
Abstract
Background
Lactic acid bacteria (LAB) play an important role in agricultural as well as industrial biotechnology. Development of improved LAB strains using e.g. library approaches is often limited by low transformation efficiencies wherefore one reason could be differences in the DNA methylation patterns between the Escherichia coli intermediate host for plasmid amplification and the final LAB host. In the present study, we examined the influence of DNA methylation on transformation efficiency in LAB and developed a direct cloning approach for Lactobacillus plantarum CD033. Therefore, we propagated plasmid pCD256 in E. coli strains with different dam/dcm-methylation properties. The obtained plasmid DNA was purified and transformed into three different L. plantarum strains and a selection of other LAB species.
Results
Best transformation efficiencies were obtained using the strain L. plantarum CD033 and non-methylated plasmid DNA. Thereby we achieved transformation efficiencies of ~ 109 colony forming units/μg DNA in L. plantarum CD033 which is in the range of transformation efficiencies reached with E. coli. Based on these results, we directly transformed recombinant expression vectors received from PCR/ligation reactions into L. plantarum CD033, omitting plasmid amplification in E. coli. Also this approach was successful and yielded a sufficient number of recombinant clones.
Conclusions
Transformation efficiency of L. plantarum CD033 was drastically increased when non-methylated plasmid DNA was used, providing the possibility to generate expression libraries in this organism. A direct cloning approach, whereby ligated PCR-products where successfully transformed directly into L. plantarum CD033, obviates the construction of shuttle vectors containing E. coli-specific sequences, as e.g. a ColEI origin of replication, and makes amplification of these vectors in E. coli obsolete. Thus, plasmid constructs become much smaller and occasional structural instability or mutagenesis during E. coli propagation is excluded. The results of our study provide new genetic tools for L. plantarum which will allow fast, forward and systems based genetic engineering of this species.
Similar content being viewed by others


Background
Lactobacillus plantarum and many other lactic acid bacteria (LAB) are “generally regarded as safe” (GRAS) organisms and possess the ability to efficiently secrete recombinant proteins directly into the culture medium. Thus, they are recognized as emerging candidates for the expression of recombinant proteins as well as for genetic and metabolic cell engineering, both in the fields of medical and industrial biotechnology[1, 2]. Lactococcus lactis is to date the most widely used LAB strain for recombinant protein expression and was engineered to express cytokines[3], bacterial and viral antigens[4, 5], membrane proteins[6] and enzymes[7]. L. plantarum plays an important role in many processes of animal feed industry[8]. For example, preservation of crop silage is often improved by adding starter cultures containing L. plantarum and other LAB. During the past decade the interest in developing new and genetically engineered LAB strains with improved properties has been continuously growing[9–13], e.g. to produce cellulose degrading enzyme activity[14]. L. plantarum has also been proved being a feasible expression host, recently expressing a ß-galactosidase from Lactobacillus delbrueckii[15] and a chitinase from Bacillus licheniformis[16].
Several vector systems exist[17–19], and different plasmid backbones and promoters are available for cloning and protein expression in LAB. Unlike for recombinant protein expression in Escherichia coli, the availability of different engineered LAB expression strains is limited. Often protein yields are very low, constraining the system to applications where small amounts are sufficient, e.g. to produce cellulose degrading enzyme activity[14]. In order to adapt a host’s metabolic capacities to the needs in biotechnology, the approach to use genetic engineering has been shown to successfully enhance product yield and quality in E. coli[20], Saccharomyces cerevisiae[21] and Pichia pastoris[22]. Also for L. plantarum, genetic engineering was shown to improve the metabolic performance, when mutations in sigma-factor (rpoD) conferred resistance against low pH conditions[13]. Often, these approaches are based on the generation of a great variety of genetically different clones and the subsequent screening for the desired phenotype. The strategy of cell engineering on a systems molecular level requires the possibility of high-throughput screening of a heterogenic pool of mutants. Thus, the generation of diverse genetic libraries is a prerequisite for fast and efficient host engineering. Also for protein engineering such as the adaptation of enzymatic activities to environmental conditions within a certain cell system, requires library based systems. While gene libraries can be generated in the size of 1010 in E. coli, and in the range of 107 in S. cerevisiae, for LAB low transformation efficiencies are often a limiting factor. During the last decades many transformation protocols for LAB strains have been published. The successful introduction of plasmid DNA into LAB is dependent on strain specific features such as cell wall structure and composition plasmid size and the origin of replication.
