
Abstract
Background
Rubinstein-Taybi syndrome (RSTS) is a rare autosomal dominant neurodevelopmental disorder characterized by broad thumbs and halluces. RSTS is caused by mutations in CREBBP and in EP300 genes in 50–60% and 8%, respectively. Up to now, 76 RSTS-EP300 patients have been described. We present the clinical and molecular characterization of a cohort of RSTS patients carrying EP300 mutations.
Methods
Patients were selected from a cohort of 72 individuals suspected of RSTS after being negative in CREBBP study. MLPA and panel-based NGS EP300 were performed.
Results
Eight patients were found to carry EP300 mutations. Phenotypic characteristics included: intellectual disability (generally mild), postnatal growth retardation, infant feeding problems, psychomotor and language delay and typical facial dysmorphisms (microcephaly, downslanting palpebral fissures, columella below the alae nasi, and prominent nose). Broad thumbs and/or halluces were common, but angulated thumbs were only found in two patients. We identified across the gene novel mutations, including large deletion, frameshift mutations, nonsense, missense and splicing alterations, confirming de novo origin in all but one (the mother, possibly underdiagnosed, has short and broad thumbs and had learning difficulties).
Conclusions
The clinical evaluation of our patients corroborates that clinical features in EP300 are less marked than in CREBBP patients although it is difficult to establish a genotype-phenotype correlation although. It is remarkable that these findings are observed in a RSTS-diagnosed cohort; some patients harbouring EP300 mutations could present a different phenotype. Broadening the knowledge about EP300-RSTS phenotype may contribute to improve the management of patients and the counselling to the families.
Similar content being viewed by others
Background
Rubinstein-Taybi syndrome (RSTS; OMIM #180849, #613684) is a rare (1:125000) neurodevelopmental disorder. It affects equally males and females. This syndrome is characterized by a well-defined clinical features group including: variable degree of intellectual disability (ID), distinctive facial features (downslanting palpebral fissures, convex nasal bridge, columella below alae nasi, etc), skeletal abnormalities (broad and angulated thumbs and halluces, or duplication of distal phalanxes), growth retardation, microcephaly and behavioural problems. Broad and angulated thumbs and halluces are considered hallmarks in clinical diagnosis [1, 2].
The diagnosis of RSTS is essentially clinic based on frequent clinical characteristics such as: broad short thumbs and halluces, downslanting palpebral fissures, broad nasal bridge, hypotonia, gastrointestinal problems and recurrent infections, among others. Usually the diagnosis is made at an early stage, even at birth. However, there are some cases with milder features that lack diagnosis until adulthood [3, 4].
The first gene associated with RSTS is CREBBP, located on chromosome 16p13.3, that encodes a CREB-binding protein (CBP) [5]. Mutations in the gene EP300 were detected in individuals clinically diagnosed of RSTS, setting up this gene as an alternative cause of RSTS [6]. EP300 maps to 22q13.2 and encodes E1A-associated protein p300. CBP and p300 are ubiquitously expressed nuclear proteins, have intrinsic lysine acetyltransferase (KAT) activity and act as transcriptional coactivators in the regulation of gene expression mediating many of the same signalling pathways. They have been defined as “writers”. By acetylating histones, they loosen up the contact between histones and DNA, causing the relaxation of the chromatin and faciliting the access of transcription factors (TFs) and the basal transcriptional machinery to specific DNA sequences. RSTS is therefore classified as a disorder of the histone machinery. The fact that a defect in either CREBBP or EP300 leads to the same syndrome might indicate that both histone acetyltransferases are targeted to an overlapping set of genes [7, 8]. 50–60% of RSTS cases are caused by mutations of the CREBBP gene, and by EP300 gene mutations in around 8%. Thereby, in approximately one third of the RSTS patients the molecular cause of the syndrome remains unknown [9].
To date, about 230 causative mutations have been reported in CREBBP, in more than 200 patients. However, to the best of our knowledge, only 76 RSTS patients with EP300 mutations have been described [10,11,12,13,14].
In this report we present the clinical and molecular characterization of a cohort of 8 RSTS patients carrying EP300 mutations identified from a group of 72 RSTS patients. The description of more RSTS patients, and specifically EP300-cases may contribute to better understand the range of phenotypes providing clinical pointers that would improve earlier detection and diagnosis of these patients.
