
Background
Iron (Fe) is essential micronutrient for plants and its deficiency as well as toxicity is a serious agricultural problem. The mechanisms of Fe deficiency are reasonably understood, however our knowledge about plants response to excess Fe is limited. Moreover, the regulation of small open reading frames (sORFs) in response to abiotic stress has not been reported in rice. Understanding the regulation of rice transcriptome in response to Fe deficiency and excess could provide bases for developing strategies to breed plants tolerant to Fe deficiency as well as excess Fe.
Results
We used a novel rice 110 K microarray harbouring ~48,620 sORFs to understand the transcriptomic changes that occur in response to Fe deficiency and excess. In roots, 36 genes were upregulated by excess Fe, of which three were sORFs. In contrast, 1509 genes were upregulated by Fe deficiency, of which 90 (6%) were sORFs. Co-expression analysis revealed that the expression of some sORFs was positively correlated with the genes upregulated by Fe deficiency. In shoots, 50 (19%) of the genes upregulated by Fe deficiency and 1076 out of 2480 (43%) genes upregulated by excess Fe were sORFs. These results suggest that excess Fe may significantly alter metabolism, particularly in shoots.
Conclusion
These data not only reveal the genes regulated by excess Fe, but also suggest that sORFs might play an important role in the response of plants to Fe deficiency and excess.
Similar content being viewed by others


Background
Iron (Fe) is an essential micronutrient for all higher organisms, and its deficiency causes a serious nutritional problem in both humans and plants. Although mineral soils are rich in Fe (>5%), various factors such as a high soil pH and the presence of sodium carbonate adversely affect the availability and uptake of Fe through plant roots (Marschner [1995]; Mori [1999]). In contrast, a low soil pH and anaerobic conditions, such as in a paddy field, lead to the reduction of Fe3+ to Fe2+, which can result in increased absorption and conditions of excess Fe (Neue et al. [1998]; Quinet et al. [2012]). Fe toxicity can occur in flooded soils with a pH below 5.8 under aerobic conditions, and at a pH below 6.5 under anaerobic conditions (Fageria et al. [2008]). Fe toxicity is a serious agricultural problem, particularly when plants are grown in acidic soils (Guerinot and Ying [1994]; Quinet et al. [2012]). Developing plants that can grow in problematic soils requires an understanding of the molecular mechanisms of Fe uptake, transport, and storage in plants under conditions of varying Fe availability (Bashir et al. [2013a]). The molecular mechanisms of Fe uptake from soil have been extensively studied (Bashir et al. [2010]; Bashir et al. [2011b]; Bashir et al. [2013a]; Guerinot [2010]; Guerinot and Ying [1994]; Ishimaru et al. [2011b]; Ishimaru et al. [2011a]; Kobayashi and Nishizawa [2012]; Marschner [1995]). Plants are divided into two broad categories (strategies I and II) based on how they uptake Fe from the soil (Marschner [1995]; Marschner and Römheld [1994]). Rice is a strategy II plant, and secretes 2'-deoxymugineic acid (DMA) to acquire soil Fe. The genes involved in DMA synthesis have been cloned and characterized (Bashir et al. [2006]; Bashir and Nishizawa [2006]; Inoue et al. [2003]; Inoue et al. [2008]; Nozoye et al. [2004]; Suzuki et al. [2006]; Suzuki et al. [2008]; Suzuki et al. [2012]; Takahashi et al. [1999]). Specifically, L-methionine is converted to nicotianamine (NA) by NA synthase 1-3 (OsNAS1-3), and is then converted to 3'-keto acid by NA aminotransferase 1 (OsNAAT1) and finally DMA synthase (OsDMAS1) converts this 3'-keto acid to DMA (Bashir et al. [2006]; Bashir and Nishizawa [2006]; Bashir et al. [2010]; Inoue et al. [2003]; Inoue et al. [2008]; Ma et al. [1995]; Ma et al. [1999]; Mori and Nishizawa [1987]; Nozoye et al. [2014a]; Nozoye et al. [2014b]). DMA is then secreted to the rhizosphere via the mugineic acid transporter (OsTOM1) Nozoye et al. [2011]. In the rhizosphere, DMA binds to Fe(III), and the resulting DMA-Fe (III) complex is taken up by OsYSL15 (Inoue et al. [2009]; Lee et al. [2009]). Rice also uses OsIRT1 to uptake ferrous Fe under paddy field conditions, and secretes phenolics to solubilize apoplasmic Fe (Bashir et al. [2011b]; Ishimaru et al. [2011a]; Ishimaru et al. [2011b]). Once Fe is absorbed through roots, it is translocated to the aerial parts of the plant. The genes involved in root-to-shoot translocation and the transport of Fe to subcellular organelles have also been characterized (Aoyama et al. [2009]; Bashir et al. [2011a]; Bashir et al. [2011c]; Bashir et al. [2013b]; Ishimaru et al. [2009]; Ishimaru et al. [2010]; Ishimaru et al. [2011a]; Ishimaru et al. [2011b]; Ishimaru et al. [2012]; Kakei et al. [2012]; Koike et al. [2004]; Lee et al. [2012]; Yokosho et al. [2009]; Zhang et al. [2012b]).
Plants can accumulate varying levels of Fe and the response of rice to Fe toxicity was recently summarized after comprehensive transcriptomic and physiological analyses (Quinet et al. [2012]). In the current study, our main objective was to understand the transcriptomic response of rice to different conditions of Fe availability. We therefore performed a microarray analysis of plants accumulating high, yet not physiologically toxic, levels of Fe. Although the rice genome has been sequenced (Kawahara et al. [2013]), the identification of small open reading frames (sORFs) typically consisting of fewer than 100 codons was not addressed in plants until recently (Hanada et al. [2013]; Hanada et al. [2010]; Hanada et al. [2007]). These sORFs play a critical role in morphogenesis in Arabidopsis thaliana (Hanada et al. [2013]; Hanada et al. [2010]; Hanada et al. [2007]). Although the potential role of sORF in rice is recently discussed (Okamoto et al. [2014]) their regulation in response to different abiotic stresses has not been assessed in rice. In this study, we used a novel 110 K rice microarray that, along with previously identified genes, includes ~48,620 sORFs to identify transcriptional changes in response to Fe deficiency and excess in rice roots and shoots. This will allow a better understanding of the response of plants to these stresses, and suggests the involvement of sORFs in Fe metabolism under different conditions of Fe availability.
Results
Morphological responses to Fe deficiency and excess Fe
When plants were grown under Fe-deficient conditions, the root and shoot length as well as the chlorophyll content decreased significantly compared with plants grown in the presence of 100 μM Fe-EDTA (Figure 1a-c). In contrast, when plants were grown under conditions of excess Fe, the root length was reduced, but no significant differences were observed in plant height or chlorophyll content compared with wild-type plants (Figure 1a-c). In the shoots of Fe-deficient plants, the concentrations of Fe were 50% lower than in plants grown with 100 μM Fe, whereas plants grown under conditions of excess Fe accumulated two-fold more Fe in their leaves (Figure 1d).

Morphological characteristics and metal profiling of plants grown under conditions of Fe deficiency and excess. a) Root length (cm). b) Shoot length (cm). c) Chlorophyll content. d) Shoot Fe. e) Shoot Zn. f) Shoot Mn. g) Shoot Cu. h) Root Fe. i) Root Zn. j) Root Mn. k) Root Cu (μg/g dry weight). Vertical bars followed by different letters are significantly different from each other, according to the Tukey-Kramer test (p < 0.05; n = 4).
