
Abstract
Natural killer (NK) cell is a specialized immune effector cell type that plays a critical role in immune activation against abnormal cells. Different from events required for T cell activation, NK cell activation is governed by the interaction of NK receptors with target cells, independent of antigen processing and presentation. Due to relatively unsophisticated cues for activation, NK cell has gained significant attention in the field of cancer immunotherapy. Many efforts are emerging for developing and engineering NK cell-based cancer immunotherapy. In this review, we provide our current understandings of NK cell biology, ongoing pre-clinical and clinical development of NK cell-based therapies and discuss the progress, challenges, and future perspectives.
Similar content being viewed by others

Background
Natural killer (NK) cells are an essential part of tumor immunosurveillance, evidenced by higher cancer susceptibility and metastasis in association with diminished NK activity in mouse models and clinical studies [1,2,3]. Using an array of germline-encoded surface receptors, NK cells are able to recognize and rapidly act against malignant cells without prior sensitization. Upon activation, NK cells release cytotoxic granules containing perforin and granzymes to directly lyse tumor cells, in a similar fashion to activated cytotoxic T cells. NK cells are also potent producers of chemokines and cytokines such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) and thereby are essential in modulating adaptive immune responses. Due to their innate ability to eliminate tumor cells, NK cell-based immunotherapies against cancer have been investigated for decades. Early clinical trials have demonstrated the overall safety of NK cell infusion, even in the allogeneic setting [4,5,6,7]. The feasibility of utilizing allogeneic NK cells, the established safety profiles, and the fast-acting nature of NK cells largely have led to the emerging effort to develop “off-the-shelf” NK cell-based cancer immunotherapy. However, there are many challenges to overcome, such as difficulty to meet clinical-grade ex vivo expansion, limited in vivo persistence, limited infiltration to solid tumors, and tumor editing to evade NK cell activity. Various strategies are being employed to overcome these challenges to improve the efficacy of NK cell-based therapy, such as ex vivo pre-conditioning with cytokines and/or small molecular drugs, engineering an “off-the-shelf” or iPSC-differentiated chimeric antigen receptor (CAR)-NK. There has been an explosion of NK-based immunotherapies in pre-clinical development and clinical development. Herein, we will provide an updated overview of the emerging endeavors for developing NK cell-based cancer immunotherapy from pre-clinical conceptual development, clinical grade expansion, and ongoing clinical development.
Main text
NK cell biology
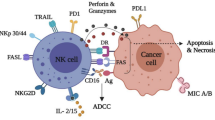
NK cells were identified over four decades ago as lymphocytes with innate ability to lyse tumor cells without the need for prior sensitization [8,9,10]. NK cells can trigger target cell death by releasing cytotoxic granules containing granzymes and perforin and through death receptor-mediated pathways (e.g., FasL/Fas) [11]. NK cells also play immunomodulatory functions by secreting chemokines and cytokines, such as RANTES and IFN-γ [12, 13].
In humans, NK cells are traditionally identified by the absence of CD3 and the presence of CD56 on their surface as characterized by flow cytometry. In mouse, the lack of CD3 and the presence of NK1.1 are canonical criterion for distinguishing NK cells. In mouse strains lacking NK1.1 expression (e.g., BALB/c), CD49b is used for NK cell identification. The natural cytotoxic receptor NKp46 is also often used to identify mouse and human NK cells in combination with the absence of CD3 expression. Notably, certain tissues such as the mucosal barriers possess subsets of recently identified innate lymphoid cells (ILCs) that also share canonical markers of NK cells. For example, a subset of IL-22 secreting human ILC3s is CD56+NKp46+CD3− [14]. Additional markers such as lack of c-kit can be used to distinguish human NK (c-kit-) from ILC3s (c-kit +) [14]. In mouse, NK and ILC1 are NK1.1+CD3− but can be further characterized by CD49a and Eomes expression. NK cells are CD49a−Eomes+, whereas ILC1s are CD49a+Eomes− [15, 16].
NK cells are found both in blood at levels of 5%-15% of circulating lymphocytes and in various lymphoid and non-lymphoid organs such as the spleen, lung, and liver [17, 18]. Based on characterization of NK cells in the peripheral blood, human NK cells are conventionally sub-divided into two major subsets: CD56brightCD16dim/− and CD56dimCD16+, with the former classically believed to be less mature and a potent cytokine producer and the latter more mature and the most cytotoxic [17, 19]. Most NK cells in the blood are CD56dim, whereas the CD56bright subset only represents less than 15% of total circulating NK cells [17]. The relative proportion of CD56bright and CD56dim NK cells in tissues can be very different from that observed in the peripheral blood [17]. Notably, many tissue-resident NK subsets are now shown to be phenotypically and functionally distinct from conventional peripheral blood NK cells [20]. For example, uterine NK cells, which constitute the majority of lymphocyte in the uterus during the first trimester, are CD56super bright and play important roles in pregnancies by regulating placental vascular remodeling [17, 20].
Conventional NK cells are short-lived innate lymphocytes that lack antigen specificity. Recent studies revealed that subset of mature NK cells can elicit long-lived “adaptive”-like nature in the specific context of CMV infection [21]. It is now understood that the “adaptive”-like NK cells express the activating receptor NKG2C and that the “adaptive” nature of NKG2C+ NK cells attributes to the non-classical MHC I molecule HLA-E presenting CMV-specific viral peptide to NKG2C [22]. The NKG2C+NK cells can be found in the circulation of HCMV-seropositive individuals [22].
NK cells originate from CD34+ hematopoietic stem cells. Bone marrow is considered to be the primary site of NK development. More recent evidence had indicated that NK cells can also develop and mature at secondary lymphoid organs, including tonsils, spleen, and lymph nodes [19]. NK cell progenitors progress through distinct developmental stages and gradually acquire the expression of surface receptors that define NK cell identity such as NK1.1 and CD56 and/or regulate their effector functions such as CD16 and NKp46 [19]. Distinct from T cells, there is no master transcriptional factor that controls NK cell development. Instead, a combination of transcription factors, including T-bet, Eomes, E4BP4, Id2, and BLIMP, was identified to instruct NK development and maturation [23]. Common gamma chain cytokines such as IL2, IL-7, and IL15 and their receptor components including CD122 and CD127 have been shown to play essential roles in NK development and homeostasis [19]. Despite decades of work, the ontogeny of NK cells in humans is still not fully elucidated. The conventional linear model suggests that mature NK cells arise from common lymphoid progenitors (CLPs) by progressing through a linear continuum [24]. The linear model proposes that CD56 marks a transition from immature into a more mature status and that immature CD56bright NK cells further differentiate into mature CD56dim populations in human [19, 24]. Recent evidence has challenged this model and suggests a possibility of more branched development in the form of both CLPs and common myeloid progenitors (CMPs) giving rise to NK cell progenitors. The branched model also proposes that distinct precursor populations independently develop into different mature NK subsets [24].
