
Abstract
Background
Fusarium wilt of banana (Musa spp.) caused by the fungal pathogen Fusarium oxysporum f. sp. cubense (Foc) is a typical soilborne disease, that severely devastates the banana industry worldwide, and soil microbial diversity is closely related to the spread of Fusarium wilt. To understand the relationship between microbial species and Fusarium wilt, it is important to understand the microbial diversity of the Fusarium wilt-diseased and disease-free soils from banana fields.
Results
Based on sequencing analysis of the bacterial 16S rRNA genes and fungal internal transcribed spacer (ITS) sequences, Foc abundance, fungal or bacterial richness and diversity were higher in the diseased soils than in the disease-free soils. Although Ascomycota and Zygomycota were the most abundant fungi phyla in all soil samples, Ascomycota abundance was significantly reduced in the disease-free soils. Mortierella (36.64%) was predominant in the disease-free soils. Regarding bacterial phyla, Proteobacteria, Acidobacteria, Chloroflexi, Firmicutes, Actinobacteria, Gemmatimonadetes, Bacteroidetes, Nitrospirae, Verrucomicrobia and Planctomycetes were dominant phyla in all soil samples. In particular, Firmicutes contributed 16.20% of the total abundance of disease-free soils. At the bacterial genus level, Bacillus, Lactococcus and Pseudomonas were abundant in disease-free soils with abundances of 8.20, 5.81 and 2.71%, respectively; lower abundances, of 4.12, 2.35 and 1.36%, respectively, were found in diseased soils. The distribution characteristics of fungal and bacterial genera may contribute to the abundance decrease of Foc in the disease-free soils.
Conclusion
Unique distributions of bacteria and fungi were observed in the diseased and disease-free soil samples from banana fields. These specific genera are useful for constructing a healthy microbial community structure of soil.
Similar content being viewed by others

Background
Microorganisms play an important role in the formation and maintenance of healthy soil, improve soil conditions through many enzymatic activities that regulate the biogeochemical cycle, reorganization and mineralization of organic matter [1,2,3,4,5,6]. The microbiota present in soil are affected by climate, cultivation methods, soil nutrients, pathogens and soil management practices [7,8,9,10]. Rhizosphere microorganisms comprise a dynamic community with a complex interaction with plants [11]. Some intrinsic microbial communities or specific sub-populations have potential functions to suppress soilborne diseases caused by fungi, oomycetes, bacteria and nematodes [5, 6, 11]. Hence, understanding the microbial composition of the plant rhizosphere is key to controlling the spread of soilborne disease.
Banana (Musa spp.) is an important cash and food crop in the tropics and subtropics and is one of the world’s top 10 staple foods [12, 13]. Most of cultivated bananas multiplied by the vegetative propagation are prone to infection by various pests and diseases, particularly Fusarium wilt of banana. The disease is caused by the soilborne fungus Fusarium oxysporum f. sp. cubense (Foc), which is one of the major constraints to banana production worldwide [14]. Foc infects the xylem through the roots, and causes extensive necrosis leading to of plant deaths [14]. Currently, management of the Fusarium wilt disease mainly involves rotation, selection of resistant varieties, and chemical or biological methods. Nonetheless, there is no an effective control for Fusarium wilt of banana, as evidenced by the continuous spread among continents, countries and regions [15, 16]. In general, biocontrol is considered as a safe, environmentally friendly and cost-effective method for disease control [5, 8, 16].
Multiple pathogen-plant systems have been widely used to suppress different innate diseases in the agricultural soil [6, 17,18,19]. Once attacked by root pathogens, plants can exploit microbial consortia from soil for protection against pathogen infection. Comparative microbiome analysis indicated that the relative abundance of bacterial and fungal diversity was significantly higher in the suppressive soil than that in the conducive soil [6]. High richness and diversity indices of bacterial communities were also detected in the soil naturally suppressive to Fusarium wilt of banana [18]. In addition, known plant-beneficial rhizobacteria such as Azospirillum, Gluconacetobacter, Burkholderia, Comamonas and Sphingomonadaceae were more prevalent in soil suppressive to tobacco black root rot disease [19]. Furthermore, application of Paenibacillus polymyxa NSY50 reduced the abundance of Fusarium and increased the population of beneficial microbes, including Bacillus, Actinobacteria, Streptomyces, Actinospica, Catenulispora and Pseudomonas genera [20]. These beneficial microorganisms inhibit pathogen infection through metabolites such as pyoverdins and iron-chelating siderophores [21]. Hence, novel bacterial and fungal taxa can serve as indicators of disease suppression in soil-quality assessments. A previous study revealed that microbial species composition and abundance, which are also significantly influenced by the chemical properties of soil, are vital for favorable microecology [22]. However, how soil microbial species are affected by mutual constraints among disease occurrence, pathogenic abundance, soil chemical properties and beneficial microorganisms are still an open question.
In the present study, we performed a comparative microbiome analysis of the Fusarium wilt-diseased and Fusarium wilt-suppressive (disease-free) rhizosphere soils. Based on 454-pyrosequencing of the fungal internal transcribed spacer (ITS) region and the bacterial 16S rRNA gene, changes in dominant bacterial and fungal species were investigated in both types of soils. Additionally, correlations between soil chemical properties and microbial distributions were analyzed. Our results offer new insight into identifying fungal and bacterial genera associated with Fusarium wilt disease-free soils and suggest the microbial genera and mechanisms involved in Fusarium wilt suppressiveness.
