
Abstract
Background
This study was to report a novel CREBBP mutation and phenotype in a child with Rubinstein–Taybi syndrome.
Methods
Case report of a 9-year-old boy.
Results
We described the patient’s clinical manifestations in detail, and found that in addition to the typical systemic manifestations of the syndrome, the outstanding manifestation of the child was severe intellectual deficiency and prominent ocular abnormalities. Whole-exome sequencing and sanger sequencing were performed on the patient and his parents, a large intragenic deletion, covering the exon 1 region and part of the intron 1 region of the TRAP1 gene, and the entire region from intron 27 to exon 30 of the CREBBP gene (chr16:3745393-3783894) was identified on the patient. This mutation affected the CREBBP histone acetyltransferase (HAT) domain.
Conclusions
This findings in our patient add to the spectrum of genetic variants described in Rubinstein–Taybi syndrome and present a RSTS patient with various ocular anomalies including early onset glaucoma.
Similar content being viewed by others
Introduction
Rubinstein–Taybi syndrome (RSTS), first described in 1963 by Rubinstein and Taybi, is a rare congenital neurodevelopment disorder characterized by facial dysmorphism, mental deficiency, growth retardation, and a variety of systemic abnormalities [1, 2]. Fetal growth rate is generally unaffected in RSTS and the abnormalities are often noted at birth and during early infancy. The height, weight and head circumference rapidly drop below the fifth percentile in the first few months of life. Adult patients are usually short in stature and become overweight relative to height during puberty and adulthood [3,4,5]. The diagnosis is usually based on the clinical criteria, including the presence of mental retardation associated with three major symptoms, such as broad thumbs or first toes, thick eyebrows or long eyelash, and columella below alae nasi [6].
The inheritance pattern of RSTS is autosomal dominant with an estimated prevalence of 1 in 100,000–125,000 live births [7, 8]. The majority of cases (~ 99%) occur sporadically de novo [9] although a few familial cases have been reported [10,11,12]. In most cases, parents of RSTS patients are not affected. When parents are clinically unaffected, sibs are still presumed to be at increased risk of RSTS due to the presence of mild phenotypes in heterozygous parents or parental somatic cells and/or germ line chimeras. Individuals with RSTS rarely reproduce. Pathogenic variants of two highly evolutionarily conserved genes are associated with the incidence of RSTS, CREBBP and EP300. 50–60% of RSTS patients are caused by mutations of CREBBP [13], 80% of which are associated with pathogenic sequence variants and 20% with deletion of variable sizes [14]. Pathogenic variants of EP300 were found in 8–10% of individuals with RSTS, predominantly frameshift type, while deletional type rarely reported [15]. However, the genetic basis of the remaining 30% of RSTS patients remains unclear [16].
In Chinese RSTS patients, a higher percentage of microcephaly, micrognathia, polydactyly and syndactyly but a lower percentage in urogenital anomalies has been noted [17, 18]. Yu and colleagues reported two relative “hot spot” regions in CREBBP gene for truncated and deletion variants in a Chinese cohort (18 kids) [18]. One is a 461-nt long region (codons 1931 and 2086) in exon 31 of CREBBP gene, and another region locates in exon 2 at the 5’ end of the CREBBP [18]. In the past, most cases of RSTS were discovered and reported by pediatricians, and there were few reports on the ocular phenotype of RSTS [19]. In this manuscript, we describe a novel CREBBP mutation in a Chinese RSTS patient first diagnosed with congenital glaucoma to broaden the genetic variant spectrum of this rare disease.
Methods
The patient and his parents were recruited in accordance with the principles of the Declaration of Helsinki. The study protocol was approved by the Medical Ethics Committee of the Beijing Tongren Hospital and written informed consent was obtained from the Legal guardians of the patients (parents) for participation in this study and to publish study finding.
Clinical examinations
The ophthalmological examinations included measurement of visual acuity, tonometry, slit lamp-assisted biomicroscopy of the anterior segment of the eye. We conducted ocular biometry for the determination of axial length using optical low-coherence reflectometry (Lenstar 900 Optical Biometer, Haag-Streit; 3098 Koeniz, Switzerland). Applying a non-mydriatic fundus camera (CR6-45NM; Canon Inc., Õsta, Tokyo, Japan), we obtained 45° fundus photographs.