In some LAB, the low number or even lack of transformants obtained after electroporation, may be attributed to various restriction modification (RM) systems encoded by the host. RM systems are widely spread in bacteria and serve the protection of invading DNA such as foreign plasmids or the DNA of bacteriophages. Most of these systems consist of a restriction enzyme and a corresponding methyltransferase that blocks the restriction activity, thus, protects the genome from self-cleavage (type I and III RM systems)[23]. In contrast, the type IV RM systems produce restriction enzymes which cleave solely methylated DNA. There are several reports, mostly referring to DNA adenine methylation (dam)/ DNA cytosine methylation (dcm) that methylation pattern of plasmid DNA has a major impact on transformation efficiency and allows plasmid DNA to circumvent host restriction mechanisms[24–26].
Since gene manipulation of expression vectors is much easier in E. coli than in LAB, and in order to gain sufficient amounts of plasmid DNA, normally, shuttle vectors are used to first build and propagate the final plasmid in E. coli. After subsequent purification the desired plasmid is then transformed into LAB. Many shuttle vectors are based on cryptic plasmids derived from LAB strains[27], which have been modified to contain both, the LAB specific and E. coli specific replicative elements. Often these shuttle vectors are structurally unstable either in E. coli or in the LAB-expression hosts, maybe due to their size or their chimeric nature, e.g. differences in GC-content (50% GC for E. coli versus 30 – 40% GC for LAB). Savijoki et al.[28] used L. lactis MG1363 as an intermediate host to circumvent such problems. However, this approach is limited to origins of replication functional in L. lactis. Another strategy is to design shuttle vectors that contain replicons which replicate in Gram (+) as well as in Gram (–) bacteria, e.g. based on the origin of replication of pWV01 or pSH71[29, 30]. Yet, due to their rolling circle-replication mechanism these plasmids tend to suffer from structural and segregational instability. Often, plasmids containing large DNA inserts cannot stably be maintained[31, 32], and thus, rolling circle-replicating plasmids are only suitable for small genes. Therefore, cloning procedures would substantially improve by having plasmids available that are devoid of any E. coli derived sequences and are based on a stable origin of replication. We have previously compared several LAB strains in terms of transformation efficiency and plasmid stability. One of the tested L. plantarum (CD033) strains showed unexpectedly high transfection yields (6 x 105 colony forming units (cfu)/μg DNA)[19]. Furthermore, this strain was found to be transformable with unmethylated DNA and therefore, became an interesting organism for further examinations regarding the influence of DNA methylation on transformation efficiency.
Results and discussion
Role of plasmid methylation in transformation of L. plantarum CD033
L. plantarum CD033 was tested for its ability to be transformed by plasmid pCD256 DNA prepared from four E. coli strains differing in their genotype regarding dam/dcm-methylation. The plasmid pCD256 is based on pUC19 containing the origin of replication of p256, originally isolated from L. plantarum NC7[33], which was previously shown to be active in L. plantarum CD033[19]. A gene encoding the chloramphenicol acetyl transferase (CAT) obtained from the Staphylococcus aureus plasmid pC194 served as a selection marker. E. coli strains JM109 (dam+, dcm+), BL21(DE3) (dam+, dcm-), GM33 (dam-, dcm+) and C2925 (dam-, dcm-) were used for plasmid propagation. The state of dam/dcm-methylation was confirmed by restriction analysis using Dpn II (blocked by dam-methylation) and PspG I (inhibited by dcm-methylation) (data not shown). After purification, we performed electrotransformation of L. plantarum CD033 and determined the number of cfu. Electroporation results are summarized in Table1 and represent mean transformation efficiencies calculated from four independently performed transformations. Transformation efficiency for dam+/dcm- pCD256 was slightly higher as compared to the transformation efficiency when pCD256 was dam+/dcm+ or dam-/dcm+. However, when pCD256 was propagated in the dam-/dcm- strain E. coli C2925 transformation efficiency increased more than 1000-fold as