
Abstract
Background
Although mutated HLA ligands are considered ideal cancer-specific immunotherapy targets, evidence for their presentation is lacking in hepatocellular carcinomas (HCCs). Employing a unique multi-omics approach comprising a neoepitope identification pipeline, we assessed exome-derived mutations naturally presented as HLA class I ligands in HCCs.
Methods
In-depth multi-omics analyses included whole exome and transcriptome sequencing to define individual patient-specific search spaces of neoepitope candidates. Evidence for the natural presentation of mutated HLA ligands was investigated through an in silico pipeline integrating proteome and HLA ligandome profiling data.
Results
The approach was successfully validated in a state-of-the-art dataset from malignant melanoma, and despite multi-omics evidence for somatic mutations, mutated naturally presented HLA ligands remained elusive in HCCs. An analysis of extensive cancer datasets confirmed fundamental differences of tumor mutational burden in HCC and malignant melanoma, challenging the notion that exome-derived mutations contribute relevantly to the expectable neoepitope pool in malignancies with only few mutations.
Conclusions
This study suggests that exome-derived mutated HLA ligands appear to be rarely presented in HCCs, inter alia resulting from a low mutational burden as compared to other malignancies such as malignant melanoma. Our results therefore demand widening the target scope for personalized immunotherapy beyond this limited range of mutated neoepitopes, particularly for malignancies with similar or lower mutational burden.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is among the malignancies with the highest death toll on a global scale [1] and with very limited therapeutic options. Particularly in advanced stage, long-term survival is uncommon [2]. Although it has been shown that the microenvironment of the liver is tolerogenic and impairs immune responses [3], antigen-specific T cell responses do occur [4]. Since infiltration of HCCs with T cells [5] and spontaneous immune responses correlate with longer survival [6] but mostly prove weak and insufficient on their own, immunotherapies unleashing the immune system hold great promise.
Immune checkpoint (ICP) inhibitors demonstrating the potency and effectiveness of the immune system to fight malignancy [7] have set the stage for cancer immunotherapies. In contrast to established cytostatic treatments for cancer, this new class of drugs has enabled long-term survival in advanced and metastatic disease previously considered incurable [8]. However, although in some malignancies ICP inhibitors have proven highly effective, results for other cancers remain disappointing. One probable mode of action for ICP inhibitors is the induction and/or restoration of T cell effector functions against individual somatic tumor mutations presented by HLA molecules (i.e., mutated neoepitopes) [9]. Since these mutated HLA ligands were unacquainted to the immune system before carcinogenesis, they have been proposed as ideal tumor-specific targets [10, 11].
In malignant melanoma (Mel), where ICP inhibitors were established first, mutational load was shown to strongly correlate with survival [12]. This has been corroborated in lung cancer [13] and colorectal carcinoma, where in the latter impressive survival benefits remained strictly limited to mismatch repair-deficient carcinomas featuring very high numbers of mutations [14]. As elevated somatic mutation rates raise the odds for generating neoantigens, this supports the notion they may be critical for ICP inhibitor effectiveness [15]. Another line of evidence suggests that neoantigens recognized by T cells can generate impressive clinical effects, when identified and exploited for therapeutic purposes. This has been shown in remarkable case reports inter alia in advanced Mel [16] and metastatic cholangiocarcinoma [17].
With current affordable next-generation sequencing (NGS) and bioinformatics, an array of approaches predicting HLA-restricted neoantigens from virtually any tumor has emerged [18,19,20]. Indeed, at present most attempts are restricted to in silico analyses, lacking actual proof that the predicted neoantigens are relevant or even exist. So far, tangible evidence is scarce and mainly restricted to T cell recognition [21]. Therefore, one frequently missing link is proof of neoantigen presentation on native tumor tissue. Such an endeavor is very challenging and has been shown feasible in mouse models [22] and cell lines [23] but in human solid tumors hitherto merely in Mel at low numbers using mass spectrometry (MS), defining the current state-of-the-art [24, 25].
Since both individual cancer traits and mutational load vary strongly between different tumor entities [26, 27], these properties may ultimately restrict the foreseeable success and feasibility of neoantigen-targeted precision cancer medicine. In HCCs, only a small proportion of about 10% of patients showed mutations potentially accessible for drug therapy [28], whereas preliminary data for ICP inhibitors showed objective response rates in 15–20% of patients combined with a manageable safety profile [29], making neoantigens in principle an interesting case for precision cancer medicine and the use of NGS.
Hence, we performed unprecedented in-depth multi-omics analyses encompassing whole exome and transcriptome sequencing, combined with proteome and HLA ligandome profiling in selected HCC patients aiming to obtain evidence for the natural presentation of exome-derived mutated HLA ligands, employing various strategies.
Materials and methods
Clinical specimens
Clinical specimens from patients (n = 16; median age: 74 years; min.–max. 55–85 years; 75% men) undergoing liver resection for hepatocellular carcinomas (HCCs), encompassing both non-malignant and malignant liver tissue as well as peripheral blood, were obtained directly after surgery and cryopreserved (for patients’ tumor characteristics, see Additional file 1: Table S1). HCC diagnosis and predominant tumor fraction within samples were histologically confirmed by an expert pathologist. All included patients were negative for chronic viral hepatitis (hepatitis B and C) and without systemic pretreatment for their malignancy.
Next-generation sequencing
DNA and RNA were extracted from fresh frozen tissue and PBMCs, respectively (a sample and analysis overview is provided in Additional file 1: Table S2). After sample preparation and enrichment, paired-end whole exome sequencing (WES) and whole transcriptome sequencing were performed on an Illumina system (details are provided in Additional file 2).
HLA typing
Typing at four-digit resolution using WES data was performed by OptiType [30] for HLA class I alleles (see Additional file 1: Table S3) as previously described [31] and confirmed in selected cases by molecular HLA typing (using clinically validated LUMINEX and sequence-based typing) during clinical routines.
Isolation of naturally presented HLA ligands from tissues for HLA ligandomics
HLA class I-peptide complexes were isolated from HCC and corresponding (non-malignant) liver tissue samples by immunoaffinity purification using the pan-HLA class I-specific monoclonal antibody W6/32 [32] (produced in-house at the Department of Immunology, Tübingen, Germany) and eluted using 0.2% trifluoroacetic acid as described previously [33].
Analysis of HLA ligands by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS)
HLA class I ligand extracts were measured once or in multiple technical replicates, as described previously [33, 34]. Samples were separated by UHPLC and eluting peptides were analyzed using collision-induced dissociation (CID) in an online coupled Orbitrap mass spectrometer. In addition to data-dependent acquisition (DDA), selected ion monitoring (SIM) and parallel reaction monitoring (PRM) targeted tandem MS (tMS2) was performed for selected samples to enhance the sensitivity and robustness of neoantigenic peptide identification (details are provided in Additional file 2).
HLA ligandomics data analysis
MS data obtained from HLA immunoprecipitates was analyzed using tools of the open-source software library for LC/MS OpenMS (2.3) [35]. Identification and post-scoring were performed using the OpenMS adapters to Comet 2016.01 rev. 3 [36] and Percolator 3.1.1 [37] at a peptide spectrum match (PSM) false discovery rate (FDR) threshold of 5%. Database search was performed against a personalized version of the human reference proteome (Swiss-Prot, reviewed UP000005640), including the patient-specific mutanome without enzymatic restriction and methionine oxidation as the only variable modification.
