
Abstract
Background
Joubert syndrome (JS) is a group of rare ciliopathies, mainly characterized by cerebellar dysplasia representing the “molar tooth sign (MTS)” on neuroimaging, hypotonia, and developmental delay. Having a complicated genotype-phenotype correlation due to its rich genetic heterogeneity, JS is usually combined with other organic defects affecting the retina, kidney, and liver. This report aimed to present new cases and novel variants of JS.
Case presentation
Five unrelated patients who were diagnosed with JS, with or without typical clinical characteristics, received integrated examinations, including whole-exome sequencing (WES) and Sanger sequencing. We identified nine pathogenic variants in the TCTN2, CPLANE1, INPP5E, NPHP1, and CC2D2A genes.
Conclusion
Four novel pathogenic mutations in the TCTN2, CPLANE1, and INPP5E genes were reported. The findings broadened the genotypic spectrum of JS and contributed to a better understanding of genotype-phenotype correlation.
Similar content being viewed by others

Background
Joubert syndrome (JS) is a rare developmental malformation, also known as a ciliopathy, which is mainly inherited in an autosomal recessive manner [1]. According to few epidemiological investigations, the prevalence of JS was roughly estimated to be between 1 and 80,000 and 1 in 100,000 [2, 3]. Because of the tremendous genetic heterogeneity of JS, multiple cilium-related genes have recently been discovered as causative in JS by booming genetic testing techniques. First described in 2004, mutations in NPHP1 and AHI1 associated with JS came to light [4, 5]. So far, over 40 genes have been linked to JS, and the related pathogenic variants account for more than 60% of the identified variants in different JS cohorts [6]. JS-causative genes encode proteins participating in the formation and function of the basal body and transition zone (TZ) of primary cilium [7], which not only function as cell sensors in almost all types of cells but also control several developmental signaling pathways, including Sonic Hedgehog (Shh), Wnt, and planer cell polarity [2]. The aberrant primary cilia caused by pathogenic variants in these genes lead to abnormal embryonic development. Aside from the commonly observed autosomal recessive pattern of inheritance, an X-linked recessive manner is particularly associated with pathogenic variants in the OFD1 gene, and recently an autosomal dominant pattern is unveiled by causative variants in the SUFU gene [8, 9].
The classical features of JS include the “molar tooth sign (MTS)” on neuroimaging, hypotonia with subsequent ataxia, and developmental delay (DD) or intellectual disability [10]. The canonical MTS characterized by cerebellar abnormalities portraying cerebellar vermis hypoplasia, thickened, elongated, and horizontally oriented superior cerebellar peduncles, and fourth ventricle enlargement [11]. Clinical evidence of MTS is necessary for the diagnosis of JS [12] and it could be mild in some patients found with thinner cerebellar peduncles and slight vermis dysplasia [5, 13]. In addition to the characteristics mentioned, abnormal respiratory patterns, such as alternating tachypnea or apnea after delivery, and abnormal eye movements, including nystagmus, strabismus, and oculomotor apraxia, are frequently observed in patients [14]. Furthermore, depending on the severity of the disease, a high proportion of individuals with JS had concomitant organ abnormalities, which included retinal involvement, renal disease, and hepatic fibrosis [15]. The varying organ abnormalities in JS patients could be explained by JS-causative gene deficiencies in corresponding type of cells, including kidney epithelial cells, retinal photoreceptors, and nerve cells [16, 17].
Previously, a group of diseases with MTS and typical clinical features of JS combined with systemic involvements were named “JS-related disorders”. Such diseases were divided, based on associated comorbidities, into clinical subtypes as follows: classic or pure JS, JS with retinal disease (JS-Ret), JS with renal disease (JS-Ren), JS with oculorenal disease (JS-OR), JS with hepatic disease (JS-H), JS with oral-facial-digital features (JS-OFD), JS with acro-callosal features (JS-AC), and JS with Jeune asphyxiating thoracic dystrophy (JS-JATD). “Joubert syndrome” is now used as a general term for this group of diseases [2].