compared to JM109 derived plasmids. The transformation efficiencies of nearly 109 cfu/μg plasmids are comparable to commercially available E. coli cloning systems and succeed the efficiencies, normally achieved in yeast[34, 35]. Thus, the possibility to produce libraries, may it be for systems based genetic engineering, evolutionary based enzyme engineering or randomization of promoter active sequences, has been made available in a LAB strain. Since many regulatory functions are conserved through-out different species of LAB, results from library screenings in L. plantarum CD033 might be transferred to other LAB strains and species. In addition to L. plantarum CD033, we further tested two other strains of the same species, L. plantarum CD032 and the type strain L. plantarum DSM20174. It turned out that the type strain could not be transformed using methylated plasmid at all, and with unmethylated plasmid the transformation efficiencies were lower as compared to the other two L. plantarum strains (Table2). Also transformation efficiencies of L. plantarum CD032 were below the ones obtained with L. plantarum CD033. However, the tendency, to not only tolerate non-methylated DNA, but to give higher numbers of transformants/μg DNA than with methylated plasmids was confirmed. A reason for the tested L. plantarum strains prefering non-methylated DNA over methylated DNA could be that the latter is restricted by methyl-dependent restriction enzymes such as e.g. the Mrr type restriction proteins which belong to type IV restriction enzymes. We searched the whole genome data of L. plantarum strains available at the NCBI data base, L. plantarum WCFS1 which has just recently been resequenced and reannotated[36], the genome of L. plantarum JDM1[37] and the complete genome of L. plantarum ST-III[38], for the presence of mrr (methylated adenine recognition and restriction) like genes. Based on the annotation of L. plantarum WCFS1 we found one mrr gene (GeneID:1061548). This gene (99% sequence identity) was also present in the genome sequences of L. plantarum JDM1 and of the type strain L. plantarum DSM20174[39]; no putative mrr genes were detected in the genome of L. plantarum ST-III. BLAST analysis[40] of these genes revealed further identities (60% on amino acid level) to Lactobacillus farciminis KCTC 3681 and Lactobacillus reuteri 100-23, all other identities were below 60%. Sequence comparison revealed the presence of the conserved Mrr motifs within the N-terminal domain containing the catalytic activity[41]. Levels of homologies between lactic acid bacterial genes were found to be much higher than when comparison was performed including the E. coli mrr gene (25% identity), indicating that certain domains within the protein binding domain are conserved within the group of LAB. In L. plantarum CD033, the presence of an mrr gene was confirmed by PCR and subsequent sequencing of the obtained amplicon (data not shown). DNA sequence comparison revealed 99% identity of the L. plantarum CD033 gene with the mrr gene of L. plantarum DSM20174.
Furthermore, three other LAB-species were tested for their susceptibility to be transformed by unmethylated plasmid DNA. Lactobacillus buchneri CD034 and Enterococcus faecium CD036 were shown previously to maintain the origin of replication from plasmid p256[19], whereas for L. lactis MG1363 plasmid pCDWV01[19], containing the origin of replication from the lactococcal plasmid pWV01, had to be used in order to provide replication, which has been shown to have similar stability properties as compared to pCD256[19]. Results showed that for none of the performed electroporations colonies were received, possibly indicating restriction of non-methylated DNA (Table2). RM systems have been described for different L. lactis strains[42] and appropriate genes have also been identified in the chromosome of L. buchneri CD034[43], supporting this hypothesis. Furthermore, restriction analysis of plasmids obtained from L. plantarum CD033 indicated that no dam/dcm-methylation is present in this strain. Hence, we failed to introduce plasmid DNA deriving from L. plantarum CD033 into L. buchneri CD034 (data not shown). The suggestion arises, that insights in methylation patterns of other species might serve to in vitro-methylate ligation reactions or unmethylated plasmids in order to overcome the bottle neck of a specific host’s restriction system.
