
Abstract
Background
Porcine circovirus type 2 (PCV2) is the pathogen of porcine circovirus associated diseases (PCVAD) and one of the main pathogens in the global pig industry, which has brought huge economic losses to the pig industry. In recent years, there has been limited research on the prevalence of PCV2 in Henan Province. This study investigated the genotype and evolution of PCV2 in this area.
Results
We collected 117 clinical samples from different regions of Henan Province from 2015 to 2018. Here, we found that the PCV2 infection rate of PCV2 was 62.4%. Thirty-seven positive clinical samples were selected to amplify the complete genome of PCV2 and were sequenced. Based on the phylogenetic analysis of PCV2 ORF2 and complete genome, it was found that the 37 newly detected strains belonged to PCV2a (3 of 37), PCV2b (21 of 37) and PCV2d (13 of 37), indicating the predominant prevalence of PCV2b and PCV2d strains. In addition, we compared the amino acid sequences and found several amino acid mutation sites among different genotypes. Furthermore, the results of selective pressure analysis showed that there were 5 positive selection sites.
Conclusions
This study indicated the genetic diversity, molecular epidemiology and evolution of PCV2 genotypes in Henan Province during 2015–2018.
Similar content being viewed by others

Background
Porcine circovirus (PCV) is a kind of single-stranded circular DNA virus, including PCV1, PCV2 and PCV3. The non-pathogenic PCV1 was first discovered in 1974 [1, 2]. In the late 1990s, the first post-weaning multisystemic wasting syndrome (PMWS) outbreak was identified and soon PCV2 became endemic causing severe economic losses in the swine industry [3, 4]. Porcine circovirus type 2 (PCV2) plays a significant role in porcine circovirus associated diseases (PCVAD) [5], previously known as PMWS [6]. Many studies have reported the co-infection of PCV2 with other swine pathogens, such as porcine reproductive and respiratory syndrome virus, porcine parvovirus, swine influenza virus, Mycoplasma pneumoniae and Salmonella spp. [7]. Recently, a novel circovirus was identified by next generation sequence (NGS) analysis from aborted fetuses of sows and named porcine circovirus type 3 (PCV3) [8]. The newly discovered virus was associated with porcine dermatitis and nephropathy syndrome (PDNS), reproductive failure, cardiac and multisystemic inflammation [8,9,10,11]. In addition, PCV3 was also detected in healthy pigs without any clinical signs [12].
PCV2 encodes at least five proteins, including two major proteins, Rep and Cap, which are encoded by open reading frame 1(ORF1) and open reading frame 2(ORF2), respectively [13, 14]. ORF2 encodes the capsid, which is a unique structure protein (Cap) and main antigenic determinant of PCV2, which is also commonly used for analyzing PCV2 genetic diversity [15]. Currently, five major genotypes were distinguished, including PCV2a-e [16, 17]. PCV2a was the earliest genotype. Over time, PCV2b and PCV2d are the major genotypes widely existing in the world, and PCV2c was only found in some areas [18,19,20,21,22]. PCV2e is a new genotype that has been circulating in the United States since 2015. Compared with PCV2a-PCV2d, its ORF2 sequences have 12 or 15 additional nts [17, 23]. In addition, a few recombinants of different genotypes existed in nature [24]. PCV2 have a high evolutionary dynamics as single-stranded RNA viruses [3].
Although the current vaccines have decreased the economic impact of PCVAD in pig herds [25], PCV2 remains economically important in the global swine industry and still exists in pig herds. Continuous reports of newly emerging strains worldwide, but information on the genetic variation of PCV2 in the Henan province is limited. Henan Province is the hinterland of China, with well-developed transportation, as well as a large pig-raising province. There are many pig farms, frequent introduction and transfer of pigs, and low biosafety level. All these will bring opportunities for the transmission, variation and recombination of pathogenic microorganisms. Therefore, to understand the current molecular epidemiology of PCV2 in central China, the objective of the present work was to investigate the epidemiological and evolutionary characteristics of PCV2 in the Henan province from 2015 to 2018.