NK cell receptors and NK activation
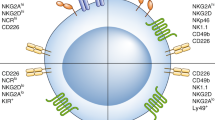
NK cells are mounted with a repertoire of inhibitory and activating surface receptors (Table 1) [25,26,27,28,29]. Distinctly different from T cell receptors, NK cell receptors are germline-encoded and “hard-wired” receptors without a requirement for “V(D)J” recombination. Upon ligation, these receptors transmit either inhibitory or activating signals to control NK activation. The integration and balance of the activating and inhibitory signals from the ligand/receptor interactions dictates the status of NK cell activation. For instance, healthy cells express no or minimal level of ligands for NK cell activating receptors, but express high levels of the major histocompatibility complex class I molecules (MHC I), also known as human leukocyte antigen (HLA), that ligates to the killer immunoglobulin-like (KIR) family inhibitory receptors on NK cells to protect them from NK attack [29]. Conversely, tumorigenic cells or virally infected cells have downregulated MHC I expression but upregulated levels of ligands for NK cell activating receptors and thus trigger NK cell activation due to the lack of inhibitory signals and/or the presence of activating signals [11]. In allogeneic transfer settings, the concept of KIR and HLA mismatches between donor and recipient was the original strategy aimed at enhancing the activation of donor NK cells to eradicate patient’s tumor cells. NK cells also play a significant role in antibody-mediated cancer therapies by utilizing the Fcγ receptor to carry out antibody-dependent cellular cytotoxicity (ADCC) [30].
Preclinical development of NK cell-based cancer immunotherapy
Current preclinical development of NK cell-based therapy was largely inspired by early clinical studies. With the understanding of how NK cells are activated, the initial NK cell-based therapy was pioneered in the clinical setting of hematopoietic stem cell transplants (HSCTs) whereby NK cells were shown to have the capacity to exert a graft versus leukemia effect. The Ruggeri group showed that KIR-mismatched alloreactive donor NK cells protected bone marrow-transplanted AML patients from AML relapse while sparing graft versus host diseases (GVHD) [31, 32]. Miller et al. further pioneered the use of NK cells in non-transplant settings. They showed that infusion of NK cells from HLA-haploidentical donors combined with subcutaneous IL-2 administration after a pre-conditioning regimen of high-dose cyclophosphamide and fludarabine resulted in successful in vivo expansion of donor NK cells and the induction of complete remission in 5 out of 19 patients with poor-prognosis acute myeloid leukemia (AML) [5]. Miller et al. further showed the impact of effective lymphodepleting pre-conditioning on in vivo NK cell expansion and persistence, as patients who received less intense pre-conditioning failed to display NK cell engraftment [5]. It is currently believed that the success of adoptive transfer requires the creation of a lymphopenic environment to provide a niche for donor cells to survive and proliferate.
Initial successes of adoptive NK cell transfer in treating hematological cancers prompted clinical endeavors in using the strategy against solid cancers. NK cells are cytotoxic against a wide range of tumor cells of solid cancer types in vitro. Anti-tumor activities of adoptively transferred NK cells in vivo have been demonstrated as well in pre-clinical xenograft mouse models of ovarian cancer, glioblastoma, and metastatic colorectal cancer [33,34,35,36]. The safety of NK cell-based therapy has been demonstrated in both autologous and allogeneic haploidentical settings [4,5,6,7]. Clinical efficacy of this strategy has proven to be thus far be limited. The following section summarizes the current pre-clinical efforts to enhance the efficacy of NK cell-based therapy.
CAR-NK cell as an alternative to CAR-T therapy
T cells equipped with CARs have been shown to provide clinical benefit for patients with select liquid cancers. Two CD19-targeting CAR-T products were approved by the FDA for treatment of B cell lymphomas and acute lymphoblastic leukemia (ALL). However, CAR-T therapy has two major challenges: (i) requirement of a substantiate length of time to generate a therapeutic dose of autologous CAR-T cells which limits its feasibility to treat patients with rapidly progressing diseases; (ii) difficulty to obtain sufficient number of autologous T cells for CAR-T cell generation from heavily pre-treated and often lymphopenic cancer patients [37]. As an alternative to CAR-T cell therapy, CAR-NK cell therapy not only circumvents these challenges but also presents additional major advantages: (i) the ability to use unlimited allogeneic NK source without concern of GVHD [6, 31, 32]; (ii) the potential to generate “off-shelf” product with NK cell line or iPSC-NK [37,38,39,40,41]; (iii) relatively shortened production time; iv) recognition and killing tumor cells through NK cell native receptors independent of the CAR engineering [42], less likely allowing disease escape through downregulation of the CAR antigens as shown with CAR-T cell therapy [37].
Similar to CAR-T cells, CAR-NK cells are genetically modified to express CARs that recognize a specific antigen uniquely expressed or overexpressed by target cells. In most pre-clinical studies, lentiviral or retroviral-based transduction was used to achieve stable and sustained CAR expression in NK cells. Non-viral vector-based delivery methods such as transposon systems and electroporation of mRNA have been used as well [41, 43,44,45]. A wide range of tumor antigens have been targeted by CAR-NK cells in pre-clinical studies for hematological malignancies and solid tumors [38, 39, 41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76]. They are also summarized in Table 2. The antigen recognition domain usually consists of a single-chain fragment (scFV) molecule derived from a monoclonal antibody, and nanobody-based constructs have been used in limited studies to date [72]. For hematological cancers, CD19 remains a major target. Antigens such EGFRvIII, mesothelin, and Her2 have been targeted by CAR-NK cells for the treatment of solid cancers including colorectal cancer, ovarian cancer, breast cancer, and glioblastoma [41, 48]. The signaling domains of CAR-NK cells are very similar to those in CAR-T cells, typically composed of the fusion of CD3ζ with one or two TCR co-stimulatory molecule (s), such as CD28, 4-1BB, 2B4, DNAM1, and NKG2D. Among these TCR cell co-stimulatory molecules, 4-1BB, DNAM1, 2B4, and NKG2D were also expressed by NK cells as native activating receptors. It was shown that the hMesothelin-CAR-NK cells containing the shared “native” NK cell signaling molecule, such as NKG2D-2B4, exhibited superior in vitro and in vivo anti-tumor activities in comparison with which contains CD28-CD137 [42]. The NKG2D-2B4 containing CAR-NK had elevated Syk and Erk1/2 phosphorylation [42].
