
Abstract
Increasing evidences indicate that porcine circovirus type 2 (PCV2) is the causative agent of the post-weaning multisystemic wasting syndrome (PMWS). In this study, the prevalence of PCV2 infection in swine herds in southeastern China was investigated by ELISA and PCR, as well as the genetic characteristics by nucleic acid sequencing. Seroprevalence of PCV2 in samples collected from 89 swine herds was significantly higher by ELISA in post-weaning (54.1%) and growing piglets (49.9%) than that of suckling pigs (33.3%) with an average rate of 46.0% (819/1779). Seventy-eight cases out of 159 diseased pigs from these herds were PCV2 positive by PCR. Furthermore, the PCV2-positve rate at herds level in 2005 and 2006 were much higher than that in 2004 (65.63% or 69.23% vs. 32.26%, respectively), indicating that PCV-2 infection expanded rapidly over the past two years. To provide new insights into the extent of genetic heterogeneity of PCV2 isolates in southeastern China, the ORF2 genes of 27 isolates from the area during January 2004–March 2007 were sequenced and aligned. While closely related to each other with identity of 98.0–100%, these isolates displayed lower homologies to those from other regions of China (90.6–100%) or to some foreign isolates (91.3–98.9%). Alignment of deduced amino acid sequences of capsid protein identified two major hyper-variable regions (positions 53–91 and 185–215) in isolates obtained in this study, which were within or close to the putative epitope domains. The substitutions consequently resulted in higher hydrophilicity of the epitope region (positions 47–85). Phylogenetic analysis revealed two clusters of 48 isolates including those from Genbank: the large cluster I consisting of two subgroups and cluster II containing most of foreign isolates owing to the residue substitutions in epitope domains (amino acid positions 80, 86, 88 and 91). While the subgroup Ib contained all the isolates with ORF2 of 705 bp in length, the 27 isolates we sequenced were clustered exclusively in subgroup Ia together with some other Chinese strains. We conclude that PCV2 isolates prevailing in southeastern China were genetically different from those of other countries.
Similar content being viewed by others

Introduction
Porcine circovirus (PCV), which belongs to the family Circoviridae, genus Circovirus [1], is one of the smallest-animal viruses with unenveloped, single-stranded circular genome and a size of 17 nm in diameter [2, 3]. Two species of PCV, PCV1 and PCV2, have been characterized [4]. PCV1 is considered to be nonpathogenic to pigs by experimental inoculation [1], and was originally been identified as a persistent tissue culture contaminant in pig kidney cell lines [5]. PCV2 has been shown to be the causative agent of post-weaning multisystemic wasting syndrome (PMWS) of pigs [6, 7]. The presence of PCV2 antigen was first demonstrated in 1997 in lesions of animals with PMWS [8], which usually occurs in pigs aged 2–3.5 months. The affected pigs show progressive weight loss, respiratory, and enlarged lymph nodes [9] and characteristic microscopic lesions including granulomatous interstitial pneumonia, lymphadenopathy, hepatitis, nephritis, and pancreatitis [10]. The role of PCV2 in PMWS is clear in terms of its association with the pathology [11] nowadays, but still less clear about its mechanisms of disease induction and pathogenesis. To date, PMWS is most commonly diagnosed on the basis of histopathology coupled with the identification of PCV infection by multiplex or type-specific PCR, immunohistochemistry (IHC), and enzyme-linked immunosorbent assay (ELISA) [11–13].
A previous study have compared the 148 worldwide PCV2 complete genomes present at the NCBI nucleotide database and divided them into two groups and eight clusters (1A to 1C and 2A to 2E), but there was no correlation regarding groups and health status being performed [14]. However, PCV2 isolates from PMWS cases around the world share a high-nucleotide sequence identity (more than 96%) and antigenic similarities, indicating the possibility of a single patho-type prevailing in the swine population [4, 15]. Nevertheless, variations in nucleotide and amino acid sequences have been described particularly in ORF2 which is considered to be type-specific with less than 65% identity between PCV1 and PCV2 [16], and closely related to the pathogenesis. Although the significance of these variations in terms of host and tissue tropism and pathogenicity remains to be determined, understanding of the extent of genetic variations among PCV2 isolates from different geographic regions is valuable for vaccines development and disease control [17, 18]. The present study was aimed to investigate the status of PCV2 infection in swine population in southeastern China by ELISA and PCR specific for PCV2. Then, we focused on the genetic diversity of PCV2 strains from southeastern China originating from PMWS cases based on the sequence of the ORF2.