Results
PCV2 detection and genomic sequences
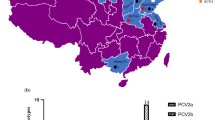
All 117 tissue samples from Henan Province during 2015–2018 were detected by PCR, of which 73 were PCV2 positive, with the infection rate as high as 62.4% (Fig. 1). 37 PCV2 genomes were selected and obtained for further analysis of PCV2 characteristics. Table 1 summarizes the strain designation, year of collection, geographic origin, genotype and GenBank accession number of these PCV2 genomes. Interestingly, 2 (HN-QY-2016 and HN-BF-2016) out of the 37 PCV2 genomes obtained in this study included 1778 nt. Apart from above two strain, the genome size of PCV2 HN-JY-2015 strain was 1766 nt and other two (HN-YY-2016 and HN-LH-2018) genome had 1768 nt, all other PCV2 genome size was 1767 nt, that is still within the size range of published PCV2 genomic sequences (1766–1768 nt). Pairwise-sequence comparisons of complete genomes revealed that the nucleotide sequence similarity between 37 PCV2 strains and 15 PCV2 reference strains varied from 93.6 to 99.9%, and the homology among 37 PCV2 strains was between 94.2 and 99.9%. In addition, the length of the ORF2 was varied from 702 to 705 nt with nucleotide homology ranged from 89.2 to 100% for ORF2. Of these, 25 sequences were 702 nt in length and 12 were 705 nt.

Geographical distribution and PCV2 test results of clinical tissue samples in Henan province from 2015 to 2018. The first number in parentheses represents the total number of samples detected in this area, and the latter number represents the number of PCV2 positive samples detected in this area. The map in this figure is my own
Phylogenetic analysis and genotype identification
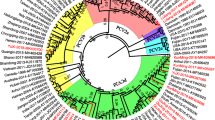
In order to get a better understanding of the genetic relationship and evolution of PCV2, a phylogenetic tree based on the ORF2 gene of the 37 PCV2 strains sequenced here was constructed together with 19 reference sequences deposited in GenBank database, which represent PCV genotypes from China and other countries (Fig. 2a). The phylogenetic tree included three groups (PCV1, PCV2 and PCV3) and showed that the 37 isolates could be divided into PCV2a, PCV2b and PCV2d genotype groups with different clustering levels. All these strains we analyzed in this study displayed genotype diversity. Among all 37 isolates, 3 fall into genotype PCV2a (8.1%), 21 belong to PCV2b (56.8%) and 13 exist in PCV2d (35.1%). Comparing the newly detected strains with the reference strains, ORF2 nucleotide homology was evaluated and varied between 92.9–95.0% (PCV2a), 96.6–99.9% (PCV2b) and 95.3–99.9% (PCV2d); and the variation at the ORF2 amino acid level was 91.1–94% (PCV2a), 95.3–100% (PCV2b) and 96.2–100% (PCV2d). Further comparative analysis of ORF2 nucleotide and amino acid sequences of each two PCV2 genotypes was performed and sequence identities varied between 86.2–97.7% and 83.0–98.7% (Table 2). In comparison with the phylogenetic tree based on the ORF2 gene of PCV2, all 56 strains have the same classification according to phylogenetic tree by complete genome (Fig. 2b). From here we see that PCV2b and PCV2d become the two predominant genotypes in the Henan province from 2015 to 2018.

Phylogenetic tree based on ORF2 (a) and genome (b). Phylogenetic trees were constructed with the MEGA 7.0.14 software using the neighbor-joining (NJ) method, with the Jukes-cantor model as a nucleotide substitution model. The reliability of the generated trees was determined with 1000 replicates of the data set. 37 isolates and 19 reference sequences were used for this analysis. The isolations detected in this study were labeled by the red star, and reference strains downloaded from NCBI weren’t labeled
Molecular characterization
The molecular characteristics of the whole genome show that the genomes of three PCV2 isolated strains (HN-JY-2015 (1766 nt), HN-YY-2016 (1768 nt) and HN-LH-2018 (1768 nt)) are inserted one nucleotide (T) at a position 1042 in the ORF2, which has no effect on ORF2. In addition, the genome of HN-JY-2015 (1766 nt) is deleted two nucleotides (G and T) at position 996–997 in the intergenic region between ORF1 and ORF2. Interestingly, eleven nucleotides (CCTCAGCAGCA) are inserted in the genome of HN-QY-2016 and HN-BF-2016 (1778 nt) at position 49–59 in the intergenic region between loop structure and ORF1.