Database matching
HLA ligandome database queries refer to the in-house database (maintained at the Department of Immunology) encompassing > 300,000 unique HLA class I-eluted peptides identified through LC-MS/MS in diverse tissues (non-malignant samples as well as samples with pathologies including malignancies). Database matching was carried out using rSQL, querying for an exact string match of a wild-type ligand (WTlig) corresponding to the respective predicted mutated neoepitope (PNE). All HLA class I allotypes of the HCC and Mel cohort were covered by respective samples in the database. Each sample containing the WTlig was counted as a separate match (further details are provided in Additional file 2). Besides neoepitopes, we additionally screened our HCC HLA class I ligandome dataset against cancer-testis antigens (CTAs) as deposited in the CTDatabase (http://www.cta.lncc.br; [38]).
Protein in-gel digestion for shotgun protein identification
Sample lysates were separated by SDS-PAGE. Coomassie-stained gel pieces were digested using trypsin. Peptides were desalted using C18 Stage tips and subjected to LC-MS/MS analysis.
Shotgun protein tandem mass spectrometry
Liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) analyses were performed on an EasyLC nano-HPLC system (Proxeon Biosystems, Roskilde, Denmark) coupled to an LTQ Orbitrap Elite mass spectrometer (ThermoFisher) (additional details are provided in Additional file 2).
Proteomic data analysis
MS data were processed with MaxQuant software suite v.1.5.2.8 [39]. Database search was performed using the Andromeda search engine [40], integrated into the MaxQuant framework. The human reference database was obtained from UniProt (taxonomy ID 9606, containing 91,646 protein entries and 285 commonly occurring laboratory contaminants) and concatenated with the patient-specific mutanome. Endoprotease trypsin was fixed as enzyme with a maximum of two missed cleavages. Oxidation of methionines and N-terminal acetylation were specified as variable modifications, whereas carbamidomethylation of cysteines was defined as a fixed modification. Initial maximum allowed mass tolerance was set to 6 ppm. Re-quantify was enabled. A FDR of 1% was applied at peptide and protein level.
Bioinformatics
Data management and bioinformatic analysis was performed through the qPortal instance at the Quantitative Biology Center (QBiC), Tübingen, if not stated otherwise [41].
Variant calling
Reads were processed using the megSAP pipeline (https://github.com/imgag/megSAP) and the ngs-bits package (https://github.com/imgag/ngs-bits) by the Department of Medical Genetics and Applied Genomics (Tübingen, Germany). Reads were mapped against the Genome Reference Consortium Human Build 37 (GRCh37) using BWA-mem [42]. Somatic variant calling was performed using Strelka and Strelka2 [43, 44] or with a proprietary software (CeGaT GmbH, Tübingen, Germany). Somatic mutations were annotated using SnpEff 4.1 k [45]. Further details are provided in Additional file 2.
Gene expression analysis
Gene expression values were calculated as fragments per kilobase of exon per million reads mapped (FPKM) of the corresponding transcripts and RNA tumor sequencing depth at the corresponding variant position. Mapping of RNA reads was done using TopHat 2 (v2.0.12) [46]. Details are provided in Additional file 2.
Protein quantification analysis of shotgun proteomics data
Label-free protein quantification was done using MaxQuant v1.5.00 [39]. Parameter groups were defined for non-malignant tissue- and tumor-derived raw files, respectively. The multiplicity was set to one. Protein N-terminal acetylation as well as oxidation of methionine residues were selected as variable modifications, whereas carbamidomethylation of cysteine residues was set as fixed modification. Trypsin was selected as protease with a specific digestion mode. Further, we specified the match type as MatchFromAndTo and set the number of MaxMissedCleavages to two. Requantification and matching between runs were enabled. As a reference, we specified the Swiss-Prot reviewed human proteome (version UP000005640, derived: 02/16/2016).
Peptide prediction
To define the sample-specific mutated peptide search space (PSS), peptides of 8–11 amino acid length were constructed by sliding a shifting window of the peptide length over the affected mutated positions. Resulting peptides were filtered against the human proteome (UniProt UP000005640, derived: 02/29/16) and the Ensembl proteome reference (release 84, 04/27/2016) to exclude peptides contained within wild-type proteins. Transcript information was retrieved via BioMart, based on the stable database version of GRCh37 (http://feb2014.archive.ensembl.org). HLA-binding prediction was performed with SYFPEITHI [47], netMHC 4.0 [48, 49], and netMHCpan 3.0 [50, 51]. The workflow was implemented using FRED2 [52] (see Additional file 2 for further details).
Differential gene expression analysis and pathway analysis
Differential gene expression analysis was performed using the R package DESeq2 [53]. Expression data of HCC datasets from TCGA were retrieved and analyzed with the recount2 package [54].
Pathway analysis was carried out using clusterProfiler [55] and Pathview [56]. Differentially expressed genes were categorized using DAVID [57]. Details are provided in Additional file 2.
Results
A multi-omics approach to detect mutated HLA ligands in HCCs
We performed analyses of malignant and non-malignant liver tissue, resected during surgery for HCCs (Additional file 1: Table S1 & Table S2), by a multi-omics approach encompassing analyses on exome (n = 16), transcriptome (n = 16), shotgun proteome (n = 7), and HLA ligandome level (i.e., HLA-presented peptides; n = 16). Multi-allelic HLA class I expression was confirmed in all patients of our HCC cohort (results are provided in Additional file 1: Table S3). The overall aim of our research was to identify individual exome-derived somatic tumor mutations resulting in natural HLA ligands presented to T cells.
Detection of somatic variants (mutations) in HCCs
On average, we detected 151 ± 40 somatic variants (Var) per HCC, including single nucleotide variants, small insertions/deletions, and frameshift variants; thereof, 44% (66 ± 19) cause changes in the amino acid sequence of the encoded protein (i.e., non-synonymous variants; Varns - a glossary of abbreviations and terminology used is provided as Table 1), when referenced against DNA from blood. From these Varns, on average about half were also detectable on transcript level (44 ± 10%; Fig. 1a). Across all patients, we observed 1039 unique Varns in total, affecting 864 different genes and 45% of them (n = 392) with additional evidence on RNA level (Varexp). This translates to an average tumor mutational burden (TMB; estimated as previously described [58]) of 1.89 ± 0.49 per megabase observed in our HCC cohort (see Additional file 1: Table S4).

