
Abstract
Background
Prior to their release in the environment, transgenic crops are examined for their health and environmental safety. In addition, transgene expression needs to be consistent in order to express the introduced trait (e.g. insecticidal and/or herbicide tolerance). Moreover, data on expression levels for GM events are usually required for approval, but these are rarely disclosed or they are considered insufficient. On the other hand, biosafety regulators do not consider epigenetic regulation (e.g. DNA methylation, ncRNAs and histone modifications), which are broadly known to affect gene expression, within their risk assessment analyses. Here we report the results of a DNA methylation (bisulfite sequencing) and transgene transcript accumulation (RT-qPCR) analysis of four Bt-expressing single transgenic maize hybrids, under different genetic backgrounds, and a stacked transgenic hybrid expressing both insecticidal and herbicide tolerance traits.
Results
Our results showed differences in cytosine methylation levels in the FMV promoter and cry2Ab2 transgene of the four Bt-expressing hybrid varieties. The comparison between single and stacked hybrids under the same genetic background showed differences in the 35S promoter sequence. The results of transgene transcript accumulation levels showed differences in both cry1A.105 and cry2Ab2 transgenes among the four Bt-expressing hybrid varieties. The comparison between single and stacked hybrids showed difference for the cry2Ab2 transgene only.
Conclusions
Overall, our results show differences in DNA methylation patterns in all varieties, as well as in transgene transcript accumulation levels. Although the detection of changes in DNA methylation and transgenic accumulation levels does not present a safety issue per se, it demonstrates the need for additional studies that focus on detecting possible safety implications of such changes.
Similar content being viewed by others


Background
The Cartagena Protocol on biosafety defines living modified organism (LMO), also known as genetically modified organism (GMO), as “any living organism that possesses a novel combination of genetic material obtained through the use of modern biotechnology” [1]. It also defines modern biotechnology as the application of “in vitro nucleic acid techniques, including recombinant deoxyribonucleic acid (DNA) and direct injection of nucleic acid into cells or organelles, or ii) fusion of cells beyond the taxonomic family and that are not techniques used in traditional breeding and selection”. This protocol acts as a regulatory document for assessing the biosafety of GMOs in an international level. The use of agricultural GMOs has been growing steadily over the last decade [2], which shows the need for adequate regulation of its use.
Domestic and international GMO regulations, such as The Cartagena Protocol, provide guidance regarding GMO risk assessment in order to ensure, among other safety aspects, the genetic stability of a transgene. Although there is no agreed operational definition for the stability concept among regulators, these measures may include the identification of any novel genotypic and phenotypic characteristics associated with the GMO that may have adverse effects on biological diversity, as well as information regarding the genetic characteristics of the inserted nucleic acid and the function it specifies, and/or characteristics of the modification introduced [1].
Plant genetic engineering was mainly achieved through Agrobacterium-mediated transformation, which takes advantage of the natural ability of the soil bacterium Agrobacterium tumefaciens to transfer a segment of its DNA, the T-DNA, into the host plant genome [3], or through particle bombardment (biobalistic or gene gun), which relies on the delivery of gold particles coated with the DNA to be inserted into the plant nuclear genome [4]. After plants have been transformed, regeneration through in vitro culture can cause rearrangements in the transgene sequence or even in the host genome [5], sometimes showing extensive genomic variations, especially epigenetic changes, in the process of micro propagation, which may or may not result in phenotypic changes [6], but once transgenic plants have been generated, it is assumed that the transgene is stable and mutations occur at the same rates as endogenous genes [7].
Routine genetic stability analyses performed by GMO developers usually rely on protein quantification (ELISA—Enzyme-Linked Immuno-Sorbent Assay) and transgene integrity (Southern blot) [8, 9]. Independent studies have also assessed genetic stability of GMO events not only with different approaches (e.g. cytogenetics and Southern Blot) [10, 11] but also with more rigorous criterions, such as seasonal and tissue-specific ELISA analysis [12] and RNA expression using Northern blot technique [11]. In addition, studies have detected transgene rearrangements, such as sequence deletions [13–15] and nucleotide addition of undesired fragments into the transgene sequence [16], and also, alterations in mRNA expression levels were also observed [17]. However, changes in DNA sequences are not the only source of alteration in transgene expression. Epigenetics—defined as the study of molecular mechanisms involved in hereditable changes—are able to regulate gene expression without changing the DNA sequence per se [18]. These variations are often associated with DNA methylation, histone modifications and non-coding RNAs (ncRNAs), which can lead to phenotypic variation and transgene silencing [19]. Both ncRNAs and histone modifications have been linked to RNAi (RNA interference), PTGS (Post-Transcriptional Gene Silencing) and TGS (Transcriptional Gene Silencing) pathways [20], and are associated with up-regulation of gene transcription or with transcription repression [21, 22].
Cytosine methylation is an epigenetic regulatory mechanism that is able to control gene expression by inhibiting protein binding to DNA and by changing chromatin structure [23]. Plant DNA sequences are known to be highly methylated, mostly 5-methylcytosine (m5C), which is located mainly in symmetrical CG sites. However, the high amount of m5C found in some plant species suggested that methylation is not restricted to the CG sequence context but also methylated in CHG (where H is A or T) and CHH sites (where H is A, C or T) [24–26]. Investigations of cytosine methylation patterns have been conducted in transgenic models such as Arabidopsis thaliana [27–29] and Petunia hybrida [30, 31]. Methylation changes have been also studied in maize, targeting endogenous genes and transposable elements [32–34]. To date, La Paz et al. [6] conducted the only study regarding epigenetic aspects of commercialized GM crops, in which the authors analysed the cytosine methylation levels of different Bt-expressing (MON810) varieties.
Therefore, in order to contribute to better understanding of epigenetic mechanisms that may impact transgene expression of GMOs, the aim of this study was to investigate the epigenetic profiles of the transgenic cassette of a double-Bt-expressing transgenic maize event (MON-89Ø34-3), as well as the transcription expression of the inserted traits. We have analysed the levels of cytosine methylation by bisulfite-sequencing technique, we have verified the transgene transcript accumulation by RT-qPCR and compared single and stacked GM maize hybrid varieties containing the MON-89Ø34-3 event. In addition, this manuscript also aims to provide relevant information and insight for reliable risk assessments of GMOs.
Methods
A schematic overview of our experimental design, sampling strategy and analytical approach is provided in Fig. 1.