Results
Quantitative analysis of Foc colonization
Fusarium wilt-diseased soil samples (XB + HB + NB) and disease-free soil samples (XJ + HJ + NJ) were collected from three farms. Abundance of Foc was analyzed in each soil sample. Our results showed that population of Foc ranged from1.73 × 103 CFU/g to 3.07 × 103 CFU/g in Fusarium wilt-diseased soils and from 0.90 × 103 CFU/g to 1.30 × 103 CFU/g in the diseased-free soils. The largest population of Foc (3.07 × 103 CFU/g) was detected in the NB sample and the smallest (0.90 × 103 CFU/g) in the XJ sample. Quantitative analysis of Foc colonization showed more than 1700 CFU/g in the diseased soil samples from the three different farms, but less than 1300 CFU/g of Foc was detected in the disease-free soils. On the same farm, the Foc population in the diseased soil samples (HB, NB and XB) were higher (p < 0.05) than those in the disease-free soil samples (HJ, NJ and XJ) (Fig. 1).

Number of the pathogen Fusarium oxysporum f. sp. cubense in different soil samples. HB, NB and XB represent the diseased soil samples collected from Huangtong, Nanbao and Xinying farms, respectively. HJ, NJ and XJ represent the disease-free soil samples collected from Huangtong, Nanbao and Xinying farms, respectively. The data in each column is the average value of three replicates (n = 10). The letter on each bar represents a significant difference at the 5% level
Microbiome data acquisition and analysis
After quality evaluation of raw sequence libraries, a pyrosequencing-based analysis was performed to detect the V3-V4 regions of the bacterial 16S rRNA genes and the fungal ITS sequences. In total, we obtained 540,711 high-quality bacterial sequences and 542,218 fungal sequences from 18 soil samples. The high-quality reads ranged from 20,119 to 38,942 for bacterial datasets and from 21,437 to 37,800 for fungal datasets. The length distribution of trimmed sequences ranged from 304 bp to 509 bp for bacterial datasets and from 201 bp to 356 bp for fungal datasets. After homogenization, a total of 444,817 high-quality bacterial 16S rRNA sequences were clustered into 3368 distinct bacterial Operational Taxonomic Units (OTUs). Regarding fungal communities, 1517 distinct OTUs were observed for 380,736 high-quality fungal ITS sequences. Rarefaction curves were drawn for the fungal and bacterial datasets according to OTUs. These data had been submitted to NCBI under accession number SRP132524.
Richness and diversity of bacterial and fungal communities
Pooled sequences from three replicates for each sample were compared at 3% dissimilarity. A similar tendency of OTUs change was found for the diseased and disease-free soil samples from the same farm (Fig. 2). The diseased soil samples (NB and HB) showed higher OUTs than those in the disease-free soil samples (NJ and HJ). Similarly, higher indices of the fungal ACE and Chao1 were observed in the diseased soil samples (Table 1). In contrast, no obvious difference (p < 0.05) in species diversity according to Shannon and Simpson indices was observed between the soil types, whereas a lower bacterial Simpson index was demonstrated in the diseased soil samples (Table 2). The disease-free soil samples owned higher indices (p < 0.05) of ACE, Chao1 and Shannon. For these bacterial sequences, the diseased soil samples exhibited higher OTUs than in the disease-free soil samples. For the fungal ITS sequences, higher OUT numbers were observed in the diseased soil samples (NB and HB) than those in the disease-free soil samples (NJ and HJ). However, opposite OUT number results were observed for XJ and XB samples.

Rarefaction analysis of the diseased and disease-free soil samples at a 3% dissimilarity level. HB, NB, XB, HJ, NJ and XJ were described in Fig. 1. Each sample had three replicates (n = 10)
Distributions of bacterial and fungal communities
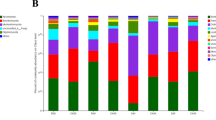
After removing singletons, 2475, 2491, 2341, 2281, 2222 and 2230 bacterial OTUs were obtained for XB, HB, NB, HJ, NJ and XJ, respectively. For fungal sequences, 633, 453, 649, 544, 411 and 621 OTUs were found in HB, NB, XB, HJ, NJ and XJ, respectively (Figs. 3, 4). In general, there were a greater number of bacterial and fungal OTUs in the diseased soil samples than in the disease-free soil samples from the same farm. Hierarchical cluster analysis revealed that the same farm presented the similar bacterial and fungal distributions (Fig. 5a and c), and bar plot analysis of the community structure further showed 25 bacterial and 32 fungal genera among the different farms (Fig. 5b and d). Although no obvious difference was observed in the number of genera between the diseased soils and the disease-free soils, the community distribution of unique genera was significantly different (p < 0.05).

Venn diagram of bacterial disease and disease-free soil samples from the three different farms. Diseased soil samples (XB + HB + NB) and disease-free soil samples (XJ + HJ + NJ) were collected from different farms. The shared and unique bacteria OTUs were shown at a 0.03 dissimilarity distance after removing singletons

Venn diagram for fungal disease and disease-free soil samples from the three different farms. Diseased soil samples (XB + HB + NB) and disease-free soil samples (XJ + HJ + NJ) were collected from different farms. The shared and unique fungus OTUs were shown at a 0.03 dissimilarity distance after removing singletons

Construction of hierarchical cluster trees using a weighted UniFace algorithm for the diseased and the disease-free soil samples. Each sample has three replicates. a hierarchical cluster tree of bacteria; b abundance of bacterial community at the level of genus; c hierarchical cluster tree of fungi; d abundance of fungal community at the level of genus. “Others” indicate the abundance of less than 2% of genus
Bacterial and fungal community composition
The top 15 dominant bacterial genera and phyla were identified according to the total relative abundances of each genus and phylum in all samples (Fig. 6a, b). At the genus level, Bacillus, Lactoccocus, JG37-AG-4, Acidobacteriaceae, Nitrospira, Anaerolineaceae, Pseudomonas, Xanthobacteraceae, Nitrosomonadaceae, DA111, Bryobacter and JG30-KF-AS9 were dominant genera with over 1% of abundance in the diseased and disease-free soils. Four dominant genera (Bacillus, Lactococcus, Pseudomonasand, Nitrosomonadaceae) exhibited different proportions in both types of soil samples (Fig. 6a). At the phylum level, Proteobacteria, Acidobacteria, Chloroflexi, Firmicutes, Actinobacteria, Gemmatimonadetes, Bacteroidetes, Nitrospirae, Verrucomicrobia and Planctomycetes were identified as the dominant phyla. Firmicutes showed a proportional difference (p = 0.00079) between both types of soil samples. (Fig. 6b).