Whole exome sequencing
Samples of peripheral venous blood (5 ml) were collected from patient and his parents for genomic DNA extraction using a QIAmp DNA Mini Blood Kit (Qiagen, Hilden, Germany). To reveal the disease causing mutation in this family, whole exome sequencing approach was performed on this family. Briefly, the libraries for whole exome sequence were established from the DNA samples using an exon capture kit (SureSelect ver. 6 + UTR, Agilent Technologies), according to the manufacturer’s instructions. The exons were sequenced as 100-basepairs paired-end reads by an Illumina HiSeq2500 (Illumina). The original sequencing data were processed by Illumina Basecalling Software 1.7 and compared with NCBI HUMAN genome DNA reference sequence (NCBI Build 37.1). Single nucleotide variation (SNV) information is analyzed using SOAP software (http://soap.Genomics.org.cn). The information related to Inserts and deletions (Indel) is analysed using BWA software (http://bio-bwa.sourceforge.net/) to obtain all the mutations occurring in the DNA sequences of the sample. The common variants (MAF > 1%) that appear in the database (DB135) are filtered out, and then those that have no effect on the structure and function of the protein are filtered out. Candidate pathogenic gene variants were obtained by filtration.
In silico analysis
The possible effects of the amino acid changes on the function was predicted by using the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), the Sorting Tolerant from Intolerant (SIFT) algorithm (http://sift.jcvi.org/www/SIFT_enst_submit.html), and the MutationTaster (http://www.mutationtaster.org/). Mutations were classified as clinically unclear when at least one of the four predictions had a benign outcome or when there was insufficient evidence of pathogenicity. When all the predicted results were pathogenic, the variation was classified as possible pathogenic variation combined with other evidence. Frameshift variation, nonsense variation and variation with experimental evidence of protein function loss are classified as pathogenic variation. Sanger sequencing was performed for intrafamilial cosegregation analysis. The allele frequencies of the mutations in the normal population were viewed in 1000 Genome (http://browser.1000genomes.org/index.html), EVS (http://evs.gs.washington.edu/EVS/) and EXAC (http://exac.broadinstitute.org/) databases.
Results
Clinical evaluation
The 9-year-old Chinese boy had low normal height, truncal obesity and low normal skull circumference (On examination, his weight was 65 kg, height was 124 cm, and head circumference was 49 cm). He had severe mental retardation (IQ estimated to be 10–20, according to Zhang et al. 2005) [20], and his psychomotor development was delayed. Shortly after the child was born, the parents found that the development of the child was slower than that of other normal children, and the time of raising his head, crawling, and standing was significantly delayed, and he was unable to perform fine operations or walked steadily. The child's language development is also obviously delayed. Until now, he can only understand some simple daily sentences, and can only speak some simple words, and cannot communicate normally. He had the characteristic facial dysmorphism and facial grimacing seen in RSTS. His distinctive features included microcephaly, high arched eyebrows, low-set ears, low and thick columella, thin upper lip, a protuberant lower lip, grimacing smile with almost closed eyes on smiling. He also had broad thumbs, persistent fetal finger pads, and broad great toes (Fig. 1).

The patient had the characteristic facial dysmorphism and facial grimacing seen in RSTS, including microcephaly, high arched eyebrows, low-set ears, low and thick columella, thin upper lip, a protuberant lower lip (A, B), broad thumbs, fetal finger pads, and broad great toes (C, D)
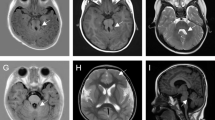
The patient was diagnosed as congenital glaucoma and underwent trabeculotomy twice at the age of 1 and 2-year-old, respectively. Combined with Latanoprost, Brinzolamide and Timolol Maleate eye drops, the intraocular pressure of both eyes have been normal since the surgery until 5 months ago, after going through the surgery of correction of lower eyelid inversion combined with ptosis. When admitted to the hospital, the patient can’t cooperate with visual acuity or other ocular examination. Examination of the eyes under general anesthesia showed a horizontal corneal diameter of 13 mm in the right eye and 15 mm in the left eye. Schiotz tonometry showed an intraocular pressure of 40.08 mm Hg in the right eye and 14.57 mm Hg in the left eye. The axial lengths were 29.7 mm in the right eye and 30.8 mm in the left eye. In his right eye, ectopia lentis was noted, and he had a cataract in the lens, corneal clouding, iris coloboma, and no light reflex. In the left eye, he had relatively clear cornea, iris atrophy with peripheral incision in the superior, pale optic disc and choroidal atrophy (Fig. 2). The patient underwent vitrectomy, lens cutting, and internal transscleral ciliary body photocoagulation in his right eye. The intraocular pressure of the child was maintained at a normal level after the surgery.