Schematic overview of the experimental design, sampling strategy and analytical approach used in the study. Leaf samples of single and stacked GM events (MON-89Ø34-3 and MON-89Ø34-3 × MON-ØØ6Ø3-6, respectively) were used for DNA methylation and transgene transcript accumulation analysis. Three biological replicates were analysed for each of the GM varieties. Four regions of the transgene cassette (FMV promoter, cry1A.105 transgene, 35S promoter and cry2Ab2 transgene) were chosen for DNA methylation analysis. The accumulation of cry1A.105 and cry2Ab2 transgenes was quantified by RT-qPCR
Plant material and growth conditions
Five maize varieties were used in this study. Four of them are considered single-event varieties because they contain only one transgenic event with resistance to some lepidopteran species: DKB240PRO and DKB350PRO (unique identifier MON-89Ø34-3 from Monsanto Company, resistance to some lepidopteran species, distributed by Dekalb) and AG8041PRO and AG9045PRO (unique identifier MON-89Ø34-3 from Monsanto Company, resistance to some lepidopteran species, distributed by Sementes Agroceres). The DKB240PRO2 (unique identifier MON-89Ø34-3 × MON-ØØ6Ø3-6 from Monsanto Company, stacked event resistant to some lepidopteran species and a class of herbicide, Dekalb) is considered a stacked event because it contains two transgenic events. The combination of the two transgenic events in a single hybrid was obtained by traditional crossing. These are named in this study as DKB240, DKB350, AG8041, AG9045 and DKB240-ST, respectively (Table 1). The used varieties are hybrid progenies of the single cross between maternal endogamous line “A” with the paternal endogamous line “B”. Thus, the hybrid variety seeds used have high genetic similarity (all seeds are AB genotype). All these five commercial varieties were produced by the aforementioned company and are commonly found in the seed market in southern Brazil.
The transformation events MON-89Ø34-3 and MON-89Ø34-3 × MON-ØØ6Ø3-6 were approved for commercial use in Brazil in 2009 [35] and 2010 [36], respectively. The MON-89Ø34-3 event expresses two insecticidal proteins (cry1A.105 and cry2Ab2 proteins derived from Bacillus thuringiensis, which are active against certain lepidopteran insect species) and the MON-ØØ6Ø3-6 event expresses the enzyme CP4-epsps (5-enolpyruvylshikimate-3-phosphate synthase) that confers tolerance against glyphosate-based herbicides.
The seeds from all five varieties were grown side-by-side in a controlled environment set to 16 h light period and 25 °C (±2 °C). Seedlings were germinated and grown in 3 L plastic pots with Plantmax HT substrate (Buschle & Lepper S.A.) and watered daily. No pesticide or fertilizer was applied. Ten plants were grown in controlled climate out of which leaf samples from 3 random plants sampled per maize variety (genotype). Therefore, each variety contained 3 biological replicates that were used for the experiment. In order to standardize the sampling, the third and fourth leaves, starting from the bottom, were sampled for each plant and kept at −80 °C until RNA and DNA extraction.
Bisulfite treatment of genomic DNA
Genomic DNA from three biological replicates for each variety was isolated from 100 mg of frozen leaf tissue using the column-based NucleoSpin® Plant II (Macherey–Nagel GmbH & Co. KG, Germany) extraction kit and further quantified in a NanoDrop 2000c spectrophotometer (Thermo Scientific, Wilmington, USA). Subsequently, 200 ng of isolated DNA was submitted to bisulfite treatment in order to convert non-methylated cytosines into uracil. The conversion was performed using the EpiTect® Bisulfite Kit (Qiagen, Hilden, Germany) following manufacturer’s recommendations.
Bisulfite PCR and fragment purification
Four regions of the MON-89Ø34-3 transgenic insert were chosen to analyse the patterns of methylation: one in the e35S promoter, one in the cry1A.105 transgene, one in the FMV promoter and one in the cry2Ab2 transgene. Four modified pairs of primers, designed to amplify the converted DNA, were designed using the MethPrimer software [37] (Table 2). The size and the amount of cytosine types—CG, CHG (where H is nitrogenous base A or T) and CHH (where H is a nitrogenous base different from G)—of each fragment are shown in Table 3. In order to differentiate the 35S promoter of the 35S:cry1A.105 construct from the 35S:EPSPS construct, the BS35S-F primer was designed to anneal in a sequence containing 8 SNPs between the promoter regions (Additional file 1). Therefore, only the 35S promoter regions from the cry1A.105 transgene are expected to be amplified.
After bisulfite treatment, the bisulfite-converted DNA was used to PCR amplify the four regions in each of the three replicates for each variety. The PCR conditions were 1× PfuTurbo Cx reaction buffer, 0.2 mM of each dNTP, 0.5 µM of forward and reverse primers, 1 U of PfuTurbo Cx hotstart DNA polymerase (Agilent Technologies, California, USA) and 60 ng of converted DNA. The steps used for the amplification were 95 °C for 3 min; 35 cycles of 95 °C for 30 s annealing temperature (specific for each primer) for 30 s, 65 °C for 2 min and 65 °C for 10 min. The reactions were carried out in a S1000™ Thermal Cycler (BioRad, California, USA).
The PCR products were stained with GelRed™ (Uniscience, Florida, USA) and resolved in 1% (w/v) agarose gel in horizontal electrophoresis for 90 min at 100 V. Bands were excised from the gel and purified using the NucleoSpin® Gel and PCR Clean-up (Macherey–Nagel) following manufacturer’s recommendations.
Cloning
Each of the four regions from the three replicates of each variety were cloned in the pCR II-Blunt-TOPO vector using the Zero Blunt® TOPO® PCR Cloning Kit (Life Technologies, California, USA), following manufacturer’s recommendations, and transformed in chemically competent Escherichia coli cells (TOP10 strain). Briefly, 2 μL of the cloning reaction were added to E. coli cells, thoroughly agitated and incubated in ice for 10 min. Following, the cells were submitted to thermal shock at 42 °C for 30 s and immediately transferred to ice. 250 μL of SOC medium (provided with the kit) were added to the cells and horizontally agitated (200 rpm) for 1 h at 37 °C. Lastly, 40 μL of the E. coli + SOC medium solution were added to a petri dish containing solid Luria–Bertani (LB) medium [38] supplemented with 50 μg/mL of kanamycin antibiotic. The plates were incubated overnight at 37 °C for the bacteria colonies to grow.
Cell multiplication and Miniprep
Previously cultivated E. coli cells were multiplied in order to increase the number of colonies to be cloned. Eight random bacteria colonies (eight different clones) for each fragment were selected and transferred to a bacteria-growing plate (96-wells) containing liquid LB medium (supplemented with 50 μg/mL of kanamycin antibiotic). The plate was sealed and agitated at 37 °C for 22 h.
After multiplication, the plate was centrifuged for 6 min (2000g) at room temperature for pellet formation. The plate was inverted to remove the supernatant. The Miniprep was performed using the NucleoSpin Plasmid kit® (Macherey–Nagel) following manufacturer’s recommendations.
Sequencing
The cloned fragments were sequenced using the M13 reverse universal primer (5′-CAGGAAACAGCTATGAC-3′) by chain-terminator technique [39] using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems®, Singapore, Singapore). The sequencing reactions were performed in a volume of 20 μL containing 1 μL of BigDye v3.1, 3 μL of BigDye v3.1 reaction buffer, 3.5 pmol of the M13 reverse universal primer and 140 ng of the plasmid containing the fragments. The steps used for the amplification were 96 °C for 5 min, 25 cycles of 96 °C for 10 s, 50 °C for 5 s and 60 °C for 4 min. The reactions were carried out in a S1000™ Thermal Cycler (BioRad). Purification of sequencing reactions and capillary electrophoresis were performed in the DNA sequencing core facility at the University Hospital of North Norway (Universitetssykehuset Nord-Norge, Tromsø, Norway) using a Genetic Analyzer 3130xl (Applied Biosystems®).