Materials and methods
Seroprevalence of PCV2 infection by ELISA
From January 2004 to December 2006, 1779 serum samples from suckling piglets (1–30-days old), post-weaning pigs (31–100-days old) and growing pigs (older than 100 days) were collected from 89 herds located in different regions of southeastern China. Serum antibodies to PCV2 were detected by ELISA as previously described by Shibata [19] with some modifications. Briefly, 100 μl purified PCV2 recombinant Cap protein (produced in our laboratory) diluted with 50 mM sodium carbonate buffer (pH9.6) to a final concentration of 50 μg/ml was added into each well of a 96-well microtiter plates (MaxiSorp, Nunc, Denmark). The plates were then incubated at 4°C for 18 h. After washing with phosphate-buffered saline (PBS) containing 0.05% Tween-20 (PBS-T), the plates were blocked with PBS-T containing 5% (w/v) skim milk for 2 h at 37°C. Serum samples diluted by 100-fold with PBS-T containing 0.5% (w/v) skim milk were added to corresponding wells and the mixtures were incubated for 1 h at 37°C. Then, 100 μl/well of goat anti-pig IgG conjugated with horseradish peroxidase (KPL, UK) was added at a dilution of 1:2000 and incubated for 1 h at 37°C. Thereafter, plates were washed and added with 100 μl/well of the substrate solution (o-phenylenediamine dihydrochloride, Sigma, USA). After incubation for 30 min at 37°C in the dark, the reaction was stopped by adding 50 μl of 2 N H2SO4. The OD492nm values were measured using spectraMax@ M2 microplate reader (Molecular devices Corp., USA).
A panel of PCV2 negative sera (n = 20) was also examined by ELISA to determine the cut-off OD492nm value (mean ± 3SD) in the study. Serum samples with an optical density at 490 nm greater than the cut-off value were considered as seropositive for PCV2.
Statistical test
Difference among periods of ages regarding seroprevalence or PCR-positive rates in different years was evaluated with the mean of Student’s t-test.
Detection of PCV2 nucleic acids in tissue samples by PCR
Tissue samples (n = 159) including lymph nodes, lungs, and livers from sick pigs originating from 89 geographically different herds in southeastern China were collected from December 2003 to March 2007. Total DNA was extracted from tissues using QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. For each sample, lysis buffer with Proteinase K (20 mg/ml) was added to mechanically homogenized tissue the mixtures were incubated at 55°C for 1 h. The final ethanol-washed DNA was eluted in 50 μl buffer and stored at −20°C until PCR analysis.
A pair of primers (P1: 5′-CACGGATATTGTAGTCCTGGTC-3′, P2: 5′-GCCGAAGTGCGCTGGTAATA-3′) were designed based on the ORF2 of a published strain that isolated from southeastern China (Accession No.: DQ195679) to provide differential detection of PCV2 genome from PCV1. The target DNA region was amplified by PCR on HybaidR PCR thermocycler (Hybaid Equipment Ltd., UK) with cycles consisted of an initial step of 94°C for 3 min, followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 57°C for 45 s, and extension at 72°C for 1 min, and a final extension at 72°C for 10 min.
Genetic analysis of PCV2 ORF2 gene
To get a better insight of the extent of genetic relationships among PCV2 isolates within different geographic regions, a fragment encompassing the full-length ORF2 gene was amplified from PCV2-positive samples originating from different regions of southeastern China from January 2004 to March 2007 using PCV2 type-specific primers: Ps7, 5′- TTTCCCCCCCATGCCCTGA-3′ and Ps8, 5′-TTTCGTTTTCAGATATGACGT-3’. The products were then cloned into pMD18-T vector (TaKaRa) for sequencing. The sequences were then multiple-aligned with representative sequences from other regions of China or from other countries published to date using CLUSTAL X (version 1.83). Phylogenetic analysis was performed with Molecular Evolutionary Genetics Analysis (MEGA, version 3.1), in which a PCV1 sequence served as outgroup. Pairwise comparison of nucleotide and amino acid sequence similarities was conducted using MEGALIGN 5.03 in the DNASTAR Lasergene software package, and hydrophilicity profile was generated by the method of Kyte and Doolittle [20] using DNASIS 2.5 software package.
