
Abstract
Background
B. napus (oilseed) is an important source of edible vegetable oil, and its nutritional and economic value is determined by its fatty acid composition and content.
Results
Using the Brassica 60 K SNP array, we performed a genome-wide association study of fatty acid composition in a population of 520 genetically diverse oilseed accessions. Using the PCA + K model in TASSEL 5.2.1, we identified 62 genomic regions that were significantly associated with the composition of seven fatty acids, and five consensus regions that mapped to the A2, A8, A9, C1, and C3 chromosomes, respectively, of the Brassica napus Darmor-bzh genome. We then identified 24 orthologs of the functional candidate genes involved in fatty acid biosynthesis, excluding BnaA.FAE1 and BnaC.FAE1 on the A8 and C3 homologous genome blocks, which are known to have critical roles in the fatty acid biosynthesis pathway, and potential orthologs of these genes (e.g., LACS9, KCR1, FAB1, LPAT4, KCS17, CER4, TT16, and ACBP5).
Conclusions
Our results demonstrate the power of association mapping in identifying genes of interest in B. napus and provide insight into the genetic basis of fatty acid biosynthesis in B. napus. Furthermore, our findings may facilitate marker-based breeding efforts aimed at improving fatty acid composition and quality in B. napus.
Similar content being viewed by others

Background
Oilseed rape (Brassica napus L., genome AACC, 2n = 38) is the most important source of edible vegetable oil and protein-rich meal in the Chinese diet and the second most important oilseed crop in the world after soybean [1]. Moreover, the fatty acid composition of Brassica oil determines its physical, chemical, and nutritional qualities [2, 3]. Rapeseed oil has a lower saturated fatty acid content than most other vegetable oils, consisting of about 60% oleic acid [C18:1], 4% palmitic acid [C16:0], and 2% stearic acid [C18:0], and its fatty acid composition is considered by many nutritionists to be ideal for human nutrition and superior to that of many other plant oils [4]. However, Brassica species oils also contain high levels of erucic acid and glucosinolate, which are toxic, so double-zero rapeseed breeding is the most important breeding objective in rapeseed. Therefore, there is much interest in improving the fatty acid profile of B. napus.
In plants, fatty acids have essential roles in maintaining membrane function and cell growth and development, and oilseed species such as B. napus synthesize storage oils (TAGs, triacylglycerols) from fatty acids. Previous research showed that the de novo synthesis of fatty acids primarily occurred in the plastids of plants [5, 6]. In rapeseed, however, numerous important quality traits are typical quantitative traits with complex underlying genetic mechanisms. In the past decade, quantitative trait locus (QTL) mapping has yielded a vast amount of information on complex traits of rapeseed, such as oil content [7–13] and fatty acid composition [2–4, 14, 15], and the underlying QTLs were mapped to all 19 linkage groups of B. napus. Nevertheless, few of the detected QTLs have been successfully used in rapeseed breeding programs [10].
Recently, association mapping, also known as linkage disequilibrium (LD) mapping, which is a population-based survey technique used to identify trait-marker relationships based on LD in plants [16–18], significantly increased the precision of the estimated QTL localization [19]. Moreover, association mapping has been extensively used to dissect agronomic trait-marker relationships in plants, including Arabidopsis thaliana, Oryza sativa (rice), Zea mays (maize), and B. napus [17, 18, 20–24]. B. napus, an allopolyploid species, has a complex genome structure, with high levels of similarity with the A- and C-subgenomes and both homologous and non-homologous exchange between the A- and C-subgenomes [25, 26], rendering high-throughput discovery of high-quality molecular markers for genome-wide association studies (GWAS) challenging. Association mapping via genome-wide approaches has been performed using the Brassica 60 K SNP BeadChip Array in B. napus [27–31]. Additionally, these association studies make much broader use of available germplasms, thereby ensuring a more comprehensive and precise mapping of QTLs in B. napus.
In this study, a large panel of 520 rapeseed accessions were genotyped using the Brassica 60 K Infinium® SNP array [32]. The plant materials were collected from major rapeseed breeding institutes in China and overseas (German, Sweden, Denmark, Canada, and United States). Then we analyzed variations of the fatty acid composition in a diverse set of B. napus accessions and performed a GWAS for fatty acid composition, uncovering large numbers of loci that had not been reported previously. We further validated the highly promising candidate loci based on new molecular markers in an independent bi-parental breeding population. These findings will enhance our understanding of the processes of fatty acid metabolism in B. napus..
Methods
Plant materials and DNA preparation
In total, 520 genotypes of rapeseed cultivars and breeding materials, including a range of morphological types and derived from various geographical origins, were collected from the major breeding institutes across China. 497 accessions (113, 255, 88 and 41 are widely distributed on the upper, middle and lower of Yangtze, and the northern growing areas, respectively) are selected in China, and 23 accessions are introduced from different countries (Additional file 1: Table S1). According to the information from providers and observations during the experimental period, the accessions grow normally under the winter-growth conditions of the major rapeseed regions in Beibei, Chongqing, China, in the growing seasons of 2012–2013 and 2013–2014 (designated 2013Cq and 2014Cq, respectively). A randomized complete block design was used with three replications in the field experiments. For each accession, 10–12 plants were grown per row. The field management essentially followed standard agronomic procedures. At maturity, self-pollinated seeds from each accession were harvested for seed quality trait analysis.
Total gDNA was extracted from bulked young leaf tissue of each accession using a standard CTAB extraction protocol [33] with some modifications. DNA samples were quantified by visual comparison to λ DNA standards on ethidium bromide-stained agarose gels, and the concentration and purity were calculated using a GeneSpecl spectrophotometer at 260 and 280 nm. Qualified DNA samples were then used for SNP analysis.
Fatty acid extraction and GC analysis
Bulked seed samples (200 mg) were analyzed for their fatty acid composition by gas liquid chromatography according to a published method [34], and analyzed on a Perkin Elmer Gas Chromatograph Model GC-2010 (Shimazu, JAP) equipped with a fused silica capillary column DB-WAX (30 m × 0.246 mm × 0.25 um) film thickness. The oven, injector, and detector temperature were 185 °C, 250 °C, and 250 °C, respectively. The carrier gas was nitrogen, hydrogen and air, at a speed of 60 ml/min, 40 ml/min, and 400 ml/min, respectively. Two microlitres of sample was injected at a split rate of 1:70. The value of each fatty acid was expressed as a percentage of the total amount of fatty acids identified.
Phenotypic analysis, including the mean, standard deviation, correlation coefficient, and minimum and maximum values of per fatty acid from 520 accession were calculated and analyzed using SPSS15.0. The mean values of the target traits from each accession grown in two environments were used for association analysis. Variations in fatty acid at each sample was analyzed by analysis of variance (ANOVA).
