
Abstract
Background
Polyploidization is a prominent process in plant evolution, whereas the mechanism and tempo-spatial process remained poorly understood. Oryza officinalis complex, a polyploid complex in the genus Oryza, could exemplify the issues not only for it covering a variety of ploidy levels, but also for the pantropical geographic pattern of its polyploids in Asia, Africa, Australia and Americas, in which a pivotal genome, the C-genome, witnessed all the polyploidization process.
Results
Tracing the C-genome evolutionary history in Oryza officinalis complex, this study revealed the genomic relationships, polyploid forming and diverging times, and diploidization process, based on phylogeny, molecular-clock analyses and fluorescent in situ hybridization using genome-specific probes. Results showed that C-genome split with B-genome at ca. 4.8 Mya, followed by a series of speciation of C-genome diploids (ca. 1.8-0.9 Mya), which then partook in successive polyploidization events, forming CCDD tetraploids in ca. 0.9 Mya, and stepwise forming BBCC tetraploids between ca. 0.3-0.6 Mya. Inter-genomic translocations between B- and C-genomes were identified in BBCC tetraploid, O. punctata. Distinct FISH (fluorescent in situ hybridization) patterns among three CCDD species were visualized by C-genome-specific probes. B-genome was modified before forming the BBCC tetraploid, O. malampuzhaensis.
Conclusion
C-genome, shared by all polyploid species in the complex, had experienced different evolutionary history particularly after polyploidization, e.g., inter-genomic exchange in BBCC and genomic invasion in CCDD tetraploids. It diverged from B-genome at 4.8 Mya, then participated in the tetraploid formation spanning from 0.9 to 0.3 Mya, and spread into tropics of the disjunct continents by transcontinentally long-distance dispersal, instead of vicariance, as proposed by this study, given that the continental splitting was much earlier than the C-genome species radiation. We also find reliable evidence indicated that an extinct BB diploid species in Asia was presumptively the direct genomic donor of their sympatric tetraploids.
Similar content being viewed by others


Background
Polyploidization is a prominent process in the evolution of high plants. Between 50% and 70% of angiosperm species were identified as polyploids by intensive screening, while recent studies estimated that up to 100% of angiosperms underwent genome duplication at least once in their evolutionary history [1, 2]. The commonity of polyploidy suggests a potential advantage of polyploids to survive better in harsh environments than diploids [3, 4]. Thronged facts related to polyploidy were discovered, e.g., genomic divergence in allopolyploids by diploidization, rapid genomic changes, and inter-genomic invasion [5–9]. However, more evidence is needed to reveal the mechanism and tempo-spatial process of polyploidization. Polyploid complex, a group of species with a variety of ploidy levels, could be an ideal model to address the problems. Oryza officinalis complex is an excellent example, not only because it contains diploids and heterochronically formed polyploids, but also it has a "pivotal genome" [10], the C-genome, which participated in all the polyploid formation, potentially as an inner criterion to trace polyploid evolution. Moreover, geographic patterns of the polyploids distributed pantropically to isolated continents within a rather recent period, have remained mysterious [11–15].
With agricultural importance, the genus Oryza comprises 23 species including cultivated rice, combined into four species complexes [16–19]. In the last decades, molecular methods have been used to reconstruct species phylogeny and to trace evolution process in Oryza [14, 15, 20–26]. Ten distinctive genomes were identified on the basis of DNA sequences [14], or genomic in situ hybridization (GISH) [27–29]. Particularly in O. officinalis complex, the largest in Oryza, genomic relationships were found extraordinarily complicated, e.g., the BBCC tetraploid species formed independently with different parenthood by three polyploidization events, in which O. eichingeri was the maternal parent of tetraploid O. punctata while diploid O. punctata was that of tetraploids O. malampuzhaensis and O. minuta [14, 22, 30, 31]. Furthermore, three tetraploid species with CCDD genomes were assumed to be formed by one polyploidization event, where the CC genome progenitor served as the maternal parent [14, 21, 22, 32, 33]. Additionally, it seems much intriguing that the C-genome diploids and tetraploids are distributed across Asian, African and American tropics. And the CCDD tetraploids are entirely endemic to Central and South Americas where no extant diploid with C- or D-genome was found [11, 12]. Therefore, the questions arose: (a) How was the C-genome, as the pivotal genome in all the tetraploids, differentiated after polyploidizating? (b) When were the tetraploids formed and how did they spread transcontinentally? (c) Whether inter-genomic interaction, e.g., exchange or invasion, happened in the allopolyploids?