In his right eye, ectopia lentis was noted, and he had a cataract in the lens, corneal clouding, iris coloboma, and no light reflex (A). Fundus photography showed optic disc pale and choroidal atrophy in the right eye (B). In the left eye, he had relatively clear cornea, iris atrophy with peripheral incision in the superior (C), pale optic disc and choroidal atrophy (D)
This patient was born to a healthy nonconsanguineous Chinese couple. Anterior segment and fundus examination was unremarkable in this couple. The family history was otherwise noncontributory.
Mutation analysis and in silico analysis
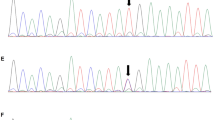
Chromosome analysis in peripheral lymphocytes showed a normal male karyotype (46,XY) at a banding resolution of 400 bands per haploid genome. Whole exome sequencing analysis was performed on the patient, a large heterozygous copy number deletion on CREBBP gene, from exon 29 to exon 31 (chr16 3778026-3781885) was identified on the patient, this mutation was de novo and not found on his parents by sanger sequencing (Fig. 3). We used long PCR and Sanger sequencing to determine the location of the breakpoint on the patient, and found that the upstream breakpoint position of the heterozygous copy number deletion is chr16:3745392, and the downstream breakpoint position is chr16:3783895. It is confirmed that the size of the heterozygous copy number deletion is 38503 bp, and a sequence of nearly 300 bp has been inserted at the breakpoint. It might be caused when DNA double-strand break damage is repaired (Fig. 3). This heterozygous copy number deletion region chr16:3745393-3783894 covers the exon 1 region and part of the intron 1 region of the TRAP1 gene, and the entire region from intron 27 to exon 30 of the CREBBP gene. In general, this pathogenic heterozygous deletion is the causative mutation for the disease phenotype in this family.

Pedigree of affected family (A). Whole exome sequencing analysis identified a large heterozygous copy number deletion on CREBBP gene, from exon 29 to exon 31 (chr16 3778026-3781885) on the patient, this mutation was de novo and not found on his parents by sanger sequencing (B). Long PCR and Sanger sequencing showed that the heterozygous copy number deletion region chr16:3745393-3783894 covers the exon 1 region and part of the intron 1 region of the TRAP1 gene, and the entire region from intron 27 to exon 30 of the CREBBP gene (C, D)
Discussion
Rubinstein–Taybi syndrome is a rare genetic disorder characterized by postnatal growth retardation, moderate to severe mental retardation, and multiple characteristic malformations. Despite its prominent facial features, broad thumbs and hallux, the diagnosis of RSTS is sometimes challenging due to the high variability of phenotypes and genotypes [21]. Whole exome sequencing is a reliable and rapid diagnostic tool for suspected genetic diseases and has been strongly applied in medical practice [18].
To date, 500 pathogenic variants have been described in CREBBP [9], and a pathogenic variant in CREBBP is identified in 50–60% of all RSTS cases. It has been reported that the mutation spectrum included 80.2% of point mutations, of which truncating mutations accounted for 55.2% of all point mutations, followed by large rearrangements (18.8%), missense mutations (16.8%), and splice mutations (9.2%) [9]. CREBBP has no real hotspot mutation sites, and its mutation spectrum is distributed along all of the 31 exons [9]. However, studies have found some recurrent mutations, such as about 52% of missense mutations are located in the location of histone acetyltransferase (HAT domain) [9]. In up to 30% of patients with suggestive clinical manifestations, the genetic etiology of RSTS remains unknown [22], possibly due to unrecognized genetic variants.