SNP genotyping and physical position analysis
Genotyping for association mapping was performed using the Brassica 60 K Illumina® Infinium SNP array [32] according to the manufacturer’s protocol (https://www.illumina.com/techniques/microarrays.html) in the National Key Laboratory of Crop Genetic Improvement, National Subcenter of Rapeseed Improvement in Wuhan, Huazhong Agricultural University, 430070 Wuhan, China. Illumina BeadStudio genotyping software was used for SNP data clustering and calling according to a previously described protocol [31]. SNPs with a call frequency of <0.9 and a minor allele frequency (MAF) of ≤0.05 were excluded in this research. The remaining SNPs were scrutinized visually and those SNPs that were resolved as three clearly separated clusters (AA, AB, and BB) in the tested B. napus material were used for further research. In addition, to identify the physical position of SNP markers, the source sequences for designing SNP probes of the Brassica 60 K SNP arrays were used to perform a BlastN search against the B. napus ‘Darmor-bzh’ reference genome (version 4.1, http://www.genoscope.cns.fr/brassicanapus/data/) [26]. Only the top BLAST hits (E values of < “1.0 E−10”) were considered to be mapped in the genome, while BLAST matches to multiple loci with the same top identity were not considered to be mapped [31, 35].
Population structure, genetic relatedness, and relative kinship analysis
After filtering the inefficient SNPs, a subset of 11,368 SNPs (MAF > 0.05) evenly distributed (1 SNP for every 50 kb) across the entire genome were used for population structure and genetic relatedness analysis. The population structure of 520 accessions was evaluated by the Bayesian model-based clustering method performed in STRUCTURE 2.1 [36]. The parameters used for association mapping were previously described [35, 37]. Briefly, the number of subgroups (K) was set from 1 to 10. Five runs for each K were performed by admixture and correlated allele frequencies model with a burn-in length and the iteration number of the Markov Chain Monte Carlo (MCMC) repetitions was set to 100,000 [38]. The optimum number of subgroups (K) was selected based on the log probability of the data [LnP(D)] using STRUCTURE 2.1 output and an ad hoc statistic ∆K method proposed by Evanno et al. [39]. The highest value of ∆K for the 520 B. napus accessions was K = 2 (Fig. 2a). The genetic diversity of 520 accessions was estimated based on all of the SNPs from the Brassica 60 K Illumina® Infinium SNP array, the neighbor-joining (NJ) phylogenetic tree among individuals was constructed based on Nei’s genetic distance [40], and principal component analysis (PCA) was performed using the GCTA tool [41]. The relative kinship coefficients (K-matrix) among 520 accessions were estimated using the software package SPAGeDi [42], and negative values were set to zero, according to previous research [43].
Haplotype block construction and linkage disequilibrium
The haploid of each chromosome was determined using haploview software [44]. The parameters were set as previously described, with a Hardy Weinberg P value cutoff of 0.001, an MAF of 0.05, and a maximum Mendel error rate of 1. Haplotype blocks were generated using the four gamete rule with default parameters. Linkage disequilibrium (LD) was calculated using TASSEL 5.2.1 (http://www.maizegenetics.net/), based on the squared allele frequency correlations (r 2) between all pairs of SNP markers. Only SNPs with an allele frequency of 5% or greater were included in the association analysis. Then the polymorphism information content (PIC) of the SNP markers was estimated using PowerMarker version 3.25 [45].
Genome-wide association study (GWAS) and candidate gene annotation
Six models were evaluated in the trait-SNP association analysis, including the naïve, Q, K, PCA, K + Q, and K + PCA model, respectively. To detect association signals, the naïve, Q, K, and PCA model were performed, using the general linear model (GLM) method, and the K + Q and K + PCA models were performed with a mixed linear model (MLM) method in TASSEL 5.2.1 software, respectively. The MLM analysis was optimized using compression and performed in TASSEL 5.2.1. The significance of the association between SNPs and traits was assessed based on the threshold P < P = 1/N (where N is the total number of SNPs in this study). Quantile-quantile (QQ) plots were shown with –log10(P) of each SNP and expected P value, and Manhattan plots were displayed using TASSEL 5.2.1. False discovery rates (FDRs) were generated as previously described methods [35]. We then summarized all marker-trait associations for each trait, we supposed that these markers (within 200 kb) could be identified the same putative QTLs [46].
The significant association regions were anchored to the the B. napus ‘Darmor-bzh’ reference genome (version 4.1, http://www.genoscope.cns.fr/brassicanapus/data/) [26], which were assigned by the peak SNP markers and haplotype block analysis. Subsequently, the significant association region within two adjacent markers where a QTL was detected or the 200-kb flanking region of the peak markers linked with a QTL was scanned for selecting the putative genes according to the B. napus reference genome (http://www.genoscope.cns.fr/brassicanapus/data/) [26]. Then the predicted gene and its orthologous sequences were annotated by BLAST analysis against the A. thaliana database (http://www.arabidopsis.org/index.jsp).
Results
Phenotypic variation of fatty acid content
Descriptive statistics of fatty acid contents, including palmitic acid [C16:0], stearic acid [C18:0], oleic acid [C18:1], linoleic acid [C18:2], linolenic acid [C18:3], eicosenoic acid [C20:1], and erucic acid [C22:1] for 520 accessions, are summarized in Table 1. Continuous and wide phenotypic variations were observed in fatty acid content among the accessions in 2013Cq and 2014Cq (Fig. 1 and Table 1). The contents of palmitic acid, stearic acid, linoleic acid, linolenic acid, and eicosenoic acid was normally distributed, but the contents of oleic acid and erucic acid had multimodal distributions in these accessions (Fig. 1). The content of seven fatty acids displayed great variation in the 520 accessions grown in different environments (Table 1). The palmitic acid [C16:0] content ranged from 2.47 to 5.91% in 2013Cq and 2.47 to 7.27% in 2014Cq, with an average of 4.09 and 4.13%, respectively. Stearic acid [C18:0] content ranged from 0.70 to 3.98% in 2013Cq and 0.42 to 3.47% in 2014Cq, with an average of 1.84 and 1.45%, respectively. Moreover, oleic acid [C18:1] content, which is usually used as a standard to assess the feeding value of oilseeds, ranged from 10.72 to 76.03% in 2013Cq and 9.56 to 76.47% in 2014Cq, with an average of 56.28 and 52.85%, respectively. In addition, considerable quantitative variation was found for the contents of linoleic acid [C18:2], linolenic acid [C18:3], eicosenoic acid [C20:1], and erucic acid [C22:1], ranging from 8.38 to 34.88%, 2.15 to 15.44%, 1.31 to 21.37%, and 0.00 to 53.45% in 2013Cq, and 9.56 to 34.07%, 1.21 to 16.56%, 0.88 to 18.89%, and 0.00 to 59.18% in 2014Cq, respectively (Table 1). Moreover, the largest CV (coefficient of variation) was found between the oleic acid and erucic acid in different environments (Table 1), indicating extensive variation in the panel of accessions. In addition, Pearson’s correlation coefficient of fatty acid content in accessions grown in 2013Cq and 2014Cq ranged from 0.1428 to 0.836 (Additional file 2: Figure S1). The ANOVA showed that genotype and environment have significant effects on the fatty acid contents of B. napus (P <0.01) (Table 1).