Relative quantification analysis of transgene transcripts
Total RNA was extracted from leaf tissues using the column-based RNeasy Plant Mini Kit (Qiagen). Reverse-transcription quantitative PCR (RT-qPCR) assay was adapted from previously developed assays for the specific detection of MON-89Ø34-3 × MON-ØØ6Ø3-6 transgenes [40] to hydrolysis ZEN—Iowa Black® Fluorescent Quencher (ZEN/IBFQ) probe chemistry (Integrated DNA Technologies, INC Iowa, USA). Following quantification, cDNA was synthesized, and amplification of each target gene was performed using the QuantiTect Probe RT-PCR Kit (Qiagen) according to the manufacturer’s instructions.
RT-qPCR experiment was carried out in technical triplicates using StepOne™ Real-Time PCR System (Applied Biosystems). Each 20 μL reaction volume comprised 10 μM of each primer and probe and 50 ng of total RNA from each sample. The amplification efficiency was obtained from relative standard curves provided for each primer and calculated according to Pfaffl equations [41]. The choice of the endogenous reference genes and the selection of the two best genes were based on the previous work of Agapito-Tenfen et al. [42], using NormFinder (Molecular Diagnostic Laboratory, Aarhus University Hospital Skejby, Denmark) statistical algorithms [43].
The leunig and membrane protein PB1A10.07c genes were used to normalize cry1A.105 and cry2Ab2 mRNA data due to their best stability value (SV for best combination of two genes 0.025, Additional file 2). Conventional samples were also analysed in order to check for PCR and/or seed contaminants. Primer and probe sequences used, as well as Genebank ID of target genes, are provided in Table 4. The primers and probes were assessed for their specificity with respect to known splice variants and single-nucleotide polymorphism positions documented in transcript and single-nucleotide polymorphism databases. The normalized relative quantity (NRQ) was calculated for both single and stacked transgenic events samples relative to one of the three DKB240 samples according to the Pfaffl equations [41].
Statistical analysis
For DNA methylation analysis, quality of the sequences were accessed using the Sequence Scanner 2 software (Applied Biosystems®), DNA methylation levels (%) in CG, CHG and CHH cytosine types were assessed using the CyMATE web tool [44], and statistical analyses were performed with R language and statistical environment [45] using in-house scripts. To calculate the percentage (%) of methylated cytosines in each fragment, the number of methylated cytosines was divided by the number of total cytosines (available in Table 2) for each analysed fragment sequence. Wilcoxon-Mann–Whitney test for non-parametric data (p < 0.05) was used for the comparative analysis of the single event (MON-89Ø34-3) versus stacked event (MON-89Ø34-3 × MON-ØØ6Ø3-6) and Kruskal–Wallis test for non-parametric data (p < 0.05) was used for the comparative analysis among the single events (MON-89Ø34-3).
For the transcript quantification, normalized gene expression data were obtained using the Pfaffl method for efficiency correction [41], which is based on a mathematical model for relative quantification in real-time PCR where the efficiency of each primer is considered as a normalizer factor from the results. Cq average from each technical replicate was calculated for each biological replicate and used to make a statistical comparison of varieties based on the standard deviation. Due to non-normal distribution, the fold change data were log10 transformed before statistical tests. The fold change means obtained for the single varieties, as well as for single versus stacked GM event, were compared using ANOVA/Tukey-test and T test, respectively, at p < 0.05 (R language) [45]. Information on real-time data for this study has followed guidelines from the minimum information for publication of quantitative real-time PCR experiments [46].
Results
Cytosine methylation levels in Bt-expressing transgenic maize
MON-89Ø34-3 is a genetically modified maize containing two transgenes: one chimeric cry1 delta-endotoxin (CRY1A.105) is inserted under the regulation of a Cauliflower Mosaic Virus promoter (CaMV 35S) and a Figwort Mosaic Virus (FMV) 35S promoter driving cry2Ab delta-endotoxin (CRY2AB2) expression. In order to study the DNA methylation levels, three types of cytosines—CG, CHG and CHH—were analysed in four different regions of the transgenic insert: (i) FMV promoter; (ii) 35S promoter; (iii) cry1A.105 transgene; and (iv) cry2Ab2 transgene, in two experimental setups.
The first experiment setup analysed the transgenic regions among four different commercial available varieties (DKB350, AG9045, AG8041 and DKB240) containing the MON-89Ø34-3 event under different genetic backgrounds. The analysis of the FMV promoter region showed significant differences between the varieties for the CG cytosine type, according to the Kruskal–Wallis test for non-parametric data (p = 0.007), while there was no statistical difference in methylation patters among CHG and CHH types. The AG8041 variety showed 2.08% of methylated CG, DKB240 showed 1.04% and DKB350 and AG9045 showed no methylation in this region (Fig. 2a).

Cytosine methylation levels among MON-89Ø34-3 maize varieties in four regions of the transgenes. The levels of cytosine methylation were measured in CG, CHG and CHH residues. a FMV promoter region; b cry2Ab2 transgene region; c 35S promoter region; d cry1A.105 transgene region. Vertical bars indicate standard errors. Means followed by different letters in the same cytosine type are significantly different according to the Kruskal–Wallis test (p < 0.05). Exact p values for the statistical test of each of the cytosine comparisons are shown on the right upper corner
The cry2Ab2 region also showed significant differences, according to the Kruskal–Wallis test (p = 0.046), for the CG residues, but not for the other cytosine types. DKB350 and AG8041 varieties showed 2.46% of methylated cytosines, AG9045 showed 1.38% and DKB240 showed 0.77% (Fig. 2b). The cry1A.105 and 35S promoter regions did not show any significant differences for the cytosines residues analysed using Kruskal–Wallis test (p > 0.05) (Fig. 2c, d).
The second experimental setup compared the methylation pattern of transgenic regions on single and stacked transgenic events in the same genetic background (DKB240). The analysis was performed by the comparison of the DKB240 single GM maize (MON-89Ø34-3) with the DKB240 stacked GM maize (MON-89Ø34-3 × MON-ØØ6Ø3-6). Significant differences were found in the CG type for the 35S promoter region according to the Wilcoxon-Mann–Whitney test for non-parametric data (p = 0.010). The single event showed 3.40% of methylated cytosines, while the stacked showed 0.38% (Fig. 3c). Furthermore, the cry1A.105 transgene, FMV promoter and cry2Ab2 transgene regions did not show significant differences for the methylation analysis (Fig. 3a, b, d). The distribution of methylated and unmethylated cytosines for all samples in all the regions is shown in the Additional files 3, 4, 5 and 6.

Cytosine methylation levels of single and stacked maize varieties in four regions of the transgenes. The levels of cytosine methylation were measured in CG, CHG and CHH residues. a FMV promoter region; b cry2Ab2 transgene region; c 35S promoter region; d cry1A.105 transgene region. Vertical bars indicate standard errors. Means followed by different letters in the same cytosine type are significantly different according to the Wilcoxon-Mann–Whitney test (p < 0.05). Exact p values for the statistical test of each of the cytosine comparisons are shown on the right upper corner
Transgene transcript accumulation in Bt-expressing maize varieties
We have further analysed the levels of transgene mRNA accumulation in the leaves of single and stacked maize varieties containing the cry1A.105 and cry2Ab2 transgenes by RT-qPCR in order to investigate if different methylation levels would result in differential mRNA accumulation. The DKB240 single GM maize was used as a reference sample for the transcript relative quantification analysis.