Beyond directing CAR-NK cytotoxicity against tumor cells by targeting tumor antigens, it has been proposed that CAR-NK cells can be used to eliminate immunosuppressive immune cells in the tumor microenvironment that include myeloid-derived suppressor cells (MDSCs) and M2 tumor-associated macrophages (TAMs) [77, 78]. Parihar and colleagues designed CAR-NK cells expressing the activating receptor NKG2D as the antigen recognition to target human MDSCs. They demonstrated in vitro and in a mouse xenograft model grafted with human neuroblastoma and MDSCs that NKG2D CAR-NK cells can reduce human MDSCs efficiently [77]. Moreover, the CAR-NK cells secrete pro-inflammatory cytokines and chemokines which may improve the infiltration and functions of subsequently infused CAR-T cells in the mouse model [77]. It has been proposed that CAR-NK cells may be able to be combined with T cell-based therapies for solid tumors [77].
Killer cell engagers unleash NK cytotoxicity against tumor cells
Although CAR-NK cells hold great promise as a future “off-the-shelf” drug, there are NK cell-specific challenges, such as potential loss of viability and/or activity with freeze–thaw process and lack of in vivo persistence. To circumvent these challenges, Bi- and tri-specific killer engagers, BiKEs and TriKEs, are in development as a complemental approach. The BiKEs or TrikEs are composed of two or three linked single-chain antibody variable fragments of different antigen specificities [79]. The current in-development BiKEs or TrikEs simultaneously engage with CD16 and tumor antigens, thereby inducing the formation of immune synapses and NK-mediated ADCC [79]. In pre-clinical studies, various BiKEs and TriKEs have been designed to target a number of tumor antigens: CD19, CD20, and CD33 for hematological cancers, HER2, EGFR, and EpCAM for solid tumors [79]. Cytokines, such as IL15, have been incorporated into killer engagers to further enhance NK cell functions [80]. Vallera et al. reported that a CD16/IL-15/CD33 TriKE not only enhances NK-mediated immunity against CD33+ targets, but also promotes the in vivo persistence, activation, and survival of NK cells by delivering IL-15 [80]. This TriKE design (GTB-3550) is currently in clinical trial for treating high-risk hematological malignancies (NCT03214666). Tri-functional NK cell engagers (NKCEs), which crosslink both NKp46 and CD16, have demonstrated superior in vitro and in vivo anti-tumor activities compared to conventional monoclonal antibodies targeting the same antigens [81].
NK cells “priming” strategies
While the CAR technology seeks to enhance NK cell functions by genetically directing their target specificity, there are active investigations on other strategies to effectively “prime” NK cells ex vivo or in vivo for optimal anti-tumor functions after their infusion. It has been shown that freshly isolated, resting NK cells are generally less lytic as compared to NK cells primed via various strategies [82].
Cytokine-mediated activation is frequently employed and currently under extensive investigation. In many studies, IL-2 or IL-15 is supplemented during ex vivo expansion of NK cells and sometimes after NK cell infusion as well [5]. ALT-803, an IL-15/IL-15Ra fusion complex, was shown to enhance NK cell functions both in vitro and in vivo [83]. Short-term (18–20 h) pre-activation with ALT-803 augmented cytotoxicity and ADCC of NK cells in vitro. In a xenograft mouse model of lymphoma, co-administration of ALT-803 with NK cells significantly enhanced anti-CD20-triggered, NK cell-mediated ADCC effects [83]. Recently, several groups demonstrated that mouse and human NK cells pre-activated with a cocktail of IL-12/15/18 had enhanced and sustained anti-tumor effector functions in vitro and in vivo after infusion [84,85,86]. It was proposed that the cytokine pre-activated NK cells were “memory-like” with an enhanced response to cytokine or activating receptor re-stimulation weeks or months after the initial pre-activation [84]
In addition to cytokine-only strategies, Cichocki and colleagues demonstrated that pharmacologic inhibition of GSK3 kinase with CHIR99021 drives late-stage maturation of ex vivo-expanded human peripheral blood NK cells and enhanced their anti-tumor effector functions both in vitro and in vivo [87]. Human NK cells expanded with IL-15 in the presence GSK3 inhibition had increased expression of the NK cell maturation marker CD57 and transcription factors associated with late-stage NK cell maturation including T-bet, ZEB2, and BLIMP-1 as compared to those expanded with IL-15 alone [87]. The expanded human NK cells showed potent ADCC activities in vitro and superior tumor control in a mouse xenograft model of ovarian cancer when combined with Herceptin, an anti-HER2 antibody [87]. This strategy of GSK3 inhibition was used to generate FATE-NK100, which is being evaluated in an ongoing phase 1 clinical trial either as a monotherapy or in combination with monoclonal antibodies in patients with advanced solid tumors (NCT03319459).
While the majority of investigation of ex vivo NK priming strategies rely on one or more cytokines, some studies indicate that cytokine supplement is not as critical for NK cell priming. One study showed that overnight co-incubation with a leukemia cell line CTV-1 or its lysate without cytokines was sufficient to prime potent NK cells in vitro cytotoxicity against otherwise NK-insensitive tumor cells but not normal hemopoietic cells [88]. The mechanism of CTV-1-mediated NK priming is unclear. It was suggested that induced expression of CD69 on NK cells is important for the cytotoxicity of the CTV-1 tumor cell-activated NK [88]. Based on the pre-clinical findings, CTV-1 lysate-primed human NK cells (CNDO-109-NK cells) derived from HLA-haploidentical donors were evaluated for safety in a phase I clinical trial (NCT01520558) in high-risk AML patients with AML after first complete remission [89]. CNDO-109-NK cells were found well tolerated without occurrence of dose-limiting toxicities [89]. Three out of 12 patients had durable complete remissions [89], providing evidence for further clinical evaluation of this approach.