The frequency distribution for fatty acid composition of 520 rapeseed accessions in 2013Cq and 2014Cq. Percentage indicates the proportion of the total dry weight of the seed represented by the fatty acid of interest. Cq indicates the growing region, Chongqing, China
SNP screening and LD analysis
The genotypes of 520 accessions were analyzed using the Brassica 60 K SNP array, which included 52,157 SNPs. A total of 2,934 SNPs were excluded, because they were not mapped to the reference genome [26]. Then, 17,754 SNPs with a call frequency of <90% or a MAF of <0.05 were removed and the remaining 31,468 SNPs were used to assess population structure, relative kinship LD, and association analysis. The 31,468 SNPs were distributed on 19 chromosomes, ranging from 886 SNPs on C9 to 2,886 SNPs on C4, and the mean density of SNPs ranged from 1 SNP/11.55 kb (A10) to 1 SNP/54.74 kb (C9), indicating that SNPs were not evenly distributed across the entire genome (Table 2). However, all PIC (Polymorphism Information Content) values of SNPs were uniform in each linkage group, varying from 0.279 (C9) to 0.312 (C2).
In addition, the LD decay rate was measured as the chromosomal distance at which the average pairwise correlation coefficient (r 2) dropped to half of its maximum value, and the LD decay of the genomes and subgenomes of 520 accessions are displayed (Additional file 2: Figure S2). The average distance at which r 2 decreased to half of its maximum value was 0.05-0.10 Mb for the A subgenome and 1.25-1.50 Mb for the C subgenome (Table 2). Further, LD decay varied greatly among the 19 chromosomes.
Population structure and relative kinship
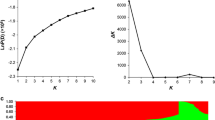
We next inferred the population structure and genetic relatedness of the 520 accessions using the model-based program STRUCTURE v2.2 [36], with a subset of SNPs (11,368 SNPs; 1 SNP/50 kb) evenly distributed throughout the genome. The results showed that the most significant change of log likelihood value [LnP(D)] occurred when K increased from 1 to 2 (Fig. 2a), and the highest ∆K showed a sharp peak at K = 2 (Fig. 2b). The 520 accessions were classified into two sub-populations (P1 and P2) with Q matrix values of ≥0.7 (Fig 2b), and a mixed sub-population with a Q matrix value of <0.7. P1 was composed of 325 accessions, 4 of which were from overseas, and the remaining accessions were selected by different breeding institutes of China; P2 contained 52 accessions, 35 of which were spring rapeseed, and 15 of which were from Canada, Denmark, German, and Sweden; and 143 lines were classified as a mixed sub-population of P1 and P2 (Q matrix values of <0.7), most of which were semi-winter rapeseed (Additional file 1: Table S1, Additional file 2: Figure S3). The F st value between the P1 and P2 subpopulations, which exhibited much variation among the accessions, was 0.36, and PCA revealed that the first two principal components could explain 31 and 25% of the genetic variance (Fig. 2d).

Analysis of the population structure and relative kinships of the 520 rapeseed accessions. a Estimated LnP(D) of possible clusters (K) from 1 to 10. b ΔK based on rate of change of LnP (D) between successive K values. c Model-based Bayesian clustering performed using STRUCTURE 2.1 for K = 2 subpopulations. Each individual is indicated by a vertical bar, partitioned into colored segments with the length of each segment representing the proportion of the individual’s genome. Green indicates subpopulation P1 genotypes and red represents subpopulation P2 genotypes. d The sub-populations in a population of 520 B. napus accessions, suggested by principal coordinate analysis (PCA). e Distribution of relative kinship coefficients across the 520 accessions. Only kinship coefficients ranging from 0 to 0.5 are shown
In addition, the average relative kinship value was 0.0869 between any two accessions. Further, approximately 51.0% of the kinship coefficients were equal to 0, and 23.9% of the kinship coefficients ranged from 0 to 0.2 (Fig. 2e). These results showed that most accessions included in this study have no or weak kinship.
Association analysis
To effectively and rapidly identify the association signals, 11,368 SNPs that were evenly distributed throughout the B. napus genome were used for the association analysis. To control for false positives, we first used six separate models (naïve, Q, K, PCA, K + Q, and K + PCA) to perform association mapping of fatty acids in the 2013Cq and 2014Cq accessions. The QQ plot showed that the K + PCA model had the lowest number of false positives and was most suitable for association mapping in this research (Additional file 2: Figure S4 and S5). Hence, we used the K+ PCA model, with a P-value of <8.8 × 10−5, to identify association signals. The results of the association analysis for the seven fatty acids combined over two environments are given in Table 3 and Fig. 3, and are summarized below.

Manhattan plots of marker-trait association analysis using the PCA + K model for the fatty acid composition of 520 accessions in 2013Cq and 2014Cq. a and b palmitic acid; c and d stearic acid; e and f oleic acid; g and h linoleic acid; i and j linolenic acid; k and l eicosenoic acid; and (m and n) erucic acid. Cq indicates the growing region, Chongqing, China. The dashed horizontal line represents the Bonferroni-adjusted significance threshold (P < 8.8 × 10−5)
Palmitic acid [C16:0]
In 2013Cq and 2014Cq, 35 and 31 significant association signals for palmitic acid were identified by the K+ PCA model, explaining 4.43 to 11.52% and 4.70 to 9.41% of the phenotypic variance, respectively. Importantly, two significant regions mapped to the A8 and C3 chromosomes, covering 3.68 and 7.47 Mb in pseudo-molecules of the B. napus ‘Darmor-Bzh’ reference genome (Table 3, Fig. 3a, b). However, significant signals were also detected in the different environments. For instance, significant regions mapped to A9 and A10 in 2013Cq, but to A2, A5, A6, C1, and C9 in 2014Cq, explaining 4.68 to 4.52% and 4.52 to 4.68% of the phenotypic variance, respectively (Table 3), indicating that palmitic acid was a classical quantitative trait, controlled by minor genes and affected by environmental factors.