Characterization of somatic variants and their potential for HLA presentation in HCC. a Numbers of somatic variants across HCC patients (n = 16). Numbers are shown for all variants passing initial filtering (Var), coding non-synonymous variants (Varns), and coding non-synonymous variants with RNA level evidence (Varexp). Boxplots show means ± SD. b Varexp shared among HCC patients. Varexp affecting identical genes in ≥ 3 patients are displayed in gray. Varexp observed at identical genomic positions are displayed in red (the shown HLA-DR variants should be cautiously interpreted as potential artifacts). c Correlation between Varns and predicted HLA-binding neoepitopes (PNE) (left; blue). Correlation between Varexp and expressed PNE (PNEexp) (right; orange). d Scatter plot of numbers of Varns and PNE in HCC patients (blue) and a benchmarking dataset of melanoma (Mel) patients (red) as previously described by Bassani-Sternberg et al. [24]
Assessing mutational hotspots, we observed alterations (Varexp) in β-catenin (CTNNB1; 50%) and in neuroblastoma breakpoint family, member 1 (NBPF1; 38%), but also in genes encoding proteins typically expressed in the liver, such as albumin (ALB; 19%), apolipoprotein b (APOB; 13%), and γ-glutamyltransferase (GGT1; 19%) (Fig. 1b). Varexp frequently affected the HLA class II loci HLA-DRB1 (6%), HLA-DQA1 (13%), and HLA-DRB5 (19%). However, due to the highly polymorphic nature of the HLA locus [59], variant detection in these regions is particularly error-prone and results should be cautiously interpreted as potential artifacts. For HLA class I loci, suitable computational pipelines for mutation detection are available [60], whereas for HLA class II to the best of our knowledge this is not the case. Overall, only 1.5% (6/392) of Varexp-containing genes were shared among > 2 patients and only one single mutation (in NBPF1; Chr. 1:16891365 G>T) reoccurred identically in three patients. Considering established driver mutations included in the Cancer Gene Census ( [61]; https://cancer.sanger.ac.uk/census), we observed respective Varns in most of the HCCs (n = 9; 1–3 Varns per patient), foremost the previously mentioned gene CTNNB1 (n = 8) but also the androgen receptor, mediator complex subunit 12 (MED12), nuclear receptor corepressor 1 (NCOR1), neurogenic locus notch homolog protein 1 (NOTCH1) (all n = 2), and NOTCH2/PIK3CA (n = 1). Nevertheless, except from CTNNB1, Varns comprised in the Cancer Gene Census appeared rather infrequently among the examined HCCs.
Discovery of mutation-derived HLA ligands on different omics levels
Exome
In a first step, we sought to assess the number of neoepitopes (PNE) per patient predicted to bind to each individual set of HLA class I alleles, using established binding predictions. On average, 244 ± 77 PNE per HCC patient were predicted from 66 ± 19 Varns, exceeding the respective binding thresholds (Fig. 1c; left panel). The observed increase in PNE numbers compared to Varns is explained by the fact that Varns may give rise to multiple PNE due to the shifting window approach used with different peptide lengths (8–11 amino acids) as well as the HLA-binding prediction for up to six individual HLA alleles. Comparing the numbers of PNE to the numbers of protein-altering variants (Varns), this resulted in a very weak correlation (Pearson’s correlation coefficient r = 0.38).
Transcriptome
When accounting for supplemental evidence for PNE on RNA level, numbers of predicted peptides (PNEexp) decreased by half (49 ± 8% of PNE), yielding an average of 118 ± 40 PNEexp per patient. The correlation between expressed protein-changing genomic variants (Varexp) and PNEexp also remained moderate (Pearson’s correlation coefficient r = 0.50) (Fig. 1c; right panel).
Proteome
In order to gain additional protein level evidence for PNEexp, we annotated all PNE with log2-intensities from shotgun proteome data (n = 7) of HCCs. In this way for a total of 159 PNE (17 ± 14% of PNEexp), supportive protein level data was available (on average for 23 ± 21 PNE per patient), mapping to various source proteins (see Additional file 1: Table S5). Only in one patient, no evidence for PNE corresponding to any detected source protein was found (HCC034), yet on average a fraction of 10% (9.8 ± 8.6%) of PNE were supported by additional evidence for occurrence of their source proteins (n = 33) on shotgun proteome level.
HLA ligandome
To directly assess the presence of mutated HLA ligands, we used the well-established technique of UHPLC-coupled MS/MS to identify naturally presented HLA ligands from HCCs and non-malignant liver tissues. These analyses yielded on average 1403 ± 621 HLA class I-associated peptides from HCC and 1159 ± 525 peptides from non-malignant liver tissue (FDR 5%, length 8–11 amino acids; see also Additional file 3: Figure S1). On average, 51 ± 11% of these peptides were shared between matching malignant and non-malignant liver tissue. When predicting HLA class I binding affinities and filtering for MS-detected peptides exceeding the respective binding threshold for the patients’ HLA allotypes (see Additional file 1: Table S3), on average 1026 ± 451 peptides per tumor (73 ± 10%) and 867 ± 450 peptides per non-malignant liver sample (72% ± 11%) showed HLA-binding properties. This filtering step was performed to enrich for high probability HLA class I ligands, excluding contaminant peptides from downstream analyses. On average, 58 ± 12% of those peptides occurred both in matched malignant and non-malignant liver tissues.
Importantly, we did not find any evidence for naturally presented mutated HLA ligands (NElig) in HCCs, independent of filtering criteria. However, in two HCC patients, we identified one wild-type sequence HLA ligand (WTlig) each, corresponding to a PNE.
Benchmarking HCC and melanoma (Mel) HLA ligandomics datasets
To demonstrate the high sensitivity of our neoepitope identification pipeline, we additionally processed a publicly available dataset of somatic variants from five Mel patients as a reference [24]. The numbers of Varns and PNE in Mel (Fig. 1d; red dots) showed remarkable differences from our HCC dataset (blue dots). Whereas in two cases, Mel samples showed comparable properties to the HCCs analyzed with respect to the numbers of Varns and resulting PNE (Mel8, Mel16), these counts were substantially higher in the majority of Mel samples (Mel5, Mel12, Mel15). This resulted in an average number of 531 Varns in Mel in comparison to only 66 Varns in HCC, corresponding to an eightfold increased mutated peptide search space (PSS) in Mel. Derived predicted neoepitopes amount to an average of 243 PNE in HCC in contrast to 1550 PNE in the Mel data (Fig. 2a), resulting from a tenfold increased TMB in Mel (on average: 19.06 ± 13.97 per megabase; see Additional file 1: Table S4).

Numbers of predicted neoepitopes with evidence on different omics levels. a Numbers of somatic variants and non-synonymous somatic variants (Var and Varns), respectively; peptide search space (PSS), predicted HLA-binding neoepitopes (PNE), and PNE on the different available omics levels: expressed PNE (PNEexp), PNE with evidence on shotgun proteome level (PNEprot), and neoepitopes observed as natural HLA ligands (NElig) are shown for the HCC dataset (left; n = 16) and the Mel dataset (right; n = 5) published previously by Bassani-Sternberg et al. [24]. Numbers are given as mean ± SD. b Numbers of peptides after processing with our neoepitope identification pipeline are shown on a per patient basis according to the different omics levels as observed in the HCC dataset (left) as well as the Mel dataset (right). For each patient, total counts of predicted peptides (PSS) are annotated in black, numbers of NElig for Mel patients are shown in red (median = 1.0)
On a per patient basis (Fig. 2b), the HCC dataset proved much more homogenous (PSS: ~ 2500 to 10,000; PNE: 111 to 382) than the Mel data, where the PSS ranged from 4000 to 84,000 (PNE: 169 to 3717).