In this study, we described five patients with or without the typical clinical symptoms of JS, and using whole-exome sequencing (WES), we pointed out four novel pathogenic mutations in the TCTN2, CPLANE1, and INPP5E genes that enriched the genotypic spectrum of JS.
Case presentation
Clinical manifestations
Patient 1 was a 3-month-old boy admitted to our department due to hypotonia. The patient, who was the third child in the family, was delivered vaginally at term and weighed 3280 g at birth. His parents insisted on the delivery, despite of the fetal brain MRI performed at 28 weeks of gestation suggesting the cerebellar vermis was absent. The patient also had a hearing impairment and failed to control his head with a soft neck. Gesell scores for the patient indicated a moderate to severe DD: 40 in gross motor, 54 in fine motor, 40 in adaptability, 40 in language ability, and 34 in social ability. His cranial MRI records revealed a typical MTS, an enlarged fourth ventricle, and cerebellar vermis dysplasia. However, the images could not be recovered due to a data disaster. Other routine physical exams and biochemical tests were all normal. His parents and the 6-year-old sister were healthy. The 12-year-old sister, showing claw-like hands with slender, curving fingers and intellectual disability, was unable to walk until she was five years old and fell frequently. We failed to follow up on this patient and his family members due to the COVID-19 outbreak in 2019.
Patient 2, female, the only child, was admitted to our department at 9 months due to delayed developmental milestones. She had hypotonia, sitting unsteadily and making incomprehensible sounds. Her mother was treated with levothyroxine because of hypothyroidism during pregnancy, though, the patient was born at term with a birth weight of 3000 g and with a normal thyroid function. However, she had poor visual fixation and esotropia after birth. At the age of 10 months, her Gesell assessment revealed a moderate to severe DD: 35 in gross motor, 35 in fine motor, 35 in adaptability, 40 in language ability, and 40 in social ability. Cranial MRI showed a typical MTS and “batwing” appearance of fourth ventricle (Fig. 1A-C). No abnormalities were detected in routine physical exams and biochemical tests. The phenotypes of her parents were normal and there was no relevant family history. The patient has been receiving regular rehabilitation therapies in our department and her Gesell scores at the age of two has improved to 37, 57, 55, 57, and 51, indicating mild to moderate DD. By the time she was three years old, she had a better visual fixation and chasing ability, despite the limited abduction of her left eye. She was also able to understand straightforward instructions, talk in simple words, laugh, and develop fair hand functions. However, hypotonia remained and she could not stand or walk alone.

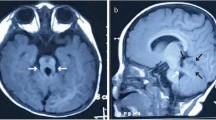
MRI results of four patients. (A-C) Patient 2.(A) Axial T2-weighted image shows cerebellar vermis hypoplasia, a deepened interpeduncular fossa and thickened, elongated superior cerebellar peduncles (arrow), presents a “molar tooth sign”; (B) Enlargement of the fourth ventricle, “bat-wing”(arrow); (C) Parasagittal T1-weighted image showed thickened, elongated, and horizontally oriented superior cerebellar peduncles (arrow); (D-F) Patient 3. (D) Axial T1-weighted image (7 months) shows a blurry deepened interpeduncular fossa and thickened, elongated superior cerebellar peduncles (arrow); (E) Axial T1-weighted image (3 years) shows vermian hypoplasia, thickened, elongated superior cerebellar peduncles (arrow); (F) Axial T2-weighted image (3 years) shows a “bat-wing” of the fourth ventricle; (G-I) Patient 4. (G) Axial T1-weighted image shows slight vermian dysplasia and thin but elongated cerebellar peduncles (arrow); (H) Axial T2-weighted image shows thickened cortex, sparse gyri, and shallow sulci especially in the bilateral frontotemporal lobes (arrow); (I) Parasagittal T1-weighted image shows cerebellar hypoplasia, thin, elongated and horizontally oriented cerebellar peduncles (arrow); (J-L) Patient 4. (J-K) Axial T1 and T2-weighted image shows thin and elongated cerebellar peduncles (arrow); (L) The subarachnoid space is widened bilaterally in the frontotemporal regions (arrow), the ventricular system is enlarged, and the volume of white matter is less than peers, suggesting white matter dysplasia