Stearic acid [C18:0]
Using the PCA + K model method, a total of 7 and 7 significant association signals for stearic acid were identified in 2013Cq and 2014Cq, respectively, which were located at A5, A8, A10, and C4 in 2013Cq, and A1, A8, A10 C3, and C5 in 2014Cq (Table 3, Fig. 3c, d). The GWAS peaks barely contributed to 3.90 to 4.77% of the phenotypic variance in 2013 Cq and 4.34 to 6.57% of the phenotypic variance in 2014Cq (Table 3).
Oleic acid [C18:1]
A total of 136 and 124 SNPs, distributed on A6, A8, A9, C1, C3, C4, C8, and C9 in 2013Cq and 2014Cq, were associated with oleic acid, and 8 SNPs located on C2 were only detected in 2013Cq (Table 3). Moreover, each of the significantly associated SNPs contributed to 2.47% to 14.48% of the phenotypic variance in 2013 Cq and to 2.91 to 11.26% of the phenotypic variance in 2014Cq, respectively (Table 3). In the two environments, however, most of the SNPs were located at four significant regions (A8, A9, C1, and C3), covering 5.94, 1.57, 6.50, and 7.40 Mb in the corresponding pseudo-molecules of the B. napus ‘Darmor-Bzh’ reference genome, respectively (Table 3, Fig. 3e, f).
Linoleic acid [C18:2]
In 2013Cq, a total of 53 SNPs were associated with linoleic acid and located on ten different chromosomes (A2, A3, A6, A8, A9, C2, C3, C5, C7, and C8), and accounted for 4.16 to 13.74% of the phenotypic variance; but in 2014 Cq, 24 significant SNPs were identified and mapped on eight chromosomes (A2, A6, A8, A9, A10, C2, C3, and C8), and explained 4.53 - 10.58% of the phenotypic variance. Importantly, two associated regions located at 9.49 and 0.73 Mb of A8 and C3 were detected in 2013Cq and 2014Cq, respectively (Table 3, Fig. 3g, h).
Linolenic acid [C18:3]
For linolenic acid, a total of 20 and 21 significant signals were identified and distributed on chromosome A1, A2, A5, A6, A8, A9, C2, C4, and C8, and each of these significantly associated SNPs explained 5.27 to 9.53% and 3.79 to 9.45% of the phenotypic variance in 2013Cq and 2014Cq, respectively (Table 3, Fig. 3i, j). However, one significantly associated region in A2 was identified that spanned 14.5 Mb in the B. napus ‘Darmor-Bzh’ reference genome.
Eicosenoic acid [C20:1]
In 2013Cq and 2014 Cq, 100 and 64 significant SNPs for eicosenoic acid were identified mainly on twelve different chromosomes, and each of these contributed to 3.34 to 16.10% and 3.91 to 13.10% of the phenotypic variance, respectively (Table 3, Fig. 3k, l). Moreover, four common regions were detected at 8.79 Mb of A8, 1.70 Mb of A9, 6.49 Mb of C1, and 22.87 Mb of C3, respectively (Table 3, Fig. 3k, l). Two peak SNPs were located on chromosomes A8 and C3 that explained 15.52 and 16.10% of the phenotypic variance in 2013Cq, and 11.44 and 13.10% of the phenotypic variance in 2014 Cq (Table 3), revealing that some important adaptation-related genes may control eicosenoic acid production near the association signal.
Erucic acid [C22:1]
In 2013Cq and 2014 Cq, 24 and 40 identified significant SNPs were observed for erucic acid and distributed on chromosomes A8, A9, A10, C3, C8, and C9, and each of these explained 2.79 to 10.97% and 2.79 to 13.29% of the phenotypic variance, respectively (Table 3, Fig. 3m, n). Two common significant regions were located at 5.93 and 5.21 Mb of chromosomes A8 and C3 in 2013Cq and 2014 Cq (Table 3; Fig. 3m, n). Hence, these regions could be used to compare the GWAS results obtained in this research with the findings of published works [15, 31, 47].
In total, we detected 62 association regions were significantly associated with fatty acid that distributed on 18 chromosomes of B. napus in 2013Cq and 2014Cq, respectively (Additional file 1: Table S2). Each of these could be explained 2.31 to 14.48% of the phenotypic variance, respectively (Table 3). In the present study, we identified five common significantly associated regions for fatty acid content in 2013Cq and 2014Cq, distributed on chromosomes A2, A8, A9, C1, and C3, respectively (Table 3, Fig. 3), Moreover, the chromosomal regions were delineated by haplotype blocks with trait-associated SNPs, which included 3, 1, 4, 3, and 3 haplotype blocks, respectively (Fig. 4). Importantly, two significantly associated regions located on chromosomes A8 and C3 were consistent with previous mapping results for fatty acid analysis [15, 31, 47]. These findings will provide insight into the candidate genes for genetic basis of fatty acid biosynthesis in B. napus.