In plants grown under Fe-deficient conditions, the concentrations of zinc and copper (Cu) increased in the shoots, whereas the manganese (Mn) concentrations were comparable to plants grown in the presence of Fe (Figure 1e-g). In contrast, plants grown in the presence of excess Fe accumulated more Mn in their shoots compared to plants supplied with 100 μM Fe (Figure 1f). In the roots of plants grown under Fe-deficient conditions, the concentrations of Fe and Mn decreased significantly, whereas the concentration of Cu increased compared to plants grown with 100 μM Fe (Figure 1h-k).
Genes upregulated by Fe deficiency and downregulated by excess Fe in roots
Several studies reported the upregulation of genes in response to Fe deficiency in rice (Bashir et al. [2013c]; Bashir and Nishizawa [2013]; Ishimaru et al. [2009]; Nozoye et al. [2011]), however little attention is paid to identify genes regulated by excess Fe. Before carrying out our microarray analysis, we used RT-PCR to assess the expression of OsDMAS1 and OsVIT2 to confirm the effects of excess Fe and deficiency treatments. OsDMAS1 is upregulated by Fe deficiency, while expression of vacuolar Fe transporter OsVIT2 is reported to be upregulated by excess Fe (Bashir et al. [2011c]; Zhang et al. [2012b]; Bashir et al. [2013b]). In our study, OsDMAS1 was upregulated by Fe deficiency in both roots and shoots, and was downregulated by excess Fe. As expected, the expression of OsVIT2 was upregulated by excess Fe in both shoots and roots (Additional file 1: Figure S1). In general, the transcriptomic changes in roots were clearer in response to Fe-deficiency as compared to excess Fe. On the other hand in shoot tissue, the expression of secondary metabolism related genes was more significantly altered by excess Fe compared to Fe deficiency. Our microarray results revealed the upregulation of 1509 genes in response to Fe deficiency in roots (Figure 2a, e and Additional file 2: Table S1), of which 90 (6%) were sORFs. In addition, 116 genes were downregulated by excess Fe (Figure 2a, Additional file 2: Table S2). Of the 1509 genes upregulated by Fe deficiency, 43 were downregulated by excess Fe (Figure 2a, Table 1). The genes presented in Table 1 are therefore highly responsive to Fe availability in roots. Consistent with previous microarray reports, the genes upregulated by Fe deficiency included those involved in the synthesis of DMA such as OsNAAT1, and OsDMAS1, those involved in Fe-NA or DMA complex transport (OsYSL2 and OsYSL15), and the DMA efflux transporter (OsTOM1) (Table 1 (Ishimaru et al. [2009])). In addition, OsIRO2 and two other basic helix loop helix (bHLH)-type transcription factors were upregulated by Fe deficiency (Tables 1 and S1). Two ABC transporters that are upregulated by excess Cu (Lin et al. [2013]), were upregulated by Fe deficiency as were two amino acid transporters, of which Os02g0788800 is also upregulated by excess Cu (Lin et al. [2013]). MapMan analysis revealed that many metabolic genes were upregulated or downregulated in response to Fe deficiency, and many of these were upregulated in response to excess Fe in roots (Additional file 1: Figure S2). Changes in the expression of OsDMAS1 and sORF chr9_-_4113943-4114041 were also confirmed through real time PCR and the data was in line with microarray analysis (Figure 3).

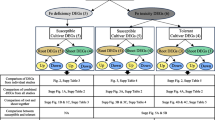
Venn diagram representing the transcriptional changes in response to Fe deficiency and excess. a) Number of genes upregulated by Fe deficiency and downregulated by excess Fe in roots. b) Number of genes downregulated by Fe deficiency and upregulated by excess Fe in roots. c) Number of genes upregulated by Fe deficiency and downregulated by excess Fe in shoots. d) Number of genes downregulated by Fe deficiency and upregulated by excess Fe in shoots. e) Genes upregulated by Fe deficiency both in roots and shoots. f) Genes upregulated by excess Fe both in roots and shoots. g) Genes downregulated by Fe deficiency both in roots and shoots. h) Genes downregulated by excess Fe both in roots and shoots.

Expressionanalysis of selected genes in response to varying Fe availability. Expression of a, f) OsDMAS1. b, g) chr9_-_4113943-4114041. c) chr6_ + _23392831-23392944. d) chr6_ + _29900249-29900395. e) Os01g0127000. h) OsFRO2. i) Os07g0142100. j) chr7_-_23991237-23991350. a-e) Root. f-g) Shoot. The graph shows mean ± s.d. relative to the expression of α-tubulin. Vertical bars followed by different letters are significantly different from each other, according to the Tukey-Kramer test (p < 0.05; n = 3).
Genes upregulated by excess Fe and downregulated by Fe deficiency in roots
In roots, 36 genes were upregulated by excess Fe, of which three were sORFs (Table 2), while 2655 genes were downregulated by Fe deficiency, of which 1225 (46%) were sORFs (Additional file 2: Table S3). However, only nine genes were upregulated by excess Fe and downregulated by Fe deficiency. The genes upregulated by excess Fe included four peroxidases, multi-Cu oxidase (Os01g0127000), and alcohol dehydrogenase, suggesting that excess Fe causes oxidative stress. Three cytochrome P450 family proteins, which may play a role in electron transport, were also upregulated, as was the expression of one subtilase family gene. Five uncharacterized proteins and three sORFs genes were also upregulated by excess Fe (Table 2). Changes in the expression of multicopper oxidase Os01g0127000 and two sORFs chr6_ + _29900249-29900395 and chr6_ + _23392831-23392944 were also confirmed through real time PCR (Figure 3).
Most of the genes downregulated by Fe deficiency (1225; 46%) were categorized as sORFs. Other downregulated genes include 15 Zn finger proteins, two WRKY transcription factors, 11 peptidase, eight heme peroxidases, and genes involved in the ethylene response and other metabolic pathways such as methionine metabolism (Additional file 2: Table S3).
Genes upregulated by Fe deficiency and downregulated by excess Fe in shoots
In shoots, 258 genes were upregulated by Fe deficiency, of which 35 genes were also downregulated by excess Fe (Figure 2c). Consistent with previous reports, genes involved in DMA synthesis and transport (such as OsNAS1-2 and OsDMAS1, OsTOM1), Fe-NA or DMA complex transport (OsYSL2) were upregulated by Fe deficiency (Additional file 2: Table S4). Other genes regulated by Fe deficiency included OsIRT2, OsIDS1, OsIRO2 and OsFRO2. OsIDS1 is a metallothionein (MT) gene highly responsive to Fe deficiency (Itai et al. [2013]). Of the genes upregulated by Fe deficiency in shoots, 50 (19%) were sORFs, but only two of these were also downregulated by excess Fe, whereas 1655 genes were downregulated by excess Fe (Additional file 2: Table S5). The genes downregulated by excess Fe include NADPH-dependent oxidoreductases and peroxidases. Two bHLH transcription factors, a cyclin-like F-box domain-containing protein, a protein kinase, and two sORFs (chr6_ + _7967232-7967441 and chr9_-_4113943-4114041) were upregulated by Fe deficiency and downregulated by excess Fe (Table 3). A total of 74 genes were upregulated by Fe deficiency in both roots and shoots (Figure 2e), but only 17 genes were downregulated by excess Fe in both roots and shoots (Figure 2h).