Considering that ORF2 carries the highest level of genetic variability in the PCV2 genome, the deduced amino acid sequences of 37 ORF2 sequences were aligned with 15 PCV2 strains reference (Fig. 3). Examination of the typical motifs of different genotypes in ORF2 (aa 86–91 and aa 190/191/206/210) indicated that amino acid residues at positions 86–91 were SNPLTV in PCV2d strains as well as PCV2c, and SNPR(H)SV or SNPLTV in PCV2b strains and TNK(E)ISI in PCV2a strains, while amino acid residues at positions 190/191/206/210 were TGI(K)D(E) in PCV2d strains, TGIE or AGIE in PCV2b strains. Furthermore, differed from PCV2a, a lysine extension at the C-terminus was observed in PCV2d capsid protein, which resulting a 705 nt ORF2 instead of 702 nt. Although HN-YS-2015 and HN-TH-2018 belong to PCV2d, their ORF2s don’t have an additional amino acid lysine. In addition, eight sites, Y8F, F53I, I57V, A68N, S121 T, T134 N, S169G/R and V215I, were mutated in PCV2d ORF2 when compared with PCV2a, PCV2b, and PCV2c.

Sequence alignment of multiple sequence amino acids in PCV2 Cap protein. This alignment included deduced amino acid sequences of ORF2 from 37 PCV2 isolates and 15 PCV2 reference strains. The diagram was plotted by EPSprint 3.0. The antibody recognition domains (A (51–81), B (113–134), C (161–208) and D (228–233)) and immunodominant decoy epitope E (168–180), were demarcated by the colored boxes, in bright green, blue, white, black and dark red, respectively
Selection pressure analysis of PCV2 ORF2 sequences
Selection pressure on the ORF2 coding region of Chinese PCV2 was estimated. Among the individual codons, 6 codons (sites 30, 63, 89, 90, 190 and 215) were considered to be under positive selection through the FEL approach (p < 0.1) and 5 codons (sites 30, 63, 89, 90 and 190) were identified as under positive selection through the MEME (p < 0.1). Similarly, positive selection was also found in the 4 codons (sites 63, 89, 90 and 190) through the SLAC approach (p < 0.1) and FUBAR approaches (Pr > 0.9) (Table 3). Finally, 5 individual codons (sites 30, 63, 89, 90 and 190) were confirmed under positive selection by at least two methods.
Discussion
Since PCV2 was detected in early 1990s, it has caused considerable economic loss in the swine industry and become one of the most extensively investigated viral agents of pigs globally. The genetic diversity of PCV2 is continuously increasing, and novel subtypes are still emerging [4, 26]. As everyone knows, the pork industry in China contributed to nearly half of worldwide pork production in 2018. During the last 30 years, pork production in China has increased two-fold [27]. Furthermore, Henan is a major pig-raising province which located in the central part of China. To some extent, the infection and molecular epidemiology of PCV2 in Henan could represent that of China. In this study, we collected 117 tissue samples between 2015 and 2018 in Henan to investigate infection and molecular epidemiology of PCV2.
The prevalence of PCV2 among swine populations in China has considerably increasing since 2000 [28,29,30]. PCV2a was the earliest and predominant genotype in China [31]. Despite a wide variety of vaccines ranging from inactivated vaccines to baculovirus-expressed recombinant capsid protein-based vaccines have been widely used since 2009 [32, 33], clinical and subclinical infections of PCV2 still existed in China. Our results show that the positive rate of PCV2 was about 62.4% in Henan. High positive rates of PCV2 have been reported in different areas of China [34,35,36]. Subsequently, a shift from PCV2a to PCV2b occurred in China, suggesting that PCV2b become the predominant genotype in China [37,38,39]. Recently, a genotype shift to PCV2d has recently occurred on a nationwide scale [4, 28]. The genotype shift of PCV2 may have an impact on virulence that may related to vaccination, pathogenesis and diagnosis [40, 41]. Consistent with previous results, our investigation indicates that 56.8% (21 of 37) of isolated PCV2 strains were the PCV2b genotype, 35.1% (13 of 37) belonged to the PCV2d genotype, and 8.1% (3 of 37) were classified to the PCV2a genotype. Previous studies also demonstrated that PCV2c and recombinant strains had existed in China [19, 42]. However, two genotypes above weren’t found in this research, which maybe because of relative small sample size and limited sampling scope (only in Henan). Interestingly, 37 PCV2 newly detected strains could be divided into four clusters (1766 nt (2.7%), 1767 nt (86.5%), 1768 nt (5.4%) and 1778 nt (5.4%)) based on genome size. Normally, genome size of PCV2 is 1767 or 1768 nt. PCV2 strains with a genome of 1766 nt have also been reported in previous investigations [37, 43]. However, 1778 nt genome strain was rarely reported [18, 44, 45].