Direct cloning in L. plantarum CD033
In order to see whether a transformation efficiency of up to almost 109 cfu would be sufficient for direct cloning, a ligation reaction consisting of the vector backbone and a target gene was directly transformed into L. plantarum CD033 by electroporation. First, the vector pCD256 was propagated in E. coli C2925 (dam-/dcm-), purified and digested. The gene for the human trefoil factor 1 (hTFF1) served as the model target gene, as it is of small size (213 bps) and encodes a protein that being expressed in a food grade organism would be of high benefit. The codon optimized, synthetic hTFF1 gene was amplified by PCR, cleaved and ligated into pCD256 to be expressed under control of the P2083 promoter. The ligation mix was directly transformed into L. plantarum CD033 by electroporation resulting in 10 colonies. PCR screening revealed that all clones contained the hTFF1 gene. Thus, the step of propagating the LAB expression plasmid first in E. coli could be dismissed, making the requirement for an E. coli/L. plantarum shuttle vector obsolete. In order to proof this, the E. coli specific elements were deleted by PCR amplification maintaining only the Lactobacillus specific sequences of pCD256, resulting in a 1690 bp DNA fragment. Self-ligation of this DNA fragment resulted in plasmid pCD256ΔEc, which now was 3100 bp smaller than pCD256 (Figure1). Transformation into L. plantarum CD033 yielded 2,5 x 105 cfu/250 ng vector DNA used in ligation mix. Given the fact that the intramolecular ligation reaction of linear fragments is very efficient, this high number of cfu is not surprising. Now, in order to test the feasibility of this approach also for direct cloning of a target gene, we linearized pCD256ΔEc by PCR and after digestion performed ligation with the synthetic hTFF1 expression cassette (Figure1C) digested with the same enzymes. Direct transformation of the ligation reaction into L. plantarum CD033 yielded 5380 cfu/ligation reaction. This is a markedly higher number than when a vector backbone containing E. coli specific elements such as the origin of replication and the ß-lactamase gene was used. Identity of the plasmid and its structural integrity were confirmed by PCR amplification of overlapping plasmid fragments and subsequent DNA sequencing. So, it was shown that L. plantarum CD033 is the most suitable strain of the as yet tested LAB strains for direct cloning of recombinant expression vectors, circumventing the intricate design of shuttle vectors that are compatible with E. coli. Thus, plasmids become smaller, are devoid of unwanted sequences and occasional mutagenesis during E. coli propagation is excluded.

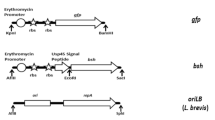
Map of pCD256, pCD256Δ Ec and the synthetic hTFF1 expression cassette. A: Map of pCD256 containing the minimal replicon (miniori) from plasmid p256[33] for replication in LAB, pMB1 origin for replication in E. coli, a chloramphenicol resistance gene (CAT) for selection in LAB and an ampicillin resistance gene (Amp) for selection in E. coli. The dam/dcm-methylation sites are indicated. B: Map of the minimal plasmid pCD256ΔEc consisting exclusively of the LAB-minimal origin from plasmid p256 and the chloramphenicol resistance gene for selection in LAB. C: Map of the synthesised hTFF1 expression cassette consisting of the optimized hTFF1-gene, N-terminally fused to the L. plantarum CD032 plnI double glycine leader sequence (GG), promoter P2083 and terminator Tldh.
Conclusions
In the present study, we examined the influence of DNA methylation patterns on transformation efficiency in LAB and developed a direct cloning approach for L. plantarum CD033. Therefore, we transformed various LAB strains with plasmid DNA exhibiting different dam/dcm-methylation patterns. Best results were obtained using non-methylated DNA, resulting in transformation efficiencies of ~ 109 cfu/μg DNA in L. plantarum CD033. Thereby, it becomes feasible to generate expression libraries, promoter libraries, etc. of sufficient size in this strain. We demonstrated direct transformation of recombinant expression vectors received from PCR/ligation reactions into L. plantarum CD033 to be feasible, making the construction of shuttle vectors obsolete. This new approach allowed the construction of minimal plasmids consisting exclusively of a LAB-origin of replication and a selection marker. Besides providing smaller expression vectors, this method excludes any structural instability or mutagenesis which is occasionally associated with plasmid propagation in E. coli. The results of our study provide new genetic tools for L. plantarum which allow faster and facilitated cloning procedures as well as systems based genetic engineering based on library techniques.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains used in this work are listed in Table3. LAB were cultured in De Man-Rogosa-Sharp (MRS) medium[44] at 37°C for L. buchneri CD034, L. lactis MG1363 and E. faecium CD036 or at 30°C for L. plantarum strains. E. coli strains were grown in Luria-Bertani (LB) medium under continuous agitation at 37°C. For selection, the medium was supplemented with the appropriate antibiotics used at the following concentrations: ampicillin 100 μg/ml for E. coli, chloramphenicol 5 μg/ml for the LAB strains.
