Y, Valadarsky L, et al. Cell composition analysis of bulk genomics using single-cell data. Nat Methods. 2019;16(4):327–32.
Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11(1):34.
Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR, et al. Towards error-free profiling of immune repertoires. Nat Methods. 2014;11(6):653–5.
Li B, Li T, Wang B, Dou R, Zhang J, Liu JS, et al. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA-seq data. Nat Genet. 2017;49(4):482–3.
Redmond D, Poran A, Elemento O. Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med. 2016;8(1):80.
Afik S, Yates KB, Bi K, Darko S, Godec J, Gerdemann U, et al. Targeted reconstruction of T cell receptor sequence from single cell RNA-seq links CDR3 length to T cell differentiation state. Nucleic Acids Res. 2017;45(16): e148.
Eltahla AA, Rizzetto S, Pirozyan MR, Betz-Stablein BD, Venturi V, Kedzierska K, et al. Linking the T cell receptor to the single cell transcriptome in antigen-specific human T cells. Immunol Cell Biol. 2016;94(6):604–11.
Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67(11–12):641–50.
Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132.
Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394.
O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 2018;7(1):129-32.e4.
Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: a genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8(1):11.
Acknowledgements
Authors would like to thank Mr. Bryan Tutt, Scientific Editor, Research Medical Library, The University of Texas MD Anderson Cancer Center for the editorial support.
Funding
Funding was provided by NCI Grant R01 CA218287 to SYL.
Author information
Authors and Affiliations
Contributions
AT and SYL established the review idea and finalized the framework. AT, RT and SYL were involved in writing and editing of the review. All authors read and approved the final version of manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tiwari, A., Trivedi, R. & Lin, SY. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J Biomed Sci 29, 83 (2022). https://doi.org/10.1186/s12929-022-00866-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00866-3