Analysis of abundance differences between the diseased soil samples and the disease-free soil samples at the bacterial genus. a richness differences of bacterial genus; b richness differences of bacterial phylum; c richness differences of fungal genus; d richness differences of fungal phylum. The y-axis represents the classification levels of species, and the x-axis represents the percentage values of species abundance in each sample. The blue and red columns represent the average results in the diseased and disease-free soil samples, respectively. Significant differences were showed according to the Wilcoxon rank-sum test (*: 0.01 < P < =0.05, **: 0.001 < P < =0.01, ***: P < =0.001)
For the top 15 dominant fungal genera and phyla (Fig. 6c, d), Mortierella, Fusarium, Pseudallescheria, Nectriaceae, Chloridium, Chaetomium, Humicola, Trichoderma, Ascomycota were the dominant fungal genera in all soil samples (Fig. 6c), with relative abundance of more than 1%. Higher proportions of Mortierella, Fusarium, Pseudallescheria, Nectriaceae, Aspergillus and Penicillium were demonstrated. At the phylum level, Ascomycota and Zygomycota were considered to be dominant phyla and showed an obvious difference between the diseased soil samples and the disease-free soil samples (Fig. 6d).
Comparison of fungal and bacterial community characteristics
For fungi, Mortierella, Fusarium, Pseudallescheria and Nectriaceae with over 0.1% of relative abundance were enriched in the diseased and disease-free soil samples. An obvious difference was observed in their relative abundance between both types of soils. In contrast, Fusarium, Pseudallescheria and Nectriaceae were enriched in the diseased soils with the abundances of 16.39, 8.71 and 9.00%, respectively, whereas only 8.30, 2.62 and 1.20% were detected in the disease-free soils, respectively. The most abundant genus Mortierella (34.64%) was detected in the disease-free soils, with only 15.44% abundance in the diseased soils. In addition, we found that the relative abundances of Aspergillus and Penicillium were 3.30 and 3.01%, respectively, in the disease-free soil samples, and 0.34 and 0.52%, respectively, in the diseased soil samples (Table 3).
Significant differences between the diseased and disease-free soils were found for 15 bacterial genera, with a relative abundance of more than 0.1%. Compared with the diseased soils, Bacillus, Lactococcus, Pseudomonas, Sphingomonas, Psychrobacter, Oceanobacillus, Brochothrix, Carnobacterium, Flavisolibacter, Streptococcus and Burkholderia were abundant in the disease-free soils. Bacillus, Lactococcus and Pseudomonas exhibited abundances of 8.20, 5.81 and 2.71% in the disease-free soils, respectively, and 4.12, 2.35 and 1.36% in the diseased soils. The abundance of Streptococcus in the diseased and disease-free soils was also significantly different (Table 3).
Effects of soil environmental variables on phylum abundance
To understand the effects of soil environmental variables on the phylum abundances of bacteria and fungi, we measured total organic carbon (TOC), total organic nitrogen (TON), pH, available phosphorus (AP) and available potassium (AK) in the different soil samples. For a given farm, the disease-free soils had higher pH and AP (p < 0.05), while concentrations of TON, TOC and AK in the diseased soils were higher (p < 0.05) (Table 4).
Redundancy analysis (RDA) was employed to evaluate the soil environmental variables and phylum abundance. Our data showed that the first and second RDA components explained 30.24% of the total fungal phylum variation (Fig. 7a) and 57.32% of the total bacterial phylum variation (Fig. 7b). The diseased and disease-free soil samples were obviously differentiated by the results of fungal and bacterial RDA1 (Fig. 7a and b). For the distribution of microbial species, Fusarium showed negative correlations with pH and AP but positive correlations with TOC and TON. Bacillus had positive correlations with pH and AP but negative correlations with TOC and TON.

Analysis of the correlation between microorganisms and environmental variables in both types of soil samples at the level of genus. Mircoorganisms at the level of genus. a bacteria; b fungi. Environmental variables: pH,AP,AK,TOC,TON
Discussion
Understanding of microbial species and distributions is essential for controlling plant diseases [23]. In our study, lower colonization of Foc was detected in the disease-free soils, suggesting that the severity of banana Fusarium wilt accompanied by the increase of Foc colonization in the field. Previous reports have indicated that 1000 CFU/g can be considered a critical index of Foc diagnosis in the pot experiments [23, 24]. However, we revealed that lower than 1300 CFU/g of Foc colonization will not cause Fusarium wilt of banana in the field. It was supported by that over 1700 CFU/g of Foc colonization were detected in the diseased soils. This difference may be due to the complex field environment.
Although the ACE index was much higher in the diseased soils than in the disease-free soils, the species diversity based on the analysis of Shannon and Simpson had no significant differences, indicating that the species structure was similar in both types of soil samples. Higher fungal OTUs in the diseased soils may be due to an increase of Foc colonization, which might provoke the changes of microbial species and distributions. Thus far, a few reports have demonstrated the relationship between the incidence of fungal disease and the type of dominant genera. In our study, the dominant fungal phyla were also similar in the diseased and disease-free soils. Ascomycota and Zygomycota were the most abundant fungal phyla, accounting for 93.46 and 88.59% of total fungal ITS sequences. The abundance of Ascomycota in the diseased soils was much higher (p = 0.002) than that in the disease-free soils. It was consistent with previous study that the decrease of Proteobacteria and Ascomycota were associated with some fungal diseases suppression [25,26,27].