Putative functional candidate genes involved in fatty acid composition within haplotype blocks of significantly associated regions of B. napus chromosomes. The degree of significant association indicates by R 2 value with different shades. The significant haplotype blocks are denoted by black triangles
Genes underlying GWAS peaks
Based on the physical position of trait-associated SNPs in B. napus reference genome [26], sequences of identical chromosome regions were extracted and investigated the candidate genes for fatty acid composition. The results showed that significant SNPs on chromosome A8 were located in a haplotype block of 437 kb, from 10.1 Mb to 10.5 Mb, and covered three key genes, KCS18 (FAE1, BnaA08g11130D), KCS17 (BnaA08g11140D), and CER4 (BnaA08g11440D). Furthermore, three haplotype blocks were identified at 10.0 Mb of chromosome C3, and three key genes, KCS18 (FAE1, BnaC03g65980D), KCS17 (BnaC03g66040D), CER4 (BnaC03g66380D), were also confirmed at a haplotype block of 500 kb from 55.6 Mb to 56.2 Mb (Fig. 4, Table 4). The key genes BnaA.FAE1 (BnaA08g11130D) and BnaC.FAE1 (BnaC03g65980D) had also been reported to be associated with erucic acid content [31, 48, 49], indicating that our association genetics approach was successful. Moreover, B. napus BnaA08g11140D and BnaC03g66040D are orthologous to A. thaliana KCS17 [50], and B. napus BnaA08g11440D and BnaC03g66380D are orthologous to A. thaliana CER4 encoding an Alcohol-Forming Fatty Acyl-Coenzyme A Reductase involved in the synthesis of very-long-chain fatty acids [51]. In addition, other ten independent association haplotype blocks were aligned to the A2, A9 and C1 chromosomes, which covered about 5.4 Mb length of chromosome A2, 2.5 Mb length of chromosome A9, and 5.3 Mb length of chromosome C1, respectively (Fig. 4, Table 3, Additional file 1: Table S2). On chromosome A2, four candidate genes were annotated within association haplotype blocks, which were orthologous to A. thaliana long‐chain acyl‐CoA synthetase 9 (LACS9), β-ketoacyl-CoA reductase (KCR1), fatty acid biosynthesis 1 (FAB1), and lysophosphatidyl acyltransferase 4 (LPAT4) (Table 4). The significant associations regions on chromosome A9 harbored three ortholog of the A. thaliana gene TRANSPARENT TESTA 16 (TT16), holocarboxylase synthase 1 (HCS1), and acyl-CoA oxidase 2 (ACX2), respectively (Table 4). However, Bntt16 had been reported that could enhance the oil production and influence the fatty acid composition of B. napus [52]. Another significant association peak for fatty acid was localized on chromosome C1 (Table 3, Additional file 1: Table S2). This region includes six candidate genes (Table 4, Fig. 4) that were annotated in the B. napus ‘Darmor-bzh’ reference genome [26]. BnaC01g24110D, BnaC01g26460D, and BnaC01g26550D are ortholog of A. thaliana gene implicated in fatty acid biosynthesis, acyl-activating enzyme 17 (AAE17), fatty acid biosynthesis 1 (FAB1), and acyl-CoA binding protein 5 (ACBP5), respectively (Table 4, Fig. 4). In B. napus, these candidate genes have not been characterized previously in linkage analyses, all except BnaA.FAE1 (BnaA08g11130D) and BnaC. FAE1 (BnaC03g65980D), which may be important QTLs associated with fatty acid composition. Hence, further studies should be verified by further analysis in the future.
Discussion
Selecting high quality rapeseed, with increased oil content and improved edible oils with a modified fatty acid composition, is an important breeding goal for rapeseed. Moreover, palmitic, stearic, oleic, linoleic, linolenic, eicosenoic, and erucic acids are important fatty acids in B. napus, which determines the flavor and nutritional quality of B. napus. Of which erucic acid [C21:1] is hardly decomposed and absorbed by human body, hence the lower erucic adid is the important goal in rapeseed production. Because the fatty acid compositions are typical quantitative trait controlled by polygenic inheritance and also by interaction of the genotype and environment [3, 4, 7]. In this study, we found that the accessions exhibited a largely normal distribution for fatty acid compositions (Fig. 1), indicating that they might be associated with their complexity genetic networks in B. napus. So far, numerous of QTLs for fatty acid composition were found to be distributed on most of the chromosomes of B. napus [3, 4, 10, 14, 15, 53, 54], but the fatty acid underlie polygenic control that can only be elucidated by quantitative genetics approaches. Based on the marker density, experimental population size and statistical methods, GWAS could be efficiently resolved the associated allelic with the particular trait, which had been widely applied in B. napus [22, 24, 55, 56]. With advances in genome sequencing and computational technologies, high-throughput SNP genotyping platforms (Brassica 60 K SNP array, including 52,157 SNPs) have been developed [32], and can be widely used for high-resolution GWAS in B. napus [15, 28, 31, 35, 57]. Additionally, association mapping relies on the decay of LD initially present in a population, at a rate determined by the physical distance between loci and the number of generations since LD arose [58]. In this study, we evaluated genome- and subgenome-wide LD in 520 accessions, and found that LDs (r 2) decayed to half of its maximum value within 0.05-0.10 Mb at the A subgenome and 1.25-1.50 Mb at the C subgenome, respectively (Table 2, Additional file 2: Figure S2), in accordance with the fact that interspecific hybridization resulted in widespread genetic recombination in the A subgenome between B. napus and B. rapa [59, 60]. Although our research showed that the LD decays in B. napus are higher than the decays of 250 kb in A. thaliana [61], and the decays of 100 kb-1 Mb in rice [62], which are lower than that in B. napus [57]. Moreover, a high resolution can be obtained due to the low level of LD in association mapping [47].
In addition, the efficiencies of association analysis is determined by population size and maker density [63]. So the Brassica 60 K SNP array was performed for the genotype of 520 rapeseed accessions. In the present study, 520 accessions were classified into two sub-populations, but it is difficult to completely classify accessions into an association panel according to their origins and self-type (Fig. 2c, d, Additional file 1: Table S1 and Additional file 2: Figure S3), indicating that they have undergone inter-specific crosses and hybridization to adapt to the local environments and improve the quality in different countries [47, 60, 64]. Significant natural phenotypic variation in fatty acid composition was observed in 520 rapeseed germplasms, indicating that the fatty acid contents were influenced by genotype and environment factors in B. napus [65] (Table 1, Additional file 2: Figure S1). Moreover, most accessions have no or weak kinship in this panel, with an average relative kinship value of 0.0869 (Fig. 2e), suggesting that this panel with 520 accessions is suitable for association analysis. False-positive associations are indispensable when different models are used to detect genotype-phenotype associations in plant GWAS [31, 47, 66, 67]. Therefore, we compared six models for their ability to reduce the number of false positives in association mapping of this panel (Additional file 2: Figure S4), and found that the observed P-values fit best to the expected P-values using the K + PCA model (Additional file 2: Figure S5), suggesting that the K + PCA model was most efficient at controlling for false-positives, in accordance with previous reports [43, 47, 68, 69].