Genes upregulated by excess Fe and downregulated by Fe deficiency in shoots
In shoots, 2480 genes were upregulated by excess Fe, of which 1076 (43%) were sORFs (Additional file 2: Table S6). The genes upregulated by excess Fe included a 2-oxoglutarate (OG)-Fe(II) oxygenase domain-containing protein, and an ATPase, and 17 transporter genes belonging to different families. These transporters include two putative plasma membrane ABC transporter domain-containing proteins [a putative subfamily B ABC-type transporter and an MRP-like ABC transporter], two putative amino acid transporters, and two transporters belonging to the multidrug and toxic compound extrusion (MATE) transporter family, which transports small organic compounds (Omote et al. [2006]). The MATE transporter Os03g0571700 is highly homologous to rice phenolics efflux zero 1, which transports phenolics to solubilize apoplasmic Fe (Ishimaru et al. [2011b]; Ishimaru et al. [2011a]). Additional transporters that putatively transport Cu, magnesium, phosphate or other anions, and oligopeptides were also upregulated (Additional file 2: Table S7). Other genes upregulated by excess Fe include those that participate in cellular metabolic processes, gene expression and translation, and the generation of precursor metabolites and energy (Table 4).
MapMan analysis revealed that many metabolic related genes were upregulated or downregulated in response to Fe deficiency, and many of these were upregulated in response to excess Fe in shoots (Additional file 1: Figure S3). A total of 43 genes were upregulated by excess Fe and downregulated by Fe deficiency (Figure 2d), of which 9 (21%) were sORFs (Table 5). In shoots, 318 genes were downregulated by Fe deficiency (Additional file 2: Table S7). Interestingly, only one gene (belonging to the cytochrome family) was upregulated in both roots and shoots in response to excess Fe (Figure 2f), whereas 53 genes were downregulated in response to Fe deficiency in both roots and shoots (Figure 2g). Genes that were downregulated by Fe deficiency included Fe sulfur [4Fe-4S] cluster assembly factor, mitochondrial substrate carrier family protein, heavy metal transporters, ferredoxin domain-containing proteins, a bHLH domain-containing protein, heme peroxidases, isocitrate dehydrogenase, OsNAS3, a ferritin gene, OsZIP7 and OsZIP10, six peroxidases, and 43 sORFs (Additional file 2: Table S7). A summary of the transcriptomic changes in chloroplasts in response to Fe deficiency and excess is shown in Additional file 1: Figure S4. The expression of photosystem II genes was either unchanged or downregulated during Fe deficiency, whereas photosystem I genes were both upregulated and downregulated. In contrast, almost all of the genes involved in ATP synthesis, PS1, and PSII were upregulated in response to excess Fe.
Discussion
Both Fe deficiency and toxicity cause significant losses in crop yield and quality. In plants, Fe is essential for various cellular processes, as it serves as a cofactor for a range of plant enzymes, including cytochromes, catalase, peroxidase isozymes, ferredoxin, and isozymes of superoxide dismutase (Marschner, [1995]). It was therefore expected that the expression of these genes would be downregulated by Fe deficiency. Genes upregulated during Fe deficiency-associated stress in graminaceous crops have been described extensively (Bashir et al. [2010]; Ishimaru et al. [2009]; Ishimaru et al. [2011b]; Kobayashi et al. [2005]; Nagasaka et al. [2009]; Negishi et al. [2002]; Nozoye et al. [2007]), and our microarray data are consistent with those of previous reports. We have therefore not discussed these genes in detail. Similarly the morphological changes in response to Fe availability as well as the effects of availability of Fe on accumulation of other metals have been widely reported in rice (Ishimaru et al. [2009]; Bashir et al. [2011c]).
Microarray analyses were performed after one week of Fe deficiency and excess treatment and at this point, plants correspond to a new transcriptomic/metabolic steady state. Many genes upregulated by Fe deficiency are also upregulated by other stresses such as cadmium (Egan et al. [2007]) toxicity (Nakanishi et al. [2006]; Takahashi et al. [2011]). Consistent with this, we observed the upregulation of several genes (Additional file 2: Table S1) that are also regulated by other abiotic stresses, including Cd toxicity (Takahashi et al. [2011]) (OsNRAMP1), Cu toxicity (Lin et al. [2013]) (Os04g0588700, Os02g0208300, and Os04g0512300), and heat stress (amino acid transporter and heat shock proteins). Many genes reported to be regulated by disease pathogenesis are also upregulated by Fe deficiency (Additional file 2: Table S1). The expression of symbiotic hemoglobin 2 (rHb2; Os03g0226200), which plays an important role in plant adaptation to unfavorable environment (Zhang et al. [2012a]), was also upregulated by Fe deficiency. These results suggest that Fe-deficient plants undergo oxidative stress, since oxidative stress is common during times of biotic or abiotic stress.
The expression of 1-aminocyclopropane-1-carboxylate oxidase 1 (Os09g0451400) was significantly upregulated in Fe-deficient shoots. This gene encodes an intermediate during the formation of ethylene, which plays a role in abiotic stress signaling (Lingam et al. [2011]). Auxins interact with ethylene metabolism, and the expression of four auxin-responsive genes (two auxin-responsive SAUR protein family proteins, one auxin-induced gene, and indoleacetic acid-induced protein 18) was also upregulated by Fe deficiency (Additional file 2: Table S1). These results suggest that ethylene signaling and the reprograming of plant metabolism may be an important strategy of rice in response to Fe deficiency.
Transcriptomic changes in response to Excess Fe
The expression of several genes was upregulated by excess Fe in roots and shoots. In roots, members of the cytochrome family, oxidases, alcohol dehydrogenase, a protein kinase, a Zn finger domain-containing protein, and a heavy metal transporter were all significantly upregulated. Many of these genes are also regulated by other stresses. For example, the cytochrome_P450_family gene Os01g0803800 is upregulated by diclofop methyl (Qian et al. [2012]), Os11g0138300 is regulated by ionizing radiation (Kim et al. [2012]), a heavy metal transporter is regulated by excess silicon and rice blast (Brunings et al. [2009]). One laccase gene that plays a role in lignin formation and two peroxidases (Os03g0369000 and Os07g0531400) are also upregulated by Fe toxicity (Quinet et al. [2012]). These results suggest that under conditions of excess Fe, the generation of reactive oxygen species (ROS) increases, as ROS production is common during times of abiotic or biotic stress. However, it is unknown if the generation of ROS is a direct effect of increased Fe concentrations or is the result of an increased metabolic rate, as suggested by our MapMan analysis.
In shoots, the expression of OsLhcb1.3 (Os01g0720500) was significantly upregulated after treatment with excess Fe. The photosynthetic apparatus of barley adapts to Fe deficiency by remodeling its PSII antenna system, in which the expression of two Hvlhcb1 genes (HvLhcb1.11 and HvLhcb1.12) is upregulated, and four genes (HvLhcb1.6-9) are downregulated by Fe deficiency (Saito et al. [2010]). Although it was not assessed experimentally, it is possible that these downregulated genes would be upregulated in response to excess Fe. Additional genes related to PSII were also upregulated, suggesting that the rate of photosynthesis is increased due to the increased availability of Fe.