By contrast, Mortierella, Fusarium, Pseudallescheria, Nectriaceae, Chloridium, Chaetomium, Humicola, Trichoderma and Ascomycota were the dominant genera in both types of soil samples. Although the soil conditions or plant varieties could cause different distributions of the dominant fungal genera, Fusarium is frequently considered as one of the most dominant genera in some studies [6, 18, 28]. Nonetheless, the dominant genera Trichoderma and Mortierella were enriched in the disease-free soils, whereas Fusarium, Pseudallescheria, Nectriacea, Chaetomium, Humicola, Sordarionmyetes and Gibellulopsis demonstrated a decreasing tendency. Trichoderma has been widely used as a biological control agent against various pathogens [29], and although Mortierella has not been as a biological control agent, some antifungal and antibacterial metabolites proved to be produced by isolates of Mortierella [30,31,32,33]. Considering the only 8.30% of Fusarium abundance in the disease-free soils, we speculated that Trichoderma and Mortierella may be associated with suppression of banana Fusarium wilt disease. However, whether the regulation mechanism is through direct antagonism or resource competition, especially in the case of Mortierella is currently unknown.
Our results indicated that the higher abundance and diversity of bacteria were detected in the diseased soils. However, opposite results showed that a large number of OTUs were observed in the Fusarium wilt disease-free soils [25]. No significant difference of bacterial community distribution and diversity was exhibited in the rhizosphere soil samples of diseased and disease-free apple tree [34]. It may be caused by the field sampling time or soil microorganism changes in the presence of pathogens. Although many studies has shown that increased microbial diversity played an important role in the control of diseases [35,36,37], the relative abundance of several bacterial taxa is a more important indicator of disease suppression than the exclusive presence of specific bacterial taxa [17, 38]. It was supported by that most of abundant phyla such as Proteobacteria, Acidobacteria, Chloroflexi, Firmicutes, Actinobacteria, Gemmatimonadetes, Bacteroidetes, Nitrospirae, Verrucomicrobia and Planctomycete were abundant in both types of soil samples. Proteobacteria and Firmicutes were the most dominant phyla, which were similar to the previous studies on diseases infected by Rhizoctonia solani [38, 39]. It might be related to the different growth rate of bacteria [40, 41]. In addition, the higher abundance of Firmicutes in the disease-free soils supported that the disease incidence was negatively correlated with the richness of Firmicutes [18]. Similarly, the abundances of Acidobacteria and Firmicutes in a wheat rhizosphere were positively correlated with disease suppression [42]. Moreover, the abundances of Bacillus, Lactococcus and Pseudomonas in the disease-free soils were two folds than those in the diseased soils (Table 3). Bacillus and Pseudomonas were proved to be responsible for natural suppression of Fusarium wilt disease [25, 43,44,45,46,47]. Streptococcus was also enriched in disease-free soils probably because of the antagonist relationship between Streptococcus and Fusarium oxysporum [48], suggesting that Streptococcus may participate resistance to Fusarium wilt disease. Actually, the complex phenomenon of disease suppression in soils cannot be simply ascribed to a single bacterial taxon or group, but was governed by microbial consortia [38]. Plants and microbiota established a microenvironment on the roots with a complex interaction. A model of seven-species community evidenced that beneficial microbes inhibited the infection of phytopathogenic fungus Fusarium verticillioides in plant roots [11]. This model system research provides a useful method for future research on the banana-microbe interaction. However, we should fully recognize the effect of beneficial species on Fusarium suppression before assembling model community on banana. Comparisons of bacterial communities from banana Fusarium wilt diseased and diseased-free soils will prove to be essential for constructing disease suppressive soil in the future.
Based on the results of RDA analysis, disease status may accelerate soil environmental variables that affect the distribution profiles of bacterial and fungal communities [25, 49,50,51]. Higher AP and pH were detected in the disease-free soil samples. In addition, AP or pH exhibited a negative correlation with abundance of Fusarium, but was a positive correlation with abundance of Bacillus. A previous study reported that a high pH enhanced the suppression of Fusarium wilt disease [52]. Similarly, high soil AP was also associated with the inhibition incidence of stem rot disease caused by Rhizoctonia solani [53]. Therefore, the soil management strategies were related to the inhibition of Fusarium wilt disease of banana [54]. Our findings provided an evidence that higher pH and AP may enhance the inhibition of banana Fusarium wilt disease. It may be related to directly increased populations of beneficial genera or indirectly to altered soil nutrient availability to host plants. Additionally, TOC and TON showed significant differences between disease and diseased-free soils, with a positive correlation with the fungal taxon Fusarium and a negative correlation with the bacterial taxon Bacillus. Previous study demonstrated that bacterial and fungal communities were mainly driven by soil organic matter [55]. Indeed, due to the resource use preference of different microbes, contents of TOC and TON may alter the microbial community. For example, the soil microorganisms responded differently to the inputs of inorganic and organic fertilizers in paddy and banana soils [56,57,58]. Based on these findings, we can effectively suppress Fusarium wilt disease in practice by optimizing the structure of fertilization.