Patient 3, male, was admitted to our department due to abnormal ocular movements at 7 months. He suffered from downward-limited eye movement, horizontal nystagmus, binocular exotropia, and head tremors. The patient was the second child of the family and born at term with a birth weight of 3100 g. On the ninth day after birth, he developed pathological jaundice, which was improved after exchange transfusion and phototherapy. At 7 months, a Gesell assessment was performed due to delayed motor and linguistic milestones, including neck weakness, wobbly sitting, and incomprehensible pronunciation. The results suggested mild DD: 46 in gross motor, 65 in fine motor, 46 in adaptability, 78 in language capacity, and 65 in social ability. His brainstem auditory evoked potential test indicated moderate hearing impairment. Cranial MRI revealed MTS, which was characterized by the deepening of interpeduncular fossa and the thickening and lengthening of superior cerebellar peduncles (Fig. 1D-F). Regular physical examinations and biochemical tests were normal. The patient’s sister likewise suffered from hypotonia, strabismus, severe myopia, and nystagmus. Follow-up showed that even though the patient turned 3 years old, he could only speak, crawl, and stand with assistance while the ocular abnormalities still remained.
Patient 4 was a 5-year-old boy admitted to our department due to delayed motor and language milestones. He was unable to stand and walk alone and he had poor hand functions and unconscious vocalization. The patient was one of the two living births in a family that had 5 unexplained miscarriages and his older brother was healthy. He was delivered via cesarean section at full term, weighing 2800 g. At the age of 26 months, his Gesell scores showed a severe DD: 48 in gross motor, 33 in fine motor, 31 in adaptability, 33 in language ability, and 38 in social ability. He had seizures at 28 months and the seizure symptoms disappeared after taking sodium valproate. Besides, broad forehead, slightly depressed nasal bridge, low-set ears, micrognathia, adduction deformity of thumb, and slender toes were noticed, along with hypertonia and limited function of the right limbs. Now his EEG result was normal, but blood tests showed a decreased level of red cells, hemoglobin, hyperuricemia, and increased level of alkaline phosphatase. Cranial MRI revealed thickened cortex, sparse gyri, shallow sulci especially in the bilateral frontotemporal lobes, thin but elongated cerebellar peduncles and slight vermis dysplasia (Fig. 1G-I). The follow-up to the age of 7 showed that his DD and hypertonia were not improved.
Patient 5, male, the family’s first child, was admitted to our department due to delayed developmental milestones. He was delivered vaginally at term and weighed 4090 g at birth. Nystagmus and impaired eye movement were discovered during a physical examination. At 7 months, he was unable to sit alone and make comprehensible sounds. Gesell scores showed mild DD: 70 in gross motor, 80 in fine motor, 80 in adaptability, 70 in language ability, and 80 in social ability. Cranial MRI exhibited MTS with a deepened interpeduncular fossa, lengthened superior cerebellar peduncles, white matter abnormalities, and enlarged ventricular system (Fig. 1J-L). Abdominal ultrasonography showed he had an enlarged liver that the lower margin of the liver extended 29 mm beyond the lower margin of the right costal arch. Other physical examinations and biochemical tests were normal. The patient’s uncle had a history of seizures. A follow-up examination revealed that the patient could play and sit independently at 11 months old; however, his motor and linguistic skills fell behind those of his peers, and he still had horizontal nystagmus.
The details of the clinical characteristics of all five patients were provided in Table 1.