The role of ethylene signaling in abiotic stress, including Fe deficiency, has been discussed extensively (Lingam et al. [2011]). Ethylene may also play a significant role in signaling under conditions of excess Fe, since two rice ethylene response factor-3 (OsERF3) genes which regulate ethylene synthesis (Zhang et al. [2013]) were upregulated in shoots in the presence of excess Fe. The expression of OsRab8A5, which may be involved in signal transduction, was also upregulated. Upregulation of LONELY GUY, a cytokinin-activating enzyme that regulates activation pathways in rice shoot meristems (Kurakawa et al. [2007]), transcription factors such as OsMADS18 and OsMADS56 involved in regulating long-day-dependent flowering (Ryu et al. [2009]) suggest that plant growth and cell division are significantly increased in shoots under conditions of excess Fe. In addition, our MapMan analysis suggested that genes that regulate metabolism are also upregulated in shoots in the response to excess amounts of Fe.
The activity and expression of glutathione reductase (GR) is already reported to change in response to Fe deficiency (Bashir et al. [2007]), while in present experiment upregulation of OsGR1 was observed in response to excess Fe. Similarly, the expression of NADPH HC toxin reductase, which is reported to be regulated by Cu toxicity (Lin et al. [2013]), also increased by excess Fe. Genes involved in brassinosteroids synthesis were also upregulated. In rice, brassinosteroids regulate multiple developmental processes and modulate several important traits such as height, leaf angle, fertility, and seed filling (Wang et al. [2010]). These results further support the hypothesis that plant metabolism and growth are stimulated under conditions of excess Fe.
The expression of OsWSL2, which is associated with the elongation of very long-chain fatty acids, and Os9BGlu32 was significantly upregulated by excess Fe. Although the function of Os9BGlu32 is unknown, it is a close homolog of Os9BGlu31, which equilibrates the levels of phenolic acids and carboxylated phytohormones and their gluco-conjugates (Luang et al. [2013]). The role of phenolic transport in Fe deficiency has been reported (Bashir et al. [2011b]; Ishimaru et al. [2011b]; Ishimaru et al. [2011a]; Jin et al. [2007]), and it is possible that these phenolics act as antioxidants in the presence of excess Fe. Although the microarray analysis indicates that metabolic rate may increase in response to excess Fe, plants still retain many responses common to different biotic and abiotic stresses. Despite the increased metabolic rate, excess Fe cannot therefore be considered optimal for rice plants, at least under the current growth conditions.
The expression of one 2OG-Fe(II) oxygenase (Os10g0559500) was upregulated by Fe deficiency, whereas one gene (Os08g0392100) was upregulated by excess Fe. In plants, 2OG-Fe(II) oxygenase are involved in the synthesis of phytosiderophores (Nakanishi et al. [2000]) and numerous other biosynthesis pathways. It was recently suggested that plant 2OG-Fe(II) oxygenases play a role in Fe sensing and metabolism reprograming under Fe-deficient conditions (Vigani et al. [2013]). The upregulation of different 2'-OG dioxygenases by opposing conditions of Fe deficiency and excess suggests that these genes are involved in Fe sensing during altered Fe availability.
Changes in expression of sORFs in response to Fe deficiency and Excess
In roots, three sORF genes were upregulated by Fe deficiency and downregulated by excess Fe. Our co-expression analysis revealed that these three sORFs are not only positively co-regulated with each other, but also with several other genes presented in Table 1. Specifically, OsIRO2, MIR, OPT, eight conserved hypothetical protein genes, and two sORF genes showed a strong positive correlation (r < 0.8) when co-expression analysis was carried out for the third sORF (chr9_-_4113943-4114041). Seven sORF genes (chr1_ + _43772594-43772752, chr12_-_7456469-7456567, chr4_-_24346205-24346330, chr4_-_5708578-5708748, chr5_ + _27469071-27469241, chr6_ + _7967232-7967441, and chr9_-_4113943-4114041) were upregulated by Fe deficiency in both roots and shoots (Additional file 2: Table S8). The upregulation of several sORFs was also confirmed through real time PCR analysis (Figure 3). Among these, very high expression of chr9_-_4113943-4114041 was observed particularly in shoot tissue (Figure 3b, g). Among these sORFs, the expression of chr1_ + _43772594-43772752 is not regulated by any other known stresses, according to HanaDB-OS (http://evolver.psc.riken.jp/seiken/OS/index.html), whereas the expression of chr9_-_4113943-4114041 is significantly downregulated in roots in response to other abiotic stresses such as drought, heat, and salt. It is therefore possible that these sORFs play a significant role (e.g., signalling) during Fe deficiency. Further characterization of these sORFs will help clarify their role in abiotic stress responses.
Conclusion
Transcriptomic and physiological changes that occur in response to short- and long-term Fe toxicity have been reported (Quinet et al. [2012]). However, our aim was to study the response to excess Fe, and to understand the specific responses of rice to varying Fe concentrations in roots and shoots. Our microarray analysis revealed that cellular metabolism was significantly reprogrammed in response to Fe deficiency and upregulated by excess Fe in shoots even though no morphological changes were observed in shoots under conditions of excess Fe. In addition to the upregulation of genes involved in various metabolic processes, our data suggest increased production of flavonoids and phenols, which may act as antioxidants. The expression of various transporters was also significantly upregulated, which suggests that these transporters coordinate the metabolic changes. Although the responses to Fe deficiency and excess share components with other stress responses, it does not significantly overlap with one particular stress. Moreover, our data reveal that the expression of several sORFs changes with varying Fe availability and that sORFs are co-regulated with other genes involved in Fe deficiency response, suggesting that they are involved in the response to Fe deficiency and/or excess in rice plants. However, the precise function of these sORFs is unclear. Because the products of these sORFs do not contain any characterized domains, it will be challenging to assess their function in response to different abiotic stresses.
It should be noted that the changes in the transcriptome are not specific to Fe, because the concentrations of Cu, Zn, and Mn changed in shoots with perturbations in the Fe level: Cu and Zn were increased during Fe deficiency, while Mn and Cu were increased with excess Fe (Figure 1). As a result, the observed changes in the transcriptome also represent changes in the availability of other metals. Indeed many of the genes reported to be regulated by metal deficiencies such as Zn deficiency changes in response to varying Fe availability (Ishimaru et al. [2012]; Suzuki et al. [2012]; Bashir et al. [2012]; Takahashi et al. [2012]). These analyses also reveal significant information about the regulation of sORFs in response to Fe deficiency and excess. Despite the rapid progress in genomics, uncharacterized and hypothetical genes still represent a large proportion of the rice genome. Understanding the role of these uncharacterized genes, including sORFs, is an important step in comprehensive understanding of the plants' response to different abiotic stresses (Hanada et al. [2007]; Hanada et al. [2010]).
Methods
Plant materials and growth conditions
Rice seeds (Oryza sativa L. cv. Nipponbare) were germinated for one week at room temperature on paper towels soaked with distilled water. After germination, the seedlings were transferred to a saran net floating on nutrient solution in a glasshouse for one week. Two-week-old plants were transferred to a 20 L plastic box containing nutrient solution with the following composition: 0.7 mM K2SO4, 0.1 mM KCl, 0.1 mM KH2PO4, 2.0 mM Ca(NO3)2, 0.5 mM MgSO4, 10 μM H3BO3, 0.5 μM MnSO4, 0.2 μM CuSO4, 0.5 μM ZnSO4, 0.05 μΜ Na2MoO4, and 100 μΜ Fe-EDTA as described previously (Suzuki et al. [2006]) and grown for one more week. Plants were grown at 25°C for 14 h of light at 320 μmol photons m-2-s-1; and at 20°C for 10 h in dark. The nutrient solution was adjusted daily to pH 5.5 with 1 M HCl and was renewed weekly. 30 plants were grown per box (2 plants per hole) and two boxes were prepared for each treatment. For the Fe deficiency and excess treatments, four-week-old plants were transferred to nutrient solution containing 0 (Fe deficiency), 100 (Control), or 500 (excess Fe) μM Fe-EDTA and cultivated for one week. The pH of the nutrient solution was adjusted daily to 5.5, and was renewed weekly. The plants were harvested at noon.