Furthermore, ORF2 was widely used for phylogenetic analysis and genotype identification of PCV2. The typical motifs in ORF2 can be used to identify different genotypes. For the PCV2a, the motif is TNKISI (aa 86–91); For PCV2b, the motifs are SNPRSV (aa 86–91) and AGIE (aa 190/191/206/210) [46, 47]; For PCV2d, the motifs are SNPLTV (aa 86–91) and TGID (aa 190/191/206/210) [4]. However, SNPLTV (aa 86–91) has also been found in PCV2c strains. Compared to typical motifs, different ORF2 motif of PCV2 strain in this study were found that TNEISI (aa 86–91) for PCV2a, SNPLTV(aa 86–91) for PCV2b, TGIE (aa 190/191/206/210) for PCV2b and TGKD or TGIE (aa 190/191/206/210) for PCV2d. Several amino acid residues changes occurred in the previously reported antibody recognition regions, including (A (51–81), B (113–134), C (161–208) and D (228–233)) and immunodominant decoy epitope E (168–180) of the Cap protein [41, 48]. From the results of amino acid alignment in this research, Y8F, F53I, I57V, A68N, S121T, T134 N, S169G/R and V215I, were found in PCV2d ORF2. Recent study revealed that nuclear localization signal (NLS) located in N-terminus of the PCV2 Cap could function as a cell-penetrating peptide (CPP) being capable of entering cells [49]. In addition, the aromatic side chains of tyrosine (Y) and phenylalanine (F) in NLS may insert into and anchor NLS-A on membranes. Because both F and Y include a bulky aromatic ring, Y8F may not affect cell penetration of NLS. Other amino acid mutations are possibly responsible for the immunogenicity change of the Cap protein. Further studies are required to determine the effects of these mutations on viral properties.
At present, the detection rate of PCV2 is still very high in the case of widespread use of vaccines, which is related to the genetic variation of PCV2. Conversely, it is this pressure that promotes the evolution(1.2 × 10− 3) of the virus, which makes PCV2 to have an evolutionary dynamics that is much closer to single-stranded RNA (ssRNA) viruses rather than ssDNA viruses [3]. In this experiment, we found five positive selection sites (30, 63, 89, 90 and 190) in PCV2 ORF2 of China under selection pressure. All positively selected sites were related to the main capsid epitope regions: Position 30 is involved the core epitope (residue 26–36) located in the nuclear localization signal region [50]; Position 63 is located in immunorelevant epitopes at residues 51–81 [41, 48]. Position 89 and 90 are located in the typical motifs of different PCV2 genotypes [46, 47]. Position 190 are important for determining neutralization activity [51, 52]. Probably, these mutations are associated with virus escape from the host immune system.
Conclusions
In conclusion, we investigated the infection of PCV2 in Henan province from 2015 to 2018 and found that the infection rate was 62.4%. 37 new isolates of PCV2 were sequenced. Through alignment and phylogenetic tree analysis, it was found that the prevalent genotypes were mainly PCV2b and PCV2d. Surprisingly, two rare 1778 nt PCV2 strains were found. Some new amino acid mutation sites were found by ORF2 amino acid alignment. Five positive selection sites in ORF2 of PCV2 in China were found by selective pressure analysis. The results of this study will lay a foundation for understanding PCV2 infection, molecular epidemics and evolution in Henan Province.
Methods
Sample collection
A total number of 117 clinical samples (lung, lymph node, tonsil and spleen) were collected from Henan Province, in central China between 2015 and 2018. The pigs suspected to have clinical symptoms of PMWS and/or PDNS came from different farms. After collecting tissue samples, the pigs were treated innocuously. Tissue samples were suspended in sterile phosphate buffered saline (PBS) and homogenized. These homogenized samples were centrifuged at 8000 rpm for 5 min. Supernatant were aliquoted and stored at − 80 °C for further analysis.