Genetic tests
After obtaining prior written consent of the parents, venous blood samples from all patients and their parents were collected and sent to the Chigene (Beijing) Translational Medical Research Center Co. Ltd. (Beijing, China) for trio-based WES. Sanger sequencing and the American College of Medical Genetics and Genomics (ACMG) clinical practice guidelines [18] were applied for variants interpretation in this study. To be specific, Patient 1 carried a compound heterozygous variation in the TCTN2 gene consisting of 2 variants, c.916 C > T (p.Q306*) and c.1147G > T (p.E383*) (NM_024809.5) (Fig. 2A-C); Patient 2 had two variants in the CPLANE1 gene, one is c.1819_1820insT(p.Y607Lfs*12) (NM_023073.4), the other is a haploid replication of exon 34–41 (NM_023073.4), detected by fluorogenic quantitative PCR (Fig. 2D-F); Patient 3 carried a complex heterozygous variation in the INPP5E gene consisting of 2 variants, c.669_670delGC (p.A223Afs*66) and c.1393G > A (p.V465I) (NM_019892.6) (Fig. 2G-I); Patient 4 had a homozygous deletion in the NPHP1 gene (exon 1–20) (NM_000272.5), while both his parents sheltered a heterozygous deletion in the NPHP1 gene (exon 1–20) respectively (Fig. 2J-K); Patient 5 carried a compound heterozygous variation in the CC2D2A gene composed of 2 variants, c.2728 C > T (p.R910*) and c.4238G > A (p.C1413Y) (NM_001080522.2) (Fig. 2L-N). Overall, four novel variants, TCTN2 Q306* and E383*, CPLANE1 Y607Lfs*12, INPP5E A223Afs* were identified. (Table 2)

Pedigree diagrams and genetic variants of the patients. (A-C) patient 1. (A) The pedigree diagram of the family, the proband’s parents and the younger sister were phenotypically normal; the proband’s elder sister carried the same variation with the proband. (B) Sanger sequencing confirmed heterozygous c.916 C > T and (C) heterozygous c.1147G > T in the TCTN2 gene; (D-F) patient 2.(D) The pedigree diagram; (E) Sanger sequencing confirmed a heterozygous c.1819_1820insT variant in the CPLANE1 gene; (F) Fluorogenic quantitative PCR result for heterozygous CPLANE1 exon 34–41 duplication, the ALB gene was used as the reference gene; (G-I) patient 3.(G) The pedigree diagram of the family, the proband’s sister carried the same variation with the proband; (H) Sanger sequencing result shows a heterozygous c.669_670delGC variant in the INPP5E gene; (I) Sanger sequencing confirmed a heterozygous c.1393G > A variant in the family; (J,K) patient 4.(J) The pedigree diagram; (K) Schematic representation of capture efficiency for homozygous NPHP1 exons 1–20 deletion; (L-N) patient 5.(L) The pedigree diagram; (M) Sanger sequencing confirmed a heterozygous c.2728 C > T variant and (N) a heterozygous c.4238G > A variant in the CC2D2A gene
Discussion
The diagnosis of JS takes MTS as a necessary criterion, classically characterized by thickened and elongated superior cerebellar peduncles, with the major manifestations including hypotonia and DD. As one of the ciliopathies, JS also has overlapping symptoms with Meckel syndrome, nephronophthisis or other diseases, especially the extra-neuro symptoms such as polydactyly, retinal dystrophy, polycystic kidney, and renal/liver fibrosis [7]. In this study, all five patients showed cerebellar hypoplasia with MTS and varying degrees of DD. After excluding other malformations of cerebellar development based on neuroimaging, JS was initially diagnosed. However, milder forms of MTS were discovered in patients 4 and 5, along with dysplasia of the cortex or white matter. They also suffered from hypertonia, rather than hypotonia in typical JS, and Patient 4 in addition had seizures. It raises a possibility that white matter dysplasia or seizures triggered hypertonia in these patients. At the same time, three patients manifested ocular abnormalities (Patient 2, 3, 5), which are common symptoms of JS. Besides, we did not find any other organ defects. To further build the connections between phenotype and genotype, we identified five variations in the TCTN2, CPLANE1, INPP5E, NPHP1, and CC2D2A genes, confirming the JS diagnosis.