The first experimental setup analysed the transgene transcript accumulation among four different single GM varieties. For the cry1A.105 transcript analysis, AG8041 and AG9045 varieties showed a higher transcript accumulation (1.67- and 1.19-fold change, respectively), while the DKB350 variety showed a lower transcript accumulation (0.87-fold change). Statistically, AG8041 and DKB350 significantly differ between themselves, while DKB240 and AG9045 did not differ from any of the other varieties (Fig. 4a). For the accumulation of cry2Ab2 transcripts, compared to the DKB240 variety, AG8041 and DKB350 varieties showed higher transcript accumulation levels (1.52- and 1.24-fold change, respectively), while the AG9045 showed a behaviour similar to the reference (0.99-fold change). Statistically, AG8041 significantly differ from DKB240 and AG9045, while DKB350 did not differ from any of the other varieties (Fig. 4b).

mRNA accumulation levels of transgenic transcripts among MON-89Ø34-3 maize varieties. a cry1A.105 transgene; b cry2Ab2 transgene. Vertical bars indicate standard deviation. Different letters above the bars indicate statistical differences among the samples at p < 0.05. Exact p value for the statistical test is shown on the right upper corner
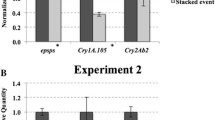
The second experimental setup analysed the transgene transcript accumulation between a single GM variety and a stacked GM variety. The stacked event showed lower accumulation levels for the cry1A.105 transgene (0.94-fold change), as well as for the cry2Ab2 transgene (0.83-fold change) (Fig. 5a, b respectively). However, only the cry2Ab2 transgene showed statistically significant differences (p = 0.0067).

mRNA accumulation levels of transgenic transcripts between single and stacked maize varieties. a cry1A.105 transgene; b cry2Ab2 transgene. Vertical bars indicate standard deviation. Asterisk above the bar indicates statistical difference between the samples at p < 0.05. ‘ns’ above the bar indicates non-significant statistical difference (p > 0.05) between samples. Exact p value for the statistical test is shown on the right upper corner
Discussion
Cytosine methylation and its correlation with transgene transcript accumulation
When a transgenic event is approved for commercial use, it is required that the transgenic trait, or locus, is introgressed into local varieties. In such cases, the same transgenic event can be introgressed by traditional backcrosses into as many maize varieties as it is desirable. It is then expected that the transgenic insert is stable in all varieties (i.e. regarding its protein and transcript expression), in all available genetic background.
Epigenetic phenomena, such as DNA methylation, are known to affect transgene expression [47–49]. La Paz et al. [7] conducted the only study regarding epigenetic aspects of commercialized GM crops, in which they assessed the cytosine methylation levels of seven single GM maize varieties containing the MON810 transgenic event construct and did not find statistical differences in the 35S promoter and cry1Ab transgene regions. In contrast, our results show statistical differences between GM varieties in three of the four transgenic regions analysed, including the 35S promoter (when comparing single vs stacked varieties). However, La Paz et al. [7] results might have been hampered by a limited statistical power (caused by small sampling size), in which they analysed only one plant per variety and five bacteria colonies per plant, while our study analysed three plants per variety and eight bacteria colonies.
On the other hand, our results indeed showed high standard deviation values in the methylation analysis for all regions, which is explained by the high variation observed among the 8 selected clones. This variation might have impacted the significance of statistical analysis. Although bisulfite sequencing of single clones has been regarded as the gold standard of DNA methylation analysis for the past few years [50], there is no consensus of how many clones are adequate to provide reliable results. Distinct studies have used five [7, 51], six [52, 53] or ten clones [54, 55]; other studies do not even mention it within their “Methods” section [56, 57].
Transgene methylation has been long associated with TGS and PTGS, both in promoter [30] and coding sequence regions [58]. DNA methylation of promoter regions is generally associated with reduced expression of the regulated genes [59], whereas DNA methylation within gene bodies has a more complex association with gene expression, varying among species and level of gene expression [29, 59–61]. Although DNA methylation is not necessary for gene silencing [56], promoter methylation plays an important role in transgene silencing in vitro and in vivo [62]. Several studies have shown a correlation between gene expression and methylation levels of either promoter or gene regions [51, 52, 57, 63, 64].
Regarding the comparison between single and stacked GM varieties in our studies, the correlation between 35S promoter and cry1A.105 transgene methylation and cry1A.105 transcript accumulation was not robust, unlike the results in the previously mentioned studies. However, we did verify a correlation for the FMV:cry2Ab2 transgene, where both FMV promoter and cry2Ab2 sequences showed higher levels of CG and CHG methylation in the stacked event accompanied by a lower expression of the cry2Ab2 transgene. The FMV (Figwort Mosaic Virus) is a double-stranded DNA virus with a genome of 8 kilobase pairs [65] and a genetic organization similar to that of cauliflower mosaic virus (CaMV) [66]. Therefore, due to the high similarity of those promoters, it is expected that the FMV promoter behaves similarly to the 35S, thus acting in the regulation of transgene expression.
Transcript accumulation in Bt-expressing maize varieties
Our results show variation in the transgene transcript accumulation among single GM varieties, which was assessed by RT-qPCR analysis. One of the few studies performed with single GM maize has analysed cry1Ab mRNA accumulation in two MON810 varieties during several plant development stages, with the results showing certain levels of variation, but in the range of the natural variation [7]. Environmental conditions such as temperature and water accessibility also play an important role in transgene expression rate. This is further complicated by different genetic backgrounds in different GM varieties (even with the same event), which respond differently to the same environmental change with regards to transgenic mRNA expression [67].
Wang et al. [68] observed a variation in the levels of CRY1AB protein, measured by ELISA, among different cultivars of GM cotton. Luo et al. [69] submitted two Bt cotton cultivars, expressing the CRY1AC protein, under salinity stress. The levels of CRY1A protein, measured by ELISA, varied between cultivars, with one of the cultivars showing lower CRY1A content earlier than the other in comparison to the controls. Other studies with Bt cotton have also reported variation of transgenic protein among cultivars containing the same GM construct [70, 71], which indicates that genetic background might influence the levels of transgene expression.
Nevertheless, changes in transgene expression levels due to differences in genetic background may have an impact in the safety and utility of GM plants. Koul et al. [72] showed a correlation between transcript accumulation and Bt protein in different transgenic tomato lines expressing the cry1Ab transgene. Olsen et al. [73] found that the developmental decline in bioefficacy (against Helicoverpa armigera) in field-grown plants was associated with reduced cry1Ac transcript levels and Bt toxin levels in postsquaring cotton. The authors were also able to corroborate the relationship between the decline in efficacy and reduced mRNA levels with plants grown in contention. The biggest safety concern with the variation of transgenic transcript and protein accumulation is related to the possibility of field-evolved resistance. Cases of resistance of target organism to Bt toxins have been report [74, 75], and they were usually related to a low adherence of farmers in cultivating a refuge area with non-GM varieties [76]. However, variation on the toxin doses can also be linked as one of the causes for the appearance of resistant insects [77, 78]. The variation of Bt toxins has been previously linked to environmental conditions and genetic background [79, 80], but the reason remains unclear and might be linked to mRNA instability, variation in promoter activity, reduction in protein expression and protein–protein interactions [42, 73].