Methods
Patients
RSTS patients who underwent EP300 analysis were selected from a cohort of 72 individuals with suspected diagnosis of RSTS after being negative in CREBBP study (Multiplex ligation-dependent probe amplification (MLPA) and next generation sequencing (NGS) of the entire gene.
Clinical data, samples and photographs were obtained after written informed consent. This work has been approved by the Committee for Ethics in Clinical Research in La Rioja (CEICLAR).
Molecular analyses
Blood samples from probands and their parents, when possible (in two cases it was not possible), were collected in EDTA tubes. DNA was extracted using QIAamp DNA Mini Kit (QIAGEN) following the manufacture’s protocol. MLPA of EP300 was performed (P333 Kit, MRC-Holland). If negative, NGS of EP300 gene was carried out. Briefly, libraries encompassing exons and introns of EP300 gene were prepared using the SureSelectXT2 Custom kit (Agilent) and sequenced to generate 150 bp single reads. The resulting reads were mapped to the human genome hg19 using BWA (version 0.7.1 2). Sequence variants were called using the Genome Analysis Toolkit (GATK) version 3.3 and called variants were annotated with Annovar. Pathogenicity of the detected variants was predicted by in silico analysis with bioinformatics tools such as Sorting Intolerant From Tolerant (SIFT), Mutation Taster, Polyphen-2, and Human Splicing Finder (HSF 3.0). ExAC browser of Broad Institute, 1000 Genomes database and dbSNP138, as well as, the Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD) and ClinVar databases were checked to assess the presence/absence of detected alterations in variations repositories.
All the pathogenic variants detected were corroborated by Sanger sequencing.
Results
Phenotype of EP300-RSTS patients
From the initial cohort of 72 patients, 8 of them harboured mutations in EP300 gene, representing 11%. Phenotypic characteristics of these patients are summarized in Table 1 (for patients #47 and #57 there were not data available about some of these features since they were only 3 and 6 months old at the moment of the evaluation). The group of RSTS-EP300 individuals included 5 males and 3 females aged from 3 months to 21 years old. Diagnosis was made at early stage in 2 cases, pediatric in 6 and adulthood in one (one of the probands´ mother) [4]. According to their phenotype, RSTS was clinically suspected in all these patients.
Only in six cases it was possible to retrieve information regarding the prenatal period: preeclampsia was found in one (case #47) and mild hypertension in other (#27); prenatal growth retardation in cases #27 and #45. Postnatal growth retardation was found in cases #67 and #45, as well as in her mother. Infant feeding problems were recorded in two cases (#11 and #27).
Psychomotor delay was observed in four cases (#11, #27, #38 and #42) and ID was present in all cases, being mild in 3/7, moderate in 4/7 and severe in 1/8. Language delay was present in three cases (#27, #38 and #42). Concerning behavioral problems, autism/autism-like was reported in three cases (#27, #42 and #67) and another one presented stereotypes.
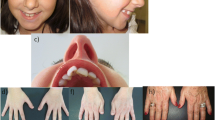
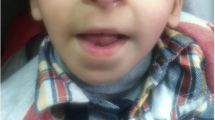
Microcephaly and typical facial dysmorphisms including downslanting palpebral fissures, columella below the alae nasi and prominent nose were found in almost all cases (Table 1, Fig. 1). Distinctive RSTS skeletal characteristics, broad thumbs and/or halluces were common, but angulated thumbs were only found in two patients (#11 and #42) (Table 1, Fig. 1).

Photographs of face, hands and feet of EP300 patients: #11 (a), #27 (b), #42 (c), #45 (d), #67 (e). For patient #45 photographs showing normal thumbs and halluces of the proband, short and broad but not angulated thumbs of her mother and her grandmother as well as detail of mother’s talon cusp at an upper incisor are shown
Features previously reported like hirsutism or keloids were only present in patients #11 and #38. Other organ malformations such as dental crowding, talon cusp, strabismus, Pectus excavatum or cryptorchidism were also found (Table 1, Fig. 1).
Genotype of EP300-RSTS patients
Among our cohort of EP300 patients we identified one large deletion, from exon12 to 21; 4 frameshift mutations (3 small deletions and 1 small insertion), 1 nonsense and 1 missense mutation and 1 intronic change with splicing alteration (Table 1, Fig. 2).