Methods to enhance the infiltration and functions of infused NK cells
Beyond engineering and priming NK cells, there are ongoing efforts toward improving tumor infiltration of adoptively transferred NK cells by modifying the chemokine–chemokine receptor axis. Lee et al. developed an NK cell recruiting protein-conjugated antibody (NRPbody) containing a mesothelin-specific tumor targeting domain Meso-scFv and the chemokine CXCL16 linked by a furin cleavage sites [90]. Mesothelin is a tumor differentiation antigen that is highly overexpressed in several human cancers including malignant mesothelioma, pancreatic, ovarian, and lung adenocarcinoma [91]. It was hypothesized that once bound to mesothelin-overexpressing tumor cells, furin-mediated cleavage would release CXCL16 from the NRPbody and thereby recruit NK cells to the tumor sites [90]. The cleavable CXCL16 containing NRPbody was shown to promote NK cell migration in vitro and infiltration into the tumor sites in vivo in xenografted mouse models of orthotopic and metastatic pancreatic cancer [90]. In the xenograft models, NK cell infusion combined with intraperitoneal injection of the NRPbody significantly reduced tumor burden as compared to NK infusion combined with the non-cleavable control [90]. It remains to be determined how NRPbody will interact with a more complex and realistic immune contexture in the TME which consists of more than NK cells, as CXCL16 was shown in other tumor models to correlate with the infiltration of monocytes and M2-macrophages as well [92, 93]. Some investigations have sought to enhance NK migration toward the tumor by genetically modifying NK cells to overexpress chemokine receptors [54, 59, 94]. In one study, Ng et al. showed that CAR-NK cells genetically modified to express the chemokine receptor CXCR1 had enhanced migration in vitro and in vivo as compared to control CAR-NK cells [54]. With the subcutaneous hypopharyngeal tumor xenograft model, it was shown that CXCR1-expressing CAR-NK cells had enhanced tumor infiltration and tumor control as compared to control CAR-NK cells [54]. Targeting immunosuppressive components in the TME to re-invigorate NK functions is also under investigation. One major immunosuppressive factor in the TME is the metabolite adenosine, whose production is catalyzed in a sequential manner by the ectoenzymes CD39 and CD73 [43]. Adenosine impairs the anti-tumor functions of both T and NK cells [43]. Wang et al. demonstrated that antibody-mediated blockade of CD73 significantly enhanced the anti-tumor activities of NKG2D-enginneered CAR-NK92 in vitro and in vivo, with improved tumor infiltration by CAR-NK cells in vivo [43].
NK expansion for clinical use
Source and clinical-scale expansion of NK cells with preserved cytotoxic activity are the major challenges for developing clinical-scale NK cell-based therapy. Currently, strategies vary depending on the clinical setting and source of cells. Freshly isolated, activated, or in vitro expanded NK cell populations display phenotypic and functional differences. The differences also arise from activation approaches, such as the choice of interleukins, their combinations, type of feeder cells, and some other factors [95,96,97]. The current source and characteristics of NK cells for immunotherapies, expansion, and activation approaches are given in Tables 3 and 4.
Donor-derived NK sources
The main source of donor NK cells is peripheral blood collected by apheresis. Using allogeneic NKs for adoptive transfer without expansion after a short overnight incubation with cytokines was shown to be sufficient to activate NK cells and enhance their cell cytotoxicity against tumor targets [86]. However, the dose of NK cells being used for therapy is limited. As the activation and expansion methods are improving, it is becoming possible to prepare increasingly higher dosages of NK cells for adoptive transfer from a single-donor phlebotomy. In order to avoid critical side effects, such as GVHD caused by alloreactive T cells [98] or a passenger lymphocyte syndrome caused by donor-derived B cells [99], purification is recommended for allogeneic NK cells before the expansion to restrict contaminating total T cells to less than 1–5 × 105/Kg. The purification of NK cells is typically achieved by magnetic depletion of CD3-expressing cells and subsequent enrichment for CD56-expressing cells [100] or by fluorescence-activated cell sorting [101].
The umbilical cord blood (UCB) [102,103,104,105] or placenta [106] represents other notable sources of NK cells that have been pursued for clinical applications. Both UCB and placenta-derived cells contain some proportion of NK cell progenitors that have the capacity to differentiate into NK cells during maturation and expansion stages. Typically, a dose of UCB or placenta donor can be expended to an amount sufficient for one adoptive transfer procedure. For instance, 21-day NK culture of placenta-isolated NKs yields an average of 1.2 × 109 NK cells with around 80% viability [106] and 1.59 × 1010 NK cells with an average purity of 92.37% from UCB [107].
iPSC-NK cell source
The donor-derived NK cells have certain limitations mainly because of their variability in functional competence and expansion potential. In the clinical setting, each batch requires validation, which results in additional lag time before a patient receives infusion. Moreover, restricted expansion capacity poses a difficulty for improving the efficacy of NK cells through genetic engineering. CAR-NK is one of the approaches in development to overcome this limitation. The approach of using the modified NK cell line CAR-NK92, which can be easily expanded to large dose, has been in clinical trial. However, NK92 is a transformed cell line that has limitations associated with its tumorigenic nature, and cytogenetic abnormalities, thus requiring irradiation for clinical use, which limits its life-time activity.
A novel source of NK cells has emerged to circumvent many of the challenges associated with NK cell therapy. It is iPSC-differentiated NK cells (iPSC-NK). Conceptually, iPSC-NK can provide a homogenously differentiated NK cell population that can be expanded to clinical scale as an “off-the-shelf” supply, overcoming the limitation of the NK-92 cell line. Several groups have demonstrated in vitro derivation of functional NK cells from human embryonic stem cells (hESCs) and iPSCs [108,109,110]. Typically, the differentiation was induced in embryoid bodies or by OP9 mouse stroma co-culture [111]. Recently, the differentiation method was also established in adherent monolayer cultures [108]. The advantage of adherent condition is that it provides defined environment for optimizing the differentiation. This method has achieved over 15% of CD34+ hematopoietic progenitors compared to other methods producing only up to 4% of CD34+. The emergence of this technology provided a completely new framework for clinical-scale NK cell production by allowing for the genetic modifications and unlimited expansion to be performed at the pluripotent cell state. CAR-engineered iPSC-NK cells already have demonstrated effectiveness in targeting human tumors in preclinical studies [41]. The other important feature of iPSC-NK cell technology is the ability to manipulate the differentiation strategy, thus shaping the phenotype and functionality of the resulting product. For instance, the enhancement of the Wnt signaling pathway with GSK3b inhibitor induces definitive hematopoiesis [112, 113] NK cells developed in such conditions had more pronounced inflammatory cytokine production phenotype, whereas Wnt independent NK subsets, similar to primary fetal NK cells, formed a bias for increased cytotoxicity [110]. Such ability to alter the course of differentiation opens a possibility to pursue resident or organ-specific phenotypes of NK cells.