Focused on the questions, we reconstructed the phylogenetic relationship, dated the divergence time among the genomes in O. officinalis complex, and detected genomic changes thereafter polyploidization by FISH methods with genome-specific probes. The goal of this study is to reveal the evolution history of the O. officinalis complex, particularly the polyploidization and its genomic impact, by tracing C-genome differentiating and dispersing process.
Methods
Plant materials
Thirty eight accessions representing eleven species of O. officinalis complex were sampled, and one accession of O. granulata, a species outside the complex, was used as outgroup (Table 1). Of them, eight species with different ploidy levels and geographic origins were used for cytogenetic analysis. All the accessions used in this study, are showed in Table 1, including their species names, genome constitutions, original collection locations and GenBank accession numbers. Total DNAs were extracted from fresh leaves of individual plants by the CTAB method [34].
Primer design, PCR amplification and sequencing
Two genes, Starch debranching enzyme (SDBE) on chromosome 4 and Os02 g0125000 (Os125) on chromosome 2 of O. sativa, were chosen in the present study. SDBE is a single copy gene [35], containing 25 introns, in which the seventh was used in this study. Os125 is also identified as a single copy gene by the criterion previously reported [23], which had three introns and the second one was selected. Primers used for PCR amplifying and sequencing are listed in Table 2.
Amplification and purification of the PCR products were performed by standard methods. Purified PCR products were sequenced directly or after cloning into pGEM-T-easy vectors (Promega, Madison, WI, USA). Sequencing was performed by ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). All sequences obtained in this study have been deposited to the GenBank database under accession numbers FJ918688-FJ918822 (Table 1).
Date analysis
Sequences were aligned with CLUSTAL_X version 1.81 [36]. GC content, base frequency, pairwise divergence and the percentage of phylogenetically informative characters were calculated by MEGA4 [37].
Phylogenetic tree was built using maximum parsimony (MP) and Bayesian inference (BI) methods. MP analyses were performed using heuristic search with 1000 replicates of random stepwise addition and tree bisection-reconnection (TBR) branch swapping in PAUP version 4.0b10 [38]. Gaps were treated as missing data. Bootstrap resampling [39] was conducted to assess topological robustness with 1000 replicates. BI analyses were performed in MrBayes version 3.1.2 [40] by Metropolis-coupled Markov Chain Monte Carlo algorithm. Sequences of each gene were divided into three different partitions (exon, intron and insertion), and the combined data have six partitions. GTR+I+G model was applied for the exon of Os125, GTR+G model for the insertion of SDBE, and HKY model for the rest. Four Markov chains were conducted for 1,000,000 generations, trees were sampled every 100 generations, and then the first 2500 trees were discarded in the burn-in period. Optimal models and parameters under the Akaike Information Criterion (AIC) were determined by Modeltest 3.06 [41] for Bayesian analyses. When different alleles from heterozygotes were grouped into one clade, one of them was excluded randomly in phylogeny of the combined data, unless they were otherwise grouped into different clades. Congruence between SDBE and Os125 was evaluated using the partition homogeneity test (PHT) [42], implemented in PAUP with 1000 replicates, random taxon addition (10 replicates), and one tree saved per replicate. Results from the PHT indicated that incongruence between these two genes was P = 0.01, ten folds higher than the suggested (P < 0.001) by Cunningham [43].
Divergence times were estimated by Bayesian dating methods [44–46], using the programs Baseml [47], Estbranches [44] and Multidivtime [46]. Splitting times of O. officinalis complex from its affiliated genus Oryza and tribe Oryzeae were determined through the plastid gene matK of 11 representatives and two outgroups (Table 3). A recent report suggested that origin of Oryzeae was about 34.5 ± 6.8 Mya [48], based on newly discovered pollen fossils [49, 50] and phytoliths [51]. These dates were used as the maximum and the minimum constraints to the crown node of Oryzeae, respectively. Other settings were F84+G model [52] and 100,000 MCMC (markov chain monte carlo) iterations, with rttm and rttmsd set at 6.0, rtrate and rtratesd set at 0.02, brownmean and brownsd set at 0.16, and big time set at 100.
For divergence time estimation within the O. officinalis complex, an MP tree with 19 sequences representing the taxa of the complex (Table 1) was applied. Insert sequences were excluded due to their considerably variable lengths. Calculations were performed using the same Bayesian relaxed clock methods stated above. Dating constraints between the complex and its outgroup, and between the first clades split within the complex, were set as 13.6 ± 3.6 Mya, and 8.0 ± 2.9 Mya respectively, which were determined by the dating of Oryzeae as described above. Other specific parameters were set as follows: rttm and rttmsd set to 1.36, rtrate and rtratesd to 0.04, brownmean and brownsd to 0.7, according to preliminary dating analysis.