Conclusions
We compared the distribution of the microbial communities in Fusarium wilt-diseased and disease-free soils. The bacterial and fungal distributions in the diseased soils were different from those in the disease-free soils. Regardles, a higher abundance of Fusarium was observed in the diseased soils, whereas the Firmicutes phylum and the Bacillus, Lactococcus and Pseudomonas genera were enriched in the disease-free soils. Regarding the fungal distribution, the abundance of Ascomycota in the diseased soils was much greater than that in the diseased-free soils. Mortierella was most dominant genera in the disease-free soils. We also found that Bacillus abundance, AP concentration and pH value were positively correlated with suppression of banana Fusarium wilt disease. These results will provide a theoretical basis for evaluating soil disease-free conditions and controlling plant diseases.
Methods
Preparation of soil samples
Three soil samples were selected from three banana farms designated as Nanbao (19°47′1″N and 109°51′17″E), Xinying (19°40′51″N and 109°35′58″E) and Huangtong (19°49′58″N and 109°50′58″E) in Hainan, China, respectively. The soil type is laterite soil. Banana has been planted on more than 30 hectares (ha) at each farm for 6 years. Each farm was separated by Fusarium wilt-diseased and disease-suppressive areas (i.e., a disease-free area). Similar field management included the banana cultivar (Musa acuminate AAA Cavendish cv. Brazil), planting density (2400 seedlings per ha), fertilization and irrigation.
Disease-free and diseased rhizosphere soil samples were collected from the three farms with the oral permission of the farm owners. Briefly, 10 individual banana trees with at least 5 m spacing were randomly selected. Each soil sample was collected from four sites of a depth of 20 cm around each tree rhizosphere using a 25 mm soil auger. Finally, a total of 40 selected soil samples from ten trees were mixed as a composite soil sample. We named the disease-free and diseased soil samples as NJ and NB from the Nanbao farm, XJ and XB from the Xinying farm, HJ and HB from the Huangtong farm, respectively. After each soil sample was ground and sieved through a 2-mm sieves, it was divided into two portions: one sample was air-dried for chemical property analysis, and the other was stored at − 70 °C for DNA extraction.
Microbial community analysis
DNA extraction and PCR amplification
Microbial DNA was extracted using an E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to the standard manufacturer’s protocol. The final DNA concentration was detected using a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). The integrity of the DNA was assessed by 1% agarose gel electrophoresis.
The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified with a pair of primers 338-F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806-R (5′-GGACTACHVGGGTWTCTAAT-3′). For amplification of the fungal ITS sequences, the forward primer ITS1-F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and the reverse primer ITS1-R (5′-GGACTACHVGGGTWTCTAAT-3′) were used. The 20-μL PCR reaction systems contained 2 μL of 10× FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.2 μL of FastPfu Polymerase, 0.2 μL of BSA and 10 ng of template DNA. The PCR reaction was performed using a thermocycler PCR system (Thermal conditions) as follows: 3 min of denaturation at 95 °C, 27 cycles at 95 °C for 30s, 55 °C for 30s and 72 °C for 45 s, and a final elongation at 72 °C for 10 min. For the fungal ITS sequences, the same parameters were applied for 35 cycles.
After 2% agarose gel electrophoresis, the PCR products were eluted using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, CA, USA). The DNA fragments were quantified by QuantiFluor™-ST (Promega, WI, USA) according to the manufacturer’s protocol [59].
Illumina MiSeq sequencing
Purified DNA fragments were pooled in equimolar amounts and paired-end sequenced (2 × 300) using the Illumina MiSeq platform (Illumina, San Diego, USA) according to standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). Raw data for bacterial 16S rRNAs and fungal ITS sequences were submitted to the NCBI Sequence Read Archive (SRA) database under accession number SRP132524.
Sequence processing
Raw sequence files were demultiplexed, quality-filtered by Trimmomatic and merged by FLASH with the following criteria: (i) these reads were truncated at any site with an average quality score < 20 over a 50-bp sliding window; (ii) primers were exactly matched allowing a 2-nucleotide mismatch, and reads containing ambiguous bases were removed; (iii) sequences with an overlap longer than 10 bp were merged according to their overlapping sequences [60]. After quality filtering and chimera removal, rarefaction curves were plotted to determine the abundance of the communities and sequencing data for each sample [59, 61]. The abundance-based coverage estimator (ACE) index, Chao richness estimator (Chao1), Shannon diversity (H) and Simpson diversity (1/D) indices were calculated using the MOTHUR package (version 1.22.2 http://www.mothur.org) with Operational Taxonomic Units (OTUs) at 0.97 level [60, 61]. OTUs were clustered with a 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/), and chimeric sequences were identified and removed using the UCHIME software [61, 62]. The classification of each 16S rRNA sequence and fungal ITS sequence was analyzed by the Ribosomal Database Project (RDP) Classifier algorithm (version 2.2 http://sourceforge.net/projects/rdp-classifier/) [61] against the Silva (Release128 http://www.arb-silva.de) 16S rRNA database and the Unite (Release 6.0 http://unite.ut.ee/index.php) database using a confidence threshold of 70% [63, 64]. To examine relationships among samples, environmental variables and frequencies of genera, RDA was carried out using CANOCO for Windows [59,60,61].
Determination of soil chemical properties
The pH of a soil water suspension (1:2.5, w/v) was measured using a glass electrode meter after shaking for 30 min. TOC and TON were determined by a dry combustion method using an Element Analyzer Vario EL (Element, Hanau, Germany). The available AP in the soil was extracted with sodium bicarbonate and then determined using a molybdenum blue method [59]. The AK in the soil was extracted with ammonium acetate and determined by a flame photometry [42, 65].