This was corroborated by analyzing datasets from The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) for both entities, showing a mean number (± SD) of Varns of 90 ± 100 for HCC (n = 363) and 461 ± 761 for Mel (n = 467) (Additional file 3: Figure S2). Assessing only the fraction of tumors with > 100 Varns as suggested previously [12], this amounted to a share of 26% in HCC vs. 77% in Mel. Selecting the share of malignancies that exhibits a high TMB as defined by Goodman et al., we observed merely 1.5% of high TMB tumors among HCC vs. 32% among Mel [62] (Fig. 3).

Comparison of the mutational burden in Mel and HCC. a Number of mutations (# Varns) of TCGA cases in Mel (n = 476). b Number of mutations (# Varns) of TCGA cases in HCC (n = 363). The data were retrieved from Genomics Data Commons Data Portal (https://portal.gdc.cancer.gov/, access date: 2018-09-16). Variants were filtered for missense variants, frameshift variants, inframe deletions, inframe insertions, and coding sequence variants. Variants that were called by Mutect2 are considered. The number of mutations was assessed with respect to high tumor mutational burden (> 400 Varns, red) and the fraction of tumors with > 100 Varns (blue)
Employing our HLA ligandomics identification pipeline, we were able to reconfirm all of the NElig that had been discovered previously by Bassani-Sternberg et al. in their MS dataset (Mel5 (n = 2); Mel8 (n = 1); Mel15 (n = 8)) [24, 63]. Furthermore, we discovered one additional NElig for Mel12 and three additional NElig for Mel15 that could be validated by matching spectra from synthetic peptides (see Additional file 1: Table S6). Only one of those NElig was discovered in a sample (Mel8) with properties comparable to our HCC cohort. Importantly, all other NElig identified on MS level (10/11) were identified on Mel with high TMB (Fig. 2b). Therefore, it can be stated that our comparatively homogenous HCC cohort, for which no NElig could be discovered, differs substantially (by at least one order of magnitude concerning TMB) from the properties of Mel patients previously published [24]. This notion is supported by a thorough comparison of both datasets as shown in Fig. 2, as well as by comparisons with comprehensive TCGA datasets (Fig. 3 and Additional file 3: Figure S2).
Evidence for mutated proteins on shotgun proteome level
To obtain the best available evidence for the presence of mutated proteins, in absence of tangible data on HLA ligandome level (NElig), we employed shotgun proteomics in HCC tissue samples. To this end, we used a tryptic digest of cell lysates, aware that detection of respective variants is difficult and technology-related sensitivity limitations apply [64] that are governed by a variety of influencing factors and the fraction of genomic alterations detectable on protein level by this approach was reported to be about 2% [65]. Indeed, we discovered one somatic mutation in albumin (ALBK375E) on proteome level represented by the tryptic peptide LAETYETTLEK in HCC025 (Fig. 4a), which was corroborated on both exome (Varns) and transcriptome (Varexp) levels. Strikingly, we not only detected the tryptic wild-type peptide LAKTYETTLEK but unexpectedly also the mutation-derived peptide LAETYETTLEK in the proteome of non-malignant liver tissue. To investigate the source of this unexpected finding, we obtained two additional serum samples at different time points from the patient and performed shotgun proteomics on them. Patient HCC025 showed tumor recurrence and active disease at both time points and the mutated peptide was detected in both samples, proving that the tumor synthesized a mutated ALB protein secreted into circulation. For HCC026, a Varexp in the ATP-dependent DNA helicase Q1 (RECQL; H19R) could be verified based on an additional tryptic cleavage site introduced through the arginine gained by mutation, which resulted in the proteotypic peptide AVEIQIQELTER. This peptide was not detected in the corresponding non-malignant liver tissue (Fig. 4b).

Evidence for mutated proteins in the shotgun proteome and database matching. a Annotated spectra of albumin (ALB) showing sequences of wild-type (LAKTYETTLEK; top) and mutated (LAETYETTLEK; bottom) protein measured by LC-MS/MS. b Annotated spectra of RecQ like helicase (RECQL) showing sequences of the peptide AVEIQIQELTER resulting from an additional tryptic cleavage side added directly in front of this sequence through a mutation from histidine to arginine, evidenced in HCC tissue only. c Database matching of natural HLA ligands with wild-type peptide sequence (with diverse HLA restrictions) covering the exact position evidenced as mutated in ALB. d Database matching of natural HLA ligands with wild-type peptide sequence (with diverse HLA restrictions) covering the exact position evidenced as mutated in RECQL
Targeted mass spectrometry for discovery of mutated HLA ligands
As NElig could not be confirmed in HLA ligandomics data of HCC obtained by data-dependent acquisition mode tandem mass spectrometry (DDA-MS/MS), we adopted measures to avoid limitations by missing values and semi-random sampling inherent to this approach [66]. Thus, we attempted to corroborate the PNEprot observed in ALB and RECQL by targeted MS approaches as well as other carefully selected PNEexp in three chosen patients. We selected sets of PNE from three HCCs (HCC025–27) for a selected ion monitoring (SIM) approach using heavy isotope-labeled peptides as a reference to increase the sensitivity for the MS/MS method and improve the probability of detection (Additional file 1: Table S7). Nevertheless, we could not validate any of the candidates and comparisons of low confidence annotations with synthetic peptides did not yield evidence for peptide presentation.
Since peptides harboring the mutations confirmed by proteomics (PNEprot) seemed of particular interest (i.e., ALBK375E in HCC025 and RECQLH19R in HCC026), we additionally performed parallel reaction monitoring (PRM) targeted tandem MS (tMS2) measurements targeting the best ranking PNE as well as corresponding wild-type HLA ligands (WTlig), covering the mutation site (for details, see Additional file 2). Despite a high number of HLA class I peptides in DDA-MS/MS (HCC025 malignant: 5063; HCC025 non-malignant: 1497; HCC026 malignant: 3678; >HCC026 non-malignant: 3197), PRM tMS2 could not corroborate any of the PNEprot (Fig. 4) as naturally presented HLA ligands in HCC (Additional file 1: Table S8 & Table S9).