Plasmids, primers and synthetic expression cassette
The plasmids used in this work are listed in Table4. The gene hTFF1 encoding the human trefoilfactor 1 was codon optimized for L. plantarum WCFS1 using JCat (http://www.jcat.de). An expression cassette consisting of the optimized hTFF1-gene, N-terminally fused to the L. plantarum CD032 plnI double glycine leader sequence, promoter P2083 and terminator Tldh was designed and synthesized by GeneArt (Life technologies, USA) (Figure1 C). P2083 was derived from L.buchneri CD034. It was found to drive transcription of the CAT gene in L. plantarum CD033 in a preliminary experiment and was later identified upstream of a gene encoding a putative fumarylacetoacetate hydrolase (LBUCD034_2083) in the L. buchneri CD034 genomic sequence[43]. Terminator Tldh was derived from Lactobacillus casei BL23 (L-lactate dehydrogenase gene, LCABL-06930). Primers used in this study are listed in Table5.
PCR, restriction digestion and ligation of DNA fragments
Unless otherwise stated, DNA fragments were amplified using the Phusion High-Fidelity DNA Polymerase in HF-buffer (New England Biolabs, NEB, USA). PCRs were performed as follows: initial denaturation for 30 s at 98°C, followed by 30 cycles of 10 s at 98°C, annealing for 20 s at a melting temperature (Tm) +3°C of the lower Tm primer and extension for 25 s/kb at 72°C. Amplification was concluded with a final extension step at 72°C for 6 min. All PCRs were carried out with a T3 Thermocycler (Biometra, Germany). All restriction enzymes were purchased from NEB, restriction digests were performed according to the manufacturer’s recommendations. DNA fragments were purified from PCRs, enzyme reactions or agarose gels using the NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel, Germany). Ligation reactions were performed using T4 DNA Ligase (NEB, USA). For a 20 μl ligation reaction 250 ng digested and purified plasmid DNA was mixed on ice with 1 μl T4 DNA ligase, 2 μl of 10x T4 ligase buffer and with a 5-fold molar excess of digested and purified insert DNA. The ligation reaction was incubated at 16°C over night precipitated with isopropanol, washed with 70% v/v ethanol, air dried for 15 min, solved in sterile ddH20 and used for transformation.
Dedection of the mrr gene in L. plantarum CD033
The presence of an mrr gene was confirmed by colony PCR using the Phusion High-Fidelity DNA Polymerase using GC-buffer (NEB, USA) and the primers mrr_Lp_F / mrr_Lp_R. PCR was conducted as follows: initial denaturation at 98°C for 30 s, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 20 s and elongation at 72°C for 30 s. Cycling was completed with a final extension step at 72°C for 6 min. The amplicon was sequenced (GATC Biotech, Germany) and confirmed by BLAST analysis[40].
Construction of pCD256_hTFF1, pCD256ΔEc and pCD256ΔEc _hTFF1
The synthetic hTFF1 expression cassette was amplified by PCR using the primers hTFF1_SacI_F / Tldh_amp_R and cloned Sac I/Sal I into pCD256 resulting in the plasmid pCD256_hTFF1.
For the construction of pCD256ΔEc, a DNA fragment comprising only lactobacillus specific elements (CAT, miniori256) was amplified from pCD256 by PCR using the primers M13_R_NheI / Cat_F_NheI, thereby introducing a novel Nhe I restriction site. The obtained fragment was digested with Nhe I and subsequently self-ligated.
For the ligation of two PCR products, pCD256ΔEc and the hTFF1 expression cassette were amplified by PCR using the primers M13_R_XhoI / Cat_F_NheI and hTFF1_F_NheI / Tldh_amp_R_XhoI. The two PCR products were Xho I/Nhe I-digested and ligated one with each other, resulting in the plasmid pCD256ΔEc _hTFF1. DNA-cloning techniques in E. coli were performed according to Sambrook and Russell[46]. All vector constructs were confirmed by DNA sequencing (GATC Biotech, Germany).