DNA extraction and sequencing
Total viral DNA was extracted from 200 μL of tissue sample homogenate using MiniBEST Viral RNA/DNA Extraction Kit Ver.5.0 (TaKaRa, Dalian, China) with strict adherence to the manufacturer procedures. Initially, all samples were tested for PCV2 through polymerase chain reaction (PCR) by using the primers PCV2-F and PCV2-R (PCV2-F: AATGGCATCTTCAACACCCGCCT; PCV2-R: TTAAGGGTTAAGTGGGGGGTCTTT), which amplified a 579 base pair (bp) DNA fragment of ORF2 region. The 20 μL PCR reaction mixtures contained 10 μL of 2 × Premix Taq (Ex Taq version 2.0 plus dye) (TaKaRa, Dalian, China), 0.5 μL of each primer at a concentration of 10 μM, 2 μL of DNA template, and 7 μL of ddH2O. And the amplification was carried out as follows: 98 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, 58 °C for 30 s, 72 °C for 50 s, and 72 °C for 5 min. Subsequently, the positive samples were selected for the whole genome amplification by using two pairs of primers P1/P2 (P1: AGGGCTGTGGCCTTTGTTAC; P2: TCTTCCAATCACGCTTCTGC) and P3/P4 (P3: TGGTGACCGTTGCAGAGCAG; P4: TGGGCGGTGGACATGATGAG), which amplified a 989 bp and a 1091 bp DNA fragments respectively [53]. PCR were performed in 50 μL reaction mixtures containing 25 μL Q5 High-Fidelity 2 × Master Mix (NEB, MA, USA), 1.25 μL10 μM each pair of primers P1/P2 (or P3/P4), 2 μL DNA template of positive samples and 20.5 μL ddH2O. PCR conditions included 30 s at 95 °C, followed by 35 cycles of 98 °C for 10 s, 55 °C for 30 s, 72 °C for 40 s, and 72 °C for 5 min. PCR products were visualized by 1.5% Agarose gels stained with Ethidium bromide (EB). Then, the target bands cloned into the pEASY-Blunt Simple Cloning Vector (TransGen Biotech, Beijing, China). Positive clones were identified through PCR and sequenced twice by Sangon Biotech (Shanghai) Co. Ltd. The whole genome was further assembled with DNAStar software and submitted to the GenBank.
Phylogenetic analysis
The Multiple sequence alignments were carried out using Clustal W of the Megalign program (DNAStar software). To determine the genotype of the PCV2 isolates, 19 reference PCV genome sequences (Table 4) were downloaded from the GenBank database. Phylogenetic trees were constructed on the basis of ORF2 and full-length nucleotide sequence of the 37 isolates and 19 downloaded sequences with the MEGA 7.0.14 software using the neighbor-joining (NJ) method, with the Jukes-cantor model as a nucleotide substitution model. The reliability of the generated trees was determined with 1000 replicates of the data set. Furthermore, the amino acid sequence of all PCV2 capsid proteins were aligned using the MegAlign software and plotted by EPSprint 3.0 [54].
Selection pressure analysis
The detection of selection pressure on the ORF2 coding sequences of 37 new isolates and 104 China PCV2 strains (Additional file 1: Table S1) was performed using Datamonkey (http://www.datamonkey.org), which based on the ratios between non-synonymous and synonymous substitution rates (dN/dS) [55]. The methods used to investigate dN/dS ratio and positive codon sites at individual codons included Single-likelihood ancestor counting (SLAC), fixed-effects likelihood (FEL), mixed effects model of evolution (MEME) and fast unconstrained Bayesian approximation (FUBAR). When p-value was below 0.1 in SLAC, FEL, MEME and the posterior probability was above 0.9 in FUBAR, positive selection pressure signals were considered to acceptable. Positively selected sites were confirmed when positive selection pressure signals were identified by at least two methods.
Availability of data and materials
The PCV2 genome sequence obtained in this study had been uploaded to GenBank. The GenBank accession number of all the sequences was provided, and the relevant data can be obtained after landing. Phylogenetic data and alignments have been stored in TreeBASE with a submission ID of 24956 (Accession URL: http://purl.org/phylo/treebase/phylows/study/TB2:S24956).
Abbreviations
- PCR:
-
Polymerase chain reaction
- PCV2:
-
Porcine circovirus type 2
- PCVAD:
-
Porcine circovirus associated diseases
- PDNS:
-
Porcine dermatitis and nephropathy syndrome
- PMWS:
-
Post-weaning multisystemic wasting syndrome
References
Huang L, Lu Y, Wei Y, Guo L, Liu C. Identification of three new type-specific antigen epitopes in the capsid protein of porcine circovirus type 1. Arch Virol. 2012;157:1339–44.
Tischer I, Gelderblom H, Vettermann W, Koch MA. A very small porcine virus with circular single-stranded DNA. Nature. 1982;295:64–6.
Firth C, Charleston MA, Duffy S, Shapiro B, Holmes EC. Insights into the evolutionary history of an emerging livestock pathogen: porcine circovirus 2. J Virol. 2009;83:12813–21.