Preparation of genome specific sequences
C-genome-specific sequences (against B-genome) were isolated by a modified subtractive hybridization methods [53] as follows: genomic DNAs from O. officinalis (CC, Accession 102460) and O. punctata (BB, Accession 105607) were digested with MseI (New England Biolabs, Beverly, MA, USA) into 500 to 1000 bp fragments; and then the fragments of C-genome were ligated with adapter-C and those of B genome were ligated with biotinylated adapter-B (Table 4). Ligation efficiency was checked by PCR amplification using adapter specific primers, C-adp1 and B-adp1. The C-genome ligation was denatured and annealed together with excess B-genome ligation in a single tube. The anneal temperature was 68°C with 0.99 M sodium salt overnight, and then the supernatant containing C-genome-specific sequences was selectively recovered from the reaction mix with streptavidin-coated magnetic beads (Dynabeads, Dynal, Lake Success, NY, USA). A more round of subtracting process was necessary to enrich the genome-specific sequences. Finally, molecules containing the genome-specific sequences were amplified with C-adp1 as primer, and then used for the plasmid transformation.
The transformed plasmids were sequenced, and the sequences were BLAST searched in GenBank. Then a series of primers (Table 4) designed from the sequences were used to test whether the sequences were genome-specific or not by PCR amplification onto the related accessions, no correspond bands were seen in BB diploid species (date not shown). Further, the genome-specific sequences were labelled as probes and finally verified by fluorescent in situ hybridization (FISH). The FISH images showed no signal on the chromosomes and nuclei of BB diploid species (data not showed). All these data confirmed that the sequences were C-genome-specific.
Cytogenetics analysis
Chromosome spreads were prepared by enzymatic maceration/air-dry method [54, 55]. Total genomic, genome-specific and 45S rDNA probes were labelled by nick translation with biotin-16-dUTP (Roche Diagnostics GmbH, Mannheim, Germany) or DIG-11-dUTP (Roche Diagnostics GmbH, Mannheim, Germany), respectively. Multicolor fluorescence in situ hybridization (FISH) was performed as described [29] with slight modification. After overnight hybridization, the slides were given a stringent wash in 20% (v/v) formamide in 0.1× SSC at 42°C, resulting in 80%-85% stringency. The biotinylated-probes were detected by avidin-FITC (Roche Diagnostics GmbH, Mannheim, Germany), and the digoxigenin-labelled probes by anti-digoxigenin rhodamine conjugate (Roche Diagnostics GmbH, Mannheim, Germany). The chromosome spreads were mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA), and examined under a Leica DMRBE microscope (Leica, Wetzlar, Germany). Photographs were captured by a SPOT cooled color digital camera system (Diagnostic instruments Inc., MI, USA), then imported into Adobe Photoshop 7.0 (Adobe Systems Inc., San Jose, CA, USA) for processing.
Results
Sequence characterization
Two distinct sequences in both SDBE and Os125 genes were identified from each accession of all tetraploids. One was longer (740-2272 bp in SDBE and 871-1021 bp in Os125) and the other was shorter (420-458 bp in SDBE and 400-701 bp in Os125). Each longer sequence was highly similar to and phylogenetically grouped with the corresponding sequences of CC diploids (Figures 1, 2, 3), and thereby was named as C-like copy. Each shorter one was similar to that of BB or EE diploids, and thus was named as B- or E- like copy accordingly. Aligned sequence of SDBE (2337 bp) had 101 (4.3%) informative sites, and that of the Os125 (1112 bp) contained 164 (14.7%) informative sites. The combined sequence of these two genes was aligned to be 3449 bp in length (Table 5).

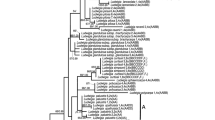
Strict consensus trees of SDBE gene from 72 most parsimonious trees. Numbers above branches: bootstrap values (only those > 50% showed), below: Posterior probability (only those > 0.5 showed). a or b: alleles of a heterozygous locus. 2× or 4×: ploidy levels. Dash lines indicated the nodes supported by Bayesian inference. Tree length = 184, Consistency index (CI) = 0.8859, Retention index (RI) = 0.9694, Bayesian inference -ln L = -4517.46 (Gray Square: C and C-like copy; White Square: B and B-like copy; Black Square: E and E-like copy).

