
Abstract
Background
KIAA0586, also known as Talpid3, plays critical roles in primary cilia formation and hedgehog signaling in humans. Variants in KIAA0586 could cause some different ciliopathies, including Joubert syndrome (JBTS), which is a clinically and genetically heterogeneous group of autosomal recessive neurological disorders.
Methods and Results
A 9-month-old girl was diagnosed as JBTS by the “molar tooth sign” of the mid-brain and global developmental delay. By whole-exome sequencing, we identified a single nucleotide variant c.3303G > A and a 1.38-kb deletion in KIAA0586 in the proband. These two variants of KIAA0586 were consistent with the mode of autosomal recessive inheritance in the family, which was verified using Sanger sequencing.
Conclusions
This finding of a compound heterozygote with a 1.38-kb deletion and c.3303G > A gave a precise genetic diagnosis for the patient, and the novel 1.38-kb deletion also expanded the pathogenic variation spectrum of JBTS caused by KIAA0586.
Similar content being viewed by others


Introduction
JBTS (JBTS, OMIM#PS213300) is a clinically and genetically heterogeneous group of autosomal recessive neurological disorders characterized by a distinctive mid-hindbrain malformation (“molar tooth sign” on brain imaging), hypotonia and developmental delay/intellectual disability [1,2,3]. KIAA0586 (also known as Talpid3) encodes a centrosomal protein that is essential for primary cilia formation and hedgehog signaling [4,5,6]. Variations in KIAA0586 could cause some different ciliopathies, such as one subtype of Joubert syndrome (JBTS23, OMIM #616,490), preaxial polydactyly and short rib thoracic dysplasia 14 (SRTD14; OMIM #616,546) [7,8,9,10]. More than 50 variations in the KIAA0586 gene were reported to be cause of JBTS in homozygous or compound heterozygous states, and the types of these variations varied, including missense/nonsense mutations, splice sites, small insertions/deletions or long-fragment genomic deletions [7, 11,12,13,14,15,16,17].
With whole-exome sequencing (WES), we identified a novel 1.38-kb deletion and a single nucleotide variant (SNV) in KIAA0586 in a Joubert syndrome patient and confirmed them using Sanger sequencing in the family.
Materials and methods
Clinical summary
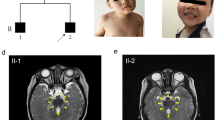
A 9-month-old girl was born after normal labor with a birth weight of 3.30 kg and height of 50 cm at 39 weeks gestation. Her parents were inconsanguineous. She is the second child of her family and has an elder sister. Her parents and sister are unaffected. The girl was hospitalized because of severe respiratory problems at birth, and then for delayed global development at 9 months. She cannot raise her head until 4 months, cannot turn over until 8 months, cannot sat all by herself, and cannot stand when supporting station. She can grasp objects on her own initiative. She can identify her mother and speak with only monosyllables, i. e. mama, baba. She often stuck out her tongue unconsciously. The girl had moderate intellectual disability, and severe retardation for large motor abilities, as evaluated by the Chinese Developmental Scale for children aged 0–6 years (WS/T 580–2017). The girl was fed with milk powder. The girl had a height of 74 cm (+ 1 SD), weight of 9 kg (median ~ + 1 SD), and occipitofrontal circumference of 45 cm (+ 1 SD) at 9 months old. Physical examination showed strabismus, hypotonia and lower myodynamia. No abnormalities were mentioned for the heart, liver, gallbladder, spleen, kidneys, ureter, or bladder. Magnetic resonance imaging of her brain revealed that the superior cerebellar peduncles were thickened and lengthened (molar tooth sign), and the shape of the cerebellar vermis was irregular and partially missing (Fig. 1). There was a link near the midline of the bilateral cerebellar hemispheres. The upper part of the fourth ventricle seemed to be bat wings. Thus, the girl was diagnosed with Joubert syndrome.