The high proliferation capacity of pluripotent stem cells allows for the introduction of various genetic modifications and for the development libraries of off-the-shelf haplotype-specific cells for treating a range of diseases. There are a number of ongoing clinical trials for cancer immunotherapy using engineered iPSC-NK cells, which are summarized in the following sections.
Cytokine-induced human NK expansion and activation
Cytokines are the critical components of NK maintenance system and activation as discussed earlier. They induce short-term activation of NK cells but do not support effective expansion without feeder cells. Interleukin (IL)-2 (IL-2) is one of the first and most important cytokines used for NK maintenance and is integral for NK cell survival. It is one of the two cytokines, IL-2 and interferon alpha (IFN-α), approved by the FDA for the treatment of several malignant diseases [114]. IL-2 was used to induce lymphokine-activated killer (LAK) cells, a heterogeneous population of cells consisting primarily of NK, NKT, and T cell for autologous killer cell-based cancer therapy decades ago [115]. The anti-tumor response of LAK cells was shown attributed mainly to NK cells [116]. Noteworthy, IL-2 primarily activates NK cytotoxicity, while supporting proliferation of both NK and T cells. The expansion of NK cells using IL-2 alone is relatively modest and typically results in only several cell divisions in medium containing 1000 U/mL of IL-2 [100]. IL-21 has various effects on human NK cells. On the one hand, it was shown to enhance interferon gamma (IFN-γ) production, cytotoxic functions, and antibody-dependent cellular cytotoxicity (ADCC) responses [117]. On the other hand, it has limited viability support and can trigger proliferative arrest and apoptosis of NK cells at higher dosage (50 ng/mL) [118]. IL-21 has cumulative activation effect in combination with IL-2 [119] or IL-15. It was shown that of CD3-depleted peripheral blood mononuclear cells (PBMC) with IL-21 and IL-15 for 13–20 days resulted in 3.7-fold expansion of NK cells with clinical activity in delaying leukemia progression [120]. The combination of IL-2/IL-15/IL-21 can support up to eightfold expansion of NK cells [121]. Efforts are still ongoing to identify the most optimal cytokine combination for NK cell expansion. Most recent studies showed that ex vivo stimulation of human NK cells with the combination of IL-15/IL-18/IL-27 can achieve 17-fold expansion [122], and that the combination of IL-2 with IL-18 can achieve approximately 500-fold expansion over two-week period [123]. The presence of autologous feeder cells (typically CD3-depleted PBMCs) in culture additionally facilitates NK expansion [124].
It is believed that membrane-bound interleukins are able to stimulate the expansion of NK cells more effectively than the soluble form. A study by Campana and coworkers has shown that stimulation of NK cells with gene-modified K562 expressing the NK-stimulatory molecules 4-1BB ligand and IL-15 induced a median 21.6-fold expansion during a 7-day culture period. It yielded a greater than 1000-fold expansion of NK cells after 3 weeks of culture [38, 125, 126]. An even greater expansion of NK cells, of over 30,000-fold in a period of 3 weeks, was achieved with K562 membrane-bound IL-21 and 4-1BB ligand [127,128,129,130]. This protocol created a possibility to generate a substantially higher number of NK cells from a single dose of peripheral blood [131] and is currently in phase I/II clinical trial (NCT01787474) with expanded haploid-identical NK cells for treating relapsed or refractory AML. Moreover, highly cytotoxic NK cells derived using such method are capable of producing endogenous cytokines that improve their survival, proliferation, and function [132].
Other methods to induce human NK cell expansion
Beyond cytokines, other stimulants, including tumor cells, allogeneic PBMCs, antibodies, and microbiol derivatives, have been explored for enhancing ex vivo NK cell expansion. Exposure of NK to unmodified NK cell-sensitive leukemia cells (K562) stimulates expansion and short-term proliferation [133]. Over 100-fold expansion was achieved with Wilms tumor HFWT [134] and immortalized T lymphocyte Jurkat cell lines [135]. Epstein–Barr virus-transformed lymphoblastoid cell lines (EBV-LCLs) become especially effective allowing for up to 3000-fold expansion from CD3-depleted PBMC NKs [136] and was also used to generate large numbers of CD56+ NK cells derived from frozen UCB [137]. Chemical stimuli such as Concanavalin A (ConA) [138], Phytohemagglutinin (PHA), and ionomycin [139] were also used in combination with irradiated allogeneic PBMCs to facilitate the activation. Although lethal irradiation of feeder cells before use is required, for safety concern, the residual contamination of feeder cells should be assessed. The release criteria should be developed with unique signature to distinguish the feeder cells from expanded NK cells to ensure no feeder cell contamination. For instance, in the use of CD19-modified K562 to propagate NK cells, the contamination was assessed by flow cytometry detection of surface expression of the NK cell endogenous molecule CD32 and the K562 transgene CD19 to distinguish NK cells from the feeder cells [140]. Other methods may include transgene of suicide gene or expression of a fluorescent marker in the feeder cell. Thus, feeder-free approaches are an alternative or a more desirable method to avoid safety concerns associated with the clinical application of cancer cell-derived feeder cells. One approach to address the safety concern is the use of lysed cell product. For example, using the membrane particles of K562 cells with membrane-bound IL-21 and 4-1BB ligand as the feeder has achieved the activation and 250-fold expansion of NK cells after approximately two weeks of ex vivo culture [141].