Variation on the levels of transgene expression is widely known, but its cause has not been fully understood yet [81]. An active transgenes might be silenced through the introduction in its genome of a second transgene regulated by the same promoter [82]. In addition, there are reports showing that gene silencing is frequently associated with multiple transgene integrations due to homology between the transgenes [83–85]. In fact, DNA homology has long been suggested as a mechanism for two trans-inactivation systems comprising a silencing transgene locus and a target transgene locus that share homology only in promoter regions [86, 87]. In the NOS promoter-based system, silencing phenomena was observed with a promoter homology of about 300 bp [88]. In the 35S promoter-based system, however, silencing was observed even when promoter homology comprised only 90 bp [86]. Even though 90 bp seems a short sequence for direct DNA–DNA pairing, it still does not preclude the recognition of DNA homology [89]. Mishiba et al. [52] report that gentian plants showed no expression of bar and GtMADS genes, under the regulation of the 35S promoter, and that the possible cause for the silencing phenomenon might be homology-dependent gene silencing involving the promoter regions.
Agapito-Tenfen et al. [42] also found reduced transgene expression in stacked transgenes with the same GM event, but with different varieties. The authors suggested that these reductions might be related to the high energetic demand of the cell. In this aspect, evidences support the idea that constitutive promoters involve a high energetic cost, leading to a penalty in transgenic plants [90–92]. Moreover, the authors have also performed a proteomic profiling of single and stacked GM, and some metabolic pathways, such as energy/carbohydrate metabolism and detoxification metabolism pathways, were differentially modulated. These results suggest that the insertion of a new transgene in a single GM plant may alter the expression of endogenous genes, especially those related to energetic metabolism, since the transgene are being expressed constantly in the plant tissues and, therefore, demand high levels of energy.
Even though a quantification of transgenic proteins was not performed in our study, the transgene analysis (in our case cry1A.105 and cry2Ab2 transgenes) of transcripts accumulation showed some variation among varieties. Therefore, additional studies could be performed in order to further investigate the correlation of transgene transcripts and levels of Bt toxin production, and even the bioefficacy of the transgenic proteins against the target organisms, and thus the biological meaning behind it. Taken together, our results are in agreement to that of our previous work that showed reduced transgene expression in stacked transgenes with the same GM event, but with different varieties [42]. We have observed a significant reduction of the expression of the cry2Ab2 transgenes in the stacked event. Safety implications are related to cases in which the expression level of an introduced/modified trait in a GM stacked event falls outside the range of what was determined in the parental line, and a re-evaluation of the environmental aspects might be necessary [93].
Contributions to risk assessment of GM crops
Regulatory practice within the European Union (EU) consider stacked events as new GM organisms, and additional information on the stability of transgene insertions, expression levels and potential antagonistic or synergistic interactions should be provided prior to marketing [93–95]. Overall, there is a few available data on transgene expression levels in both stacked and single transgene GM crops in the scientific literature. Although data on expression levels for stacked GM events are required for approval according to EU regulations (No 503/2013), these are rarely disclosed or they are considered insufficient [42, 96, 97]. Recent discussions about potential risks of stacked events, as well as the opinion of the European Food Safety Authority (EFSA) on those issues, have highlighted the lack of consensus with regard to whether such GMOs should be subject to specific assessments [98].
As for the DNA methylation data, there is also a lack of scientific literature for any GM crop, and the regulations do not consider yet the need for this type of analysis. Data related to possible epigenetic variations of GM crops, such as DNA methylation levels and ncRNAs, could help to extend our knowledge on the safety of such organism and, therefore, may be taken in account in risk assessments. While there is a lack of specific investigation of cytosine methylation levels in commercialized transgenic crops, the literature records show that methylation of promoter regions and coding sequences may also result in decreased levels of transgene transcription [51, 57, 63]. The same can be applied to ncRNAs, where studies have reported the existence of gene silencing mediated by 35S promoter homology between transgenes; however, whether the silencing is mediated by 35S promoter siRNAs produced from complex transgene inserts is still not fully known [99]. Moreover, the 35S promoter can also be silenced and methylated by the production of homologous siRNAs [100, 101]. Since both epsps and cry1A.105 transgenes present in the stacked line used in this study are controlled by homologous 35S promoters, TGS, PTGS or other processes being involved in transgene transcript modulation in the stacked line cannot be ruled out.
So far, no other study has compared the methylation levels of transgene sequences of this particular GM event in both single and stacked varieties, and its possible correlation with transgene transcript accumulation. It is important to emphasize that the findings of the present study are restricted to the set of varieties used. We cannot extrapolate this information to other single or stacked GM varieties without testing it. Hence, there is a lack of this kind of data that might be important in order to reliably assess the safety of stacked and single GM events.
Conclusions
In conclusion, our results showed that DNA methylation levels of transgenic sequences vary among single GM maize varieties and between single and stacked GM maize varieties. We also observed that the accumulation of transgene transcript showed variation among single GM varieties, which indicates that genetic background might have some influence in the levels of transgene expression. Likewise, accumulation of transgene transcript also varied between single and stacked GM varieties, with the stacked one showing a statistically significant reduction for the cry2Ab2 transgene.
These conclusions arose from the statistically different levels of DNA methylation in the FMV promoter and cry2Ab2 gene among single GM varieties and in the 35S promoter for the comparison of single and stacked GM varieties. In addition, transgenic transcript accumulation levels demonstrate a high variation between single GM varieties, with samples showing an increase up to 1.67-fold change and a decrease of 0.87-fold change in accumulation compared to the reference variety (for the cry1A.105 transgene). Moreover, transgenic transcript accumulation levels in the stacked GM variety showed a reduction of about 0.94- and 0.87-fold change (cry1A.105 and cry2Ab2 transgenes, respectively) when compared to parental single event.
Similar results of transgenic transcript accumulation have been reported in our previous study. However, this is one of the first studies that assessed and verified changes in methylation levels of transgenic sequences of single and stacked GM crops. Although the detection of changes in DNA methylation levels and transgenic accumulation levels does not present a safety issue per se, it demonstrates the need for additional studies that address the biological relevance and the possible safety implications of such changes.