Brain magnetic resonance imaging (MRI) findings for the Joubert syndrome individual. Molar tooth sign with lengthening and thickening of superior cerebellar peduncles as indicated by white arrows in a and b, and the shape of cerebellar vermis was irregular and partially missing as indicated by black arrows in b
Genetic investigations
A 2 mL EDTA anticoagulant venous blood sample was obtained from the proband, her parents and sister. Genomic DNA was extracted from whole blood using QIAamp® DNA Blood mini Kit (QIAGEN, Germany) according to the manufacturer’s protocol. Whole-Exome Sequencing (WES) and SNVs/InDel analysis were performed for the proband as previously described [18]. Splice variants were evaluated using the online tool SpliceAI (https://spliceailookup.broadinstitute.org). Identification and annotation of genetically mobile domains and the analysis of domain architectures were performed by SMART (a Simple Modular Architecture Research Tool) (http://smart.embl-heidelberg.de/).
The WES data were further analyzed for copy number variants (CNVs). We made a virtual gene panel for JBTS and other ciliopathies using Phenolyzer [19] and OMIM (https://omim.org/) (There were a total of 169 related genes, data not shown). We obtained the chromosomal position information of exons of the classical transcripts from UCSC (https://genome.ucsc.edu/) for all candidate genes, and generated a bed file for all exons. Based on the hypothesis that exon deletion/duplication rarely occurs in JBTS, we performed CNV assessment using a read depth-based CNV detection algorithm [20]. Three WES data at the same sequencing batch, including the proband and two unaffected individuals were involved in CNV analysis. The mean depth of each exon was obtained with Samtools (https://anaconda.org/bioconda/samtools). Using Integrative Genomics Viewer (IGV, http://software.broadinstitute.org/software/igv/), the called exon deletions/duplications were confirmed manually.
SNV verification
PCR was performed using specific primer pairs for candidate SNV validation screened from WES in the proband and her family members. The Additional file 1: Table S1 lists the primer sequences.
CNV verification and precise breakpoint determination
To verify the CNV, we firstly carried out a relative quantitative PCR to analyze genome relative copy number of exon 15 of KIAA0586 (NM_001244189.1) in the family. Triplicate quantitative PCR for genomic DNA was performed using SYBR Green qPCR Master Mix (Life Technologies, USA) on ABI7500 (Life Technologies, USA). ALB (albumin) was used as an internal control. The delta-delta CT value analysis method was used to evaluate the relative copy number of genome KIAA0586 exon 15 for all samples. Target and internal control gene primer pairs are listed in Additional file 1: Table S1. Then, we performed a long PCR to determine the precise breakpoint of the deletion. Based on the predicted PCR products length, genomic DNA was amplified following the long PCR program: 94 °Cfor 3 min; 98 °Cfor 10 s, 68 °Cfor 3 min (30 cycles); 72 °Cfor 10 min. The long PCR primer sequences are shown in Additional file 1: Table S1.
RNA extraction and reverse transcription
The Tempus™ Spin RNA Isolation Kit (Invitrogen, USA) was used for RNA extraction from whole blood cells of the proband and her parents and sister. One microgram RNA was reverse transcribed into cDNA using a SuperScript™ IV First-Strand Synthesis System Kit (Invitrogen, USA). The primer sequences for investigating the effect of deletion in the transcript are listed in Additional file 1: Table S1.
The PCR amplification products from the verification of SNVs and CNVs were analyzed by agarose gel electrophoresis, and then Sanger sequencing was performed on an ABI3730xl Genetic Analyzer (Life Technologies, USA) following the manufacturer's protocol.