Xiao CT, Halbur PG, Opriessnig T. Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J Gen Virol. 2015;96:1830–41.
Chae C. A review of porcine circovirus 2-associated syndromes and diseases. Vet J. 2005;169:326–36.
Ellis J, Hassard L, Clark E, Harding J, Allan G, Willson P, Strokappe J, Martin K, McNeilly F, Meehan B, et al. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can Vet J. 1998;39:44–51.
Ouyang T, Zhang X, Liu X, Ren L. Co-infection of swine with porcine Circovirus type 2 and other swine viruses. Viruses. 2019;11:185.
Palinski R, Pineyro P, Shang P, Yuan F, Guo R, Fang Y, Byers E, Hause BM. A novel porcine Circovirus distantly related to known Circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. J Virol. 2017;91:e01879-16.
Phan TG, Giannitti F, Rossow S, Marthaler D, Knutson TP, Li L, Deng X, Resende T, Vannucci F, Delwart E. Detection of a novel circovirus PCV3 in pigs with cardiac and multi-systemic inflammation. Virol J. 2016;13:184.
Jiang H, Wang D, Wang J, Zhu S, She R, Ren X, Tian J, Quan R, Hou L, Li Z, et al. Induction of porcine dermatitis and nephropathy syndrome in piglets by infection with porcine circovirus type 3. J Virol. 2019;93:e02045-18.
Ouyang T, Niu G, Liu X, Zhang X, Zhang Y, Ren L. Recent progress on porcine circovirus type 3. Infect Genet Evol. 2019;73:227–33.
Kedkovid R, Woonwong Y, Arunorat J, Sirisereewan C, Sangpratum N, Lumyai M, Kesdangsakonwut S, Teankum K, Jittimanee S, Thanawongnuwech R. Porcine circovirus type 3 (PCV3) infection in grower pigs from a Thai farm suffering from porcine respiratory disease complex (PRDC). Vet Microbiol. 2018;215:71–6.
Lv QZ, Guo KK, Zhang YM. Current understanding of genomic DNA of porcine circovirus type 2. Virus Genes. 2014;49:1–10.
Ren L, Chen X, Ouyang H. Interactions of porcine circovirus 2 with its hosts. Virus Genes. 2016;52:437–44.
Olvera A, Cortey M, Segales J. Molecular evolution of porcine circovirus type 2 genomes: phylogeny and clonality. Virology. 2007;357:175–85.
Yang S, Yin S, Shang Y, Liu B, Yuan L, Zafar Khan MU, Liu X, Cai J. Phylogenetic and genetic variation analyses of porcine circovirus type 2 isolated from China. Transbound Emerg Dis. 2018;65:e383–92.
Harmon KM, Gauger PC, Zhang J, Pineyro PE, Dunn DD, Chriswell AJ. Whole-genome sequences of novel porcine Circovirus type 2 viruses detected in swine from Mexico and the United States. Genome Announc. 2015;3:e01315-15.
Dupont K, Nielsen EO, Baekbo P, Larsen LE. Genomic analysis of PCV2 isolates from Danish archives and a current PMWS case-control study supports a shift in genotypes with time. Vet Microbiol. 2008;128:56–64.
Liu X, Wang FX, Zhu HW, Sun N, Wu H. Phylogenetic analysis of porcine circovirus type 2 (PCV2) isolates from China with high homology to PCV2c. Arch Virol. 2016;161:1591–9.
Franzo G, Cortey M, de Castro AM, Piovezan U, Szabo MP, Drigo M, Segales J, Richtzenhain LJ. Genetic characterisation of porcine circovirus type 2 (PCV2) strains from feral pigs in the Brazilian Pantanal: An opportunity to reconstruct the history of PCV2 evolution. Vet Microbiol. 2015;178:158–62.
Franzo G, Cortey M, Segales J, Hughes J, Drigo M. Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol Phylogenet Evol. 2016;100:269–80.
Guo L, Fu Y, Wang Y, Lu Y, Wei Y, Tang Q, Fan P, Liu J, Zhang L, Zhang F. A porcine circovirus type 2 (PCV2) mutant with 234 amino acids in capsid protein showed more virulence in vivo, compared with classical PCV2a/b strain. PLoS One. 2012;7:e41463.
Davies B, Wang X, Dvorak CM, Marthaler D, Murtaugh MP. Diagnostic phylogenetics reveals a new porcine circovirus 2 cluster. Virus Res. 2016;217:32–7.