As an excellent model for association analysis, rapeseed developed extensive architectural variation across its native range and diverse germplasm collections through artificial selection. In the present study, we identified 673 significant SNPs associated with fatty acid composition (palmitic, stearic, oleic, linoleic, linolenic, eicosenoic, and erucic acids) using the K + PCA model. Some of these SNPs were simultaneously detected in both 2013Cq and 2014Cq, whereas others were detected only in one of these (Additional file 1: Table S2). Ideally, published genome information of B. napus ‘Darmor-Bzh’ should be combined with the relative physical positions of SNPs on pseudo-molecules of rapeseed for association mapping [26]. Accordingly, 62 significant association regions for fatty acid compositions were localized in 18 chromosomes of B. napus in 2013Cq and 2014Cq (Table 3, Additional file 1: Table S2). Among them, five common significantly associated regions were identified on chromosomes A2, A8, A9, C1, and C3, respectively (Fig. 4). Further, identical chromosome regions on A8 and C3 were repeatedly detected in previously research and included two homoeologues genes (namely BnaA.FAE1 and BnaC.FAE1), which had been responsible for controlling erucic acid content [31, 48, 49], indicating that our results are credible in this study. In additon, two homologous genes KCS17 (BnaA08g11140D and BnaC03g66040D) and CER4 (BnaA08g11440D and BnaC03g66380D) were predicted in common regions of chromosomes A8 and C3, respectively (Table 4, Fig. 4). These genes are involved in the biosynthesis of saturated fatty acids [70], and encode an alcohol-forming fatty acyl-coenzyme A reductase (FAR), respectively [51]. In an association block on chromosome A2, four genes were ascribed to fatty acid within the asscociated regions (Table 4). For example, the A. thaliana gene LACS9 could catalyzes the formation of acyl-CoA from fatty acids, ATP, and CoA, which is involved in Arabidopsis seed oil biosynthesis [71], KCR1 is a functional KCR isoform involved in a multiprotein membrane-bound fatty acid elongation system [72], FAB1 could be involved in the elongation of C16:0-ACP to C18:0-ACP, which is the key step for fatty acid synthesis [73], and LPAT4 is the member of LPAT gene family, which is an essential candidate for oil composition and increase the seed oil [74]. Moreover, the another orthologous gene FAB1 (BnaC01g26460D) was also identified in the block on chromosome C1 (Table 4). On the other hand, three candidate genes, transparent testa 16 (tt16), holocarboxylase synthetase 1 (HCS1), and acyl-CoA oxidase 2 (ACX2) were localized in chromosome A9 blocks, which had been confirmed to be associated with the fatty acid biosynthesis (Table 4) [52, 75–77]. Moreover, previous results showed that Bntt16 promotes oil production and influences the fatty acid composition of B. napus [52], and ACX2 encodes an enzyme that catalyzes the first step of peroxisomal fatty acid β-oxidation and is optimally active on long-chain saturated and unsaturated acyl-CoAs [77]. Furthermore, six genes associated with lipid biosynthesis and metabolism were detected in C1 blocks, including genes encoding acyl-activating enzyme 17 (AAE17), fatty acid biosynthesis 1 (FAB1), and acyl-CoA binding protein 5 (ACBP5) [78, 79]. Although we were able to identify key genes involved in fatty acid biosynthesis through trait-marker associations in our panel of 520 oilseed accessions, the genetic mechanisms that control fatty acid composition remain unclear. Therefore, further studies are necessary to reveal the DNA variation of these candidate genes in B. napus.
Taken together, these findings provided important insights into the understanding of the important genes affecting the fatty acid metabolism in B. napus.
Conclusion
We identified SNP-trait associations through association mapping in B. napus. In total, 62 significant association regions, distributed throughout the genome, were detected for fatty acid composition using the PCA + K model. Importantly, five common significantly associated regions, located on chromosomes A2, A8, A9, C1, and C3, respectively, were identified. In addition, 24 orthologs of the functional candidate genes involved in fatty acid composition were identified based on the B. napus Darmor-bzh reference genome sequences. Our results provide a basis for deciphering the mechanism underlying the determination of fatty acid composition in B. napus. Moreover, the SNP markers identified here demonstrate that marker-assisted selection is a powerful strategy for identifying genes of interest in B. napus and can be used in breeding programs aimed at optimizing fatty acid profiles in oilseed.
Abbreviations
- ANOVA:
-
Analysis of variance
- CV:
-
Coefficient of variation
- FDRs:
-
False discovery rates
- GLM:
-
General linear model
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- LOD:
-
Logarithm of odds
- MAF:
-
Minimum allele frequency
- MCMC:
-
Markov Chain Monte Carlo
- MLM:
-
Mixed linear model
- PCA:
-
Principal component analysis
- PIC:
-
Polymorphism Information Content
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
References
Kimber D, McGregor D: The species and their origin, cultivation and world production. Brassica Oilseeds-Production and Utilization DS Kimber and DI McGregor, eds CAB International, Oxon, UK 1995:1–8
Bauer B, Kostik V, Gjorgjeska B. Fatty acid composition of seed oil obtained from different canola varieties. Farmaceutski glasnik. 2015;71(1):1–7.
Wen J, Xu J, Long Y, Xu H, Wu J, Meng J, Shi C. Mapping QTLs controlling beneficial fatty acids based on the embryo and maternal plant genomes in Brassica napus L. J Am Oil Chem Soc. 2015;92(4):541–52.
Zhao J, Dimov Z, Becker HC, Ecke W, Möllers C. Mapping QTL controlling fatty acid composition in a doubled haploid rapeseed population segregating for oil content. Mol Breed. 2008;21(1):115–25.
Rawsthorne S. Carbon flux and fatty acid synthesis in plants. Prog Lipid Res. 2002;41(2):182–96.
Thelen JJ, Ohlrogge JB. Metabolic engineering of fatty acid biosynthesis in plants. Metab Eng. 2002;4(1):12–21.
Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W. Oil content in a European × Chinese rapeseed population: QTL with additive and epistatic effects and their genotype-environment interactions. Crop Sci. 2005;45(1):51–9.
Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W. Conditional QTL mapping of oil content in rapeseed with respect to protein content and traits related to plant development and grain yield. Theor Appl Genet. 2006;113(1):33–8.
Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, Specel S, Hanneton L, Dheu J, Deschamps M. Genetic control of oil content in oilseed rape (Brassica napus L.). Theor Appl Genet. 2006;113(7):1331–45.
Qiu D, Morgan C, Shi J, Long Y, Liu J, Li R, Zhuang X, Wang Y, Tan X, Dietrich E. A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet. 2006;114(1):67–80.
Chen G, Geng J, Rahman M, Liu X, Tu J, Fu T, Li G, McVetty PB, Tahir M. Identification of QTL for oil content, seed yield, and flowering time in oilseed rape (Brassica napus). Euphytica. 2010;175(2):161–74.
Jiang C, Shi J, Li R, Long Y, Wang H, Li D, Zhao J, Meng J. Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.). Theor Appl Genet. 2014;127(4):957–68.
Yan XY, Li JN, Fu FY, Jin MY, Chen L, Liu LZ. Co-location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus L. Euphytica. 2009;170(3):355–64.
Burns M, Barnes S, Bowman J, Clarke M, Werner C, Kearsey M. QTL analysis of an intervarietal set of substitution lines in Brassica napus:(i) Seed oil content and fatty acid composition. Heredity. 2003;90(1):39–48.
Lee S, Jang M-S, Jeon E-J, Yun K-Y, Kim S: QTL Analysis for Erucic Acid and Oleic Acid Content in Brassica napus Using F2 Population. In: Plant and Animal Genome XXIII Conference: 2015: Plant and Animal Genome; 2015
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics. 2004;168(1):435–46.
Yan JB, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One. 2009;4(12):e8451.