TCTN2 localizes to the outer compartments of TZ [19] and is crucial in the regulation of ciliogenesis and embryonic development, especially the development of the nervous system [20]. Shown by genetic tests, Patient 1 had a compound heterozygous variation in the TCTN2 gene comprising of previously unreported mutations c.916 C > T (p.Q306*) and c.1147G > T (p.E383*). Compared to other JS-associated genes, TCTN2 gene mutations are more likely to result in intellectual disability and less likely to result in renal, hepatic, or retinal involvement [21]. Similarly, Patient 1 had a severe DD with no other organ defects.
A component of TZ, CPLANE1 is commonly associated with JS cases, of which over 125 mutations were identified, accounting for about 8–14% of all JS patients [22, 23]. These mutations in CPLANE1 lead to dysfunction of TZ, abnormalities of ciliogenesis and Shh signal transduction, causing defects in cerebellar developmental [23]. From Patient 2, the haploid replication of exons 34–41 in the CPLANE1 gene is the latest discovery. The other genomic alteration c.1819_1820insT in the CPLANE1 gene is novel, but the amino acid change p.Y607Lfs*12 (rs777686211) shares the same result with a previously reported case (c.1819delT, p.Y607Lfs*6) [22].
NPHP1 and CC2D2A similarly function as essential components of TZ, playing roles in ciliary development. In terms of Patient 4, the homozygous deletion in the NPHP1 gene (exon 1–20) results in loss of function of the protein, which has been observed in several other JS patients [24, 25]. Notably, as the unusual MRI finding of Patient 4, NPHP1-associated JS cases have repeatedly been reported to exhibit milder “MTS” [5, 13, 25], suggesting that NPHP1 defects may have less impact on cerebellar development. Nonetheless, seizures have not been reported in NPHP1-associated JS, while such symptom have been linked to other JS-related genes, such as the CC2D2A and AHI1. The relationship between JS genotype and seizure is ambiguous, but seizure management is similar for JS and non-JS individuals [26]. As for Patient 5, c.4238G > A (p.C1413Y) in the CC2D2A gene was previously reported in JS [25] and nephronophthisis [27], and c.2728 C > T (p.R910*) was also found in a case of prenatally diagnosed JS [28].
Unlike the previously mentioned proteins, INPP5E localizes to various subcompartments of primary cilium in a TZ-dependent manner and regulates ciliogenesis through phosphoinositide 3-kinase signaling pathway [29]. The variant c.1393G > A (p.V465I) in the INPP5E gene was previously detected in a patient with inherited retinal degenerations [30], and c.669_670delGC (p.A223Afs*66) is firstly found in JS.
Since there is no curative therapy for patients with JS, only regular follow-up and symptomatic treatment are practicable. Although a gene-targeted therapy for JS has entered the early stage of research, the extreme genetic heterogeneity of JS and the differences between human and animal models make the research exceedingly difficult [31]. Prenatal screening accordingly becomes crucial, especially for families having a JS-related history. Routine prenatal ultrasound and fetal brain MRI can be used for early screening for cerebellar malformations, but they may not work well due to the inability to detect a typical MTS, which needs genetic tests to confirm the prenatal diagnosis [8, 32, 33]. Careful follow-up and rehabilitation training may enhance the patients’ daily activities for individuals who receive their diagnosis in the early stage of life. Regarding possible involvement in several organs, such as retinal atrophy and renal/liver disorders, it is necessary to monitor the renal status and renal/liver function of the patients for a long period [31]. To date, no patient in this study has shown extra organic involvement. All patients received individual, scientific, and regular rehabilitation, including transcranial magnetic/ultrasound stimulation, physical therapy, occupational therapy, speech therapy and traditional Chinese medicine treatment, such as acupuncture, moxibustion and massage. Follow-ups showed that Patient 2 has made considerable progress, while Patient 4 and 5 improved slowly due to a complex encephalodysplasia and a history of seizures.