Results
Seroprevalence of PCV2 infection
The optimal dilutions of purified protein (50 μg/ml) and serum samples (1:100) in ELISA were determined in the preliminary assays. Based on the cut-off OD492 nm value of 0.3022 (mean ± 3 SD) in this assay, the average serum-positive rate was 46.0% (819/1779). Seroprevalence was significant higher in post-weaning pigs (54.1%, 223/412) and growing pigs (49.9%, 423/848) than in suckling piglets (33.3%, 173/519) (P < 0.01) (Table 1).
Detection of PCV2 nucleic acids
Among 159 PMWS suspected cases, most were with wasting or tachypnea, and enlarged inguinal lymph nodes, no other apparent symptoms were observed. But some were occasionally with paleness and diarrhea. Moreover, seventy-eight samples were PCV2-positive by PCR with an average positive rate of 49.06%. Since these cases were from 89 herds, the positive rate at the herd level was 55.05% on average during the 17 months’ period. Furthermore, there was a significant increase of PCV2-positive cases in 2005, as compared with that of the preceding period from December 2003 to December 2004 (55.10% vs. 35.19% at the case level and 65.63% vs. 32.26% at the herd level) (Table 2).
Genetic characterization of PCV2 ORF2
A total of 27 samples out of 78 PCR-positive ones, representing 27 different herds from regions of southeastern China, were again PCR-amplified targeting the full-length ORF2. In addition, nine out of them were found to be coinfected with porcine reproductive and respiratory syndrome virus (PRRSV) (Table 3). Sequence analysis showed that ORF2 gene of all the 27 PCV2 isolates were 702 nt in length. In pairwise comparisons, they were closely related to each other with high nucleotide sequence identity from 98.0% to 100%. However, they were more divergent from those of other countries or other regions of China with lower level of similarities ranging from 91.3 to 98.9% or 90.6 to 100% respectively. Further alignment of the deduced amino acid sequences indicated that the divergence at amino acid level was greater than that of the nucleotide sequence, showing high identity of 97.4–99.6% to each other. However, they had the lower similarities as compared with isolates from other regions of China (89.3–99.6%) or from other countries (90.2–98.3%) (Table 4). Although variations distributed across the whole capsid protein sequence, there were two major regions (located at positions 53–91 and 185–215) of more substitutions in our isolates, especially at positions 57, 59, 63, 80, 86, 89, 90, 91, 121, 151, 190 and 191. Interestingly, these regions were almost situated in putative epitope domains of PCV2 capsid protein [21, 22]. In contrast, the N-terminus of the protein encompassing the nuclear localization signals (NLS) was found to be rather conserved (Fig. 1).

Pairwise alignment of amino acid sequences of the putative capsid protein obtained from this study with those from Genbank. The residues that are consistent with 05-HZh (EF190931) are indicated with dots. The variable regions were included in frames and the residues that were prone to variation in isolates prevailing in southeastern China were indicated by asterisks. The reported main putative epitope domains were marked by arrows above the alignment
The schematic distribution of amino acid diversities across the ORF2 protein was analyzed for all 48 sequences included in Table 1 (Fig. 2). The hydrophilicity profile of ORF2 protein was calculated from the arbitrarily deduced consensus sequence of the isolate 05 HZh in which all of the amino acid residues coincided with those prevailing in the isolates sequenced in this study. Notably, the hydrophilicity of the putative epitopes differed considerably among the isolates described herein and those from Genbank, especially in the region of putative epitope A (positions 47–85) where the variability and the hydrophilicity of southeastern China isolates were much higher than those from other regions.