Yan J, Warburton M, Crouch J. Association mapping for enhancing maize (L.) genetic improvement. Crop Sci. 2011;51(2):433–49.
Meuwissen T, Goddard M. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics. 2000;155(1):421–30.
Agrama H, Eizenga G, Yan W. Association mapping of yield and its components in rice cultivars. Mol Breed. 2007;19(4):341–56.
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet. 2002;30(2):190–3.
Hasan M, Friedt W, Pons-Kühnemann J, Freitag N, Link K, Snowdon R. Association of gene-linked SSR markers to seed glucosinolate content in oilseed rape (Brassica napus ssp. napus). Theor Appl Genet. 2008;116(8):1035–49.
Qu C, Hasan M, Lu K, Liu L, Zhang K, Fu F, Wang M, Liu S, Bu H, Wang R, et al. Identification of QTLs for seed coat colour and oil content in Brassica napus by association mapping using SSR markers. Can J Plant Sci. 2014;95(2):387–95.
Zou J, Jiang C, Cao Z, Li R, Long Y, Chen S, Meng J. Association mapping of seed oil content in Brassica napus and comparison with quantitative trait loci identified from linkage mapping. Genome. 2010;53(11):908–16.
Cai G, Yang Q, Yi B, Fan C, Edwards D, Batley J, Zhou Y. A complex recombination pattern in the genome of allotetraploid Brassica napus as revealed by a high-density genetic map. PLoS One. 2014;9(10):e109910.
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–3.
Delourme R, Falentin C, Fomeju BF, Boillot M, Lassalle G, André I, Duarte J, Gauthier V, Lucante N, Marty A. High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genomics. 2013;14(1):120.
Hatzig SV, Frisch M, Breuer F, Nesi N, Ducournau S, Wagner M-H, Leckband G, Abbadi A, Snowdon RJ. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front Plant Sci. 2015;6:221.
Luo X, Ma C, Yue Y, Hu K, Li Y, Duan Z, Wu M, Tu J, Shen J, Yi B. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics. 2015;16(1):379.
Lu G, Harper AL, Trick M, Morgan C, Fraser F, O’Neill C, Bancroft I. Associative transcriptomics study dissects the genetic architecture of seed glucosinolate content in Brassica napus. DNA Res. 2014;21(6):613–25.
Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper AL, Trick M, Liu S, Gao G. Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. 2014;21(4):355–67.
Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C, Khedikar Y, Batley J, Edwards D, Meng J, Li R, et al. A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor Appl Genet. 2016;129(10):1887–99.
Doyle JJ. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–5.
Rücker B, Röbbelen G. Impact of low linolenic acid content on seed yield of winter oilseed rape (Brassica napus L.). Plant Breed. 1996;115(4):226–30.
Wei L, Jian H, Lu K, Filardo F, Yin N, Liu L, Qu C, Li W, Du H, Li J. Genome‐wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol J 2015;14:1368–80.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Qu C-M, Li S-M, Duan X-J, Fan J-H, Jia L-D, Zhao H-Y, Lu K, Li J-N, Xu X-F, Wang R. Identification of candidate genes for seed glucosinolate content using association mapping in Brassica napus L. Genes. 2015;6(4):1215–29.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Nei M. Genetic distance between populations. American naturalist. 1972;106(949):283–92.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82.
Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2(4):618–20.
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38(2):203–8.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5.
Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–9.
Mohammadi M, Blake TK, Budde AD, Chao S, Hayes PM, Horsley RD, Obert DE, Ullrich SE, Smith KP. A genome-wide association study of malting quality across eight US barley breeding programs. Theor Appl Genet. 2015;128(4):705–21.
Xu J, Long Y, Wu J, Xu H, Zhao Z, Wen J, Meng J, Shi C. QTL identification on two genetic systems for rapeseed glucosinolate and erucic acid contents over two seasons. Euphytica. 2015;205(3):647–57.
Wu G, Wu Y, Xiao L, Li X, Lu C. Zero erucic acid trait of rapeseed (Brassica napus L.) results from a deletion of four base pairs in the fatty acid elongase 1 gene. Theor Appl Genet. 2008;116(4):491–9.
Wang N, Wang Y, Tian F, King GJ, Zhang C, Long Y, Shi L, Meng J. A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol. 2008;180(4):751–65.
Joubès J, Raffaele S, Bourdenx B, Garcia C, Laroche-Traineau J, Moreau P, Domergue F, Lessire R. The VLCFA elongase gene family in Arabidopsis thaliana: phylogenetic analysis, 3D modelling and expression profiling. Plant Mol Biol. 2008;67(5):547.
Rowland O, Zheng H, Hepworth SR, Lam P, Jetter R, Kunst L. CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol. 2006;142(3):866–77.
Deng W, Chen G, Peng F, Truksa M, Snyder CL, Weselake RJ. Transparent testa16 plays multiple roles in plant development and is involved in lipid synthesis and embryo development in canola. Plant Physiol. 2012;160(2):978–89.
Hu X, Sullivan-Gilbert M, Gupta M, Thompson SA. Mapping of the loci controlling oleic and linolenic acid contents and development of fad2 and fad3 allele-specific markers in canola (Brassica napus L.). Theor Appl Genet. 2006;113(3):497–507.
Javed N, Geng J, Tahir M, McVetty P, Li G, Duncan RW. Identification of QTL influencing seed oil content, fatty acid profile and days to flowering in Brassica napus L. Euphytica. 2016;207(1):191–211.
Lu K, Xiao Z, Jian H, Peng L, Qu C, Fu M, He B, Tie L, Liang Y, Xu X, Li J. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in Brassica napus. Scientific reports. 2016;6:36452.
Rezaeizad A, Wittkop B, Snowdon R, Hasan M, Mohammadi V, Zali A, Friedt W. Identification of QTLs for phenolic compounds in oilseed rape (Brassica napus L.) by association mapping using SSR markers. Euphytica. 2011;177(3):335–42.
Qian L, Qian W, Snowdon RJ. Sub-genomic selection patterns as a signature of breeding in the allopolyploid Brassica napus genome. BMC Genomics. 2014;15(1):1.
Mackay I, Powell W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007;12(2):57–63.
Qian W, Meng J, Li M, Frauen M, Sass O, Noack J, Jung C. Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus L.), with emphasis on the evolution of Chinese rapeseed. Theor Appl Genet. 2006;113(1):49–54.
Donini P, Chen S, Nelson M, Ghamkhar K, Fu T, Cowling W. Divergent patterns of allelic diversity from similar origins: the case of oilseed rape (Brassica napus L.) in China and Australia. Genome. 2007;51(1):1–10.
Rafalski A. Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol. 2002;5(2):94–100.
Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2:467.