References
Convention on Biological Diversity (CBD) (2000) Cartagena Protocol on biosafety to the convention on biological diversity: text and annexes. Secretariat of the Convention on Biological Diversity, Montreal
James C (2014) Global Status of commercialized biotech, GM Crops:2014. ISAAA Brief 49. ISAAA, Ithaca
Gelvin SB (2003) Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol R 67(1):16–37
Kohli A, Twyman RM, Abranches R, Wegel E, Stoger E, Christou P (2003) Transgene integration, organization and interaction in plants. Plant Mol Biol 52(2):247–258
Kohli A, Griffiths S, Palacios N, Twyman RM, Vain P, Laurie DA et al (1999) Molecular characterization of transforming plasmid rearrangements in transgenic rice reveals a recombination hotspot in the CaMV 35S promoter and confirms the predominance of microhomology mediated recombination. Plant J 17:591–601
Wu Y, Wu R, Zhang B, Jiang T, Li N, Qian K et al (2012) Epigenetic instability in genetically stable micropropagated plants of Gardenia jasminoides Ellis. Plant Growth Regul 66:137–143
La Paz JL, Pla M, Papazova N, Puigdomènech P, Vicient C (2010) Stability of the MON 810 transgene in maize. Plant Mol Biol 74:563–571
Comissão Técnica Nacional de Biossegurança (2007) Parecer Técnico no 1.100/2007—Liberação Comercial de Milho Geneticamente Modificado resistente a Insetos Evento MON810—Processo 01200.002995/1999-54. http://ctnbio.mcti.gov.br/en/liberacao-comercial. Accessed 04 July 2016
Comissão Técnica Nacional de Biossegurança (2008) Parecer Técnico no 1596/2008—Liberação Comercial de Milho Geneticamente Modificado Tolerante ao Glifosato, Milho Roundup Ready 2, Evento NK603—Processo no 01200.002293/2004-16. http://ctnbio.mcti.gov.br/en/liberacao-comercial. Accessed 04 June 2016
Jakowitsch J, Papp I, Moscone EA, Van Der Winden J, Matzke M, Matzke AJM (1999) Molecular and cytogenetic characterization of a transgene locus that induces silencing and methylation of homologous promoters in trans. Plant J 17:131–140
Zhao Y, Guo L, Wang H, Huang D (2011) Integration and expression stability of transgenes in hybriding transmission of transgenic rice plants produced by particle bombardment. Mol Plant Breed 2(8):48–59
Nguyen HT, Jehle JA (2007) Quantitative analysis of the seasonal and tissue specific expression of Cry1Ab in transgenic maize MON 810. J Plant Dis Protect 114:82–87
Holck A, Va M, Didierjean L, Rudi K (2002) 5′-Nuclease PCR for quantitative event-specific detection of the genetically modified Mon810 MaisGard maize. Eur Food Res Technol 214:449–453
Hernández M, Pla M, Esteve T, Prat S, Puigdomenech P, Ferrando A (2003) A specific real-time quantitative PCR detection system for event MON810 in maize YieldGard based on the 3′-transgene integration sequence. Transgenic Res 12:179–189
Rosati A, Bogani P, Santarlasci A, Buiatti M (2008) Characterisation of 3′ transgene insertion site and derived mRNAs in MON810 YieldGard maize. Plant Mol Biol 67:271–281
Windels P, Taverniers I, Depicker A, Van Bockstaele E, De Loose M (2001) Characterisation of the Roundup Ready soybean insert. Eur Food Res Technol 213:107–112
La Paz JL, Vicient C, Puigdomènech P, Pla M (2010) Characterization of polyadenylated cryIA(b) transcripts in maize MON810 commercial varieties. Anal Bioanal Chem 396:2125–2133
Bredfeldt TG, Walker CL (2010) Genetic and epigenetic determinants of susceptibility to environmental injury. In: McQueen CA (ed) Comprehensive toxicology, 2nd edn. Elsevier, New York, pp 337–355
Makarevitch I, Stupar RM, Iniguez AL, Haun WJ, Barbazuk WB, Kaeppler SM et al (2007) Natural variation for alleles under epigenetic control by the maize chromomethylase zmet2. Genetics 77:749–760
Heinemann JA, Agapito-Tenfen SZ, Carman JA (2013) A comparative evaluation of the regulation of GM crops or products containing dsRNA and suggested improvements to risk assessments. Environ Int 55:43–55
Chen LT, Luo M, Wang YY, Wu K (2010) Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J Exp Bot 61:3345–3353
Chen M, Lv S, Meng Y (2010) Epigenetic performers in plants. Dev Growth Differ 52:555–566
Osabe K, Clement JD, Bedon F, Pettolino FA, Ziolkowski L, Llewellyn DJ et al (2014) Genetic and DNA methylation changes in cotton (Gossypium) genotypes and tissues. Plos ONE 9(1):e86049
Gruenbaum Y, Naveh-Many T, Cedar H, Razin A (1981) Sequence specificity of methylation in higher plant DNA. Nature 292:860–862
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD et al (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452:215–219
Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH et al (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133:523–536
Finnegan EJ, Peacock WJ, Dennis ES (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci USA 93:8449–8454
Chan SW, Henderson IR, Jacobsen SE (2005) Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet 6:351–360
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers interdependence between methylation and transcription. Nat Genet 39:61–69
Linn F, Heidmann I, Saedler H, Meyer P (1990) Epigenetic changes in the expression of the maize A1 gene in Petunia hybrida: role of numbers of integrated gene copies and state of methylation. Mol Gen Genet 222:329–336
Meyer P, Niedenhof I, Lohuis M (1994) Evidence for cytosine methylation of non-symmetrical sequences in transgenic Petunia hybrida. EMBO J 13:2084–2088
Steward N, Ito M, Yamaguchi Y, Koizumi N, Sano H (2002) Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem 277:37741–37746
Zhao X, Chai Y, Liu B (2007) Epigenetic inheritance and variation of DNA methylation level and pattern in maize intra-specific hybrids. Plant Sci 172:930–938
Tan M (2010) Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol Bioch 48:21–26
Comissão Técnica Nacional de Biossegurança (2008) Parecer Técnico no 2052/2009—Liberação Comercial de Milho Resistente a Insetos, Milho MON 89034—Processo no 01200.003326/2008-61. http://ctnbio.mcti.gov.br/en/liberacao-comercial. Accessed 04 July 2016
Comissão Técnica Nacional de Biossegurança (2009) Parecer Técnico no 2725/2010—Liberação Comercial de Milho Geneticamente Modificado Resistente a Insetos e Tolerante a Herbicidas, Milho MON 89034 x NK 603—Processo no 01200.003952/2009-38. http://ctnbio.mcti.gov.br/en/liberacao-comercial. Accessed 04 July 2016
Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431
Sambrook J, Russell DW (2001) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sanger F, Nicklen S, Coulson R (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Savini C, Bogni A, Grazioli E, Munaro B, Mazzara M, Van den Eede G (2008) Event-specific method for the quantification of maize line MON 89034 using real-time PCR. JRC Scientific and Technical Reports
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Agapito-Tenfen SZ, Vilperte V, Benevenuto RF, Rover CM, Traavik TI, Nodari RO (2014) Effect of stacking insecticidal cry and herbicide tolerance epsps transgenes on transgenic maize proteome. BMC Plant Biol 14:346