Pathogen quantification
A soil solution obtained from a root wash was used for pathogen quantification for all samples collected in 2015. The number of Foc with colony forming units (CFU) colonizing the banana rhizosphere was quantified by plating a serial dilution of rhizosphere soil suspensions onto Petri plates containing the modified Komada’s selective medium. The basal medium, including 1 g of K2HPO4, 0.5 g of MgSO4·7H2O, 0.5 g of KCl, 0.01 g of Fe-Na-EDTA, 10 g of D-Galactose (in contrast to 20 g in Komada’s original medium), 2 g of L-asparagine and 16 g of agar, was diluted in 900 mL of distilled water. The basal medium was mixed with 100 mL of a solution containing 0.9 g of PCNB (pentachloronitrobenzene, 75% WP), 0.5 g of Na2B4O7·10H2O, 0.45 g of oxgall and 0.3 g of streptomycin sulfate. The pH was adjusted to 3.8 ± 0.2 with 10% (v/v) phosphoric acid. After plating, the plates were stored at 25 °C for 10 d before colonies were counted in triplicate for each sample.
Statistical analysis
All parameters were analyzed using a one-way analysis of variance (ANOVA). A Tukey HSD test was performed for a multiple comparison using the SPSS Statistics 20.0 software (IBM, NewYork, USA). The distributions of bacterial and fungal species in the two types of soil samples were compared using the method of the Wilcoxon rank-sum test in the SPSS software (IBM, New York, USA).
Abbreviations
- AK:
-
Available potassium
- AP:
-
Available phosphorus
- Foc :
-
Fusarium oxysporum f. sp. cubense
- ITS:
-
Internal transcribed spacer
- OTU:
-
Operational Taxonomic Units
- RDA:
-
Redundancy analysis
- TOC:
-
Total organic carbon
- TON:
-
Total organic nitrogen
References
Luan F, Zhang L, Lou Y, Wang L, Liu Y, Zhang H. Analysis of microbial diversity and niche in rhizosphere soil of healthy and diseased cotton at the flowering stage in southern Xinjiang. Genet Mol Res. 2015;14:1602–11.
Bonanomi G, Antignani V, Capodilupo M, Scala F. Identifying the characteristics of organic soil amendments that suppress soilborne plant diseases. Soil Biol Biochem. 2010;42:136–44.
Shiomi Y, Nishiyama M, Onizuka T, Marumoto T. Comparison of bacterial community structures in the rhizoplane of tomato plants grown in soils suppressive and conducive towards bacterial wilt. Appl Environ Microbiol. 1999;65:3996–4001.
Larkin RP. Soil health paradigms and implications for disease management. Annu Rev Phytopathol. 2015;53:199–221.
Van Bruggen AHC, Sharma K, Kaku E, Karfopoulos S, Zelenev VV, Blok WJ. Soil health indicators and Fusarium wilt suppression in organically and conventionally managed green house soils. Appl Soil Ecol. 2015;86:192–201.
Siegel-Hertz K, Edel-Hermann V, Chapelle E, Terrat S, Raaijmakers JM, Steinberg C. Comparative microbiome analysis of a Fusarium wilt suppressive soil and a Fusarium wilt conducive soil from the Châteaurenard region. Front Microbiol. 2018;9:568.
Dong L, Xu J, Zhang L, Cheng R, Wei G, Su H, Yang J, Qian J, Xu R, Chen S. Rhizospheric microbial communities are driven by Panax ginseng at different growth stages and biocontrol bacteria alleviates replanting mortality. Acta Pharm Sin B. 2018;8:272–82.
Huang N, Wang W, Yao Y, Zhu F, Wang W, Chang X. The influence of different concentrations of bio-organic fertilizer on cucumber Fusarium wilt and soil microflora alterations. PLoS One. 2017;12:e0171490.
Zhou X, Shen Y, Fu X, Wu F. Application of sodium silicate enhances cucumber resistance to Fusarium wilt and alters soil microbial communities. Front Plant Sci. 2018;9:624.
Wang Z, Zhang J, Wu F, Zhou X. Changes in rhizosphere microbial communities in potted cucumber seedlings treated with syringic acid. PLoS One. 2018;13:e0200007.
Niu B, Paulson JN, Zheng X, Kolter R. Simplified and representative bacterial community of maize roots. Proc Natl Acad Sci U S A. 2017;114:e2450–59.
Butler D. Fungus threatens top banana. Nature. 2013;504:195.
FAO Database. http://www.fao.org/faostat/en/#data/QC. Accessed 10 Oct 2018.
Dita M, Barquero M, Heck D, Mizubuti E, Staver C. Fusarium wilt of banana: current knowledge on epidemiology and research needs toward sustainable disease management. Front Plant Sci. 2018;9:1468.
Getha K, Vikineswary S. Antagonistic effects of Streptomyces violaceusniger strain G10 on Fusarium oxysporum f. sp. cubense race 4: indirect evidence for the role of antibiosis in the antagonistic process. J Ind Microbiol Biotechnol. 2002;28:303–10.
Wang B, Yuan J, Zhang J, Shen Z, Zhang M, Li R, Ruan Y, Shen QR. Effects of novel bioorganic fertilizer produced by Bacillus amyloliquefaciens W19 on antagonism of Fusarium wilt of banana. Biol Fertil Soils. 2013;49:435–46.
Weller DM, Raaijmakers JM, Gardener BBM, Thomashow LS. Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol. 2002;40:309–48.
Shen Z, Ruan Y, Xue C, Zhong S, Li R, Shen Q. Soils naturally suppressive to banana Fusarium wilt disease harbor unique bacterial communities. Plant Soil. 2015;393:21–33.
Kyselková M, Kopecký J, Frapolli M, Défago G, Ságová-Marečková M, Grundmann GL, Moënne-Loccoz Y. Comparison of rhizobacterial community composition in soil suppressive or conducive to tobacco black root rot disease. ISME J. 2009;3:1127.