By now, no convincing genotype–phenotype correlation in RSTS has yet been established [23], though recent studies produced insight into the role of the CREBBP and EP300 genes in neural cell and brain development, regulating precursor cell migration and neuronal plasticity [24,25,26,27]. The syndrome has been subdivided into type 1 associated with the CREBBP mutation spectrum (RSTS1; OMIM#180849) and type 2 associated with the EP300 mutation spectrum (RSTS2; OMIM#613684). There are different phenotypes of RSTS1 [28]. The classic phenotype, which caused by deletions or truncating mutations of the CREBBP gene, is characterized by intellectual disability, broad thumbs, and characteristic facial dysmorphism [29]. A very severe phenotype of RSTS1, also known as the chromosome 16p 13.3 contiguous deletion syndrome, caused by large deletions including the CREBBP gene and the 3’ adjacent genes, viz., DNASE1 and TRAP1 [28], always exhibits severe mental retardation, life-threatening infections and systemic complications, and other classic features [30]. However, more and more evidence showed that no significant correlation shown between the phenotype and the mutation type and location or deletion size for either CREBBP or EP300 genes in RSTS patients [15, 31,32,33].
In this study, we report the deletion of exon 27 to exon 30 of the CREBBP gene and the 3’ adjacent genes, TRAP1, in one patient molecularly confirming the diagnosis of RSTS. To best of our knowledge, this mutation has not been published previously. We described the patient’s clinical manifestations in detail, and found that in addition to the typical systemic manifestations of the syndrome, such as intellectual disability and facial dysmorphism, the outstanding manifestation of the child was ocular abnormalities (infantile glaucoma, ptosis and epicanthus) and significantly lower IQ (10–20). The child’s IQ was significantly lower than most other reports (average IQ was reported between 35 and 50) [34], who can only understand simple, daily sentences and single words, has no language or only a few words, and usually walks unsteadily. Since no observable genotype–phenotype relationship has been observed for other mutations in RSTS, this mutation is less likely to lead to a specific clinical RSTS subtype.
Intragenic deletion was report in 10%-23% of the CREBBP-mutated patients [33] and the reported CREBBP deletions were spread along the gene (Table 1). The deletions vary in size and genomic location, some involving the whole CREBBP gene and its flanking regions, while others only involving an intragenic portion [33, 35,36,37]. There are currently no reports in which ethnic groups or regions of the population with CREBBP gene deletions are more common [31,32,33, 35, 38,39,40,41,42,43,44], while it seems that deletions more frequently involve the HAT region [32]. In Chinese patients, CREBBP intragenic deletions account for 11–37% of CREBBP pathogenic genes [17, 18]. Ye and colleagues reports a relative “hot spot” in CREBBP for deletion, locating in exon 2 at the 5’ end of the CREBBP [18]. Half of the patients carried deletions of five exons or more [31]. Although not confirmed in other populations [33], the association between severe phenotypes and deletion of CREBBP and adjacent genes has been reported [30]. In addition, researchers did not find an association of larger deletions with disease severity [31, 45]. No correlation was observed between the location of the affected exons and the phenotype [31, 32]. However, previous studies have reported a worse cognitive phenotype in patient carriers of mutations that affect the CREBBP HAT domain [33], which also seen in our case. CREBBP is highly conserved, with 95% homology between the human and murine genes [46]. The reported mutational spectrum of CREBBP, including deletions, point mutations, and large rearrangements, distributed throughout the 31 coding exons. However, the highly conserved HAT region is believed to be particularly important for the RSTS phenotype. A large study including 93 patients with RSTS found that almost all the truncating mutations lead to premature termination of the protein before or within the HAT domain (corresponding to exons 19–30) and there was a clustering of single amino acid changes in the HAT region, indicating the importance of HAT activity in the phenotype [13, 44]. In this patient, three exons (exons 27–30) of CREBBP are deleted. This is predicted to disrupt HAT activity and supports the importance of the HAT domain in causing the phenotype of RSTS.
In our patient, another affected gene was the TRAP1 gene encoding tumor necrosis factor receptor-associated protein 1. As a mitochondrial ATP-binding protein, TRAP1 gene participants in maintaining mitochondrial function [47]. TRAP1 also acts as a molecular chaperone and is a heat shock protein 90-related protein [48]. However, the TRAP1 gene has not been associated with human disease [28] and whether the deletion of TRAP1 is associated with a more severe phenotype is also inconclusive. Rusconi et al. reported a slight increase in growth retardation in patients carrying deletions involving not only CREBBP but also other genes like TRAP1[33]. However, the presentations of two patients described by Stef et al. [37] and one patient reported by Lai et al. [44] did not support the description of a severe RSTS phenotype in patients with a large deletion involving CREBBP and contiguous genes, such as DNASE1 and TRAP1.