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-pcr data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5252
Hetzl J, Foerster AM, Raidl G, Mittelsten Scheid O (2007) CyMATE: a new tool for methylation analysis of plant genomic DNA after bisulphite sequencing. Plant J 51(3):526–536
Team RC (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M et al (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Gelvin SB, Karcher SJ, DiRita VJ (1983) Methylation of the T-DNA in Agrobacterium tumefaciens and in several crown gall tumors. Nucleic Acids Res 11(1):159–174
Matzke MA, Primig M, Trnovsky J, Matzke AJM (1989) Reversible methylation and inactivation of marker genes in sequentially transformed tobacco plants. EMBO J 8(3):643–649
Meyer P, Linn F, Heidmann I, Meyer H, Niedenhof H, Saedler H (1992) Endogenous and environmental factors influence 35S promoter methylation of a maize A1 gene construct in transgenic petunia and its colour phenotype. Mol Gen Genet 231:345–352
Zhang Y, Jeltsch A (2010) The application of next generation sequencing in DNA methylation analysis. Genes 1:85–101
Fan J, Liu X, Xu S, Xu Q, Guo W (2011) T-DNA direct repeat and 35S promoter methylation affect transgene expression but do not cause silencing in transgenic sweet orange. Plant Cell Tiss Organ Cult 107:225–232
Mishiba K, Nishihara M, Nakatsuka T, Abe Y, Hirano H, Yokoi T et al (2005) Consistent transcriptional silencing of 35S-driven transgenes in gentian. Plant J 44:541–556
Mishiba K, Yamasaki S, Nakatsuka T, Abe Y, Daimon H, Oda M et al (2012) Strict de novo methylation of the 35S enhancer sequence in gentian. Plos ONE 5(3):e9670
Yamasaki S, Oda M, Koizumi N, Mitsukuri K, Johkan M, Nakatsuka T et al (2011) De novo DNA methylation of the 35S enhancer revealed by high-resolution methylation analysis of an entire T-DNA segment in transgenic gentian. Plant Biotechnol 28:223–230
Eike MC, Mercy IS, Aalen RB (2005) Transgene silencing may be mediated by aberrant sense promoter sequence transcripts generated from cryptic promoters. Cell Mol Life Sci 62:3080–3091
Skårn M, Eike CE, Meza TJ, Mercy IS, Jakobsen KS, Aalen RB (2006) An inverted repeat transgene with a structure that cannot generate double-stranded RNA, suffers silencing independent of DNA methylation. Transgenic Res 15:489–500
Weinhold A, Kallenbach M, Baldwin IT (2013) Progressive 35S promoter methylation increases rapidly during vegetative development in transgenic Nicotiana attenuata plants. BMC Plant Biol 13:99
Ingelbrecht I, Van Houdt H, Van Montagu M, Depicker A (1994) Posttranscriptional silencing of reporter transgenes in tobacco correlates with DNA methylation. Proc Natl Acad Sci USA 91:10502–10506
Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SWL, Chen H et al (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126:1189–1201
Lauria M, Rossi V (2011) Epigenetic control of gene regulation in plants. Biochim Biophys Acta 1809:369–378
Vining KJ, Pomraning KR, Wilhelm LJ, Priest HD, Pellegrini M, Mockler TC et al (2012) Dynamic DNA cytosine methylation in the Populus trichocarpa genome: tissue- level variation and relationship to gene expression. BMC Genom 13:27
Vaucheret H, Fagard M (2001) Transcriptional gene silencing in plants: targets, inducers and regulators. Trends Genet 17(1):29–35
Wang H, Wu S, Li F, Chen T, Jiang Y, Ju M et al (2008) Transgene silencing caused by 35S promoter methylation in upland cotton (Gossypium hirsutum). Cott Sci 20(1):271–280
Gambino G, Perron I, Carra A, Chitarra W, Boccacc P, Marinoni DT et al (2010) Transgene silencing in grapevines transformed with GFLV resistance genes: analysis of variable expression of transgene, siRNAs production and cytosine methylation. Transgenic Res 19:17–27
Richins RD, Scholthof HB, Sheperd RJ (1987) Sequence of figwort mosaic virus DNA (caulimovirus group). Nucleic Acids Res 15(20):8451–8466
Franck A, Guilley H, Jonard G, Richards K, Hirth L (1980) Nucleotide sequence of cauliflower mosaic virus DNA. Cell 21:285–294
Trtikova M, Wikmark O, Zemp N, Widmer A, Hilbeck A (2015) Transgene expression and Bt protein content in transgenic Bt Maize (MON810) under optimal and stressful environmental conditions. Plos ONE 10(4):e0123011
Wang Y, Ye G, Luan N, Xiao J, Chen Y, Chen D (2009) Boll size affects the insecticidal protein content in Bacillus thuringiensis (Bt) cotton. Field Crop Res 110:106–110
Luo Z, Dong H, Li W, Ming Z, Zhu Y (2008) Individual and combined effects of salinity and waterlogging on Cry1Ac expression and insecticidal efficacy of Bt cotton. Crop Prot 27:1485–1490
Chen D, Ye G, Yang C, Chen Y, Wu Y (2005) The effect of high temperature on the insecticidal properties of Bt cotton. Environ Exp Bot 53:333–342
Chen Y, Chen Y, Wen Y, Zhang X, Chen D (2012) The effects of the relative humidity on the insecticidal expression level of Bt cotton during bolling period under high temperature. Field Crop Res 137:141–147
Koul B, Srivastava S, Sanyal I, Tripathi B, Sharma V, Amla DV (2014) Transgenic tomato line expressing modified Bacillus thuringiensis cry1Ab gene showing complete resistance to two lepidopteran pests. SpringerPlus 3(84):1–13
Olsen KM, Daly JC, Holt HE, Finnegan EJ (2005) Season-long variation in expression of Cry1Ac gene and efficacy of Bacillus thuringiensis toxin in transgenic cotton against Helicoverpa armigera (Lepidoptera: Noctuidae). J Econ Entomol 98(3):1007–1017
Storer NP, Babcock JM, Schlenz M, Meade T, Thompson GD, Bing JW, Huckaba RM (2010) Discovery and characterization of field resistance to Bt maize: Spodoptera frugiperda (Lepidoptera: Noctuidae) in Puerto Rico. J Econ Entomol 103:1031–1038
Huang F, Andow DA, Buschman LL (2011) Success of the high-dose/refuge resistance management strategy after 15 years of Bt crop use in North America. Entomol Exp Appl 140:1–16
Kruger M, Van Rensburg J, Van den Berg J (2011) Resistance to Bt maize in Busseola fusca (Lepidoptera: Noctuidae) from Vaalharts. S Afr Environ Entomol 40:477–483
Gassmann AJ (2012) Field-evolved resistance to Bt maize by western corn rootworm: predictions from the laboratory and effects in the field. J Invertebr Pathol 110:287–293
Tabashnik BE, Gould F (2012) Delaying corn rootworm resistance to Bt corn. J Econ Entomol 105:767–776
Showalter AM, Heuberger S, Tabashnik BE, Carrière Y, Coates B (2009) A primer for using transgenic insecticidal cotton in developing countries. J Insect Sci 9:22
Nguyen HT, Jehle JA (2009) Expression of Cry3Bb1 in transgenic corn MON88017. J Agric Food Chem 57:9990–9996
Stam M, Mol JNM, Kooter JM (1997) The silence of genes in transgenic plants. Ann Bot 79:3–12
Fagard M, Vaucheret H (2000) (Trans)Gene silencing in plants: how many mechanisms? Annu Rev Plant Physiol Plant Mol Biol 51:167–194
Cluster PD, O’Dell M, Metzlaff M, Flavell RB (1996) Details of T-DNA structural organization from a transgenic Petunia population exhibiting co-suppression. Plant Mol Biol 32:1197–1203
Pang SZ, Jan FJ, Carney K, Stout J, Tricoli DM, Quemada HD et al (1996) Post-transcriptional transgene silencing and consequent tospovirus resistance in transgenic lettuce are affected by transgene dosage and plant development. Plant J 9(6):899–909