RT-PCR and microarray analyses
For each treatment, RNA was extracted from six plants in duplicate (two biological replicates, each including six plants). RT-PCR was performed as described previously (Bashir et al. [2011c]), using the primers OsDMAS1 RT (forward) 5`-GCCGGCATCCCGCAGCGGAAGATCA-3' and OsDMAS1 RT (reverse) 5`-CTCTCTCTCTCGCACGTGCTAGCGT-3'. The primers used to assess osvit2 by RT-PCR (qRT-PCR) were (forward) 5`-AAGGCCTGGCTCGAATTCATG-3' and (reverse) 5`-GTGTATTAGATGTTCTGGAGGTG-3'. The α-tubulin primers used were (forward) 5`-TCTTCCACCCTGAGCAGCTC-3' and (reverse) 5`-AACCTTGGAGACCAGTGCAG-3'. Primers used for real time PCR were as follows OsDMAS1, (forward) 5`-GAGGAGGAGAGGCAGAGGAT-3' and (reverse) 5`-TCAACACGATCGTCAAGAGC-3', OsFRO2 (forward) 5`-GCCAGATGTTCGAGCTCTTC-3' and (reverse) 5`-GGGCTTTTGCAGAAGTTGAG-3', Os01g0127000 (forward) 5`-GAGAACATGACGAGCAACGA-3' and (reverse) 5`-AGCATGCAGCTCTTGAAGGT-3', Os07g0142100 (forward) 5`-CGTCTTCCTCGATAGCCAAA-3' and (reverse) 5`-AGCTGGAGCCACATCGAC-3', chr6_ + _23392831-23392944 (forward) 5`-TCGTGTGTAATAATATGGGCTGTT-3' and (reverse) 5`-GGATACAATGGGAAATGAGCA-3', chr6_ + _29900249-29900395 (forward) 5`-CACACGTGCGAGATCTACCT-3' and (reverse) 5`-AAAGGAAAGATTGCCATCCA-3', chr7_-_23991237-23991350 (forward) 5`-ATGTTCTACCCCATGCCACT-3' and (reverse) 5`-ATGTCGCTGGACACCCTAAC-3', chr9_-_4113943-4114041 were (forward) 5`-GGCCTGTGCTAGTTTTGGTG-3' and (reverse) 5`-ATGGGCGCAAATTACATCAT-3' respectively. All experiments were performed in a minimum of triplicates.
The microarray slides were custom-designed and contained 101,720, 60 mer probes. Of these, 48,620 were for sORFs, 50,962 probes represented RAP-DB, and the rest belonged to TIGR. For our microarray analysis, RNA was labelled using an Agilent Low RNA Input Linear Amplification Kit (Agilent Technologies Inc., Santa Clara, CA), following the manufacturer's instructions. The microarray analyses were performed as described previously (Hanada et al. [2013]) with the exception that two biological replicates were used. Data analysis was performed using Feature Extraction and Image Analysis software (Agilent Technologies Inc.) and Microarray Suite (Affymetrix, Santa Clara, CA), and normalized and processed as described (Hanada et al. [2013]). Those genes with a low signal intensity (<300) were filtered to focus on genes that were highly expressed under conditions of Fe deficiency and excess. For our MapMan analysis, the average log2 value of both biological replicates was calculated for individual annotations in response to Fe deficiency and excess in roots and shoots. This log2 value was then used to compare the transcriptomic changes in metabolism-related genes using MapMan 3.5.1R2 (Thimm et al. [2004]). Our gene ontology analyses were carried out at http://www.geneontology.org/. Coexpression analyses were done at http://evolver.psc.riken.jp/seiken/OS/co-express.html. This database contains microarray data of 40 different experimental conditions obtained through microarray analysis using the same custom microarray chip as described in this manuscript.
Determination of metal concentrations
Roots were washed with de-ionized water before harvesting. Leaf and root samples were dried for three days at 70°C, and then digested with 3 ml of 13 M HNO3 at 220 C for 40 min using a MARS XPRESS microwave reaction system (CEM, Matthews, NC). All samples were processed with four biological replicates. After digestion, the samples were collected, diluted to 5 ml, and analyzed by ICP-AES (SPS1200VR; Seiko, Tokyo, Japan), as described previously (Ishimaru et al. [2011b]; Ishimaru et al. [2007]).
Recording of the morphological characteristics of the plants
Root and shoot lengths were measured using a scale. The degree of chlorosis in the youngest fully expanded leaf was determined using a SPAD-502 chlorophyll meter (Minolta Co., Tokyo, Japan), as described previously (Ishimaru et al. [2012]).
Authors' contributions
KB, KH and NN designed the study, KB, MS and KH performed the research, and KB, KH, MS, HN and NN discussed the data and wrote the manuscript. All authors read and approved the final manuscript.