In addition, deletion of HAT domain of CREBBP has been reported to causing ocular abnormality [7, 31]. Various ocular features have been reported in RSTS with a high occurrence of 80% [19], particularly refractive errors (56%) and strabismus with subsequent risk of amblyopia (71%) [49]. These usually respond well to interventions. Patients with RSTS also have a higher frequency of lacrimal duct obstructions (38–47%), which may require surgical intervention. Other serious ocular abnormalities were also described, such as corneal abnormalities (19.7%, such as megalocornea without glaucoma, opacities, keretoglobus, sclerocorenea), congenital glaucoma (26.5%), congenital cataract (12.8%), and coloboma (33.3%) [49]. Peri-ocular findings adding to the characteristic facial appearance has been described: ptosis, downward slanted palpebral fissures, hypertelorism and epicanthus [49]. The presence of childhood glaucoma in the present patients corresponds to the ocular spectrum of RSTS. Brei and colleagues found 32 glaucoma and 25 corneal opacities in 614 patients with RSTS, and they concluded that the incidence of glaucoma in RSTS exceeds that of the general population and is often congenital or develops in infancy [50]. Since congenital glaucoma is a preventable cause of blindness and early treatment is critical, it is especially important to request a detailed ophthalmological examination to rule out ocular complications after diagnosis [34]. It is recommended that all children with RSTS should undergo a detailed ophthalmologic examination by a pediatric ophthalmologist shortly after diagnosis, or at 6 months of age in patients with RSTS diagnosed in the neonatal period. If an eye problem is suspected, referral to an ophthalmologist should be made as soon as possible. Depending on the examination results, regular ophthalmological visits should be performed every 12 months or less [19].
Although whole exome sequencing has the advantage of high efficiency and convenience in screening pathogenic genes, the importance of traditional sanger sequencing cannot be ignored. For example, in our research, we find a large deletion mutation of exon 29 to 31 on CREBBP gene, after verification by Sanger sequencing, it was finally confirmed to be the mutation covers the exon 1 region and part of the intron 1 region of the TRAP1 gene, and the entire region from intron 27 to exon 30 of the CREBBP gene.
Conclusions
The genetic variant found in this case was reported in this work for the first time, hence, contributing to the RSTS molecular knowledge and expanding the CREBBP genetic variant repertoire of this complex disorder. Meanwhile, this study further suggests the importance of ophthalmic examination and follow-up in patients with RSTS. Timely and correct treatment for congenital glaucoma and other ophthalmic diseases is helpful to improve the quality of life of children.
Availability of data and materials
The datasets generated during the current study are available in the DDBJ BioSample repository, the sample of the patient can be obtained from the web link: https://ddbj.nig.ac.jp/resource/biosample/SAMD00469867; the sample of the patient’s father can be obtained from the web link: https://ddbj.nig.ac.jp/resource/biosample/SAMD00469868; the sample of the patient’s mother can be obtained from the web link: https://ddbj.nig.ac.jp/resource/biosample/SAMD00469869
References
Stevens CA, Carey JC, Blackburn BL. Rubinstein–Taybi syndrome: a natural history study. A J Med Genet Suppl. 1990;6:30–7.
Bensal S, Relhan V, Grag VK. Rubinstein–Taybi syndrome: a report of two siblings with unreported cutaneous stigmata. Indian J Dermatol Venereol Leprol. 2013;79(5):714–7.
Stevens CA, Hennekam RC, Blackburn BL. Growth in the Rubinstein–Taybi syndrome. Am J Med Genet Suppl. 1990;6:51–5.
Rubinstein JH. Broad thumb-hallux (Rubinstein–Taybi) syndrome 1957–1988. Am J Med Genet Suppl. 1990;6:3–16.
Hutchinson DT, Sullivan R. Rubinstein–Taybi Syndrome. J Hand Surg Am. 2015;40(8):1711–2.
Yagi Y, Kuwatsuka Y, Asai M, Honda M, Utani A. Coexistence of keloids and pilomatricoma in a patient with Rubinstein–Taybi syndrome. Dermatol Online J. 2018;24:19–20.
Hennekam RCM. Rubinstein–Taybi syndrome. Eur J Hum Genet. 2006;14:981–5.
Belzen M, Bartsch O, Lacombe D, Peters DJM, Hennekam RCM. Rubinstein–Taybi syndrome (CREBBP, EP300). Eur J Hum Genet. 2011;19:121.
Van Gils J, Magdinier F, Fergelot P, Lacombe D. Rubinstein–Taybi syndrome: a model of epigenetic disorder. Genes (Basel). 2021;12:968–70.