Results
WES revealed a splicing variant (NM_001244189.1: c.3303G > A) and a genomic region with a 1.38-kb deletion (NC_000014.9: g.58,926,242–58,927,621del) in KIAA0586 as causes of Joubert syndrome in a 9-month-old girl (Fig. 2). The reported splicing variant c.3303G > A (NM_001244189.1) was verified in the heterozygous state in the proband, her mother and sister, which was validated by direct Sanger sequencing (Fig. 2 b). This variant might cause aberrant splicing predicted by the online software SpliceAI, remove 56 base pairs at the end of exon 23 (NM_001244189.1) and terminate early translation (NP_001231118.1: p.Pro1084LysfsTer22), which was described in detail by Bachmann-Gagescu et al. [7]. Our RNA extraction and reverse transcription experiments also confirmed this finding (data not shown). By long PCR, there were two fragments amplified on the patient and her father, only one fragment on the mother and sister (Fig. 2c and Additional file 3: Fig. S1). The precise breakpoints of the deletion were at positions 58,926,242 and 58,927,621 on chromosome 14 determined by Sanger sequencing (Fig. 2d). The 1.38-kb deletion led to the removal of part of intron 14, exon 15 and part of intron 15 of KIAA0586 (NM_001244189.1), causing exon 15 skipping in the transcript (Fig. 3), which was predicted to be a 76 amino acid deletion (position 605 to 681) encoded by exon 15 during protein translation. Compared to the mother and the sister, results of qPCR showed that the proband and her father only had relative half-fold copy for exon 15 (Additional file 2: Table S2). The Database of Genomic Variants (http://dgv.tcag.ca) and Decipher (https://decipher.sanger.ac.uk) had not previously described deletions for this genomic region. According to the ACMG guidelines, these two variants were classified as pathogenic and likely pathogenic. No additional deleterious variant of other Joubert syndrome- or ciliopathy-related genes was identified in the WES data of the proband.

Pedigree of Joubert syndrome patient and Sanger chromatograms. a Pedigree of the Joubert syndrome patient. b Sanger sequencing of KIAA0586 variant c.3303G > A. The black arrow indicates the position of variant. c Gel image for long PCR. Two fragments were obtained (2,747 bp and 1,367 bp) from the patient (II 2) and the father (I 1). d Sanger sequencing of KIAA0586 genomic deletion g.58,926,242–58,927,621del. Red arrow indicates the position of the break locus, and green arrow indicates the position of the connection locus

Sanger sequencing for the splicing effect of g.58,926,242–58,927,621del in the transcript. Exon 15 was skipped in the transcript for the patient (II 2) and the father (I 1)
Discussion
We identified a novel genomic deletion (g.58,926,242–58,927,621del) in the KIAA0586 gene combined with a splicing variant (NM_001244189.1: c.3303G > A) as a cause for JBTS. The deletion was paternally inherited, and the c.3303G > A variant was maternally inherited. These two variants were present in the heterozygous state in the affected child, which was consistent with the autosomal recessive inheritance mode.
DNA fragments of approximately > 50 bp are defined as CNVs, which include deletions and duplications [21, 22]. In addition to SNVs or small insertions/deletions, CNVs may also be the cause of some human diseases [23, 24]. Three different long fragment genomic deletions in KIAA0586 were reported previously [13,14,15]. With a functional genome-wide siRNA screen, Roosing et al. found a > 15.6 kb deletion spanning exons 12 to 20 (NM_001244189.1, chr14:?_58923420_58938997_?del; c.1413-?_2793 + ?del) in a Turkish patient, and the breakpoints were not defined[14]. In the transcript, this deletion was predicted to lead to the removal of exons 12 to 20 in KIAA0586 (NM_001244189.1), causing direct splicing of exons 11 to 21, leading to early translational termination (p.Phe472LysfsTer49). An 8.26-kb genomic deletion that led to the removal of exons 8, 9 and 10 was reported by Malicdan et al. in two Jeune and Joubert syndrome families [15]. This deletion removed a 544-bp gene fragment including exons 8, 9 and 10 in KIAA0586 (NM_001244189.1) in the transcript, and led to a frameshift and formation of a premature stop codon (p.Val249GlufsTer3) in exon 11. Sumathipala et al. reported another 8.3 kb genomic deletion in KIAA0586 using WGS data [13]. The deletion also led to the removal of exons 8, 9 and 10 and then made an early termination (p.Val249GlufsTer3) in the transcript. However, these two deletions had different breakpoints, the former was in chr14 (GRCh37):g.58,910,322–58,918,560, and the latter in chr14 (GRCh37):g.58,910,301–58,918,610. In humans, TALPID3 domain is located at amino acids 183 to 1,411 as the critical functional domain of KIAA0586, which spans exon 6 to 30. The reported KIAA0586 long genomic deletions were all located in this domain and predicted to make truncated proteins. The other single nucleotide variants (SNVs)/indels in these affected individuals also produce premature stop codon. The novel genomic deletion (g.58,926,242–58,927,621del) in our study removed exon 15 in KIAA0586 (NM_001244189.1), and divided the whole TALPID3 domain into two parts (amino acids 183 to 608 and 605 to 1335), which suggested that the changed protein could perform incomplete TALPID3 functions. The other variant (c.3303G > A, p.Pro1084LysfsTer22) produced truncated protein, which may be the reason why the patient's phenotype was milder than those previously reported. Further exploration of KIAA0586 function is still necessary.