Additional files
Abbreviations
- Fe:
-
Iron
- sORF:
-
Small open reading frames
References
Aoyama T, Kobayashi T, Takahashi M, Nagasaka S, Usuda K, Kakei Y, Ishimaru Y, Nakanishi H, Mori S, Nishizawa NK: OsYSL18 is a rice iron(III)-deoxymugineic acid transporter specifically expressed in reproductive organs and phloem of lamina joints. Plant Mol Biol 2009, 70(6):681–692. 10.1007/s11103-009-9500-3
Bashir K, Inoue H, Nagasaka S, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Cloning and characterization of deoxymugineic acid synthase genes from graminaceous plants. J Biol Chem 2006, 281(43):32395–32402. 10.1074/jbc.M604133200
Bashir K, Ishimaru Y, Nishizawa NK: Iron uptake and loading into rice grains. Rice 2010, 3(2):122–130. 10.1007/s12284-010-9042-y
Bashir K, Ishimaru Y, Nishizawa NK: Identification and characterization of the major mitochondrial Fe transporter in rice. Plant Signal & Behav 2011, 6(10):1591–1593. 10.4161/psb.6.10.17132
Bashir K, Ishimaru Y, Nishizawa NK: Molecular mechanisms of zinc uptake and translocation in rice. Plant Soil 2012, 361: 189–201. 10.1007/s11104-012-1240-5
Bashir K, Ishimaru Y, Shimo H, Kakei Y, Senoura T, Takahashi R, Sato Y, Sato Y, Uozumi N, Nakanishi H, Nishizawa NK: Rice phenolics efflux transporter 2 (PEZ2) plays an important role in solubilizing apoplasmic iron. Soil Sci Plant Nutr 2011, 57(6):803–812. 10.1080/00380768.2011.637305
Bashir K, Ishimaru Y, Shimo H, Nagasaka S, Fujimoto M, Takanashi H, Tsutsumi N, An G, Nakanishi H, Nishizawa NK (2011c) The rice mitochondrial iron transporter is essential for plant growth. Nature Commun 2:322. doi:10.1038/ncomms1326
Bashir K, Nagasaka S, Itai RN, Kobayashi T, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Expression and enzyme activity of glutathione reductase is upregulated by Fe-deficiency in graminaceous plants. Plant Mol Biol 2007, 65(3):277–284. 10.1007/s11103-007-9216-1
Bashir K, Nishizawa N: Iron proteins, plant iron transporters. In Encyclopedia of metalloproteins. Edited by: Kretsinger R, Uversky V, Permyakov E. Springer, New York; 2013:1015–1023. 10.1007/978-1-4614-1533-6_356
Bashir K, Nishizawa NK: Deoxymugineic acid synthase: a gene important for Fe-acquisition and homeostasis. Plant Signal & Behav 2006, 1(6):290–292. 10.4161/psb.1.6.3590
Bashir K, Nozoye T, Ishimaru Y, Nakanishi H, Nishizawa NK: Exploiting new tools for iron bio-fortification of rice. Biotech Advances 2013, 31(8):1624–1633. 10.1016/j.biotechadv.2013.08.012
Bashir K, Takahashi R, Akhtar S, Ishimaru Y, Nakanishi H, Nishizawa NK: The knockdown of OsVIT2 and MIT affects iron localization in rice seed. Rice 2013, 6(1):1–7. 10.1186/1939-8433-6-31
Bashir K, Takahashi R, Nakanishi H, Nishizawa NK (2013c) The road to micronutrient biofortification of rice: progress and prospects. Front Plant Sci 4(15): doi:10.3389/fpls.2013.00015
Brunings AM, Datnoff LE, Ma JF, Mitani N, Nagamura Y, Rathinasabapathi B, Kirst M: Differential gene expression of rice in response to silicon and rice blast fungus Magnaporthe oryzae . Ann Appl Biol 2009, 155(2):161–170. 10.1111/j.1744-7348.2009.00347.x
Egan S, Bolger P, Carrington C: Update of US FDA's total diet study food list and diets. J Exp Sci Environ Epid 2007, 17: 573–582. 10.1038/sj.jes.7500554
Fageria NK, Santos AB, Barbosa Filho MP, Guimarães CM: Iron toxicity in lowland rice. J Plant Nutr 2008, 31(9):1676–1697. 10.1080/01904160802244902
Guerinot ML: Iron. In Cell Biology of Metals and Nutrients, vol 17. Plant Cell Monographs. Edited by: Hell R, Mendel R-R. Springer Berlin, Heidelberg; 2010:75–94. 10.1007/978-3-642-10613-2_4
Guerinot ML, Ying Y: Iron: nutritious, noxious, and not readily available. Plant Physiol 1994, 104: 815–820.
Hanada K, Akiyama K, Sakurai T, Toyoda T, Shinozaki K, Shiu SH: sORF finder: a program package to identify small open reading frames with high coding potential. Bioinform 2010, 26(3):399–400. 10.1093/bioinformatics/btp688
Hanada K, Higuchi-Takeuchi M, Okamoto M, Yoshizumi T, Shimizu M, Nakaminami K, Nishi R, Ohashi C, Iida K, Tanaka M, Horii Y, Kawashima M, Matsui K, Toyoda T, Shinozaki K, Seki M, Matsui M: Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc Natl Acad Sci 2013, 110(6):2395–2400. 10.1073/pnas.1213958110
Hanada K, Zhang X, Borevitz JO, Li WH, Shiu SH: A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res 2007, 17(5):632–640. 10.1101/gr.5836207
Inoue H, Higuchi K, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Three rice nicotianamine synthase genes, OsNAS1 , OsNAS2 , and OsNAS3 are expressed in cells involved in long-distance transport of iron and differentially regulated by iron. Plant J 2003, 36(3):366–381. 10.1046/j.1365-313X.2003.01878.x
Inoue H, Kobayashi T, Nozoye T, Takahashi M, Kakei Y, Suzuki K, Nakazono M, Nakanishi H, Mori S, Nishizawa NK: Rice OsYSL15 is an iron-regulated Iron(III)-deoxymugineic acid transporter expressed in the roots and is essential for iron uptake in early growth of the seedlings. J Biol Chem 2009, 284(6):3470–3479. 10.1074/jbc.M806042200
Inoue H, Takahashi M, Kobayashi T, Suzuki M, Nakanishi H, Mori S, Nishizawa NK: Identification and localisation of the rice nicotianamine aminotransferase gene OsNAAT1 expression suggests the site of phytosiderophore synthesis in rice. Plant Mol Biol 2008, 66(1):193–203. 10.1007/s11103-007-9262-8
Ishimaru Y, Bashir K, Fujimoto M, An G, Itai RN, Tsutsumi N, Nakanishi H, Nishizawa NK: Rice-specific mitochondrial iron-regulated gene (MIR) plays an important role in iron homeostasis. Mol Plant 2009, 2(5):1059–1066. 10.1093/mp/ssp051
Ishimaru Y, Bashir K, Nakanishi H, Nishizawa NK: The role of rice phenolics efflux transporter in solubilizing apoplasmic iron. Plant Signal & Behav 2011, 6(10):1624–1626. 10.4161/psb.6.10.17694
Ishimaru Y, Kakei Y, Shimo H, Bashir K, Sato Y, Sato Y, Uozumi N, Nakanishi H, Nishizawa NK: A rice phenolic efflux transporter is essential for solubilizing precipitated apoplasmic iron in the plant stele. J Biol Chem 2011, 286(28):24649–24655. 10.1074/jbc.M111.221168
Ishimaru Y, Kim S, Tsukamoto T, Oki H, Kobayashi T, Watanabe S, Matsuhashi S, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Mutational reconstructed ferric chelate reductase confers enhanced tolerance in rice to iron deficiency in calcareous soil. Proc Natl Acad Sci 2007, 104(18):7373–7378. 10.1073/pnas.0610555104
Ishimaru Y, Masuda H, Bashir K, Inoue H, Tsukamoto T, Takahashi M, Nakanishi H, Aoki N, Hirose T, Ohsugi R, Nishizawa NK: Rice metal-nicotianamine transporter, OsYSL2, is required for the long-distance transport of iron and manganese. Plant J 2010, 62(3):379–390. 10.1111/j.1365-313X.2010.04158.x
Ishimaru Y, Takahashi R, Bashir K, Shimo H, Senoura T, Sugimoto K, Ono K, Yano M, Ishikawa S, Arao T, Nakanishi H, Nishizawa NK (2012) Characterizing the role of rice NRAMP5 in manganese, iron and cadmium transport. Sci Rep 2:286. doi:10.1038/srep00286.
Itai RN, Ogo Y, Kobayashi T, Nakanishi H, Nishizawa NK (2013) Rice genes involved in phytosiderophore biosynthesis are synchronously regulated during the early stages of iron deficiency in roots. Rice 6:16. doi:10.1186/1939–8433–6-16.