Bartsch O, Kress W, Kempf O, Lechno S, Haaf T, Zechner U. Inheritance and variable expression in Rubinstein–Taybi Syndrome. Am J Med Genet A. 2010;152A:2254–61.
López M, Seidel V, Santibáñez P, Cervera-Acedo C, Castro-de Castro P, Domínguez-Garrido E. First case report of inherited Rubinstein–Taybi Syndrome associated with a novel EP300 Variant. BMC Med Genet. 2016;17:1–5.
Hennekam RC, Lommen EJ, Strengers JL, Van Spijker HG, Jansen-Kokx TM. Rubinstein–Taybi Syndrome in a mother and son. Eur J Pediatr. 1989;148:439–41.
Schorry EK, Keddache M, Lanphear N, Rubinstein JH, Srodulski S, Fletcher D, Blough-Pfau RI, Grabowski GA. Genotype–phenotype correlations in Rubinstein–Taybi syndrome. Am J Med Genet A. 2008;146A:2512–9.
Spena S, Milani D, Rusconi D, Negri G, Colapietro P, Elcioglu N, Bedeschi F, Pilotta A, Spaccini L, Ficcadenti A, et al. Insights into genotype-phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein Taybi syndrome patients. Clin Genet. 2015;88:431–40.
Fergelot P, Van Belzen M, Van Gils J, Afenjar A, Armour CM, Arveiler B, Beets L, Burglen L, Busa T, Collet M, et al. Phenotype and genotype in 52 patients with Rubinstein–Taybi syndrome caused by EP300 mutations. Am J Med Genet A. 2016;170:3069–82.
Bartsch O, Schmidt S, Richter M, Morlot S, Seemanová E, Wiebe G, Rasi S. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein–Taybi syndrome (RSTS) and in another patient with imcomplete RSTS. Hum Genet. 2005;117:485–93.
Yu PT, Luk HM, Lo IFM. Rubinstein–Taybi syndrome in Chinese population with four novel mutations. Am J Med Genet. 2021;185A:267–73.
Yu S, Wu B, Qian Y, Zhang P, Lu Y, Dong X, Wang Q, Zhao X, Liu R, Zhou W, et al. Clinical exome sequencing identifies novel CREBBP variants in 18 Chinese Rubinstein–Taybi Syndrome kids with high frequency of polydactyly. Mol Genet Genomic Med. 2019;7:e1009.
Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein–Taybi Syndrome medical guidelines. Am J Med Genet A. 2003;119A:101–10.
Zhang X, Snijders A, Segraves R, Zhang X, Niebuhr A, Albertson D, Yang H, Gray J, Niebuhr E, Bolund L, et al. High-resolution mapping of genotype-phenotype relationships in Cri du Chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005;76:312–26.
Spena S, Gervasini C, Milani D. Ultra-rare syndromes: the example of Rubinstein–Taybi syndrome. J Pediatr Genet. 2015;4(3):177–86.
Stevens CA. Rubinstein–Taybi syndrome. In: Adam A, Ardinger H, Pagon R, Wallace S, Bean L, Stephens K, Amemiya A, editors. Gene Reviews. 9th ed. Seattle, WA: University of Washington, USA; 2019. p. 1–21.
Tekendo-Ngongang C, Owosela B, Fleischer N, Addissie Y, Malonga B, Badoe E, Gupta N, Moresco A, Huckstadt V, Ashaat EA, et al. Rubinstein–Taybi sundrome in diverse populations. Am J Med Genet A. 2020;182(12):2939–50.
Chatterjee S, Angelakos CC, Bahl E, Hawk JD, Gaine ME, Poplawski SG, Schneider-Anthony A, Yadav M, Porcari GS, Cassel JC, et al. The CBP KIX domain regulates long-term memory and circadian avtivity. BMC Biol. 2020;18:155.
Schoof M, Launspach M, Holdhof D, Nguyen L, Engel V, Filser S, Peters F, Immenschuh J, Hellwig M, Niesen J, et al. The transcriptional coactivator and histone acetyltransferase CBP regulates neural precursor cell development and migration. Acta Neuropathol Commun. 2019;7:199.