Lu S, Du N, Shu S, Sun J, Li S, Guo S. Paenibacillus polymyxa NSY50 suppresses Fusarium wilt in cucumbers by regulating the rhizospheric microbial community. Sci Rep. 2017;7:41234.
Larkin RP, Fravel DR. Efficacy of various fungal and bacterial biocontrol organisms for control of Fusarium wilt of tomato. Plant Dis. 1998;82:1022–9.
Manici LM, Caputo F. Fungal community diversity and soil health in intensive potato cropping systems of the east Povalley, northern Italy. Ann Appl Biol. 2009;155:245–58.
Xue C, Penton RC, Shen ZZ, Zhang RF, Huang QW, Li R, Ruan YZ, Shen QR. Manipulating the banana rhizosphere microbiome for biological control of Panama disease. Sci Rep. 2015;5:11124.
Biju VC, Fokkens L, Houterman PM, Rep M, Cornelissen BJC. Multiple evolutionary trajectories have led to the emergence of races in Fusarium oxysporum f. sp. lycopersici. Appl Environ Microbiol. 2017;83:e02548–16.
Shen Z, Ruan Y, Xue C, Zhang J, Li R, Shen Q. Rhizosphere microbial community manipulated by 2 years of consecutive biofertilizer application associated with banana Fusarium wilt disease suppression. Biol Fertil Soils. 2015;51:553–62.
Yao H, Wu F. Soil microbial community structure in cucumber rhizosphere of different resistance cultivars to Fusarium wilt. FEMS Microbiol Ecol. 2010;72(3):456–63.
Monkai J, Hyde KD, Xu J, Mortimer PE. Diversity and ecology of soil fungal communities in rubber plantations. Fungal Biol Rev. 2017;31:1–11.
Wang Y, Xu J, Shen JH, Luo YM, Scheu S, Ke X. Tillage, residue burning and crop rotation alter soil fungal community and water-stable aggregation in arable fields. Soil Tillage Res. 2010;107(2):71–9.
FEO I, Monday U. Root zone microflora is responsible for suppressiveness of the white root rot disease in Akwete rubber plantations. J Plant Pathol Microbiol. 2012;3:151.
Kendrick A, Ratledge C. Lipids of selected molds grown for production of n-3 and n-6 polyunsaturated fatty acids. Lipids. 1992;27:15–20.
Botha A, Paul I, Roux C, Kock JL, Coetzee DJ, Strauss T, Maree C. An isolation procedure for arachidonic acid producing Mortierella species. Antonie Van Leeuwenhoek. 1999;75:253–6.
Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, Karpinets T, Uberbacher E, Tuskan GA, Vilgalys R. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol. 2011;77:5934–44.
Soman AG, Gloer JB, Wicklow DT. Antifungal and antibacterial metabolites from a sclerotium-colonizing isolate of Mortierella vinacea. J Nat Prod. 1999;62:386–8.
Zhang Z, Feng H, Xiao X, Li H. Influence of bio-fertilizers on control of banana wilt disease and soil microbial diversity. J Fruit Sci (in Chinese). 2010;27:575–9.
Niu J, Rang Z, Zhang C, Chen W, Tian F, Yin H, Dai L. The succession pattern of soil microbial communities and its relationship with tobacco bacterial wilt. BMC Microbiol. 2016;16:233.
Qiao J, Yu X, Liang X, Liu Y, Borriss R, Liu Y. Addition of plant-growth-promoting Bacillus subtilis PTS-394 on tomato rhizosphere has no durable impact on composition of root microbiome. BMC Microbiol. 2017;17:131.
Ling N, Deng K, Song Y, Wu Y, Zhao J, Waseem R, Huang Q, Shen Q. Variation of rhizosphere bacterial community in water-melon continuous mono-cropping soil by long-term application of a novel bioorganic fertilizer. Microbiol Res. 2014;169:570–8.
Mendes R, Kruijt M, De Bruijn I, Dekkers E, Van der Voort M, Schneider JHM, Piceno YM, De Santis TZ, Andersen GL. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 2011;332:1097–100.
Li JG, Ren GD, Jia ZJ, Dong YH. Composition and activity of rhizosphere microbial communities associated with healthy and diseased greenhouse tomatoes. Plant Soil. 2014;380:337–47.
De Angelis KM, Brodie EL, De Santis TZ, Andersen GL, Lindow SE, Firestone MK. Selective progressive response of soil microbial community to wild oat roots. ISME J. 2009;3:168–78.
Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–64.
Sanguin H, Sarniguet A, Gazengel K, Moënne-Loccoz Y, Grundmann GL. Rhizosphere bacterial communities associated with disease suppressiveness stages of take-all decline in wheat monoculture. New Phytol. 2009;184:694–707.
Douriet-Gámez NR, Maldonado-Mendoza IE, Ibarra-Laclette E, Blom J, Calderón-Vázquez CL. Genomic analysis of Bacillus sp. strain B25, a biocontrol agent of maize pathogen Fusarium verticillioides. Curr Microbiol. 2018;75:247–55.
Naik PR, Raman G, Narayanan KB, Sakthivel N. Assessment of genetic and functional diversity of phosphate solubilizing fluorescent pseudomonads isolated from rhizospheric soil. BMC Microbiol. 2008;8:230.
Kejela T, Thakkar VR, Thakor P. Bacillus species (BT42) isolated from Coffea arabica L. rhizosphere antagonizes Colletotrichum gloeosporioides and Fusarium oxysporum and also exhibits multiple plant growth promoting activity. BMC Microbiol. 2016;16:277.
Castiglioni B, Sutton DA, Rinaldi MG, Fung J, Kusne S. Pseudallescheria boydii (anamorph Scedosporium apiospermum). Infection in solid organ transplant recipients in a tertiary medical center and review of the literature. Medicine. 2002;81:333–48.