Schematic depiction of genetic variability of PCV2 capsid proteins. (a) The hydrophilicity profile of the representative reference sequence 05-Hzh which showed high homology to those isolates sequenced in this study. The x-axis represents the amino acid residue numbers and the average hydrophilicity scores are shown on y-axis. Hydrophilic regions appear below the middle line. (b) The amino acid positions of isolates from other geographic regions showing lower level of hydrophilicity in epitope domain A. (c) All the PCV2 sequences in Table 3 were calculated for the number of PCV2 isolates with amino acid variation in each residue compared with 05-HZh. The y-axis and the x-axis represent the number of sequences differing from deduced consensus sequence or amino acid positions, respectively. The reported two main epitope domains (A and B) are marked by arrows
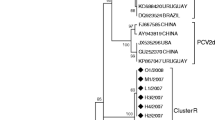
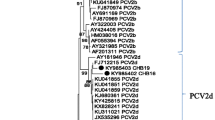
Phylogenetic analysis of PCV2 isolates from different geographic regions
Phylogenetic analysis revealed that there were two major clusters within the main genotype of PCV2 distinct from PCV1 (Fig. 3, I and II). All the foreign isolates except for the French (AF201311) and Netherlandish (AF484410) ones and some isolates from other geographic regions of China were included in cluster II, as shown by great bootstrap values compared with cluster I. Within cluster I, all of PCV2 sequences obtained in this study were clustered in subgroup Ia irrespective of the years or geographic regions of isolation, together with some isolates also from China. In contrast, most of isolates from other regions of China and foreign isolates were distributed in either cluster II or cluster Ib that represented those isolates with 705 bp in length of ORF2.

Phylogenetic analysis depicting genetic relationships between 27 PCV2 strains in this study (indicated by▴) and isolates from other regions of China or those from other countries based on the major structural gene of ORF2 (702 bp or 705 bp). (a) The tree was constructed using neighbor-joining algorithm based on the Kimura two-parameter distance estimation method by MEGA 3.1. Bootstrap values are indicated as a percentage for 1000 replicates. Two main clusters of PCV2 isolate (I and II) and the subgroups (Ia and Ib) were indicated. A PCV1 sequence was rooted as ourgroup. (b) The residues essential for clustering of cluster II or subgroup Ib among the most variable positions in capsid protein were highlighted in gray
Discussions
The etiology and pathogenesis of PMWS appear to be complicated, since other viral or non-viral agents are considered as co-factors [10, 23–25]. However, increasing evidences suggest that PCV2 is the principal causative agent [4, 6, 10, 16, 26]. Nevertheless, infection of a herd with PCV2 often results in subclinical infection only [26], in which the pigs, however, could also act as potential viral shedders [9, 13]. Thus, it is necessary to monitor PCV2 infection in herds based on serological study or PCR method. The prevalence of PCV2 infection could significantly increase at weaning, reaching a peak in excess of 65% in 3–4-month old pigs [19] and more than half of the tested diseased pigs were detected as PCV2 positive in field surveys by qualitative PCR [27]. Consistently, seroprevalence of PCV2 infection status investigated in this study increased markedly from 33.3% in suckling piglets to 54.1% in post-weaning pigs, and almost maintained at this level in growing pigs, indicating that PCV2 infection in pig populations was prevalent in the area (Table 1). Further testing found 78 cases out of 159 PMWS-suspected cases with sign of wasting as PCV2-positive by PCR and IFA (data not shown). Most of these 159 cases were seropositive of PCV2, but unfortunately, there were also some cases without serological test since they were dead at the time of necropsy and some with only tissues submitted. Furthermore, the PCV2-positive herds were increased significantly in the past years, which indicated that PCV2 infections spread rapidly within the herds in southeastern China.
Boisseson et al. [9] reported that the variations among the PCV2 genomic sequences were mainly due to variability within ORF2. As capsid protein is the major structural protein responsible for viral pathogenecity, the ORF2 gene is a reliable phylogenetic and epidemiological marker since it was not affected by recombination and could be used to reconstruct the same tree as the whole-viral genome [14]. We were interested to determine whether variations of the capsid gene of PCV2 in southeastern China and other areas have actually occurred. Pairwise comparisons revealed a notable diversity of nucleotide and deduced amino acid sequences between 27 isolates representing different herds in southeastern China and those from other regions worldwide with the lowest nucleotide sequence identity at 90.6%. However, phylogenetic analysis revealed that all the examined isolates could be divided into two major groups with three subgroups due to several marker positions. The most striking observation was that all of the isolates we sequenced were exclusively clustered in the subgroup Ia together with only some other southeastern Chinese strains regardless the different herds or years of isolation (Fig. 3). The finding was consistent with the result by Olvera et al. [14], in which most of southeastern China isolates were clustered in group 1A.