Schematic representation of p300 protein and EP300 gene, including their functional and structural domains and their localization. Distribution of the variants found in EP300 gene in our cohort are showed. Symbols represent the mutation types as indicated. The different domains of the p300 protein are represented with different colours, and the exon codifying these domains are showed in the same colour: pink (NLS, nuclear localization signal), green (TAZ1, transcriptional-adaptor zinc-finger domain 1), purple (KIX, kinase inducible domain of CREB interacting domain), orange (Br, bromodomain), yellow (HAT, histone acetyltransferase domain), blue (ZZ, bromodomain), green (TAZ2, transcriptional-adaptor zinc-finger domain 2), light pink (IBiD, IRF3-binding domain). Numbers indicate aminoacid position in the protein and the exon in the gene
Large deletion was detected thorough MLPA analysis in patient #57. It displayed a multiexonic heterozygous deletion, from exon 12 to 21. It was not possible to confirm its de novo origin because of the absence of parents samples (Fig. 2).
Five novel heterozygous-inactivating mutations were identified: in patient #67 it was found a TC deletion (c.70_71del) which generates a premature stop codon, leading to an early truncated protein (p.(Ser24Glyfs*14)). This variant was not present in her parents, confirming its de novo origin. Patient #11 harboured a de novo small duplication (c.4954_4957dup) that gives place to a premature protein truncation (p.(Cys1653Tyrfs*21)). A transition from C to G was detected in patient #38, which converts an arginine triplet into a stop codon (c.3163C > T; p.(Arg1055*)); in this case there was not possibility of studying parents samples. In patient #27 an intronic variant was found, c.3728 + 5G > C. This change was predicted to alter the splicing removing the Natural Splice Site according to several predictors. RNA analysis corroborated that 22 nucleotides, belonging to the intron 21 sequence, were introduced between exons 21 and 22 leading to a premature stop codon. This variant was not present in parent samples.
Patient #42 has a de novo deletion in exon 31 (c.6627_6638del) which generates a deletion of five aminoacids, Asn-Gln-Phe-Gln-Gln, and a insertion of Lys (p.(Asn2209_Gln2213delinsLys)).
In the case of patient #47 a de novo missense variant located in exon 28 was identified (c.4511 T > G; p.(Phe1504Cys)). This point variation was predicted as deleterious, damaging, probably damaging and disease causing according to Provean, SIFT, Polyphen-2 and Mutation Taster, respectively. Although missense mutations in EP300 have been hardly reported, the variant found in patient #47 (c.4511 T > G; p.(Phe1504Cys)) appear to be the causative of RSTS. It has been classified as pathogenic by different in silico analysis, have a Grantham score of 204.39 and a class C65 of align GVGD. Futhermore, this de novo variant is located in a functionally significant and conserved amino acid.
Analysis of the sample of patient #45 revealed a novel heterozygous frameshift mutation in exon 31 (c.7222_7223del; p.(Gln2408Glufs*39)). The deletion generates a frameshift that leads to loss of the original stop codon and results in a prolonged protein 31 aminoacids longer. However, the variant, considered to be pathogenic/likely pathogenic, according to ACMG interpretation, was present also in her mother, who also had learning difficulties and was found to have short and broad thumbs. These results were previously published [4].
All the variants detected were found in heterozygosity, as it is pointed out by the number of reads (Table 1). Moreover, all were confirmed by Sanger sequencing. To the best of our knowledge, these variants are novel (although two of them were previously described [14], Table 1), and are not included in ClinVar, HGMD or LOVD. They have been submitted to ClinVar (SCV266471, SCV000297724-SCV000297728, SCV000301482) and LOVD databases (variants #0000096186, #0000127944–0000127949, #0000130337). Furthermore, none of the detected variants was found in 100 healthy controls and are not present in 1000G, ExAC and dbSNP, pointing out that they are not common in population; with the exception of c.6627_6638del in patient #42 in which the role of this mutation is not clear.
Discussion
In this report we present the genetic and clinical characterization of 8 new cases of RSTS associated to mutations in EP300 gene. These results enlarge the number of EP300-RSTS cases to 84, broadening the knowledge about clinical presentation of these patients.
All the patients included in this study presented typical signs pointing to RSTS. The characteristics in EP300 mutated patients are less marked than in CREEBP ones, in general. Even so, microcephaly and typical facial anomalies of RSTS were generally present in our cohort: downslanting palpebral fissures, columella below the alae nasi, and prominent nose; although other RSTS dysmorphic signs were only partially found among our patients (low set and rotated ears, grimacing smile, etc), supporting previously reported data (Table 2) [10, 12, 14].