Alari V, Russo S, Terragni B, Ajmone PF, Sironi A, Catusi I, Calzari L, Concolino D, Marotta R, Milani D, et al. iPSC-derived neurons of CREBBP- and EP300-mutated Rubinstein–Taybi syndrome patients show morphological alterations and hypoexcitability. Stem Cell Res. 2018;30:130–40.
Ateca-Cabarga JC, Cosa A, Pallarés V, López-Atalaya JP, Barco Á, Canals S, Moratal D. Brain size regulations by cbp haploinsufficiency evaluated by in-vivo MRI based volumetry. Sci Rep. 2015;5:16256.
AI-Qattan MM, Rahbeeni ZA, AI-Hassnan ZN, Jarman A, Rafique A, Mahabbat N, Alsufayan FAS. Chromosome 16p13.3 contiguous gene deletion syndrome including the SLX4, DNASE1, TRAP1, and CREBBP genes presenting as a relatively mild Rubinstein-Taybin Syndrome phenotype: a case report of a Saudi boy. Case Rep Genet. 2020;2020:6143050.
AI-Qattan MM, Jarman A, Rafique A, AI-Hassnan ZN, AI-Qattan HM. Rubinstein–Taybi syndrome in a Saudi boy with distinct features and variants in both the CREBBP and EP300 genes: a case report. BMC Med Genet. 2019;20(1):12.
Bartsch O, Rasi S, Delicado A, Dyack S, Neumann LM, Seemanová E, Volleth M, Haaf T, Kalscheuer VM. Evidence for a new contiguous gene syndrome, the chromosome 16p13.3 deletion syndrome alias severe Rubinstein–Taybi syndrome. Hum Genet. 2006;120(2):179–86.
Pérez-Grijalba V, García-Oguiza A, López M, Armstrong J, García-Miñaur S, Mesa-Latorre JM, O’Callaghan M, Marfa MP, Ramos-Arroyo MA, Santos-Simarro F, et al. New insights into genetic variant spectrum and genotype-phenotype correlations of Rubinstein–Taybi syndrome in 39 CREBBP-positive patients. Mol Genet Genomic Med. 2019;7(11): e972.
Cross E, Duncan-Flavell PJ, Howarth RJ, Hobbs JI, Thomas NS, Bunyan DJ. Screening of a large Rubinstein–Taybi cohort identified many novel variants and emphasizes the importance of the CREBBP histone acetyltransferase domain. Am J Med Genet. 2020;182(11):2508–20.
Rusconi D, Negri G, Colapietro P, Picinelli C, Milani D, Spena S, Magnani C, Silengo MC, Sorasio L, Curtisova V, et al. Characterization of 14 novel deletions underlying Rubinstein–Taybi syndrome: an update of the CREBBP deletion repertoire. Hum Genet. 2015;134(6):613–26.
Candan S, Ornek C, Candan F. Ocular anomalies in Rubinstein–Taybi syndrome: a further case report and review of the literature. Clin Dysmorphol. 2014;23(4):138–42.
Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, Papadia F, Bacina CA, den Dunnen JT, van Ommen GJ, Breuning MH, et al. Genetic heterogeneity in Rubinstein–Taybi syndrome: muatations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76(4):572–80.
Coupry I, Monnet L, Attia AA, Taine L, Lacombe D, Arveiler B. Analysis of CBP (CREBBP) gene deletions in Rubinstein–Taybi syndrome patients using real time quantitative PCR. Hum Mutat. 2004;23(3):278–84.
Stef M, Simon D, Mardirossian B, Delrue MA, Burgelin I, Huber C, Marche M, Bonnet F, Gorry P, Longy M, et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein–Taybi syndrome patients. Eur J Hum Genet. 2007;15(8):843–7.
Choi N, Kim HY, Lim BC, Chae JH, Kim SY, Ko JM. Genetic and clinical heterogeneity in Korean patients with Rubinstein–Taybi syndrome. Mol Genet Genomic Med. 2021;9: e1791.
Breuning MH, Dauwerse HG, Fugazza G, Saris JJ, Spruit L, Wijnen H, Tommerup N, van der Hagen CB, Imaizumi K, Kurodi Y, et al. Rubinstein–Taybi syndrome caused by submicroscopic deletions within 16p13.3. Am J Hum Genet. 1993;52:249–54.
Mogensen M, Skjørringe T, Kodama H, Silver K, Horn N, Møller LB. Exon duplications in the ATP7A gene: frequency and transcriptional behaviour. Orphanet J Rare Dis. 2011;6:73.