Compared to SRTD14 and other forms of Joubert syndrome, the phenotype of JBTS23 is relatively mild, and most organ systems are generally unaffected. We diagnosed the 9-month-old girl with Joubert syndrome basically for hindbrain malformation and other characteristics according to the criteria of JBTS diagnosis. The other symptoms in our patient included breathing problems, strabismus, hypotonia and lower myodynamia, but no abnormalities for other organ systems. We identified pathogenic compound heterozygous variants in KIAA0586 by WES, which was compatible with the reported genotype–phenotype for JBTS caused by KIAA0586. Thus, a genetic diagnosis of JBTS23 was given to the girl. Abnormal neurological, skeletal, and renal phenotypes are often observed in JBTS patients with KIAA0586 variants [7]. This suggests it is necessary to pay attention to the development of these systems in medical examinations for this girl.
In our study, the finding of a compound heterozygote with a 1.38-kb deletion and c.3303G > A variant gave a precise molecular diagnosis for the Joubert syndrome patient, and was helpful for the doctors and parents providing quality care to her. Meanwhile, the novel 1.38-kb deletion also expanded pathogenic variation spectrum of JBTS caused by KIAA0586. Further functional validation is still necessary for clarify of the pathogenic mechanism of the KIAA0586 gene in Joubert syndrome.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the human reference 8 genome (GRCh37/hg19, http://genome.ucsc.edu), NM_001244189.1 (https://www.ncbi.nlm.nih.gov/nuccore/NM_001244189.1).
References
Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12(7):423–30.
Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15(5):511–21.
Poretti A, Snow J, Summers AC, Tekes A, Huisman T, Aygun N, Carson KA, Doherty D, Parisi MA, Toro C, et al. Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause. J Med Genet. 2017;54(8):521–9.
Yin Y, Bangs F, Paton IR, Prescott A, James J, Davey MG, Whitley P, Genikhovich G, Technau U, Burt DW, et al. The Talpid3 gene (KIAA0586) encodes a centrosomal protein that is essential for primary cilia formation. Development. 2009;136(4):655–64.
Bangs F, Antonio N, Thongnuek P, Welten M, Davey MG, Briscoe J, Tickle C. Generation of mice with functional inactivation of talpid3, a gene first identified in chicken. Development. 2011;138(15):3261–72.
Nagase T, Ishikawa K, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O. Prediction of the coding sequences of unidentified human genes. IX. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1998;5(1):31–9.
Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, Boyle EA, Wilson M, Marques Lourenco C, Arslan M, Shendure J, et al. KIAA0586 is mutated in Joubert syndrome. Hum Mutat. 2015;36(9):831–5.
Alby C, Piquand K, Huber C, Megarbane A, Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G, Bessieres B, et al. Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am J Hum Genet. 2015;97(2):311–8.
Cocciadiferro D, Agolini E, Digilio MC, Sinibaldi L, Castori M, Silvestri E, Dotta A, Dallapiccola B, Novelli A. The splice c.1815G>A variant in KIAA0586 results in a phenotype bridging short-rib-polydactyly and oral-facial-digital syndrome: a case report and literature review. Medicine. 2020;99(8):e19169.
Wang T, Xuan Z, Dou Y, Liu Y, Fu Y, Ren J, Lu L. Identification of novel mutations in preaxial polydactyly patients through whole-exome sequencing. Mol Genet Genomic Med. 2019;7(6):e690.