Prioritizing predicted mutated HLA ligands in absence of HLA ligandome evidence
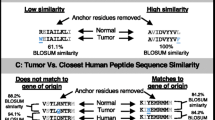
Lacking detection of mutated HLA ligands (NElig) does not equal their absence due to several reasons: inter alia (1) detection limits of the LC-MS/MS instrumentation, (2) lacking ionizability of respective peptides, (3) particularly strongly hydrophilic and hydrophobic peptides may be missed by the UHPLC method, (4) unknown temporal dynamics of the HLA ligandome [67]. As one way for PNE prioritization, we propose a knowledge-based approach using previously measured wild-type HLA ligands (WTlig). Hence, we assumed that the more frequently a WTlig was already detected as a natural HLA ligand by MS the more likely its corresponding NElig counterpart should exist, provided that the mutation does not negatively impact its HLA-binding affinity, or the respective HLA allele was lost. To this end, we compared the number of database matches of all WTlig in HCC and Mel to an in-house database of HLA ligands measured over the last decades (Fig. 5, Additional file 1: Table S10 & Table S11). Almost all of the malignancies assessed carry at least one mutation (Varns) that could potentially give rise to a PNE whose corresponding WTlig was previously measured multiple times as an HLA-eluted ligand by MS. Interestingly, four of the 15 MS-detected NElig in Mel support this approach, since also their corresponding WTlig produced multiple hits in our database, including GA-binding protein alpha chain (GABPA; 20 matches), synaptotagmin like 4 (SYTL4; 8 matches), nucleoporin 153 (NUP153; 2 matches), and outstandingly septin 2 (SEPT2; 298 matches). Moreover, the two pinpointed PNEprot in ALB (59 matches) and RECQL (17 matches) give rise to the two most frequently contained WTlig of the respective patients in our database. In addition, WTlig tensin 1/3 (TENS1/3; 54 matches; HCC027) and SPECC1L-ADORA (33 matches; HCC028) were detectable in the respective tumor HLA immunoprecipitates, proving that at least the wild-type sequence peptide is processed and presented on HLA. Speculatively, this might favor the presentation of their NElig counterparts on HLA ligandome level, although it could not be detected by us. The four mentioned WTlig from HCC (ALB; RECQL; TENS1/3; SPECC1L-ADORA) and two from Mel (SEPT2; SYTL4) have also been documented in the immune epitope database (https://www.iedb.org/ access date: September 2018), which might also guide the way. Ultimately, these results may question HLA ligandome level detection depth and call for establishing large community-based HLA peptidomics databases [67, 68], since individual attempts do not seem reasonable, given the extent of this task.

Number of database matches of wild-type ligands (WTlig) corresponding to predicted mutated neoepitopes (PNE). PNE with additional evidence in HCC and Mel [24] are highlighted: (1) black: wild-type sequence of PNE contained in database; (2) yellow: wild-type sequence peptide corresponding to PNE confirmed in autologous tissue as natural HLA ligand by mass spectrometry; (3) blue: mutated protein confirmed by shotgun proteomics - PNEprot; (4) red: PNE confirmed as natural HLA ligand by mass spectrometry - NElig
Narrowing the scope on alternative (immunological) targets
As alternative targets among HLA ligands with potential therapeutic relevance, we screened our HCC dataset for proteins previously described as cancer-testis antigens (CTA) and found eight different HLA class I ligands mapping to six CTA. These few CTA encompass ARMC3 (Q5W041), ATAD2 (Q6PL18), MAEL (Q96JY0), PRAME (P78395), proteins of the SSX family, and TFDP3 (Q5H9I0) (Table 2).
Further, we identified a limited number of CTA among different patients on shotgun proteome level (Additional file 1: Table S12).
Additionally, gene expression analysis revealed 213 differentially expressed (DE) genes, resulting from comparison of autologous tumor and non-malignant tissues. All but one DE gene showed downregulation when compared to matching non-malignant liver (Additional file 3: Figure S3). Respective results indicate apparent differences in the underlying gene expression patterns of tumor and non-malignant liver samples, corroborating the separation of the tumor and non-malignant liver samples in the PCA (on principal component one level; Additional file 3: Figure S4). Visualization by heatmaps and accompanying dendrograms clearly support these observations and show that tumor and non-malignant tissue samples mostly separate in two distinct clades. This separation is seen in most tumor samples except for six patients (HCC024/ 028/ 034/ 035/ 043/ 045), which rather group with the non-malignant tissue samples on the heatmap. To benchmark results from DE expression analysis to publicly available RNA-Seq datasets of HCC, we used recount2, a multi-experiment resource of analysis-ready RNA-Seq datasets with the R package recount. We performed a simple pairwise comparison of the TCGA dataset between non-malignant (n = 50) and tumor (n = 374) samples using DESeq2. We identified 6044 genes that were DE, based on a statistical significance that was determined by a multiple-testing adjusted p value < 0.05 and log2 fold-change > 1 or < − 1. From the 213 DE genes observed in our HCC cohort, about half (n = 105) were also found differentially regulated in the TCGA dataset.
Gene functional classification analysis (using DAVID [69, 70]) pointed to mono-oxygenases (CYP450 enzymes) as most prominently inhibited class among DE genes. This gene list was mapped to unique Entrez IDs (n = 115), which were mapped to 14 significantly enriched pathways in return (Additional file 1: Table S13).
Finally, we assessed mutations evidenced in our HCC cohort on transcriptome level (Varexp) regarding their potential druggability. As previously published [28], also in our HCC patient cohort, mutations druggable by approved pharmaceuticals were missing. Instead, we found one mutation (PIK3CAE542K; HCC041; https://www.mycancergenome.org/content/disease/lung-cancer/pik3ca/7/) that has been implicated with lacking drug response to anti-epidermal growth factor (EGFR) antibodies [71].
Discussion
Neoepitopes, i.e., unique peptides derived from tumor-specific mutations presented as natural HLA ligands and recognized by T cells, have been suggested as highly attractive targets for cancer immunotherapy. It is undisputable that there is mounting (indirect) evidence to suggest that increased numbers of mutations may render malignancies more immunogenic through their neoantigenic repertoire (i.e., mutated HLA ligands) and ultimately more amenable to immunotherapies [9]. Particularly for tumors that are characterized by a high tumor mutational burden (TMB), a correlation with benefits of ICP inhibition has been shown [12,13,14, 62].
One of the greatest challenges in understanding and ultimately harnessing this neoantigenic repertoire of cancers is the selection and validation of suitable targets from an array of predicted neoepitopes (PNE) derived from computational algorithms [72]. In this connection, it is very plausible to assume that most PNE are irrelevant and would ultimately fail to make an impact on treatment outcomes of individual patients. On the other hand, the selection of a single suitable neoepitope may have unprecedented therapeutic consequences [17, 73] and such a single neoepitope has already been shown to be a target of T cells induced by ICP inhibition [22]. Certainly, this notion is not limited to neoepitopes, but it also applies to tumor-associated antigens, which can possess a comparable immunogenicity [74]. Consequently, non-mutated tumor-specific or highly tumor-associated antigens should be considered prime choice for personalized immunotherapy, when they can be individually validated [75]. Although many assumptions regarding mutated neoepitopes are theoretically and bio-mechanistically plausible [15], there is a fundamental lack of knowledge concerning the precise immunological underpinnings behind tumor specificity [76] and therapeutic implications.
Moreover, biomarkers predicting response to ICP inhibitors with higher precision than TMB [62] are sought-after [77]. A respective biomarker might not only assess the odds for ICP therapy success but may simultaneously allow the development of tailored neoantigen-targeted immunotherapies.
In contrast to the vast array of data available relating to PNE [78, 79], often derived from data of consortia like the International Cancer Genome Consortium (ICGC) or TCGA, current physical evidence for exome-derived mutated HLA ligands (NElig) seems anecdotal (reviewed in [63]) and positive examples for finding this proverbial needle in the haystack are scarce. Hence, to be able to benchmark our results obtained in HCC, we used the best evidence available to us, provided by a dataset from Bassani-Sternberg et al. [24]. Even though this dataset from malignant melanoma (Mel) differs fundamentally from HCCs in a variety of aspects, including inter alia a tenfold increased average TMB and a sixfold higher PNE count, this approach enabled benchmarking our pipeline against a dataset containing the required targets (NElig). This notion was also confirmed on a larger scale by TCGA data, corroborating that the average mutation numbers were typically fivefold increased in Mel vs. HCC and the proportion of tumors with high TMB (< 100 Varns) was elevated from 1.5% in HCC to 32% in Mel.