Respecting skeletal malformations, 88% of EP300-individuals displayed broad thumbs and only two cases also angulated, confirming previous results that showed the uncommon presence angulated thumbs in EP300-RSTS patients (Table 2) [7, 14].
Milder RSTS features overall were recorded in this group, in comparison with the phenotype of CREBB-patients (prenatal growth retardation, delayed growth, infant feeding problems, hirsutism and keloids) were absent or detected in a low percentage. In line with literature, neurological affectation was generally mild, ID was generally mild and most of patients did not show psychomotor or speech delay. In addition, despite the recent description of high burden of behavioral difficulties in RSTS2 by Hamilton et al. [10], we have not detected enrichment in this problem among our cohort. In the same vein, even if several studies have set up an association between maternal pre-eclampsia and fetal EP300-RSTS only one of our cases registered this clinical fact (another one suffered mild hypertension). Nevertheless, the number of cases is not significative and these data could not be collected in all of them [7, 10,11,12, 14, 15].
Even if the number of cases in our study is not representative, it agrees in the most common type of variant found, small insertions and deletions that generate a frameshift. Our results subscribe with the last data reported by Spena et al. [9] who shows in a recent review that 53.5% of mutations in EP300 are frameshift type, 25% nonsense, 14% large deletions, 3.6% missense and 3.6% splicing type (intronic).
Taking into account the location of mutations within EP300, it is important to point out that those found towards the 3′-end of the protein may result in milder phenotypes [3, 10, 15]. Furthermore, the number of mutations in exon 31 is lower, maybe because the phenotype in these patients is different and they are therefore not studied and lacked. In our study, patients #42 and #45 present mutations in this region. In the case of #45 this fact is corroborated, since the patient and her mother presented a mild phenotype with well preserved intelligence (in fact, the mother was not diagnosed before); however, patient #42 showed severe intellectual disability. Nevertheless, the role of this mutation is controversial, since it was present in ExAC, and the possibility of a second mutation separate to EP300 making a significant contribution to his phenotype has not been entirely excluded.
It is remarkable that the only missense mutation found in this work was located in HAT domain. Although this kind of mutation is not commonly found as causative of RSTS (only a single missense mutation listed in LOVD), our variant is certainly pathogenic, because it is located in the HAT domain, it affects a highly conserved amino acid and in silico analysis predicted its pathogenicity. Accordingly, other authors found similar cases of apparently pathogenic missense mutations in EP300 (Hamilton et al. 2016). In this regard, some authors described that probably missense mutations that affects HAT domain could lead to classical RSTS [14].
It has not been possible to establish a clear correlation between the genotype, type and location of mutations in EP300, with the phenotype, neither in our study, nor in the previous publications. This could be due to the low number of cases described and the wide spectrum of clinical features detected, emphasizing on the clinical variability of this syndrome. It is important to remark that our findings are observed in a RSTS-diagnosed cohort; some patients harbouring EP300 mutations present a different phenotype as it has been demonstrated by the detection of EP300-mutated individuals through “hypothesis free” approaches (microarray or exome sequencing) without a previous clinical diagnosis of RSTS [10, 13, 16]. These cases without an initial RSTS diagnosis and “typical” EP300-phenotype plus other uncommon attributes, support the wide clinical spectrum of this syndrome. Possibly, EP300 mutations may be more prevalent than we suppose, but the phenotype is different or overlaps with other syndromes, and as a result, molecular testing for RSTS is not offered.
The description of recurrent mutations in patients sharing the same defects and specific clinical signs as well as the description of other EP300-patients without RSTS diagnosis will help to set possible genotype-phenotype associations. Despite this fact, the clinical evaluation of our patients corroborates that clinical features in EP300 are less marked than in CREBBP patients, being severe ID as well as angulation of thumbs and halluces, rare.
Conclusions
In summary, it is difficult to establish a genotype-phenotype correlation, although alterations in HAT domain are proposed to cause classical RSTS and mutations not affecting this domain could explain the milder phenotypes. However, it is hampered by the low number of EP300-RSTS cases described worldwide and the clinical heterogeneity found, even among the few cases described with the same mutation. Broadening the knowledge about EP300-RSTS phenotype may contribute to improve the management of patients and the counselling to the families.