Stimulating antibodies is typically used along with irradiated allogeneic PBMC to further promote NK activation and expansion. OKT3, an anti-CD3 mAb, has been commonly added to the irradiated autologous PBMC feeder, which can promote the expansion of NK cells up to over 1000-fold [4, 142,143,144]. The anti-CD3 mAb presumably activates T cells in feeder to secret cytokines which subsequently create a milieu favorable for NK cell expansion [145].
Masuyama et al. reported an approximately 1500-fold expansion of NK cells after PBMC stimulation with a combination of anti-CD3 and anti-CD56 mAbs [146]. Using irradiated autologous PBMCs and anti-CD16 mAb, Lee et al. demonstrated a more than 500-fold NK expansion with over 98% purity within 2 weeks and a greater than 5000-fold NK expansion over a 3-week period [147].
A more simplified NK cell expansion method has been used by combining group A streptococcus and zoledronate with IL-2 to stimulate UCB-derived mononuclear cells. This method resulted in a 1,560-fold expansion of NK cells with a purity of 92.37% after 21 days of ex vivo culture [107]. This method was advantageous in that it did not require magnetic cell sorting, feeder cells, or multiple cytokines, potentially lowering the cost of production. Furthermore, IL-2, streptococcus A group, and zoledronate have all been approved for human use. Clinical evaluation of safety and efficacy of NK cells under this expansion is warranted.
Clinical development of NK cell-based cancer therapy
The “off-shelf” NK cell therapeutic product oNKord, the allogeneic partial HLA-matched NK cells derived from UCB-CD34+ progenitors, has received an orphan drug designation from EMA and FDA for treating AML patients who were not eligible for allogeneic stem cell transplantation. This approval was based on clinical study demonstrating that oNKord improves survival in year 1 of 80% vs. 35% in the control arm. Recently, the FDA has approved the investigational new drug (IND) for the use of placenta-expanded NK cells (CYNK-001) against glioblastoma (GBM). The success has encouraged many ongoing clinical investigations of NK cell-based cancer therapy alone or in combination with other regimes. Clinicaltrials.gov currently lists over 100 clinical trials of NK cell-based cancer immunotherapy. Herein we highlight current evaluations for hematological malignancies and solid tumors.
NK cell-based clinical trial for hematological malignancies
The safety and efficacy of allogeneic or autologous donor-derived NK cell-based therapy for treating hematological malignancies, such as AML, have been well established [5, 29, 148]. With the ongoing effort to improve the treatment response and new methods of generating more feasible clinical scales of NK cells, emerging clinical trials are being designed to evaluate these new modalities and to expand their indications. A first-in-human clinical trial of CAR NK-92 cells in 3 patients with relapsed or refractory AML showed that CAR NK-92 can be infused at doses up to 5 billion cells per patient without causing significant adverse effects [149]. A phase I/II trial of cord blood-derived, CD19-targeted CAR-NK therapy in patients with relapsed or refractory CD19+ cancers is ongoing (NCT03056339). The interim results showed that 8 out of 11 patients had an objective response to treatment without development of major toxic effects [150]. Table 5 summarizes the clinical trials of NK cell-based therapy for hematological malignancies to date.
NK cell-based clinical trial for solid tumors
There are great numbers of clinical trials on NK cell immunotherapy to treat solid tumors. For example, there is an ongoing phase 2 clinical trial to evaluate the safety and efficacy of human HLA-haploidentical hematopoietic cell transplantation (HCT) followed by an early, post-transplant infusion of donor NK cells (NCT02100891). The subjects of the study were patients with high-risk solid tumors, including Ewing Sarcoma, Neuroblastoma, Rhabdomyosarcoma, Osteosarcoma, and CNS tumors. There are also many ongoing clinical trials to evaluate the safety and efficacy of tumor-targeting CAR-NK cells, including using HER2-specific CAR-NK cell to treat advanced or metastatic HER2-expressing solid tumors (NCT04319757), ROBO1-specific CAR-NK cell to treat a broad spectrum of solid tumors (NCT03940820), and MUC1-specific CAR-pNK to treat patients with MUC1-positive relapsed or refractory solid tumor (NCT02839954). MUC1-specific CAR-pNK therapy presented good safety profile and preliminary efficacy in preventing both tumor relapses and graft versus host disease [151]. There are numerous ongoing clinical trials exploring the safety and efficacy of NK-based therapy in combination with other modalities. Phase I/II clinical trials are ongoing to evaluate the safety and efficacy of NK cell combined with nimotuzumab to treat late-stage malignancies (NCT03554889) and of NK cell infusion on patients with advanced malignant tumors following multi-line therapies (NCT03619954). Table 6 summarizes updated clinical trials of NK cell-based therapy for all solid tumors.
Conclusions
NK cell represents a specialized immune effector cell population equipped with fast-acting and potent anti-tumor capacity. The concept of adoptive NK cell cancer immunotherapy was proven a decade ago from pioneering clinical studies against hematological malignancies. The difficulty to obtain large quantity of NK cells, to expand to clinical scale ex vivo, and to sustain in vivo survival and activity of infused NK cells has encumbered the progress. With the new tools of iPSC-NK and genetic engineering approach as well as new understandings of NK cell biology, it is the time to re-explore the therapeutic potentials of NK cells. Although challenges exist, perspectives are enticed by currently approved NK cell-based therapies and emerging pre-clinical and clinical studies.
Availability of data and materials
Not applicable.
Abbreviations
- NK:
-
Natural killer
- MHC I:
-
Major histocompatibility complex I
- CAR:
-
Chimeric antigen receptor
- IL:
-
Interleukin
- ADCC:
-
Antibody-dependent cell cytotoxicity
- AML:
-
Acute myeloid leukemia
- iPSC:
-
Induced pluripotent stem cell
- LAK:
-
Lymphokine-activated killer
References
Guerra N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28(4):571–80.
Imai K, et al. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356(9244):1795–9.
Lopez-Soto A, et al. Control of metastasis by NK Cells. Cancer Cell. 2017;32(2):135–54.
Sakamoto N, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. 2015;13:277.
Miller JS, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7.
Rubnitz JE, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28(6):955–9.
Iliopoulou EG, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother. 2010;59(12):1781–9.
Kiessling R, Klein E, Wigzell H. Natural" killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5(2): 112–7.
Lanier LL, et al. Natural killer cells: definition of a cell type rather than a function. J Immunol. 1986;137(9):2735–9.
Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. 1975; 16(2): 216–29.
Smyth MJ, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42(4):501–10.
Roda JM, et al. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66(1):517–26.
Bottcher JP, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018; 172(5): 1022–1037.
Spits H, et al. Innate lymphoid cells: a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–9.
Gao Y, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18(9):1004–15.
Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. 2018;48(6):1104–17.
Freud AG, et al. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47(5):820–33.
Bjorkstrom NK, Ljunggren HG, Michaelsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. 2016;16(5):310–20.
Abel AM, et al. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869.
Hashemi E, Malarkannan S. Tissue-resident NK cells: development, maturation, and clinical relevance. Cancers (Basel), 2020; 12(6).
Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16(2):112–23.
Hammer Q, et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol. 2018;19(5):453–63.
Luevano M, Madrigal A, Saudemont A. Transcription factors involved in the regulation of natural killer cell development and function: an update. Front Immunol. 2012;3:319.
Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. 2019;10:2078.
Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55–61.
Zingoni A, et al. NKG2D and DNAM-1 activating receptors and their ligands in NK-T cell interactions: role in the NK cell-mediated negative regulation of T cell responses. Front Immunol. 2012;3:408.
Parodi M, et al. NKp44-NKp44 ligand interactions in the regulation of natural killer cells and other innate lymphoid cells in humans. Front Immunol. 2019;10:719.
Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. 2019;10:909.
Handgretinger R, Lang P, Andre MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. 2016;127(26):3341–9.
Wang W, et al. NK Cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6:368.
Ruggeri L, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333–9.
Ruggeri L, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100.
Gras Navarro A, et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J Immunol. 2014;193(12):6192–206.
Veluchamy JP, et al. In vivo efficacy of umbilical cord blood stem cell-derived NK cells in the treatment of metastatic colorectal cancer. Front Immunol. 2017;8:87.
Geller MA, et al. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy. 2013;15(10):1297–306.
Lee SJ, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011.
Rezvani K, et al. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–81.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Liu E, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Ni Z, et al. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells. 2014;32(4):1021–31.
Li Y, et al. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181-192.e5.
Li Y, et al. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018; 23(2): 181–192.
Wang J, et al. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136.
Chang YH, et al. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–86.
Shimasaki N, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40.
Xu Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49.
Schnalzger TE. et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J, 2019; 38(12).
Schonfeld K, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8.
Romanski A, et al. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med. 2016;20(7):1287–94.
Zhang Q, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:4263520.
Chen X, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7(19):27764–77.
Oelsner S, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–49.
Chu J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Ng YY, Tay JCK, Wang S. cxcr1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85.
Topfer K, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201–12.
Liu B, et al. Development of cMET-specific chimeric antigen receptor-engineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol Med Rep. 2019;20(3):2823–31.
Huang Y, et al. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Genssler S, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2016;5(4):e1119354.
Muller N, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother. 2015;38(5):197–210.
Zhang C., et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016; 108(5).
Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61.
Oelsner S, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145(7):1935–45.
Muller S, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2019;10:3123.
Klapdor R, et al. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28(10):886–96.
Esser R, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16(3):569–81.
Ueda T, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–90.
You F, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64–78.
Lu C, et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200–6.
Shiozawa M, et al. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018;19(1):27.
Uherek C, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100(4):1265–73.
Zhang Q, et al. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. 2017;2017:6915912.
Hambach, J., et al., Targeting CD38-expressing multiple myeloma and Burkitt lymphoma cells in vitro with nanobody-based chimeric antigen receptors (Nb-CARs). Cells, 2020; 9(2).
Pinz KG, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8(68):112783–96.
Hu Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci Rep. 2020;10(1):2815.
Cao B, et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem Biophys Res Commun. 2020;524(1):96–102.
Han J, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
Parihar R, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. 2019;7(3):363–75.
Zhang P, et al. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy. 2018;10(11):935–49.
Felices M, et al. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol. 2016;1441:333–46.
Vallera DA, et al. IL15 Trispecific Killer Engagers (TriKE) Make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440–50.
Gauthier, L., et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell, 2019; 177(7): 1701–1713.
Bryceson YT, et al. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–66.
Rosario M, et al. The IL-15-based ALT-803 complex enhances FcgammaRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin Cancer Res. 2016;22(3):596–608.
Ni J, et al. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–65.
Romee, R., et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016; 8(357): p. 357ra123.
Leong JW, et al. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transpl. 2014;20(4):463–73.
Cichocki F, et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017;77(20):5664–75.
North J, et al. Tumor-primed human natural killer cells lyse NK-resistant tumor targets: evidence of a two-stage process in resting NK cell activation. J Immunol. 2007;178(1):85–94.
Fehniger TA, et al. A phase 1 trial of CNDO-109-activated natural killer cells in patients with high-risk acute myeloid leukemia. Biol Blood Marrow Transpl. 2018;24(8):1581–9.
Lee J, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res. 2019;7(2):219–29.
Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44(1):46–53.
Allaoui R, et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat Commun. 2016;7:13050.
Kim MJ, et al. CXCL16 positively correlated with M2-macrophage infiltration, enhanced angiogenesis, and poor prognosis in thyroid cancer. Sci Rep. 2019;9(1):13288.
Carlsten M, et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7:105.
Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program. 2013;2013(1):234–46.
Granzin M, et al. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. 2017;8:458.
Chabannon C, et al. Manufacturing natural killer cells as medicinal products. Front Immunol. 2016;7:504.
Ferrara JL, et al. Graft-versus-host disease. Lancet. 2009;373(9674):1550–61.
Skeate R, et al. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion. 2013;53(2):419–23.
Koehl U, et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol. 2013;3:118.
Siegler U, et al. Good manufacturing practice-compliant cell sorting and large-scale expansion of single KIR-positive alloreactive human natural killer cells for multiple infusions to leukemia patients. Cytotherapy. 2010;12(6):750–63.
Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Front Med (Lausanne). 2015;2:93.
Nham T, et al. Ex vivo-expanded NK cells from blood and ascites of ovarian cancer patients are cytotoxic against autologous primary ovarian cancer cells. Cancer Immunol Immunother. 2018;67(4):575–87.
Sarvaria A, et al. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. 2017;8:329.
Spanholtz J, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS ONE. 2011;6(6):e20740.