Jin CW, You GY, He YF, Tang C, Wu P, Zheng SJ: Iron deficiency-induced secretion of phenolics facilitates the reutilization of root apoplastic iron in red clover. Plant Physiol 2007, 144(1):278–285. 10.1104/pp.107.095794
Kakei Y, Ishimaru Y, Kobayashi T, Yamakawa T, Nakanishi H, Nishizawa NK: OsYSL16 plays a role in the allocation of iron. Plant Mol Biol 2012, 79(6):583–594. 10.1007/s11103-012-9930-1
Kawahara Y, de la Bastide M, Hamilton J, Kanamori H, McCombie WR, Ouyang S, Schwartz D, Tanaka T, Wu J, Zhou S, Childs K, Davidson R, Lin H, Quesada–Ocampo L, Vaillancourt B, Sakai H, Lee SS, Kim J, Numa H, Itoh T, Buell CR, Matsumoto T (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6(1):4. doi:10.1186/1939-8433-6-4
Kim SH, Song M, Lee K, Hwang SG, Jang C, Kim JB, Kim S, Ha BK, Kang SY, Kim D: Genome-wide transcriptome profiling of ROS scavenging and signal transduction pathways in rice (Oryza sativa L.) in response to different types of ionizing radiation. Mol Biol Rep 2012, 39(12):11231–11248. 10.1007/s11033-012-2034-9
Kobayashi T, Nishizawa NK: Iron uptake, translocation, and regulation in higher plants. Ann Rev Plant Biol 2012, 63(1):131–152. 10.1146/annurev-arplant-042811-105522
Kobayashi T, Suzuki M, Inoue H, Itai RN, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Expression of iron-acquisition-related genes in iron-deficient rice is co-ordinately induced by partially conserved iron-deficiency-responsive elements. J Exp Bot 2005, 56(415):1305–1316. 10.1093/jxb/eri131
Koike S, Inoue H, Mizuno D, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: OsYSL2 is a rice metal-nicotianamine transporter that is regulated by iron and expressed in the phloem. Plant J 2004, 39(3):415–424. 10.1111/j.1365-313X.2004.02146.x
Kurakawa T, Ueda N, Maekawa M, Kobayashi K, Kojima M, Nagato Y, Sakakibara H, Kyozuka J: Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445(7128):652–655. 10.1038/nature05504
Lee S, Chiecko JC, Kim SA, Walker EL, Lee Y, Guerinot ML, An G: Disruption of OsYSL15 Leads to iron inefficiency in rice plants. Plant Physiol 2009, 150(2):786–800. 10.1104/pp.109.135418
Lee S, Ryoo N, Jeon JS, Guerinot M, An G: Activation of rice Yellow Stripe1-Like 16 ( OsYSL16 ) enhances iron efficiency. Mol Cells 2012, 33(2):117–126. 10.1007/s10059-012-2165-9
Lin CY, Trinh N, Fu SF, Hsiung YC, Chia LC, Lin CW, Huang HJ: Comparison of early transcriptome responses to copper and cadmium in rice roots. Plant Mol Biol 2013, 81(4-5):507–522. 10.1007/s11103-013-0020-9
Lingam S, Mohrbacher J, Brumbarova T, Potuschak T, Fink-Straube C, Blondet E, Genschik P, Bauer P: Interaction between the bHLH transcription factor FIT and ETHYLENE INSENSITIVE3/ETHYLENE INSENSITIVE3-LIKE1 reveals molecular linkage between the regulation of iron acquisition and ethylene signaling in arabidopsis. Plant Cell 2011, 23(5):1815–1829. 10.1105/tpc.111.084715
Luang S, Cho JI, Mahong B, Opassiri R, Akiyama T, Phasai K, Komvongsa J, Sasaki N, Yl H, Matsuba Y, Ozeki Y, Jeon JS, Ketudat Cairns JR: Rice Os9BGlu31 is a transglucosidase with the capacity to equilibrate phenolpropenoid, flavonoid and phytohormone glycoconjugates. J Biol Chem 2013, 288(14):10111–10123. 10.1074/jbc.M112.423533
Ma JF, Shinada T, Matsuda C, Nomoto K: Biosynthesis of phytosiderophores, mugineic acids, associated with methionine cycling. J Biol Chem 1995, 270(28):16549–16554. 10.1074/jbc.270.28.16549
Ma JF, Taketa S, Chang Y-C, Iwashita T, Matsumoto H, Takeda K, Nomoto K: Genes controlling hydroxylations of phytosiderophores are located on different chromosomes in barley ( Hordeum vulgare L.). Planta 1999, 207(4):590–596. 10.1007/s004250050522
Marschner H: Mineral nutrition of higher plants. Academic, London; 1995.
Marschner H, Römheld V: Strategies of plants for acquisition of iron. Plant Soil 1994, 165(2):261–274. 10.1007/BF00008069
Mori S: Iron acquisition by plants. Curr Opin Plant Biol 1999, 2(3):250–253. 10.1016/S1369-5266(99)80043-0
Mori S, Nishizawa NK: Methionine as a dominant precursor of phytosiderophores in graminaceae plants. Plant Cell Physiol 1987, 28(6):1081–1092.
Nagasaka S, Takahashi M, Itai RN, Bashir K, Nakanishi H, Mori S, Nishizawa NK: Time course analysis of gene expression over 24 hours in Fe-deficient barley roots. Plant Mol Biol 2009, 69(5):621–631. 10.1007/s11103-008-9443-0
Nakanishi H, Ogawa I, Ishimaru Y, Mori S, Nishizawa NK: Iron deficiency enhances cadmium uptake and translocation mediated by the Fe2+ transporters OsIRT1 and OsIRT2 in rice. Soil Sci Plant Nutr 2006, 52(4):464–469. 10.1111/j.1747-0765.2006.00055.x
Nakanishi H, Yamaguchi H, Sasakuma T, Nishizawa NK, Mori S: Two dioxygenase genes, Ids3 and Ids2, from Hordeum vulgare are involved in the biosynthesis of mugineic acid family phytosiderophores. Plant Mol Biol 2000, 44(2):199–207. 10.1023/A:1006491521586
Negishi T, Nakanishi H, Yazaki J, Kishimoto N, Fujii F, Shimbo K, Yamamoto K, Sakata K, Sasaki T, Kikuchi S, Mori S, Nishizawa NK: cDNA microarray analysis of gene expression during Fe-deficiency stress in barley suggests that polar transport of vesicles is implicated in phytosiderophore secretion in Fe-deficient barley roots. Plant J 2002, 30(1):83–94. 10.1046/j.1365-313X.2002.01270.x
Neue HU, Quijano C, Senadhira D, Setter T: Strategies for dealing with micronutrient disorders and salinity in lowland rice systems. Field Crop Res 1998, 56(1-2):139–155. 10.1016/S0378-4290(97)00125-1
Nozoye T, Inoue H, Takahashi M, Ishimaru Y, Nakanishi H, Mori S, Nishizawa NK: The expression of iron homeostasis-related genes during rice germination. Plant Mol Biol 2007, 64(1):35–47. 10.1007/s11103-007-9132-4
Nozoye T, Itai RN, Nagasaka S, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Diurnal changes in the expression of genes that participate in phytosiderophore synthesis in rice. Soil Sci Plant Nutr 2004, 50: 1125–1131. 10.1080/00380768.2004.10408585
Nozoye T, Nagasaka S, Bashir K, Takahashi M, Kobayashi T, Nakanishi H, Nishizawa NK: Nicotianamine synthase 2 localizes to the vesicles of iron-deficient rice roots, and its mutation in the YXXφ or LL motif causes the disruption of vesicle formation or movement in rice. Plant J 2014, 77(2):246–260. 10.1111/tpj.12383