Our HCC dataset is characterized by close to 70 amino acid-changing mutations (Varns) on average translating to a TMB of about two per megabase, numbers corresponding very well with data from a comprehensive set of resectable HCCs [80]. These mutations encompass established hotspots, and a limited number of genes was found to be recurrently mutated [80], affecting the well-established CTNNB1 primarily but also NBPF1. The latter remained the only gene with a repeat identical mutation in our patient cohort, emphasizing that in combination with an individual set of HLA class I allotypes, a neoepitope-targeted therapy needs to be strictly personalized [76]. Since in HCCs only about half of the initially 244 Varns could be corroborated by RNA level evidence (Varexp), this bisected the computationally predicted neoepitope numbers to an average of 118 expressed PNE (PNEexp). Further, the correlation of both PNE and PNEexp numbers with mutation counts, showed only a weak correlation. This may imply that there is no direct interconnection between mutation frequency and respective HLA ligands but rather a probabilistic model applies [22], which is governed by the HLA ligandome with distinct rules of presentation [81]. Since we had shotgun proteomics data available, we also assessed whether we could establish any additional physical evidence for the respective source proteins (PNEprot) constituting the immediate proteomic context of NElig, which was the case in about one fifth of PNEexp and comprised about 10% of the initial PNE pool. Nevertheless, since this neither implies the actual detection of a mutation in the proteomics dataset (only the identification of at least one tryptic peptide matching the respective protein), nor the HLA presentation of a NElig, we assessed the eluted HLA ligands and searched for any PNE with actual evidence for HLA presentation by LC-MS/MS. Although the ~ 1400 HLA-bound peptides detected on average in HCCs are generally comparable with the numbers previously published in solid cancers [34, 82], they do fall short of the considerable depth reached in Mel, particularly in one single exceptional case, for which more than 20,000 HLA-bound peptides were reported (Mel15; [24]). Since this Mel dataset was available to us and could be processed by our pipeline, we can prove that we would be able to discover NElig when MS/MS spectra are acquired. In this way, we corroborated all NElig previously reported [24], as well as four additional NElig previously unidentified, validated by matching spectra from synthetic peptides. However, in this direct comparison, it becomes particularly clear that Mel and HCC, despite both representing solid tumors, feature fundamental differences on a variety of biological levels. Those differences may imply disparities in antigenicity, determining the odds for immunotherapy success [15]. This notion is confirmed by an extensive analysis of 30 cancer types using comprehensive sequencing data from ICGC and TCGA [26], with striking differences concerning the PNE pool between HCCs and Mel or lung and colorectal cancer [79]. Indeed, we only found a single case with comparable Varns counts among Mel [24] similar to our relatively homogeneous HCC cohort, where a NElig could be verified. Hence, chances for presentation of exome-derived NElig in HCC may be commonly very low, possibly due to cancer immunoediting [83], and this limited target scope may need to be widened to better estimate the odds of neoantigen targeted immunotherapy success in HCC.
This notion is supported by our findings in two out of seven patients where we could confirm a mutation in the proteome, once directly and in the other case through the introduction of an additional tryptic cleavage site by mutation. A comparable approach has been published for rhabdomyosarcoma xenografts, claiming this might be a way to infer relevance for PNE determined by bioinformatics algorithms [78].
Searching for alternatives, we assessed cancer-testis antigens contained among HLA ligands in HCC, which was unrewarding. Furthermore, the odds for administering targeted therapies available to HCC patients in our cohort remained small as previously encountered [28], so we additionally assessed RNA expression and benchmarked data to TCGA datasets to pinpoint signaling pathways that might be harnessed for therapeutic purposes in the future.
Conclusions
We failed to confirm any exome-derived mutated HLA ligands with sophisticated (targeted) MS approaches in HCC, supporting the assumption that in malignancies with low TMB immunoediting may be a relevant driving force shaping the HLA ligand landscape [84]. Certainly, LC-MS/MS comes with specific limitations that must be considered and HLA ligands may be missed, but it remains the best tool currently available.
Paying close attention to the rules of HLA presentation is important and may support choosing suitable NElig candidates. We therefore screened our HLA ligand database and found that respective knowledge may indeed guide selection. Undoubtedly, since the HLA ligandome is very complex and our data are limited, the required knowledge needs to be generated in a community effort [85]. Even though our results do suggest there may be relevant value in this approach, the attempt will evidently not solve the underlying probabilistic issues encountered with rare NElig in HCC. As the scope of our work was primarily focused on simple NElig, derived from exome-derived low complexity variants, our analyses suggest this is only a narrow subset of potential targets that might be used for personalized immunotherapies. Among interesting avenues to be pursued in the future are non-mutated neoantigens [86] and tumor alterations influencing the HLA ligandome composition of malignancies [34], as well as RNA editing and splicing [87, 88], post-translational modifications [89, 90] and targets beyond the exome [91]. In this regard, we may curb the enthusiasm for simple NElig in HCC, simultaneously suggesting that there is a wide array of alternatives available, which is not even tapped remotely today.
Abbreviations
- ALB:
-
Albumin
- CID:
-
Collision-induced dissociation
- CTA:
-
Cancer-testis antigen
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DDA:
-
Data-dependent acquisition (MS)
- DE:
-
Differentially expressed
- FDR:
-
False discovery rate
- FPKM:
-
Fragments per kilobase of exon per million reads mapped
- HCC:
-
Hepatocellular carcinoma
- HPLC:
-
High-performance liquid chromatography
- ICP:
-
Immune checkpoint
- ID:
-
Identifier
- LC-MS/MS:
-
Liquid chromatography-coupled tandem mass spectrometry
- LTQ:
-
Linear trap quadrupole
- Mel:
-
Malignant melanoma
- MS:
-
Mass spectrometry
- MS/MS:
-
Tandem mass spectrometry
- NElig :
-
Mutated neoantigen with HLA ligandome level evidence (HLA class I)
- NGS:
-
Next-generation sequencing
- PNE:
-
Predicted mutated Neoepitope
- PNEexp :
-
Predicted mutated Neoepitope with transcript level evidence
- PNEprot :
-
Predicted mutated Neoepitope with protein level evidence
- ppm:
-
Parts per million
- PRM:
-
Parallel reaction monitoring
- PSS:
-
Peptide search space
- RECQL:
-
ATP-dependent DNA helicase Q1
- RT:
-
Retention time
- SIM:
-
Selected ion monitoring
- TCGA:
-
The Cancer Genome Atlas
- TMB:
-
Tumor mutational burden
- tMS2:
-
Targeted tandem MS
- UHPLC:
-
Ultra-high-performance liquid chromatography
- Var:
-
Somatic variant
- Varexp :
-
Expressed non-synonymous somatic variant
- Varns :
-
Non-synonymous somatic variant
- WES:
-
Whole exome sequencing
- WTlig :
-
Wild-type peptide corresponding to PNE with evidence on HLA ligandome level (HLA class I)
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
European Association for the Study of the Liver, European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–43.