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, Yeung A, Peters H, Mordaunt D, Cowie S, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090–6.
Stephen LA, Tawamie H, Davis GM, Tebbe L, Nurnberg P, Nurnberg G, Thiele H, Thoenes M, Boltshauser E, Uebe S et al: TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). Elife. 2015; 4.
Sumathipala D, Stromme P, Gilissen C, Einarsen IH, Bjorndalen HJ, Server A, Corominas J, Hassel B, Fannemel M, Misceo D, et al. Sudden death in epilepsy and ectopic neurohypophysis in Joubert syndrome 23 diagnosed using SNVs/indels and structural variants pipelines on WGS data: a case report. BMC Med Genet. 2020;21(1):96.
Roosing S, Hofree M, Kim S, Scott E, Copeland B, Romani M, Silhavy JL, Rosti RO, Schroth J, Mazza T, et al. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. Elife. 2015;4:e06602.
Malicdan MC, Vilboux T, Stephen J, Maglic D, Mian L, Konzman D, Guo J, Yildirimli D, Bryant J, Fischer R, et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J Med Genet. 2015;52(12):830–9.
Akawi N, McRae J, Ansari M, Balasubramanian M, Blyth M, Brady AF, Clayton S, Cole T, Deshpande C, Fitzgerald TW, et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat Genet. 2015;47(11):1363–9.
Summers AC, Snow J, Wiggs E, Liu AG, Toro C, Poretti A, Zein WM, Brooks BP, Parisi MA, Inati S, et al. Neuropsychological phenotypes of 76 individuals with Joubert syndrome evaluated at a single center. Am J Med Genet A. 2017;173(7):1796–812.
Shen Y, Wang H, Liu Z, Luo M, Ma S, Lu C, Cao Z, Yu Y, Cai R, Chen C, et al. Identification of two novel pathogenic variants of PIBF1 by whole exome sequencing in a 2-year-old boy with Joubert syndrome. BMC Med Genet. 2020;21(1):192.
Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015;12(9):841–3.
Fromer M, Purcell SM. Using XHMM software to detect copy number variation in whole-exome sequencing data. Curr Protoc Hum Genet. 2014;81:7–23.
Kerkhof J, Schenkel LC, Reilly J, McRobbie S, Aref-Eshghi E, Stuart A, Rupar CA, Adams P, Hegele RA, Lin H, et al. Clinical validation of copy number variant detection from targeted next-generation sequencing panels. J Mol Diagn JMD. 2017;19(6):905–20.
Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20(1):117.
Truty R, Paul J, Kennemer M, Lincoln SE, Olivares E, Nussbaum RL, Aradhya S. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med Off J Am Coll Med Genet. 2019;21(1):114–23.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–46.
Acknowledgements
We were so thankful for participation of the patient family in this study.
Funding
The work was supported by the following: National Key Research and Development Program of China (2016YFC1000307) to Xu Ma, The Non-profit Central Research Institute Fund of National Research Institute for Family Planning (2020GJZ05) to Minna Luo. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YS was the major contributor in writing the manuscript. YS and ML analyzed and interpreted the patient data. CL and TC contributed to DNA extraction and sequencing experiments. ZC and CC analyzed the sequencing data. XM and HG provided useful suggestions for writing the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the patient’s parents. This study was approved by the ethical committees of the National Research Institute for Family Planning and clearly stated that the blood samples would be used for scientific research purposes, including genetic studies. All the procedures followed were in accordance with the Helsinki Declaration.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1.
Primers for verification of candidate variants.
Additional file 2. Table S2.
Results of qPCR for the 1.38kb deletion.
Additional file 3. Fig. S1.
Original gel image for long PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, Y., Lu, C., Cheng, T. et al. A novel 1.38-kb deletion combined with a single nucleotide variant in KIAA0586 as a cause of Joubert syndrome. BMC Med Genomics 16, 4 (2023). https://doi.org/10.1186/s12920-023-01438-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01438-6