Electroporation of LAB
Electroporation was done using an ECM 630 Precision Pulse (BTX Harvard apparatus, USA). L. plantarum CD033 and L. buchneri CD034 were transformed according to[19], L. plantarum DSM20174 was transformed using the same protocol as for L. plantarum CD033, L. lactis MG1363 was transformed according to[47], E. faecium CD036 and L. plantarum CD032 were transformed as follows. An overnight culture was diluted with MRS broth to an optical density at 600 nm wavelength (OD600) of 0.2 and incubated at the appropriate temperature until an OD600 of 0.5 was reached. The cells were harvested by centrifugation at 10,000 rcf for 6 min at 4°C. The pellets were washed three times with ice cold 0.3 M sucrose. The pellet was resuspended to one hundredth of initial volume in 0.3 M sucrose and kept on ice until electroporation. Plasmid DNA (0.25-1 μg) was mixed with 40 μl cell suspension in a ice-cold electroporation cuvette (0.2 cm gap, Sigma Aldrich, Germany) and electroporated at 2.5 kV, 200 Ω and 25 μF. After the pulse, cells were resupended in 360 μl MRS broth for 2 h at the appropriate temperature. The bacteria were spread on MRS plates containing chloramphenicol and incubated for 3 days at the appropriate conditions. Proper assemble of recombinant plasmids was confirmed by colony PCR and supsequent sequencing of the obtained amplicons using the primers CAT_seq_back/p256miniori_for. In the case of pCD256ΔEc _hTFF1 which was transformed directly with a purified ligation reaction, structural integrity was proven by PCR using the primers CAT_seq_R/Tldh_amp_R and CAT_seq_back/GG_hTFF1_sense yielding overlapping DNA fragments and subsequent DNA sequencing of the obtained fragments.
References
Xu W, Huang M, Zhang Y, Yi X, Dong W, Gao X, Jia C: Novel surface display system for heterogonous proteins on Lactobacillus plantarum. Lett Appl Microbiol. 2011, 53: 641-648. 10.1111/j.1472-765X.2011.03160.x.
Nguyen TT, Mathiesen G, Fredriksen L, Kittl R, Nguyen TH, Eijsink VG, Haltrich D, Peterbauer CK: A food-grade system for inducible gene expression in Lactobacillus plantarum using an alanine racemase-encoding selection marker. J Agric Food Chem. 2011, 59: 5617-5624. 10.1021/jf104755r.
Steidler L: In situ delivery of cytokines by genetically engineered Lactococcus lactis. Antonie Van Leeuwenhoek. 2002, 82: 323-331. 10.1023/A:1020656220815.
Nouaille S, Ribeiro LA, Miyoshi A, Pontes D, Le Loir Y, Oliveira SC, Langella P, Azevedo V: Heterologous protein production and delivery systems for Lactococcus lactis. Genet Mol Res. 2003, 2: 102-111.
Wells JM, Wilson PW, Norton PM, Gasson MJ, Le Page RWF: Lactococcus lactis: high‐level expression of tetanus toxin fragment C and protection against lethal challenge. Mol Microbiol. 1993, 8: 1155-1162. 10.1111/j.1365-2958.1993.tb01660.x.
Kunji ER, Slotboom DJ, Poolman B: Lactococcus lactis as host for overproduction of functional membrane proteins. Biochim Biophys Acta. 2003, 1610: 97-108. 10.1016/S0005-2736(02)00712-5.
van de Guchte M, van der Vossen JM, Kok J, Venema G: Construction of a lactococcal expression vector: expression of hen egg white lysozyme in Lactococcus lactis subsp. lactis. Appl Environ Microbiol. 1989, 55: 224-228.
Giraffa G, Chanishvili N, Widyastuti Y: Importance of lactobacilli in food and feed biotechnology. Res Microbiol. 2010, 161: 480-487. 10.1016/j.resmic.2010.03.001.
Ladero V, Ramos A, Wiersma A, Goffin P, Schanck A, Kleerebezem M, Hugenholtz J, Smid E, Hols P: High-level production of the low-calorie sugar sorbitol by Lactobacillus plantarum through metabolic engineering. Appl Environ Microbiol. 2007, 73: 1864-1872. 10.1128/AEM.02304-06.
Rossi F, Rudella A, Marzotto M, Dellaglio F: Vector-free cloning of a bacterial endo-1,4-beta-glucanase in Lactobacillus plantarum and its effect on the acidifying activity in silage: use of recombinant cellulolytic Lactobacillus plantarum as silage inoculant. Antonie Van Leeuwenhoek. 2001, 80: 139-147. 10.1023/A:1012223220427.
Sharp R, O'donnell AG, Gilbert HG, Hazlewood GP: Growth and Survival of Genetically Manipulated Lactobacillus plantarum in Silage. Appl Environ Microbiol. 1992, 58: 2517-2522.
Helanto M, Kiviharju K, Leisola M, Nyyssölä A: Metabolic engineering of Lactobacillus plantarum for production of L-ribulose. Appl Environ Microbiol. 2007, 73: 7083-7091. 10.1128/AEM.01180-07.
Klein-Marcuschamer D, Stephanopoulos G: Assessing the potential of mutational strategies to elicit new phenotypes in industrial strains. Proc Natl Acad Sci. 2007, 105: 2319-2324.