Wang H, Gu J, Xing G, Qiu X, An S, Wang Y, Zhang C, Liu C, Gong W, Tu C, et al. Genetic diversity of porcine circovirus type 2 in China between 1999-2017. Transbound Emerg Dis. 2019;66:599-605.
Feng H, Blanco G, Segalés J, Sibila M. Can porcine circovirus type 2 (PCV2) infection be eradicated by mass vaccination? Vet Microbiol. 2014;172:92–9.
Liu J, Wei C, Dai A, Lin Z, Fan K, Fan J, Liu J, Luo M, Yang X. Detection of PCV2e strains in Southeast China. PeerJ. 2018;6:e4476.
Anderson BD, Ma MJ, Wang GL, Bi ZQ, Lu B, Wang XJ, Wang CX, Chen SH, Qian YH, Song SX, et al. Prospective surveillance for influenza. virus in Chinese swine farms. Emerg Microbes Infect. 2018;7:87.
Jiang CG, Wang G, Tu YB, Liu YG, Wang SJ, Cai XH, An TQ. Genetic analysis of porcine circovirus type 2 in China. Arch Virol. 2017;162:2715–26.
Shi J, Xu S, Fu F, Cong X, Yuan X, Peng Z, Wu J, Sun W, Du Y, Li J, et al. Phylogenetic analysis of 32 porcine circovirus type 2 isolates from Shandong, China. Arch Virol. 2015;160:465–8.
Shuai J, Wei W, Li X, Chen N, Zhang Z, Chen X, Fang W. Genetic characterization of porcine circovirus type 2 (PCV2) from pigs in high-seroprevalence areas in southeastern China. Virus Genes. 2007;35:619–27.
Bao F, Mi S, Luo Q, Guo H, Tu C, Zhu G, Gong W. Retrospective study of porcine circovirus type 2 infection reveals a novel genotype PCV2f. Transbound Emerg Dis. 2018;65:432–40.
Afghah Z, Webb B, Meng XJ, Ramamoorthy S. Ten years of PCV2 vaccines and vaccination: is eradication a possibility? Vet Microbiol. 2017;206:21–8.
Zhai SL, Chen SN, Xu ZH, Tang MH, Wang FG, Li XJ, Sun BB, Deng SF, Hu J, Lv DH, et al. Porcine circovirus type 2 in China: an update on and insights to its prevalence and control. Virol J. 2014;11:88.
Sun J, Huang L, Wei Y, Wang Y, Chen D, Du W, Wu H, Liu C. Prevalence of emerging porcine parvoviruses and their co-infections with porcine circovirus type 2 in China. Arch Virol. 2015;160:1339–44.
Qu T, Li R, Yan M, Luo B, Yang T, Yu X. High prevalence of PCV2d in Hunan province, China: a retrospective analysis of samples collected from 2006 to 2016. Arch Virol. 2018;163:1897–906.
Wei C, Zhang M, Chen Y, Xie J, Huang Z, Zhu W, Xu T, Cao Z, Zhou P, Su S, Zhang G. Genetic evolution and phylogenetic analysis of porcine circovirus type 2 infections in southern China from 2011 to 2012. Infect Genet Evol. 2013;17:87–92.
Guo LJ, Lu YH, Wei YW, Huang LP, Liu CM. Porcine circovirus type 2 (PCV2): genetic variation and newly emerging genotypes in China. Virol J. 2010;7:273.
Cai L, Ni J, Xia Y, Zi Z, Ning K, Qiu P, Li X, Wang B, Liu Q, Hu D, et al. Identification of an emerging recombinant cluster in porcine circovirus type 2. Virus Res. 2012;165:95–102.
Wang F, Guo X, Ge X, Wang Z, Chen Y, Cha Z, Yang H. Genetic variation analysis of Chinese strains of porcine circovirus type 2. Virus Res. 2009;145:151–6.
Karuppannan AK, Opriessnig T. Porcine Circovirus type 2 (PCV2) vaccines in the context of current molecular epidemiology. Viruses. 2017;9:99.
Trible BR, Rowland RR. Genetic variation of porcine circovirus type 2 (PCV2) and its relevance to vaccination, pathogenesis and diagnosis. Virus Res. 2012;164:68–77.
Hu J, Zhai SL, Zeng SY, Sun BB, Deng SF, Chen HL, Zheng Y, Wang HX, Li XP, Liu JK, et al. Identification of natural recombinants derived from PCV2a and PCV2b. Genet Mol Res. 2015;14:11780–90.