Bentivegna A, Milani D, Gervasini C, Castronovo P, Mottadelli F, Manzini S, Colapietro P, Giordano L, Atzeri F, Divizia MT, et al. Rubinstein–Taybi syndrome: spectrum of CREBBP mutations in Italian patients. BMC Med Genet. 2006;7:77.
López M, García-Oguiza A, Armstrong J, García-Cobaleda I, García-Miñaur S, Santos-Simarro F, Seidel V, Domínguez-Garrido E. Rubinstein–Taybi 2 associated to novel EP300 mutations: deepening the clinical and genetic spectrum. BMC Med Genet. 2018;19(1):36.
Elalaoui SC, Smaili W, Van-Gils J, Fergelot P, Ratbi I, Tajir M, Arveiler B, Lacombe D, Sefiani A. Clinical description and mutational profile of a Moroccan series of patients with Rubinstein Taybi syndrome. Afr Health Sci. 2021;21(2):960–7.
Lai AH, Brett MS, Chin WH, Lim EC, Ng JS, Tan EC. A submicroscopic deletion involving part of the CREBBP gene detected by array-CGH in a patient with Rubinstein–Taybi sundrome. Gene (Basal). 2012;499(1):182–5.
Lee JS, Byun CK, Kim H, Lim BC, Hwang H, Choi JE, Hwang YS, Seong MW, Park SS, Kim KJ, et al. Clinical and mutational spectrum in Korean patients with Rubinstein–Taybi syndrome: the spectrum of brain MRI abnormalities. Brain Dev. 2015;37(4):402–8.
Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. Rubinstein–Taybi syndrome caused by mutations in the transcriptional coactivator CBP. Nature. 1995;376(6538):348–51.
Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275(5):3305–12.
Chen CF, Chen Y, Dai K, Chen PL, Riley DJ, Lee WH. A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol Cell Biol. 1996;16(9):4691–9.
Van Genderen MM, Kinds GF, Riemslag FC, Hennekam RC. Ocular features in Rubinstein–Taybi syndrome: investigation of 24 patients and review of the literature. Br J Ophthalmol. 2000;84:1177–84.
Brei TJ, Burke MJ, Rubinstein JH. Glaucoma and findings simulating glaucoma in the Rubinstein–Taybi syndrome. J Pediatr Ophthalmol Strabismus. 1995;32:248–52.
Acknowledgements
We thank the patient and his family members for participating in the current study.
Funding
Supported by The Priming Scientific Research Foundation for the Junior Research in Beijing Tongren Hospital Capital Medical University (2017-YJJ-ZZL-009 and 2018-YJJ-ZZL-046); Beijing Tongren Hospital Top Talent Training Program; 2018 Beijing Outstanding Young Talents(2018000021469G210); Beijing Municipal Administration of Hospitals Incubating Program (Code: PX2019008); National Natural Science Foundation of China (82101146 and 82060183); The Beijing Nova Program (Z211100002121052); The key research and development project of Ningxia Hui Autonomous Region (2020BEG03047); The training project of the scientific innovation commanding talented person in Ningxia Hui Autonomous Region (KJT2020013); Leading Talent of "Innovative project" in Beijing Economic and Technological Development Zone.
Author information
Authors and Affiliations
Contributions
QW wrote the draft of manuscript. CW and XYS collected the clinical data. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from the Legal guardians of the patients (parents) for participation in this study and to publish the study findings. The consent form signed by the legal guardians of the patients (parents) has been explained in detail by the researchers, and the they have no questions about the terms. Ethical aspects of the studies and procedures related to clinical examination, human sample collection and genetic analysis were approved by the Medical Ethics Committee of the Beijing Tongren Hospital. All methods were performed in accordance with the relevant guidelines and regulations of the Declaration of Helsinki.
Consent for publication
The purpose of the study and its importance to patients and scientific community was explained to the parents of the affected individual by the clinicians. Written consent to publish study finding and to publish the images of the patient in an online open-access publication were obtained from legal guardians of the affected individuals. Clinicians involved in the disease diagnosis coordinated the consent to research team for genetic investigations.
Competing interests
The authors declare no competing intrests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Q., Wang, C., Wei, W.B. et al. A novel CREBBP mutation and its phenotype in a case of Rubinstein–Taybi syndrome. BMC Med Genomics 15, 182 (2022). https://doi.org/10.1186/s12920-022-01335-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01335-4