Indeed, our research had certain limitations. First, we cannot regain the MRI images of Patient 1 and lost the follow-ups, which makes it an imperfect case. Second, patients from other cities cannot receive timely rehabilitation and developmental assessments because of the COVID-19 epidemic. Third, the siblings of Patient 1 and 3 also had related symptoms but they did not receive proper diagnosis and treatment due to family reasons.
In conclusion, we diagnosed JS in five patients, and the WES data revealed four novel variants in the TCTN2, CPLANE1, and INPP5E genes. Our findings enrich the spectrum of pathogenic variants in JS and provide more practical experience in genetic diagnosis and counseling. Hoping that our reports could shed light on the clinical diagnosis of JS and trigger further studies on therapies.
Data availability
The majority of data presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Abbreviations
- JS:
-
joubert syndrome
- MTS:
-
molar tooth sign
- WES:
-
whole-exome sequencing
- TCTN2:
-
tectonic-2
- CPLANE1:
-
ciliogenesis and planar polarity effector complex subunit 1
- INPP5E:
-
inositol polyphosphate-5-phosphatase
- NPHP1:
-
nephrocystin 1
- CC2D2A:
-
coiled-coil and C2 domain-containing protein 2 A
- TZ:
-
transition zone
- Shh:
-
sonic hedgehog
- DD:
-
developmental delay
- MRI:
-
magnetic resonance imaging
- EEG:
-
electroencephalogram
- ACMG:
-
American College of Medical Genetics and Genomics
- PCR:
-
polymerase chain reaction
References
Parisi M, Glass I. Joubert Syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al. editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993–2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993.
Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12(9):894–905.
Nuovo S, Bacigalupo I, Ginevrino M, Battini R, Bertini E, Borgatti R, et al. Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology. 2020;94(8):e797–e801.
Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36(9):1008–13.
Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75(1):82–91.
Gana S, Serpieri V, Valente EM. Genotype-phenotype correlates in Joubert syndrome: a review. Am J Med Genet Part C Seminars Med Genet. 2022;190(1):72–88.
Van De Weghe JC, Gomez A, Doherty D. The Joubert-Meckel-Nephronophthisis Spectrum of Ciliopathies. Annu Rev Genom Hum Genet. 2022;23:301–29.
Zhang YW, Qu HB, Long N, Leng XY, Liu YQ, Yang Y. A rare mutant of OFD1 gene responsible for Joubert syndrome with significant phenotype variation. Mol Genet Genomics: MGG. 2021;296(1):33–40.
Serpieri V, D’Abrusco F, Dempsey JC, Cheng YH, Arrigoni F, Baker J, et al. SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum. J Med Genet. 2022;59(9):888–94.
Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genetics: EJHG. 2007;15(5):511–21.
Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. Joubert syndrome revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12(7):423–30.
Poretti A, Boltshauser E, Valente EM. The molar tooth sign is pathognomonic for Joubert syndrome! Pediatr Neurol. 2014;50(6):e15–6.
Castori M, Valente EM, Donati MA, Salvi S, Fazzi E, Procopio E, et al. NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J Med Genet. 2005;42(2):e9.
Wang SF, Kowal TJ, Ning K, Koo EB, Wu AY, Mahajan VB et al. Review of ocular manifestations of Joubert Syndrome. Genes. 2018;9(12).
Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Translational Sci rare Dis. 2019;4(1–2):25–49.
Hildebrandt F, Benzing T, Katsanis N, Ciliopathies. N Engl J Med. 2011;364(16):1533–43.
Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24(2):98–105.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Medicine: Official J Am Coll Med Genet. 2015;17(5):405–24.
Yang TT, Su J, Wang WJ, Craige B, Witman GB, Tsou MF, et al. Superresolution Pattern Recognition reveals the architectural map of the Ciliary Transition Zone. Sci Rep. 2015;5:14096.
Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145(4):513–28.
Huppke P, Wegener E, Böhrer-Rabel H, Bolz HJ, Zoll B, Gärtner J, et al. Tectonic gene mutations in patients with Joubert syndrome. Eur J Hum Genetics: EJHG. 2015;23(5):616–20.
Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, et al. Joubert syndrome: genotyping a northern european patient cohort. Eur J Hum Genetics: EJHG. 2016;24(2):214–20.
Zhang X, Shen Y, Li P, Cai R, Lu C, Li Q, et al. Clinical heterogeneity and intrafamilial variability of Joubert syndrome in two siblings with CPLANE1 variants. Mol Genet Genom Med. 2021;9(6):e1682.
Radha Rama Devi A, Naushad SM, Lingappa L. Clinical and molecular diagnosis of Joubert Syndrome and Related Disorders. Pediatr Neurol. 2020;106:43–9.
Zhang J, Wang L, Chen W, Duan J, Meng Y, Yang H, et al. Whole exome sequencing facilitated the diagnosis in four chinese pediatric cases of Joubert syndrome related disorders. Am J Translational Res. 2022;14(7):5088–97.
Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52(8):514–22.
Kang HG, Lee HK, Ahn YH, Joung JG, Nam J, Kim NK, et al. Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol Med. 2016;48(8):e251.
Wen H, Chen L, Yan K, He J. [Prenatal diagnosis of Joubert syndrome:one case report and literature review]. Zhejiang da xue xue bao Yi xue ban = Journal of Zhejiang University Medical Sciences. 2017;46(3):274–8.
Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41(9):1027–31.
Sangermano R, Deitch I, Peter VG, Ba-Abbad R, Place EM, Zampaglione E, et al. Broadening INPP5E phenotypic spectrum: detection of rare variants in syndromic and non-syndromic IRD. NPJ Genomic Medicine. 2021;6(1):53.
Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, Chen ML, D’Arrigo S, Glass IA, et al. Healthcare recommendations for Joubert syndrome. Am J Med Genet Part A. 2020;182(1):229–49.
Wang T, Liu YX, Luo FM, Dong Y, Li YL, Fan LL. A novel homozygous variant of TMEM231 in a case with hypoplasia of the cerebellar Vermis and Polydactyly. Front Pead. 2021;9:774575.
Zhu H, Chen W, Ren H, Zhang Y, Niu Y, Wu D, et al. Non-classic splicing mutation in the CPLANE1 (C5orf42) gene cause Joubert syndrome in a fetus with severe craniocerebral dysplasia. Eur J Med Genet. 2021;64(6):104212.
Acknowledgements
We thank all the patients and their families for their participation in this study and acknowledge the expertise of Chigene (Beijing) Translational Medical Research Center Co. Ltd. (Beijing, China).
Funding
This study was supported by the National Natural Science Foundation of China, Nos. 9021332203.
Author information
Authors and Affiliations
Contributions
LF and LW collected data, carried out the initial analyses, drafted the initial manuscript, prepared figures and tables, reviewed, and revised the manuscript. DW conceptualized and designed the study, reviewed, and revised the manuscript. DW, XX, and LY diagnosed all five patients. SP and DW made neurodevelopmental evaluations of the patients. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the Ethic Committee of Anhui Medical University.
Consent for publication
Written informed consent was obtained from the participants’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fang, L., Wang, L., Yang, L. et al. Novel variants identified in five Chinese families with Joubert Syndrome: a case report. BMC Med Genomics 16, 221 (2023). https://doi.org/10.1186/s12920-023-01669-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01669-7