Dhaisne A, Guellerin M, Laroute V, Laguerre S, Cocaign-Bousquet M, Le Bourgeois P, Loubiere P. Genotypic and phenotypic analysis of dairy Lactococcus lactis biodiversity in milk: volatile organic compounds as discriminating markers. Appl Environ Microbiol. 2013;79:4643–52.
Al-Rubaye AF, Hameed IH, Kamal SA. Screening of metabolites products of fusarium oxysporum and determination of its antibacterial and antifungal activity using medicinal plants extract. Indian J Public Health Res Dev. 2018;9(3):399–404.
Acosta-Martinez V, Dowd S, Sun Y, Allen V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem. 2008;40:2762–70.
Domínguez J, Negrín MA, Rodríguez CM. Aggregate water-stability, particle-size and soil solution properties in conducive and suppressive soils to Fusarium wilt of banana from Canary Islands (Spain). Soil Biol Biochem. 2001;33:449–55.
Peng HX, Sivasithamparam K, Turner DW. Chlamydospore germination and Fusarium wilt of banana plantlets in suppressive and conducive soils are affected by physical and chemical factors. Soil Biol Biochem. 1999;31:1363–74.
Senechkin IV, Overbeek VLS, Ahc VB. Greater Fusarium wilt suppression after complex than after simple organic amendments as affected by soil pH, total carbon and ammonia-oxidizing bacteria. Appl Soil Ecol. 2014;73:148–55.
Davey R, McNeill A, Gupta V, Barnett S. Rhizoctonia root rot suppression in an alkaline calcareous soil from a low rainfall farming system//B capturing opportunities and overcoming obstacles in Australian agronomy. In: Proceedings of 16th Australian agronomy conference; 2012. p. 14–8.
Chen Y, Zhou D, Qi D, Gao Z, Xie J, Luo Y. Growth promotion and disease suppression ability of a Streptomyces sp. CB-75 from banana rhizosphere soil. Front Microbiol. 2018;8:2704.
Shen Z, Penton CR, Lv N, Xue C, Yuan X, Ruan Y, Li R, Shen Q. Banana fusarium wilt disease incidence is influenced by shifts of soil microbial communities under different monoculture spans. Microb Ecol. 2018;75:739–50.
Wang L, Li J, Yang F, Yao E, Raza W, Huang Q, Shen Q. Application of bioorganic fertilizer significantly increased apple yields and shaped bacterial community structure in orchard soil. Microb Ecol. 2017;73:404–16.
Wang J, Song Y, Ma T, Raza W, Li J, Howland GJ, Huang Q, Shen Q. Impacts of inorganic and organic fertilization treatments on bacterial and fungal communities in a paddy soil. Appl Soil Ecol. 2017;112:42–50.
Fu L, Penton CR, Ruan Y, Shen Z, Xue C, Li R, Shen Q. Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biol Biochem. 2017;104:39–48.
Zhang H, Wang Y, Chen S, Zhao Z, Feng J, Zhang Z, Lu K, Jia J. Water bacterial and fungal community compositions associated with Urban Lakes, Xi’an, China. Int J Environ Res Public Health. 2018;15:469.
Zhang H, Feng J, Chen S, Li B, Sekar R, Zhao Z, Jia J, Wang Y, Kang P. Disentangling the drivers of diversity and distribution of fungal community composition in wastewater treatment plants across spatial scales. Front Microbiol. 2018;9:1291.
Zhang H, Jia J, Chen S, Huang T, Wang Y, Zhao Z, Feng J, Hao H, Li S, Ma X. Dynamics of bacterial and fungal communities during the outbreak and decline of an algal bloom in a drinking water reservoir. Int J Environ Res Public Health. 2018;15:361.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Song C, Wang B, Tan J, Zhu L, Lou D, Cen X. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol Gen Genomics. 2016;292:407–14.
Song L, Yang S, Liu H, Xu J. Geographic and environmental sources of variation in bacterial community composition in a large-scale municipal landfill site in China. Appl Microbiol Biotechnol. 2017;101:761–9.
Bao SD. Soil agro-chemistrical analysis. Beijing: China Agriculture Press; 2007. p. 268–270, 389-391.
Acknowledgments
The authors would like to thank China Academy of Tropical Agricultural Sciences and South China Agricultural University for equipment support, and the authors are highly appreciative of Zhufen Gao for the sampling. We sincerely thank Dr. Wei Wang from China Academy of Tropical Agricultural Sciences for his critical reading of the manuscript.
Availability of data and methods
Raw data for bacterial 16S rRNAs and fungal ITS sequence were deposited in the NCBI Sequence Read Archive (SRA) database under accession number SRP132524.
Funding
This study was financially supported by The National Key Research and Development Program of China (2017YFD0202105) and China Agriculture Research System (CARS-31) and Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (NO. 1630052016005). The funding agencies had no role in the design of this study, in the collection, analysis, or interpretation of the data, or in the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
DZ designed and performed the experiments, analyzed the data, and drafted the manuscript. TJ performed pathogen content determination and analyzed part of the data. YC performed sample collection, DNA extraction and PCR amplification. FW performed sample collection, and soil chemical property analysis. DQ and RF performed sample collection, DNA extraction and PCR amplification. JX designed and performed the experiments and analyzed data. HL helped design experiments, analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical approval is not applicable in the case of the study. However, the collection of the soil samples from three banana farms was verbally permitted by each farm owner.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, D., Jing, T., Chen, Y. et al. Deciphering microbial diversity associated with Fusarium wilt-diseased and disease-free banana rhizosphere soil. BMC Microbiol 19, 161 (2019). https://doi.org/10.1186/s12866-019-1531-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1531-6