Shang SB, Jin YL, Jiang XT, Zhou JY, Zhang X, Xing G, He JL, Yan Y. Fine mapping of antigenic epitopes on capsid proteins of porcine circovirus, and antigenic phenotype of porcine circovirus type 2. Mol Immunol. 2009;46:327–34.
Cai L, Han X, Hu D, Li X, Wang B, Ni J, Zhou Z, Yu X, Zhai X, Tian K. A novel porcine circovirus type 2a strain, 10JS-2, with eleven-nucleotide insertions in the origin of genome replication. J Virol. 2012;86:7017.
de Boisseson C, Beven V, Bigarre L, Thiery R, Rose N, Eveno E, Madec F, Jestin A. Molecular characterization of porcine circovirus type 2 isolates from post-weaning multisystemic wasting syndrome-affected and non-affected pigs. J Gen Virol. 2004;85:293–304.
Cheung AK. Homologous recombination within the capsid gene of porcine circovirus type 2 subgroup viruses via natural co-infection. Arch Virol. 2009;154:531–4.
Cheung AK, Lager KM, Kohutyuk OI, Vincent AL, Henry SC, Baker RB, Rowland RR, Dunham AG. Detection of two porcine circovirus type 2 genotypic groups in United States swine herds. Arch Virol. 2007;152:1035–44.
Trible BR, Kerrigan M, Crossland N, Potter M, Faaberg K, Hesse R, Rowland RR. Antibody recognition of porcine circovirus type 2 capsid protein epitopes after vaccination, infection, and disease. Clin Vaccine Immunol. 2011;18:749–57.
Yu W, Zhan Y, Xue B, Dong Y, Wang Y, Jiang P, Wang A, Sun Y, Yang Y. Highly efficient cellular uptake of a cell-penetrating peptide (CPP) derived from the capsid protein of porcine circovirus type 2. J Biol Chem. 2018;293:15221–32.
Guo L, Lu Y, Huang L, Wei Y, Liu C. Identification of a new antigen epitope in the nuclear localization signal region of porcine circovirus type 2 capsid protein. Intervirology. 2011;54:156–63.
Huang LP, Lu YH, Wei YW, Guo LJ, Liu CM. Identification of one critical amino acid that determines a conformational neutralizing epitope in the capsid protein of porcine circovirus type 2. BMC Microbiol. 2011;11:188.
Saha D, Lefebvre DJ, Ooms K, Huang L, Delputte PL, Van Doorsselaere J, Nauwynck HJ. Single amino acid mutations in the capsid switch the neutralization phenotype of porcine circovirus 2. J Gen Virol. 2012;93:1548–55.
Fenaux M, Halbur PG, Gill M, Toth TE, Meng XJ. Genetic characterization of type 2 porcine circovirus (PCV-2) from pigs with postweaning multisystemic wasting syndrome in different geographic regions of North America and development of a differential PCR-restriction fragment length polymorphism assay to detect and differentiate between infections with PCV-1 and PCV-2. J Clin Microbiol. 2000;38:2494–503.
Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4.
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–7.
Acknowledgements
The authors would like to thank Min Jiang and Junfang Hao for their help in preserving clinical samples.
Consent to publication
Not applicable.
Funding
We are grateful to the financial support from three fund projects. Provincial Budget Projects (20188117 and 20177620) supported the design of study and sample collection, the National Natural Science Funds of China (31702214) supported analysis, interpretation of data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZGM, LQX, DRG and ZGP designed the study, participated in all tests and drafted the manuscript. JQY, GZH, HHF, LDL, WX and ZYH participated in the procedure of collecting and testing samples. WFY, XGX, FH and TM conceived the study, contributed to the analysis of the results and preparation of revised manuscript versions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
During the entire experiment, all animals received humane care in compliance with good animal practice according to the animal ethics procedures and guidelines of China and its owners also agreed to collect samples. The procedure of sample collection was approved by the Animal Experiment Committee of Henan Academy of Agricultural Sciences (Approval number SYXK 2014–0007).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
ORF2 sequence of PCV2 strains in China were used for the analysis of selection pressure in this study. Table S1 contains GenBank accession numbers and years of 104 PCV2 ORF2 sequences in China. These ORF2 sequences were used for selection stress analysis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, G., Lu, Q., Wang, F. et al. Phylogenetic analysis of porcine circovirus type 2 (PCV2) between 2015 and 2018 in Henan Province, China. BMC Vet Res 16, 6 (2020). https://doi.org/10.1186/s12917-019-2193-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-2193-1