Okano K, Zhang Q, Yoshida S, Tanaka T, Ogino C, Fukuda H, Kondo A: D-lactic acid production from cellooligosaccharides and beta-glucan using L-LDH gene-deficient and endoglucanase-secreting Lactobacillus plantarum. Appl Microbiol Biotechnol. 2010, 85: 643-650. 10.1007/s00253-009-2111-8.
Nguyen TT, Nguyen HA, Arreola SL, Mlynek G, Djinović-Carugo K, Mathiesen G, Nguyen TH, Haltrich D: Homodimeric β-galactosidase from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081: expression in Lactobacillus plantarum and biochemical characterization. J Agric Food Chem. 2012, 60: 1713-1721. 10.1021/jf203909e.
Nguyen HA, Nguyen TH, Nguyen TT, Peterbauer CK, Mathiesen G, Haltrich D: Chitinase from Bacillus licheniformis DSM13: expression in Lactobacillus plantarum WCFS1 and biochemical characterisation. Protein Expr Purif. 2012, 81: 166-174. 10.1016/j.pep.2011.10.005.
Mierau I, Kleerebezem M: 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol. 2005, 68: 705-717. 10.1007/s00253-005-0107-6.
Sørvig E, Mathiesen G, Naterstad K, Eijsink V, Axelsson L: High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology. 2005, 151: 2439-2449. 10.1099/mic.0.28084-0.
Spath K, Heinl S, Egger E, Grabherr R: Lactobacillus plantarum and Lactobacillus buchneri as Expression Systems: Evaluation of Different Origins of Replication for the Design of Suitable Shuttle Vectors. Mol Biotechnol. 2012, 52: 40-48. 10.1007/s12033-011-9471-x.
Chou CH, Bennett GN, San KY: Effect of modified glucose uptake using genetic engineering techniques on high‐level recombinant protein production in escherichia coli dense cultures. Biotechnol Bioeng. 1994, 44: 952-960. 10.1002/bit.260440811.
Ostergaard S, Olsson L, Nielsen J: Metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2000, 64: 34-50. 10.1128/MMBR.64.1.34-50.2000.
Schroer K, Peter Luef K, Stefan Hartner F, Glieder A, Pscheidt B: Engineering the Pichia pastoris methanol oxidation pathway for improved NADH regeneration during whole-cell biotransformation. Metab Eng. 2010, 12: 8-17. 10.1016/j.ymben.2009.08.006.
Blumenthal RM, Cheng X: Restriction-Modification Systems. Modern Microbial Genetics. Edited by: Streips UN, Yasbin RE. 2002, New York: John Wiley & Sons, Inc, 177-225. 2
Alegre M, Rodríguez M, Mesas J: Transformation of Lactobacillus plantarum by electroporation with in vitro modified plasmid DNA. FEMS Microbiol Lett. 2004, 241: 73-77. 10.1016/j.femsle.2004.10.006.
Chen CK, Boucle CM, Blaschek HP: Factors involved in the transformation of previously non-transformable Clostridium perfringens type B. FEMS Microbiol Lett. 1996, 140: 185-191. 10.1111/j.1574-6968.1996.tb08334.x.
Macaluso A, Mettus AM: Efficient transformation of Bacillus thuringiensis requires nonmethylated plasmid DNA. J Bacteriol. 1991, 173: 1353-1356.
Shareck J, Choi Y, Lee B, Miguez CB: Cloning vectors based on cryptic plasmids isolated from lactic acid bacteria: their characteristics and potential applications in biotechnology. Crit Rev Biotechnol. 2004, 24: 155-208. 10.1080/07388550490904288.
Savijoki K, Kahala M, Palva A: High level heterologous protein production in Lactococcus and Lactobacillus using a new secretion system based on the Lactobacillus brevis S-layer signals. Gene. 1997, 186: 255-262. 10.1016/S0378-1119(96)00717-2.
Leenhouts K, Tolner B, Bron S, Kok J, Venema G, Seegers J: Nucleotide sequence and characterization of the broad-host-range lactococcal plasmid pWVO1. Plasmid. 1991, 26: 55-66. 10.1016/0147-619X(91)90036-V.
de Vos WM: Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett. 1987, 46: 281-295.
Gruss A, Ehrlich SD: The family of highly interrelated single-stranded deoxyribonucleic acid plasmids. Microbiol Rev. 1989, 53: 231-241.