Liu S, Fan C, Li J, Cai G, Yang Q, Wu J, Yi X, Zhang C, Zhou Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor Appl Genet. 2016;129(6):1203–15.
Cowling WA. Genetic diversity in Australian canola and implications for crop breeding for changing future environments. Field Crop Res. 2007;104(1):103–11.
Omidi H, Tahmasebi Z, Badi HAN, Torabi H, Miransari M. Fatty acid composition of canola (Brassica napus L.), as affected by agronomical, genotypic and environmental parameters. Comptes rendus biologies. 2010;333(3):248–54.
Zhang J, Mason AS, Wu J, Liu S, Zhang X, Luo T, Redden R, Batley J, Hu L, Yan G. Identification of putative candidate genes for water stress tolerance in canola (Brassica napus). Frontiers Plant Sci. 2015;6:1058.
Pace J, Gardner C, Romay C, Ganapathysubramanian B, Lübberstedt T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genomics. 2015;16(1):1.
Wang M, Yan J, Zhao J, Song W, Zhang X, Xiao Y, Zheng Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012;196:125–31.
Li F, Chen B, Xu K, Gao G, Yan G, Qiao J, Li J, Li H, Li L, Xiao X. A genome-wide association study of plant height and primary branch number in Rapeseed (Brassica napus). Plant Sci. 2016;242:169–77.
Tresch S, Heilmann M, Christiansen N, Looser R, Grossmann K. Inhibition of saturated very-long-chain fatty acid biosynthesis by mefluidide and perfluidone, selective inhibitors of 3-ketoacyl-CoA synthases. Phytochemistry. 2012;76:162–71.
Zhao L, Katavic V, Li F, Haughn GW, Kunst L. Insertional mutant analysis reveals that long‐chain acyl‐CoA synthetase 1 (LACS1), but not LACS8, functionally overlaps with LACS9 in Arabidopsis seed oil biosynthesis. Plant J. 2010;64(6):1048–58.
Beaudoin F, Wu X, Li F, Haslam RP, Markham JE, Zheng H, Napier JA, Kunst L. Functional characterization of the Arabidopsis beta-ketoacyl-coenzyme A reductase candidates of the fatty acid elongase. Plant Physiol. 2009;150(3):1174–91.
Chapman MA, Burke JM. Evidence of selection on fatty acid biosynthetic genes during the evolution of cultivated sunflower. Theor Appl Genet. 2012;125(5):897–907.
Chen S-L, Huang J-Q, Lei Y, Zhang Y-T, Ren X-P, Chen Y-N, Jiang H-F, Yan L-Y, Li Y-R, Liao B-S. Identification and characterization of a gene encoding a putative lysophosphatidyl acyltransferase from Arachis hypogaea. J Biosci. 2012;37(1):1029–39.
Chen X, Chou H-H, Wurtele ES. Holocarboxylase Synthetase 1 Physically Interacts with Histone H3 in Arabidopsis. Scientifica. 2013;2013:983501.
Puyaubert J, Denis L, Alban C. Dual targeting of Arabidopsis holocarboxylase synthetase1: a small upstream open reading frame regulates translation initiation and protein targeting. Plant Physiol. 2008;146(2):478–91.
Hooks MA, Kellas F, Graham IA. Long‐chain acyl‐CoA oxidases of Arabidopsis. Plant J. 1999;20(1):1–13.
Kim HU, Chen GQ. Identification of hydroxy fatty acid and triacylglycerol metabolism-related genes in lesquerella through seed transcriptome analysis. BMC Genomics. 2015;16(1):1.
Leung K-C, Li H-Y, Mishra G, Chye M-L. ACBP4 and ACBP5, novel Arabidopsis acyl-CoA-binding proteins with kelch motifs that bind oleoyl-CoA. Plant Mol Biol. 2005;55(2):297–309.
Acknowledgements
We would like to thank Dr. Kathy Farquharson for critical reading of this manuscript.
Funding
This work was supported by the Utilization of Heterosis and Selection of Strong Advantage of Hybrid(2016YFD0101300), the 973 Project (2015CB150201), the National Science Foundation of China (31401412, U1302266, 31571701), Projects in the National Science and Technology Pillar Program (2013BAD01B03-12), the 111 Project (B12006), Chongqing Basic Scientific and advanced technology Research (cstc2015jcyjBX0001, cstc2016shms-ztzx0016 and 21), and the China Scholarship Council (CSC).
Availability of data and materials
The data sets supporting the results of this article are included within the article and its additional files.
Authors’ contributions
CQ, and FF conceived of the study and drafted the manuscript. LJ, HZ, LW and YL performed the data mining, bioinformatics. RW, XX, and SL carried out reagents and the field experiments. KL and JL contributed to interpretation and modification of the data and manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional information
An erratum to this article is available at http://dx.doi.org/10.1186/s12864-017-3772-9.
Additional files
Additional file 1: Table S1.
List of oilseed accessions used in the present study. Table S2. Summary physical position of genome-wide significant association SNPs for fatty acid composition in B. napus. (ZIP 1606 kb)
Additional file 2: Figure S1.
Comparison of fatty acid content of accessions grown in two environments. The two environments are plotted against each other, with their Pearson’s coefficients indicated. Figure S2. Genome- and subgenome-wide linkage disequilibrium (LD) decay for all 520 accessions. The LD decay from the A subgenome is indicated by a black line; the LD decay from the C subgenome is indicated by a red line; and the LD decay from the A + C genomes is indicated by a green line. r 2 indicates the squared allele frequency correlations between all pairs of SNP markers. Figure S3. Diagram derived from the program Structure 2.1 showing the distribution of 520 rapeseed genotypes into two subpopulations (K = 2). Green indicates subpopulation P1 genotypes and red representa subpopulation P2. The x-axis indicates the Q matrix values, whereas the y-axis indicates the accession code. Figure S4. Quantile–quantile plots of estimated -log (p) from association analysis using six models for seven traits in 2013Cq and 2014Cq. (A and B) palmitic acid; (C and D) stearic acid; (E and F) oleic acid; (G and H) linoleic acid; (I and J) linolenic acid; (K and L) eicosenoic acid; and (M and N) erucic acid content. Cq indicates the growing region, Chongqing, China. The horizontal gray line represents the genome-wide significance threshold (−log10 (p) = 4.1). Figure S5. Quantile–quantile plots of estimated -log (p) from the association analysis using the PCA + K model for fatty acid composition in 2013Cq and 2014Cq. Cq refers to the growing region, Chongqing, China. (ZIP 1566 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qu, C., Jia, L., Fu, F. et al. Genome-wide association mapping and Identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genomics 18, 232 (2017). https://doi.org/10.1186/s12864-017-3607-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3607-8