As reported previously, the epitope domains concentrated the majority of the variations of ORF2 gene and the first two epitopes had more positively selected positions [14, 15]. Consistently, two major domains (positions 53–91 and 185–215) of PCV2 capsid protein sequenced in the present study, within or near the two main immunogenic regions [22], were identified to exhibited high polymorphism. The substitutions at positions 80, 86, 88 and 91 even resulted in a separated cluster II distinct from cluster I (Fig 3b). As compared with sequences from subgroup Ib that were 705 bp in length of ORF2 and had a single amino acid extension just before the stop codon with a prolonging eptope domain at C-terminus [22], all the isolates prevailing in southeastern China had a length of 702bp in ORF2. Moreover, due to these substitutions, the epitope region (positions 47–85) of southeastern China isolates was much more hydrophilic than that of most isolates from other regions (Fig. 2), which might result in alteration of the surface structure of the capsid protein. Because the epitope domains in some viruses are more variable of amino acid sequence to evade the immune response [14, 28], it is speculated that correlation between capsid protein mutations and pathogenicity of PCV2 may exist in terms of its variation in tissue tropism, immunogenicity or virus-host interactions [29]. However, despite the overall heterogeneity of ORF2 in this study, the N-terminal region containing nuclear localization signals was found to be strictly conserved with a high percentage of basic amino acids [30, 31]. In addition, de Boisseson et al. [15] reported that region between the two hyper-variable domains as also found in present study was a key region for virus-host interactions or virus assembly.
Although there is only one single patho-type PCV2 prevailing in the endemic and the mutation rate is low [4, 15], the variations in ORF2 did exist at different geographical scale. Since variations in ORF2 epitope domains were apparent in our isolates as compared to those from other regions, we speculate that the PCV2 prevailing in southeastern China might be different in immunogenecity and pathogenicity, by altering the tropism of PCV2 in the host or by interaction with cellular factors [32]. And the finding in genetic characteristic was valuable for vaccine development and disease control in the area. However, the exact correlations between variations and pathogenicity or immunogenecity remain to be further studied.
References
G.M. Allan, F. McNeilly, J.P. Cassidy, G.A. Reilly, B. Adair, W.A. Ellis, M.S. McNulty, Vet. Microbiol. 44, 49–64 (1995)
A.L. Hamel, L. Lin, G.P. Nayar, J. Virol. 72, 5262–5267 (1998)
W. Chun, T.S. Huang, C.C. Huang, C. Tu, M.H. Jong, S.Y. Lin, S.S. Lai, Virology 66, 469–475 (2004)
G.M. Allan, F. McNeilly, S. Kennedy, B. Daft, E.G. Clark, J.A. Ellis, D.M. Haines, B.M. Meehan, B.M. Adair, J. Vet. Diagn. Invest. 10, 3–10 (1998)
I. Tischer, H. Gelderblom, W. Vettermann, M.A. Koch, Nature 295, 64–66 (1982)