Chan T, Wiltrout RH, Weiss JM. Immunotherapeutic modulation of the suppressive liver and tumor microenvironments. Int Immunopharmacol. 2011;11(7):879–89.
Butterfield LH, Ribas A, Potter DM, Economou JS. Spontaneous and vaccine induced AFP-specific T cell phenotypes in subjects with AFP-positive hepatocellular cancer. Cancer Immunol Immunother. 2007;56(12):1931–43.
Yao W, He JC, Yang Y, Wang JM, Qian YW, Yang T, et al. The prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: a systematic review and meta-analysis. Sci Rep. 2017;7(1):7525.
Unitt E, Marshall A, Gelson W, Rushbrook SM, Davies S, Vowler SL, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol. 2006;45(2):246–53.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of Ipilimumab in Unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–94.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
Rammensee HG, Singh-Jasuja H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev Vaccines. 2013;12(10):1211–7.
Vonderheide RH, Nathanson KL. Immunotherapy at large: the road to personalized cancer vaccines. Nat Med. 2013;19(9):1098–100.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Gubin MM, Schreiber RD. CANCER. The odds of immunotherapy success. Science. 2015;350(6257):158–9.
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2(6):522–9.
Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–21.
Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124(3):453–62.
Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81.
Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6.
Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016;7:13404.
Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 2018;8(11):1366–75.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501.
Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23(9):1422–33.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30(23):3310–6.
Löffler MW, Chandran PA, Laske K, Schroeder C, Bonzheim I, Walzer M, et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J Hepatol. 2016;65(4):849–55.
Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, et al. Production of monoclonal antibodies to group a erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell. 1978;14(1):9–20.
Kowalewski DJ, Stevanovic S. Biochemical large-scale identification of MHC class I ligands. Methods Mol Biol. 2013;960:145–57.
Löffler MW, Kowalewski DJ, Backert L, Bernhardt J, Adam P, Schuster H, et al. Mapping the HLA Ligandome of colorectal Cancer reveals an imprint of malignant cell transformation. Cancer Res. 2018;78(16):4627–41.
Bertsch A, Gropl C, Reinert K, Kohlbacher O. OpenMS and TOPP: open source software for LC-MS data analysis. Methods Mol Biol. 2011;696:353–67.
Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13(1):22–4.
Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4(11):923–5.
Almeida LG, Sakabe NJ. deOliveira AR, Silva MC, Mundstein AS, Cohen T, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37(Database issue):D816–9.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72.
Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805.
Mohr C, Friedrich A, Wojnar D, Kenar E, Polatkan AC, Codrea MC, et al. qPortal: a platform for data-driven biomedical research. PLoS One. 2018;13(1):e0191603.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15(8):591–4.
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–9.
Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–7.
Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–17.
Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13.
Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8(1):33.
Schubert B, Walzer M, Brachvogel HP, Szolek A, Mohr C, Kohlbacher O. FRED 2: an immunoinformatics framework for python. Bioinformatics. 2016;32(13):2044–6.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, et al. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017;35(4):319–21.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1.
Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34.
Chaisson MJ, Wilson RK, Eichler EE. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet. 2015;16(11):627–40.
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152–8.
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608.
Freudenmann LK, Marcu A, Stevanovic S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology. 2018;154(3):331–45.
Qian GS, Kuang SY, He X, Groopman JD, Jackson PE. Sensitivity of electrospray ionization mass spectrometry detection of codon 249 mutations in the p53 gene compared with RFLP. Cancer Epidemiol Biomark Prev. 2002;11(10 Pt 1):1126–9.
Wang D, Eraslan B, Wieland T, Hallstrom B, Hopf T, Zolg DP, et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol. 2019;15(2):e8503.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207.
Gfeller D, Bassani-Sternberg M. Predicting antigen presentation-what could we learn from a million peptides? Front Immunol. 2018;9:1716.
Shao W, Pedrioli PGA, Wolski W, Scurtescu C, Schmid E, Vizcaino JA, et al. The SysteMHC atlas project. Nucleic Acids Res. 2018;46(D1):D1237–D47.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13.
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–7.
Editorial. The problem with neoantigen prediction. Nat Biotechnol. 2017;35(2):97.
Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–6.
Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356(6334):200–5.
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–5.
Brennick CA, George MM, Corwin WL, Srivastava PK, Ebrahimi-Nik H. Neoepitopes as cancer immunotherapy targets: key challenges and opportunities. Immunotherapy. 2017;9(4):361–71.
Cesano A, Warren S. Bringing the next Generation of Immuno-Oncology Biomarkers to the Clinic. Biomedicines. 2018;6(1).
Chang TC, Carter RA, Li Y, Li Y, Wang H, Edmonson MN, et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017;9(1):78.
Teku GN, Vihinen M. Pan-cancer analysis of neoepitopes. Sci Rep. 2018;8(1):12735.
Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60(6):1972–82.
Weinzierl AO, Lemmel C, Schoor O, Muller M, Kruger T, Wernet D, et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol Cell Proteomics. 2007;6(1):102–13.
Schuster H, Peper JK, Bösmüller HC, Röhle K, Backert L, Bilich T, et al. The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci U S A. 2017;114(46):E9942–E51.
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–4.
Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171(6):1272–83 e15.
Caron E, Aebersold R, Banaei-Esfahani A, Chong C, Bassani-Sternberg M. A case for a human Immuno-Peptidome project consortium. Immunity. 2017;47(2):203–8.
Marijt KA, Blijleven L, Verdegaal EME, Kester MG, Kowalewski DJ, Rammensee HG, et al. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J Exp Med. 2018;215(9):2325–37.
Fritsche J, Rakitsch B, Hoffgaard F, Romer M, Schuster H, Kowalewski DJ, et al. Translating Immunopeptidomics to immunotherapy-decision-making for patient and personalized target selection. Proteomics. 2018;18(12):e1700284.
Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun. 2018;9(1):3919.
Cobbold M, De La Pena H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med. 2013;5(203):203ra125.
Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of Glycopeptides as Posttranslationally modified Neoantigens in leukemia. Cancer Immunol Res. 2017;5(5):376–84.
Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10(470).
Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41(Database issue):D1063–9.
Löffler MW, Mohr C, Bichmann L, et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Data sets. PRIDE Archive. 2019; Data set Identifier: PXD013057.
Bassani-Sternberg M, Bräunlein E, Klar R, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Data sets. PRIDE Archive. 2017; Data set Identifier: PXD004894.
Löffler MW, Mohr C, Bichmann L, et al. Somatic variants (HCC). Data sets. figshare. 2019; https://figshare.com/s/6c09d3095a32402b4717.
Löffler MW, Mohr C, Bichmann L, et al. Gene counts (HCC). Data sets. figshare. 2019; https://figshare.com/s/c02d184d8f55a813456a.
Acknowledgements
The authors wish to thank Claudia Falkenburger, Patricia Hrstić, Ulrich Wulle, and Beate Pömmerl for production of peptides, monoclonal antibodies, and excellent technical support as well as Dr. Silvia Wagner and all the staff of the study center of the Department of General, Visceral and Transplant Surgery at the University Hospital Tübingen for their support and all the participating patients. Further, we gratefully acknowledge the proficient technical support by Silke Wahl and helpful discussions with Boumediene Soufi, PhD.