Abbreviations
- CBP:
-
CREB-binding protein
- CEICLAR:
-
Committee for Ethics in Clinical Research in La Rioja
- GATK:
-
Genome Analysis Toolkit
- HAT:
-
Histone acetyltransferase
- HGMD:
-
Human Gene Mutation Database
- ID:
-
intellectual disability
- KAT:
-
lysine acetyltransferase
- LOVD:
-
Leiden Open Variation Database
- MLPA:
-
Multiplex ligation-dependent probe amplification
- NGS:
-
next generation sequencing
- RSTS:
-
Rubinstein-Taybi syndrome
- SIFT:
-
Sorting Intolerant From Tolerant
- TFs:
-
transcription factors
References
Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006;14:981–5.
Bartholdi D, Roelfsema JH, Papadia F, Breuning MH, Niedrist D, Hennekam RC, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J Med Genet. 2007;44:327–33.
Bartsch O, Labonté J, Albrecht B, Wieczorek D, Lechno S, Zechner U, et al. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein-Taybi syndrome. Am J Med Genet. 2010;152A:181–4.
López M, Seidel V, Santibañez P, Cervera-Acedo C, Castro-de CP, Dominguez-Garrido E. First case report of inherited Rubinstein-Taybi syndrome associated with a novel EP300 variant. BMC Med Genet. 2016;17:97.
Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376(6538):348–51.
Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–80.
Negri G, Milani D, Colapietro P, Forzano F, Della MM, Rusconi D, et al. Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin Genet. 2015;87:148–54.
Fahrner JA, Bjornsson HT. Mendelian disorders of the epigenetic machinery: tipping the balance of chromatin states. Annu Rev Genomics Hum Genet. 2014;15:269–93.
Spena S, Gervasini C, Milani D. Ultra-rare syndromes: the example of Rubinstein-Taybi syndrome. J Pediatr Genet. 2015;4:177–86.
Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, Yau S, Lillis S, Hurst JA, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol. 2016;25:135–45.
Negri G, Magini P, Milani D, Colapietro P, Rusconi D, Scarano E, et al. From whole gene deletion to point mutations of EP300-positive Rubinstein-Taybi patients: new insights into the mutational Spectrum and peculiar clinical hallmarks. Hum Mutat. 2016;37:175–83.
Solomon BD, Bodian DL, Khromykh A, Mora GG, Lanpher BC, Iyer RK, et al. Expanding the phenotypic spectrum in EP300-related Rubinstein-Taybi syndrome. Am J Med Genet A. 2015;167A:1111–6.
Sellars EA, Sullivan BR, Schaefer GB. Whole exome sequencing reveals EP300 mutation in mildly affected female: expansion of the spectrum. Clin Case Rep. 2016;4:696–8.
Fergelot P, Van Belzen M, Van Gils J, Afenjar A, Armour CM, Arveiler B, et al. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am J Med Genet A. 2016;170:3069–82.
Zimmermann N, Acosta AM, Kohlhase J, Bartsch O. Confirmation of EP300 gene mutations as a rare cause of Rubinstein-Taybi syndrome. Eur J Hum Genet. 2007;15:837–42.
Jagla M, Tomasik TB, Czyz O, Krol M, van Houdt JK, Kwinta P, et al. Rubinstein-Taybi because of a novel EP300 mutation with novel clinical findings. Clin Dysmorphol. 2016; https://doi.org/10.1097/MCD.0000000000000164.
Acknowledgements
We are grateful to the patients and their families for their cooperation and their participation in the study.
Funding
NA
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Conception or design of the work: ML, EDG. Data collection: AGO, JA, IGC, SGM, FSS, VS. Data analysis and interpretation: ML, AGO, EDG. Drafting the article: ML, AGO, EDG. Critical revision of the article: ML, AGO, JA, IGC, SGM, FSS, VS, EDG. Read and approval of the version to be published: ML, AGO, JA, IGC, SGM, FSS, VS, EDG.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work has been approved by the Committee for Ethics in Clinical Research in La Rioja (CEICLAR). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants included in the study or from their parents in the case of minors.
Consent for publication
Consent Form for Publication of personal information in a scientific journal, including clinical data and image and photographs was obtained from all individual participants included in the study or from their parents in the case of minors.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
López, M., García-Oguiza, A., Armstrong, J. et al. Rubinstein-Taybi 2 associated to novel EP300 mutations: deepening the clinical and genetic spectrum. BMC Med Genet 19, 36 (2018). https://doi.org/10.1186/s12881-018-0548-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0548-2