J. Ellis, S. Krakowka, M. Lairmore, D. Haines, A. Bratanich, E. Clark, G. Allan, C. Konoby, L. Hassard, B. Meehan, K. Martin, J. Harding, S. Kennedy, F. McNeilly, J. Vet. Diagn. Invest. 11, 3–14 (1999)
C. Rosell, J. Segalés, J. Plana-Duran, M. Balasch, G.M. Rodriguez-Arrioja, S. Kennedy, G.M. Allan, F. McNeilly, K.S. Latimer, M. Domingo, J. comp.. Pathol. 120, 59–78 (1999)
E.G. Clark, in Proceedings of American Association of Swine Practitioners. 499–501 (1997)
I.M. Brunborg, T. Moldal, C.M. Jonassen, J. Virol. Methods 122, 171–178 (2004)
J. Segales, M. Domingo, Vet. Q 24, 109–124 (2002)
J. Segales, C. Rosell, M. Domingo, Vet. Microbiol. 98, 137–149 (2004)
Q. Liu, L. Wang, P. Willson, B. O’Connor, J. Keenliside, M. Chirino-Trejo, R. Melendez, L. Babiuk, Can. J. Vet. Res. 66, 225–231 (2002)
J. Segales, M. Calsamiglia, A. Olvera, M. Sibila, L. Badiella, M. Domingo, Vet. Microbiol. 111, 223–229 (2005)
A. Olvera, M. Cortey, J. Segalés, Virology 357, 175–185 (2006)
C. de Boisseson, V. Beven, L. Bigarre, R. Thiery, N. Rose, E. Eveno, F. Madec, A. Jestin, J. Gen. Virol. 85, 293–304 (2004)
I. Morozov, T. Sirinarumitr, S.D. Sorden, P.G. Halbur, M.K. Morgan, K.J. Yoon, P.S. Paul, J. Clin. Microbiol. 36, 2535–2541 (1998)
M. Fenaux, P.G. Halbur, M. Gill, T.E. Toth, X.J. Meng, J. Clin. Microbiol. 38, 2494–2503 (2000)
B. Meehan, F. McNeilly, I. McNair, I. Walker, J.A. Ellis, S. Krakowka, G.M. Allan, Arch. Virol. 146, 835–842 (2001)
I. Shibata, Y. Okuda, S. Yazawa, M. Ono, T. Sasaki, M. Itagaki, N. Nakajima, Y. Okabe, I. Hidejima, J. Vet. Med. Sci. 65, 405–408 (2003)
J. Kyte, R.F. Doolittle, J. Mol. Biol. 157, 105–132 (1982)
D. Mahe, P. Blanchard, C. Truong, C. Arnauld, P. Le Cann, R. Cariolet, F. Madec, E. Albina, A. Jestin, J. Gen. Virol. 81, 1815–1824 (2000)
P. Lekcharoensuk, I. Morozov, P.S. Paul, N. Thangthumniyom, W. Wajjawalku, X.J. Meng, J. Virol. 78, 8135–8145 (2004)
C. Chae, Vet J, 168, 41–49 (2004)
A. Rovira, M. Balasch, J. Segales, L. Garcia, J. Plana-Duran, C. Rosell, H. Ellerbrok, A. Mankertz, M. Domingo, J. Virol. 76, 3232–3239 (2002)
T. Opriessnig, E.L. Thacker, S. Yu, M. Fenaux, X.J. Meng, P.G. Halbur, Vet. Pathol. 41, 624–640 (2004)
A.S. Ladekjaer-Mikkelsen, J. Nielsen, T. Stadejek, T. Storgaard, S. Krakowka, J. Ellis, F. McNeilly, G. Allan, A. Botner, Vet Microbiol. 89, 97–114 (2002)
A.L. Hamel, L.L. Lin, C. Sachvie, E. Grudeski, G.P. Nayar, Can. J. Vet. Res. 64, 44–52 (2000)
B.T. Grenfell, O.G. Pybus, J.R. Gog, J.L. Wood, J.M. Daly, J.A. Mumford, E.C. Holmes, Science. 303, 327–332 (2004)
R. Larochelle, R. Magar, S. D’Allaire, Virus Res. 90, 101–112 (2002)
Q. Liu, S.K. Tikoo, L.A. Babiuk, Virology. 285, 91–99 (2001)
L. Heath, A.L. Williamson, E.P. Rybicki, J Virol. 80, 7219–7225 (2006)
A. Mankertz, R. Caliskan, K. Hattermann, B. Hillenbrand, P. Kurzendoerfer, B. Mueller, C. Schmitt, T. Steinfeldt, T. Finsterbusch, Vet. Microbiol. 98, 81–88 (2004)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shuai, J., Wei, W., Li, X. et al. Genetic characterization of porcine circovirus type 2 (PCV2) from pigs in high-seroprevalence areas in southeastern China. Virus Genes 35, 619–627 (2007). https://doi.org/10.1007/s11262-007-0121-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0121-0