Kiewiet R, Bron S, de Jonge K, Venema G, Seegers J: Theta replication of the lactococcal plasmid pWVO2. Mol Microbiol. 1993, 10: 319-327. 10.1111/j.1365-2958.1993.tb01958.x.
Sørvig E, Skaugen M, Naterstad K, Eijsink V, Axelsson L: Plasmid p256 from Lactobacillus plantarum represents a new type of replicon in lactic acid bacteria, and contains a toxin-antitoxin-like plasmid maintenance system. Microbiology. 2005, 151: 421-431. 10.1099/mic.0.27389-0.
Manivasakam P, Schiestl RH: High efficiency transformation of Saccharomyces cerevisiae by electroporation. Nucleic Acids Res. 1993, 21: 4414-4415. 10.1093/nar/21.18.4414.
Faber KN, Haima P, Harder W, Veenhuis M, AB G: Highly-efficient electrotransformation of the yeast Hansenula polymorpha. Curr Genet. 1994, 25: 305-310. 10.1007/BF00351482.
Siezen RJ, Francke C, Renckens B, Boekhorst J, Wels M, Kleerebezem M, van Hijum SA: Complete resequencing and reannotation of the Lactobacillus plantarum WCFS1 genome. J Bacteriol. 2012, 194: 195-196. 10.1128/JB.06275-11.
Zhang ZY, Liu C, Zhu YZ, Zhong Y, Zhu YQ, Zheng HJ, Zhao GP, Wang SY, Guo XK: Complete genome sequence of Lactobacillus plantarum JDM1. J Bacteriol. 2009, 191: 5020-5021. 10.1128/JB.00587-09.
Wang Y, Chen C, Ai L, Zhou F, Zhou Z, Wang L, Zhang H, Chen W, Guo B: Complete genome sequence of the probiotic Lactobacillus plantarum ST-III. J Bacteriol. 2011, 193: 313-314. 10.1128/JB.01159-10.
Orla-Jensen S: The lactic acid bacteria, by S. Orla-Jensen. 1919, Copenhagen: A.F. Höst og Sön
Altschul S, Gish W, Miller W, Myers E, Lipman D: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Zheng Y, Cohen-Karni D, Xu D, Chin HG, Wilson G, Pradhan S, Roberts RJ: A unique family of Mrr-like modification-dependent restriction endonucleases. Nucleic Acids Res. 2010, 38: 5527-5534. 10.1093/nar/gkq327.
Nyengaard N, Vogensen FK, Josephsen J: Restriction-modification systems in Lactococcus lactis. Gene. 1995, 157: 13-18. 10.1016/0378-1119(95)91235-R.
Heinl S, Wibberg D, Eikmeyer F, Szczepanowski R, Blom J, Linke B, Goesmann A, Grabherr R, Schwab H, Pühler A, Schlüter A: Insights into the completely annotated genome of Lactobacillus buchneri CD034, a strain isolated from stable grass silage. J Biotechnol. 2012, 161: 153-166. 10.1016/j.jbiotec.2012.03.007.
De Man JC, Rogosa M, Sharpe ME: A medium for the cultivation of Lactobacilli. J Appl Microbiol. 1960, 23: 130-135. 10.1111/j.1365-2672.1960.tb00188.x.
Gasson MJ: Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol. 1983, 154: 1-9.
Sambrook J, Russell DW: Molecular Cloning. 2001, New York, USA: Cold Spring Harbor Laboratory Press, Third
Papagianni M, Avramidis N, Filioussis G: High efficiency electrotransformation of Lactococcus lactis spp. lactis cells pretreated with lithium acetate and dithiothreitol. BMC Biotechnol. 2007, 7: 15-10.1186/1472-6750-7-15.
Acknowledgements
This work was supported by the Christian Doppler Research Association, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The work presented here was carried out in collaboration between all authors. KS, SH and RG defined the research theme and designed the experiments. KS carried out the laboratory experiments, analyzed the data, interpreted the results and prepared this manuscript with input, feedback and advice from SH and RG. All authors have contributed to, seen and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Spath, K., Heinl, S. & Grabherr, R. “Direct cloning in Lactobacillus plantarum: Electroporation with non-methylated plasmid DNA enhances transformation efficiency and makes shuttle vectors obsolete”. Microb Cell Fact 11, 141 (2012). https://doi.org/10.1186/1475-2859-11-141
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-11-141