Nozoye T, Tsunoda K, Nagasaka S, Bashir K, Takahashi M, Kobayashi T, Nakanishi H, Nishizawa NK (2014b) Rice nicotianamine synthase localizes to particular vesicles for proper function. Plant Signal Behav 9(3):e28660. doi:10.4161/psb.28660
Nozoye T, Nagasaka S, Kobayashi T, Takahashi M, Sato Y, Sato Y, Uozumi N, Nakanishi H, Nishizawa NK: Phytosiderophore efflux transporters are crucial for iron acquisition in graminaceous plants. J Biol Chem 2011, 286(7):5446–5454. 10.1074/jbc.M110.180026
Okamoto M, Higuchi–Takeuchi M, Shimizu M, Shinozaki K, Hanada K (2014) Substantial expression of novel small open reading frames in Oryza sativa. Plant Signal & Behav 9(1):e27848
Omote H, Hiasa M, Matsumoto T, Otsuka M, Moriyama Y: The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Tren Pharmacol Sci 2006, 27(11):587–593. 10.1016/j.tips.2006.09.001
Qian H, Wang R, Chen J, Ding H, Yong W, Songlin R, Fu Z: Analysis of enantioselective biochemical, physiological, and transcriptional effects of the chiral herbicide diclofop methyl on rice seedlings. J Agric Food Chem 2012, 60(22):5515–5523. 10.1021/jf301688a
Quinet M, Vromman D, Clippe A, Bertin P, Lequeux H, Dufey I, Lutts S, LefÈVre I: Combined transcriptomic and physiological approaches reveal strong differences between short- and long-term response of rice (Oryza sativa) to iron toxicity. Plant Cell Environ 2012, 35(10):1837–1859. 10.1111/j.1365-3040.2012.02521.x
Ryu CH, Lee S, Cho LH, Kim SL, Lee YS, Choi SC, Jeong HJ, Yi J, Park SJ, Han CD, An G: OsMADS50 and OsMADS56 function antagonistically in regulating long day (LD)-dependent flowering in rice. Plant Cell Environ 2009, 32(10):1412–1427. 10.1111/j.1365-3040.2009.02008.x
Saito A, Iino T, Sonoike K, Miwa E, Higuchi K: Remodeling of the major light-harvesting antenna protein of PSII protects the young leaves of barley ( Hordeum vulgare L.) from photoinhibition under prolonged iron deficiency. Plant Cell Physiol 2010, 51(12):2013–2030. 10.1093/pcp/pcq160
Suzuki M, Bashir K, Inoue H, Takahashi M, Nakanishi H, Nishizawa N (2012) Accumulation of starch in Zn-deficient rice. Rice 5(1):9. doi:10.1186/1939-8433-5-9
Suzuki M, Takahashi M, Tsukamoto T, Watanabe S, Matsuhashi S, Yazaki J, Kishimoto N, Kikuchi S, Nakanishi H, Mori S, Nishizawa NK: Biosynthesis and secretion of mugineic acid family phytosiderophores in zinc-deficient barley. Plant J 2006, 48(1):85–97. 10.1111/j.1365-313X.2006.02853.x
Suzuki M, Tsukamoto T, Inoue H, Watanabe S, Matsuhashi S, Takahashi M, Nakanishi H, Mori S, Nishizawa NK: Deoxymugineic acid increases Zn translocation in Zn-deficient rice plants. Plant Mol Biol 2008, 66(6):609–617. 10.1007/s11103-008-9292-x
Takahashi M, Yamaguchi H, Nakanishi H, Shioiri T, Nishizawa NK, Mori S: Cloning two genes for nicotianamine aminotransferase, a critical enzyme in iron acquisition (Strategy II) in graminaceous plants. Plant Physiol 1999, 121(3):947–956. 10.1104/pp.121.3.947
Takahashi R, Bashir K, Ishimaru Y, Nishizawa NK, Nakanishi H: The role of heavy-metal ATPases, HMAs, in zinc and cadmium transport in rice. Plant Signal & Behav 2012, 7(12):1605–1607. 10.4161/psb.22454
Takahashi R, Ishimaru Y, Senoura T, Shimo H, Ishikawa S, Arao T, Nakanishi H, Nishizawa NK: The OsNRAMP1 iron transporter is involved in Cd accumulation in rice. J Exp Bot 2011, 62(14):4843–4850. 10.1093/jxb/err136
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M: mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 2004, 37(6):914–939. 10.1111/j.1365-313X.2004.02016.x
Vigani G, Morandini P, Murgia I (2013) Searching iron sensors in plants by exploring the link among 2’–OG–dependent dioxygenases, the iron deficiency response and metabolic adjustments occurring under iron deficiency. Front Plant Sci 4: doi:10.3389/fpls.2013.00169
Wang F, Bai MY, Deng Z, Oses-Prieto JA, Burlingame AL, Lu T, Chong K, Wang Z-Y: Proteomic study identifies proteins involved in brassinosteroid regulation of rice growth. J Integr Plant Biol 2010, 52(12):1075–1085. 10.1111/j.1744-7909.2010.00992.x
Yokosho K, Yamaji N, Ueno D, Mitani N, Ma JF: OsFRDL1 Is a citrate transporter required for efficient translocation of iron in rice. Plant Physiol 2009, 149(1):297–305. 10.1104/pp.108.128132
Zhang H, Zhang J, Quan R, Pan X, Wan L, Huang R: EAR motif mutation of rice OsERF3 alters the regulation of ethylene biosynthesis and drought tolerance. Planta 2013, 237(6):1443–1451. 10.1007/s00425-013-1852-x
Zhang Q, Yao G, Hu G, Chen C, Tang B, Zhang H-l, Li Z-c: Fine mapping of qTGW3-1, a QTL for 1000-grain weight on chromosome 3 in rice. Journal of Integrative Agriculture 2012a, 11(6):879–887. 10.1016/S2095-3119(12)60078-1
Zhang Y, Xu YH, Yi HY, Gong JM: Vacuolar membrane transporters OsVIT1 and OsVIT2 modulate iron translocation between flag leaves and seeds in rice. Plant J 2012b, 72: 400–410. 10.1111/j.1365-313X.2012.05088.x
Acknowledgements
This work was supported by a grant from the Ministry of Agriculture, Forestry, and Fisheries of Japan (Green Technology Project IP-5003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
12284_2014_18_MOESM1_ESM.pdf
Additional file 1: Figure S1.: Expression of OsVIT2 and OsDMAS1 under Fe deficiency and excess Fe. Figure S2. Transcriptional changes in metabolism related genes in roots of Fe-deficient and excess Fe rice as predicted by MapMan 3.5.1R2. Figure S3. Transcriptional changes in metabolism related genes in shoots of Fe-deficient and excess Fe rice as predicted by MapMan 3.5.1R2. Figure S4. Summary of transcriptional changes in chloroplast of Fe-deficient and excess Fe rice shoots as predicted by MapMan 3.5.1R2. (PDF 2 MB)
12284_2014_18_MOESM2_ESM.xls
Additional file 2: Table S1.: Genes upgegulated by Fe deficiency in roots. Table S2. Genes down-gegulated by Fe excess in roots. Table S3. Genes down-regulated by Fe Deficiency. Table S4. Genes upregulated by Fe deficiency in shoots. Table S5. Genes down regulated by Fe excess in shoots. Table S6. Genes upregulated by Fe excess in shoots. Table S7. Genes down-regulated by Fe deficiency in shoots. Table S8. Genes upregulated by Fe deficiency in roots and shoots. (XLS 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Bashir, K., Hanada, K., Shimizu, M. et al. Transcriptomic analysis of rice in response to iron deficiency and excess. Rice 7, 18 (2014). https://doi.org/10.1186/s12284-014-0018-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-014-0018-1