Weld R, Heinemann J, Eady C (2001) Transient GFP expression in Nicotiana plumbaginifolia suspension cells: the role of gene silencing, cell death and T-DNA loss. Plant Mol Biol 45:377–385
Vaucheret H (1993) Identification of a general silencer for 19 and 35 s promoters in a transgenic tobacco plant: 90 bp of homology in the promoter sequence are sufficient for trans-inactivation. C R Acad Sci Paris 316:1471–1483
Matzke MA, Matzke AJM, Mittelsten-Scheid O (1994) Inactivation of repeated genes—DNA-DNA interaction? In: Paszkowski J (ed) Homologous recombination and gene silencing in plants. Dordrecht, Kluwer, Kluwer Academic Publishers, pp 271–307
Matzke MA, Matzke AJM (1993) Genomic imprinting in plants: parental effects and trans-inactivation phenomena. Annu Rev Plant Physiol Plant Mol Biol 44:53–76
Li X, Zhu Z, Feng D, Chang T, Liu X (2001) Influence of DNA methylation on transgene expression. Chin Sci Bull 46:1300–1303
Rus AM, Estañ MT, Gisbert C, Garcia-Sogo B, Serrano R, Caro M et al (2001) Expressing the yeast HAL1 gene in tomato increases fruit yield and enhances K+/Na+ selectivity under salt stress. Plant Cell Environ 24:875–880
Grover A, Aggarwal PK, Kapoor A, Katiyar-Agarwal S, Agarwal M, Chandramouli A (2003) Addressing abiotic stresses in agriculture through transgenic technology. Curr Sci India 84:355–367
Muñoz-Mayor A, Pineda B, Garcia-Abellán JO, Garcia-Sogo B, Moyano E, Atares A et al (2008) The HAL1 function on Na+ homeostasis is maintained over time in salt-treated transgenic tomato plants, but the high reduction of Na+ in leaf is not associated with salt tolerance. Physiol Plant 133:288–297
De Schrijver A, Devos Y, Van den Bulcke M, Cadot P, De Loosed M, Reheul D et al (2007) Risk assessment of GM stacked events obtained from crosses between GM events. Trends Food Sci Tech 18:101–109
European Food Safety Authority (EFSA) (2007) Guidance document of the scientific panel on genetically modified organisms for the risk assessment of genetically modified plants containing stacked transformation events. EFSA J 512:1–5
Ad Hoc Technical Expert Group (AHTEG) (2010) United Nations Environment Programme, CBD: Guidance document on risk assessment of living modified organisms
Nielsen KM (2013) Biosafety data as confidential business information. Plos Biol 11:e1001499
Kok EJ, Pedersen J, Onori R, Sowa S, Schauzu M, De Schrijver A et al (2014) Plants with stacked genetically modified events: to assess or not to assess? Trends Biotech 32:70–73
Spök A, Eckerstorfer M, Heissenberger A, Gaugitsch H (2007) Risk assessment of stacked events. http://www.bmg.gv.at/cms/home/attachments/7/7/0/CH1052/CMS1180523433481/cms1200659635294_internetversion_2_07stacked_events.pdf. Accessed 04 June 2016
Daxinger L, Hunter B, Sheikh M, Jauvion V, Gasciolli V, Vaucheret H et al (2008) Unexpected silencing effects from T-DNA tags in Arabidopsis. Trends Plant Sci 13(1):4–6
Sijen T, Vijn I, Rebocho A, van Blokland R, Roelofs D, Mol JNM et al (2001) Transcriptional and posttranscriptional gene silencing are mechanistically related. Curr Biol 11:436–440
Mourrain P, van Blokland R, Kooter JM, Vaucheret H (2007) A single transgene locus triggers both transcriptional and post-transcriptional silencing through double-stranded RNA production. Planta 225:365–379
Authors’ contributions
VV, SZAT, OW and RON designed the experiments. VV implemented and maintained the growth chamber experiment and collected samples. VV performed the methylation analysis. VV and SZAT performed the RT-qPCR experiment. VV wrote the manuscript. VV, SZAT, OW and RON assisted with data analysis. VV and SZAT conducted the statistical analysis. SZAT, OW and RON revised the draft of the manuscript. All authors revised the final manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Fredskorpset Norway (FK Norway) for financial support and scholarship provided to VV and CNPq for scholarship provided to RON. Financial support has been also provided by The Norwegian Agency for Development Cooperation (Ministry of Foreign Affairs, Norway) under the GenØk South-America Research Hub grant FAPEU 077/2012. This was a joint project between UFSC and GenØk-Center for Biosafety.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
12302_2016_97_MOESM1_ESM.xlsx
Additional file 1. Output results of NormFinder algorithm used for determining the best endogenous normalizer genes. Leunig (LUG) and Membrane protein PB1A10.07c (MEP) showed the best combination of two normalizer genes with a stability value of 0.025.
12302_2016_97_MOESM2_ESM.tiff
Additional file 2. Alignment of the 35S promoters from the CP4-epsps (Query) and cry1A.105 (Sbjct). The sequence in bold and underlined corresponds to the region where the BS35S-F primer was designed.
12302_2016_97_MOESM3_ESM.pdf
Additional file 3. Position and distribution of methylated and unmethylated cytosines for the 35S promoter sequence in all analysed hybrids. Each line corresponds to one sequenced bacteria colony (clone). Clones from the same hybrid contain the same nomenclature, ranging from 1-24. Class I: CG residue; Class II: CHG residue; Class III: CHH residue. Filled icons: methylated cytosine; Empty icons: non-methylated cytosines.
12302_2016_97_MOESM4_ESM.pdf
Additional file 4. Position and distribution of methylated and unmethylated cytosines for the cry1A.105 gene sequence in all analysed hybrids. Each line corresponds to one sequenced bacteria colony (clone). Clones from the same hybrid contain the same nomenclature, ranging from 1–24. Class I: CG residue; Class II: CHG residue; Class III: CHH residue. Filled icons: methylated cytosine; Empty icons: non-methylated cytosines.
12302_2016_97_MOESM5_ESM.pdf
Additional file 5. Position and distribution of methylated and unmethylated cytosines for the FMV promoter sequence in all analysed hybrids. Each line corresponds to one sequenced bacteria colony (clone). Clones from the same hybrid contain the same nomenclature, ranging from 1–24. Class I: CG residue; Class II: CHG residue; Class III: CHH residue. Filled icons: methylated cytosine; Empty icons: non-methylated cytosines.
12302_2016_97_MOESM6_ESM.pdf
Additional file 6. Position and distribution of methylated and unmethylated cytosines for the cry2Ab2 gene sequence in all analysed hybrids. Each line corresponds to one sequenced bacteria colony (clone). Clones from the same hybrid contain the same nomenclature, ranging from 1–24. Class I: CG residue; Class II: CHG residue; Class III: CHH residue. Filled icons: methylated cytosine; Empty icons: non-methylated cytosines.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vilperte, V., Agapito-Tenfen, S.Z., Wikmark, OG. et al. Levels of DNA methylation and transcript accumulation in leaves of transgenic maize varieties. Environ Sci Eur 28, 29 (2016). https://doi.org/10.1186/s12302-016-0097-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12302-016-0097-2