Kang L, et al. Characterization and ex vivo expansion of human placenta-derived natural killer cells for cancer immunotherapy. Front Immunol. 2013;4:101.
Mu YX, et al. A simple method for in vitro preparation of natural killer cells from cord blood. BMC Biotechnol. 2019;19(1):80.
Galat Y, et al. Cytokine-free directed differentiation of human pluripotent stem cells efficiently produces hemogenic endothelium with lymphoid potential. Stem Cell Res Ther. 2017;8(1):67.
Knorr DA, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–83.
Dege C, et al. Potently cytotoxic natural killer cells initially emerge from erythro-myeloid progenitors during mammalian development. Dev Cell. 2020;53(2):229–39.
Zeng J, et al. Generation of “off-the-shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Reports. 2017;9(6):1796–812.
Galat Y, et al. Application of small molecule CHIR99021 leads to the loss of hemangioblast progenitor and increased hematopoiesis of human pluripotent stem cells. Exp Hematol. 2018;65:38-48.e1.
Ditadi A, Sturgeon CM, Keller G. A view of human haematopoietic development from the Petri dish. Nat Rev Mol Cell Biol. 2017;18(1):56–67.
Berraondo P, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6–15.
Lotze MT, et al. Lysis of fresh and cultured autologous tumor by human lymphocytes cultured in T-cell growth factor. Cancer Res. 1981;41(11 Pt 1):4420–5.
Grimm, E.A., et al., Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med, 1982; 155(6): 1823–41.
McMichael EL, et al. IL-21 enhances natural killer cell response to cetuximab-coated pancreatic tumor cells. Clin Cancer Res. 2017;23(2):489–502.
Li Q, et al. Multiple effects of IL-21 on human NK cells in ex vivo expansion. Immunobiology. 2015;220(7):876–88.
Heinze, A., et al. The synergistic use of IL-15 and IL-21 for the generation of NK cells from CD3/CD19-depleted grafts improves their ex vivo expansion and cytotoxic potential against neuroblastoma: perspective for optimized immunotherapy post haploidentical stem cell transplantation. Front Immunol. 2019; 10(2816).
Choi I, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: a dose-escalation study. Biol Blood Marrow Transpl. 2014;20(5):696–704.
Oberschmidt O, et al. Development of automated separation, expansion, and quality control protocols for clinical-scale manufacturing of primary human NK cells and alpharetroviral chimeric antigen receptor engineering. Hum Gene Ther Methods. 2019;30(3):102–20.
Choi YH, et al. Correction to: IL-27 enhances IL-15/IL-18-mediated activation of human natural killer cells. J Immunother Cancer. 2019;7(1):211.
Tanaka Y, et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE. 2019;14(3):e0212455.
Torelli GF, et al. A good manufacturing practice method to ex vivo expand natural killer cells for clinical use. Blood Transfus. 2015;13(3):464–71.
Fujisaki H, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–7.
Szmania S, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38(1):24–36.
Denman CJ, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE. 2012;7(1):e30264.
Ojo EO, et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9(1):14916.
Shah N, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177(3):457–66.
Ciurea SO, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130(16):1857–68.
Lee DA, Verneris MR, Campana D. Acquisition, preparation, and functional assessment of human NK cells for adoptive immunotherapy. Methods Mol Biol. 2010;651:61–77.
Koehl U, et al. Advances in clinical NK cell studies: donor selection, manufacturing and quality control. Oncoimmunology. 2016;5(4):e1115178.
Phillips JH, Lanier LL. A model for the differentiation of human natural killer cells. Studies on the in vitro activation of Leu-11+ granular lymphocytes with a natural killer-sensitive tumor cell, K562. J Exp Med. 1985; 161(6): 1464–82.
Ishikawa E, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004;24(3b):1861–71.
Lim SA, et al. Ex vivo expansion of highly cytotoxic human NK cells by cocultivation with irradiated tumor cells for adoptive immunotherapy. Cancer Res. 2013;73(8):2598–607.
Granzin M, et al. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology. 2016;5(9):e1219007.
Vasu S, et al. A novel method to expand large numbers of CD56(+) natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy. 2015;17(11):1582–93.
Lister J, et al. Autologous peripheral blood stem cell transplantation and adoptive immunotherapy with activated natural killer cells in the immediate posttransplant period. Clin Cancer Res. 1995;1(6):607–14.
Luhm J, et al. Large-scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res. 2002;11(4):651–7.
Lee DA. Regulatory considerations for NK cells used in human immunotherapy applications. Methods Mol Biol. 2016;1441:347–61.
Oyer JL, et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: clinical implications for cancer treatment. Cytotherapy. 2016;18(5):653–63.
Parkhurst MR, et al. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17(19):6287–97.
Yang Y, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. 2016;4(3):215–24.
Kim EK, et al. Ex vivo activation and expansion of natural killer cells from patients with advanced cancer with feeder cells from healthy volunteers. Cytotherapy. 2013;15(2):231-241.e1.
Huijskens MJ, et al. Ascorbic acid promotes proliferation of natural killer cell populations in culture systems applicable for natural killer cell therapy. Cytotherapy. 2015;17(5):613–20.
Masuyama J, et al. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy. 2016;18(1):80–90.
Lee HR, et al. Expansion of cytotoxic natural killer cells using irradiated autologous peripheral blood mononuclear cells and anti-CD16 antibody. Sci Rep. 2017;7(1):11075.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2020.
Tang X, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Liu E, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Del Zotto G, et al. Markers and function of human NK cells in normal and pathological conditions. Cytometry B Clin Cytom. 2017;92(2):100–14.
Acknowledgements
Not applicable.
Funding
This work was supported by NIH/NCI grant 1R01CA208246, 1R01CA204021, 1R01CA212409, and DOD W81XWH-15-1-0406 (to J. D. Wu).
Author information
Authors and Affiliations
Contributions
Jennifer Wu helped in conception and design. Sizhe Liu, Vasiliy Galat, Yekaterina Galat, Yoo Kyung Annie Lee, Derek Wainwright, and Jennifer Wu wrote and reviewed the manuscript. Jennifer Wu supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors gave consent for publication of the manuscript.
Competing interest
All authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, S., Galat, V., Galat4, Y. et al. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol 14, 7 (2021). https://doi.org/10.1186/s13045-020-01014-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01014-w