Members of the HEPAVAC Consortium are :
Andrea Mayer-Mokler, Toni Weinschenk, Christian Flohr, Carsten Reinhardt, Harpreet Singh-Jasuja, Immatics Biotechnologies GmbH, Tübingen, Germany;
Roberto S. Accolla, Giovanna Tosi and Greta Forlani, Department of Medicine and Surgery, University of Insubria, Varese, Italy;
Yuk T. Ma, David Adams, NIHR Biomedical Research Unit in Liver Disease, School of Immunity and Infection, University of Birmingham, Birmingham, United Kingdom;
Danila Valmori, Tanguy Chaumette, EA3826 Institut de Recherche en Santé 2 Nantes Biotech, Université de Nantes, France;
Regina Heidenreich, CureVac AG, Tübingen, Germany;
Alfred Königsrainer, Markus W. Löffler, University Hospital of Tübingen, Department of General, Visceral and Transplant Surgery, Eberhard Karls University Tübingen, Tübingen, Germany;
Hans-Georg Rammensee, Cécile Gouttefangeas, Interfaculty Institute for Cell Biology, Department of Immunology, Eberhard Karls University Tübingen, Tübingen, Germany;
Bruno Sangro, Mercedes Iñarrairaegui, Liver Unit, Clinica Universidad de Navarra and Centro de Investigacion Biomedica en Red de Enfermedades Hepaticas y Digestivas (CIBEREHD), Pamplona, Spain;
Sven Francque, Luisa Vonghia, Division of Gastroenterology and Hepatology, Antwerp University Hospital, Edegem, Belgium;
Maria Tagliamonte, Annacarmen Petrizzo, Luigi Buonaguro, Cancer Immunoregulation Unit, Istituto Nazionale per lo Studio e la Cura dei Tumori, “Fondazione Pascale” – IRCCS, 80131, Naples, Italy.
Maria Lina Tornesello, Franco M. Buonaguro, Mol. Biol and Viral Oncogenesis Unit, Istituto Nazionale per lo Studio e la Cura dei Tumori, “Fondazione Pascale” - IRCCS, 80131 Naples, Italy.
Funding
This work was supported by the European Research Council (ERC; AdG 339842 MUTAEDITING) granted to H.G. Rammensee and by the European Union 7th Framework Program (EU FP7) Project Cancer Vaccine development for Hepatocellular Carcinoma – HEPAVAC (Grant No. 602893), as well as the German Consortium for Translational Cancer Research (DKTK). C. Mohr and O. Kohlbacher acknowledge funding from the European Union (APERIM, contract no. 633592) and funding from the German Ministry for Research and Education (BMBF) as part of the Medical Informatics Initiative (DIFUTURE, grant 01ZZ1804D). L. Bichmann and O. Kohlbacher acknowledge funding from BMBF as part of the German Network for Bioinformatics Infrastructure (de. NBI, grant 031A535A). S. Nahnsen and S. Czemmel acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) SFB/TR 209 Liver cancer. The NGS Competence Center Tübingen (NCCT) is financed by a structural grant of the DFG (INST 37/1049-1).
Availability of data and materials
The MS datasets, including proteome and ligandome data, generated and analyzed during the current study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRoteomics IDEntifications (PRIDE) database partner repository [92] with the dataset identifier PXD013057 [93]. The MS data reanalyzed during the current study are also available online through the PRIDE database (with the dataset identifier PXD004894) [94]. Somatic variant lists [95] and expression data [96] generated and analyzed during the current study are available from figshare. The whole exome and transcriptome sequencing raw data generated and analyzed for the current study are not publicly available, as participants did not provide respective informed consent for broad sharing of their data.
Author information
Authors and Affiliations
Consortia
Contributions
MWL, OR, BM, LBu, OK, SS, HEPAVAC and HGR are responsible for the study concept and design. MWL, LKF, NT, FJH, RSZ, LM, DJK, HS, HB, and AV are responsible for the acquisition of data. MWL, CM, LBi, LKF, MW, CMS, FJH, RSZ, LM, DJK, HS, MS, JM, SC, SNah, IK, KT, SNad, SB, FF, AV, BM, SPH, and HGR are responsible for the analysis and/or interpretation of data. MWL, CM, LBi, and LKF are responsible for the drafting of the manuscript. MW, CMS, NT, FJH, RSZ, LM, DJK, HS, MS, JM, OR, SC, SNah, IK, KT, SNad, SB, HB, FF, AV, BM, SPH, LBu, OK, SS, AK, and HGR are responsible for the critical revision of the manuscript for important intellectual content. MWL, SNah, OK, AK, HGR, and HEPAVAC obtained funding. MWL, MW, CMS, NT, FJH, RSZ, LM, DJK, HS, MS, JM, SC, KT, SNad, SB, HB, AV, SPH, LBu, and HEPAVAC are responsible for the administrative, technical, or material support. SNah, OR, FF, BM, OK, SS, AK, and HGR are responsible for the study supervision. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and applicable laws and regulations and has been approved by the local institutional review board at the University Hospital of Tübingen, Germany (Project No. 364/2014BO2). All participants provided written informed consent before study inclusion.
Consent for publication
Not applicable.
Competing interests
M.W. Löffler, D.J. Kowalewski, H. Schuster, S. Stevanović, and S.P. Haen are the inventors of patents owned by Immatics Biotechnologies GmbH. D.J. Kowalewski and H. Schuster are currently employees of Immatics Biotechnologies GmbH. H.G. Rammensee has ownership interest (including patents) in Immatics Biotechnologies GmbH, CureVac AG, and Synimmune GmbH. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Supplementary Tables. Table S1. Tumor characteristics. Table S2. Overview of samples and analyses. Table S3. HLA class I allotypes of HCC patients. Table S4. Coding variants and tumor mutational burden (TMB) per patient. Table S5. Source proteins of predicted mutated neoepitopes (PNE) with evidence on shotgun proteome level. Table S6. Identified mutated HLA ligands in the Mel dataset. Table S7. Predicted mutated neoepitopes (PNE) tested with selected ion monitoring (SIM) approach. Table S8. Predicted mutated neoepitopes (PNE) tested with parallel reaction monitoring (PRM) targeted tandem MS (tMS2) approach. Table S9. Parallel reaction monitoring (PRM) in HCC and non-malignant liver tissue samples of patients HCC025 and HCC026. Table S10. Database matches of peptides observed in Mel dataset. Table S11. Database matches of peptides observed in the HCC cohort. Table S12. Cancer testis antigens (CTA) characterized in HCC. Table S13. Identified pathways with differentially expressed genes. (PDF 3500 kb)
Additional file 2:
Extended Materials and Methods. Comprehensive Materials and Methods section with detailed descriptions of experiments and employed materials and tools. (PDF 516 kb)
Additional file 3:
Supplementary Figures. Figure S1. HLA ligandomics yields. Figure S2. Comparison of the tumor mutational burden in Mel and HCC. Figure S3. Differential expression heatmap. Figure S4. Principal component analysis (PCA) on principal component one level. (PDF 642 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Löffler, M.W., Mohr, C., Bichmann, L. et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med 11, 28 (2019). https://doi.org/10.1186/s13073-019-0636-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-019-0636-8