Strict consensus trees of Os125 genes from 500 most parsimonious trees. Numbers above branches: bootstrap values (only those > 50% showed), below: Posterior probability (only those > 0.5 showed). a or b: alleles of a heterozygous locus. 2× or 4×: ploidy levels. Tree length = 319, Consistency index (CI) = 0.8715, Retention index (RI) = 0.9807, Bayesian inference -ln L = -3585.67 (Gray Square: C and C-like copy; White Square: B and B-like copy; Black Square: E and E-like copy).

Strict consensus trees of combined data set from 126 most parsimonious trees. Numbers above branches: bootstrap values (only those > 50% showed), below: Posterior probability (only those > 0.5 showed). a or b: alleles of a heterozygous locus. 2× or 4×: ploidy levels. Tree length = 463, Consistency index (CI) = 0.8985, Retention index (RI) = 0.9771, Bayesian inference -ln L = -7735.44 (Gray Square: C and C-like copy; White Square: B and B-like copy; Black Square: E and E-like copy).
A ~320 bp insertion in SDBE and a ~150 bp insertion in Os125 were recognized in each of the C-genome-bearing species. BLAST searches in the TIGR rice repeated database http://www.tigr.org/tdb/e2k1/plant.repeats/ and then mask against Repbase Update http://www.girinst.org using CENSOR [56], identified the insertion of SDBE to be MITE-MDM2 (miniature inverted transposable element-MDM2), but no matching sequence of the Os125 insertion was found. The Os125 insertion was flanked by a short direct repeat (sequence: TACATGGCTCTTTC), but no terminal inverted-repeating sequence nor tRNA-related region was found, suggesting that this fragment is an unidentified retrotransposable element instead of a SINE (short interspersed repetitive element) [57].
In addition, a partial fragment (~1.5 kb) of L1-type retrotransposon family was found to insert into C-like SDBE gene in some accessions of O. alta and O. grandiglumis.
Phylogeny reconstruction based on SDBE, Os125and combineddataset
Phylogenetic analyses of SDBE, Os125 and combined dataset using maximum parsimony (MP) and Bayesian inference (BI), all yielded similar topologies. Parsimony analysis yielded 72, 500 and 126 equally most parsimonious trees, from SDBE, Os125 and combined dataset, respectively. The strict consensus trees of each dataset were showed in Figures 1, 2 and 3 with general features as follows: (a) The main clades were strongly supported by bootstrap values and Bayesian posterior probability. (b) B-, C- and E-like copies in tetraploid species formed three monophyletic clades with the corresponding sequences of BB, CC and EE diploid species, respectively. (c) In the C-genome clade, two monophyletic clades were formed, one involving O. eichingeri and the tetraploid O. punctata, and the other covering all the rest C-genome species. (d) The B-like copies of BBCC tetraploid species were divided into two clades, one including the Africa endemic O. punctata (comprising BB diploid and BBCC tetraploid), and the other including the Asian tetraploids only. We also put the SDBE and Os125 sequences of O. sativa (A-genome, GenBank Accession No. AB012915 and AP004885) into the datasets, but the positions of B-, C- and E-like copies, and the topology of the inferred cladegram, remained unchanged (data not showed).
With more informative sites, the cladograms of the complex constructed from Os125 and combined dataset were more resolvable (Figures 2, 3). O. officinalis (CC) and C-like copies of the BBCC tetraploids, i.e., O. malampuzhaensis and O. minuta, were consistently united into one clade. The clade was further grouped with C-like copies of all CCDD tetraploid species. Apart from the C-like copies of Os125 sequences, two E-like copies were isolated from CCDD species, which formed two clades in parallel and finally grouped with O. australiensis (EE) trichotomously in both MP and BI trees (Figure 2).
It is noteworthy that two alleles of the heterozygous accessions were grouped with each other, except eic_LAK1 and eic_UGA2, in which one of the alleles was clustered with that of different species (Figures 1, 2), suggesting that interspecific hybridization and introgression in those accessions occurred, as proposed by previous research [58]. For those heterozygous loci, the allele that clustered into the O. eichingeri clade, was selected in the combined dataset.
Divergence Dates
As showed in Figure 4, the O. officinalis complex was estimated to diverge from the rest of the genus Oryza at 7.9 ± 1.6 Mya, and the separation between B- and C- genomes took place at 4.8 ± 1.3 Mya. The molecular dating indicated that three C-genome diploid species radiated between ca. 0.9-1.8 Mya during Pliocene. In BBCC tetraploid species, C-like copy of O. punctata diverged from C genome of O. eichingeri at 0.3 Mya, very close to the divergent time (0.5 Mya) of the B-like copy from the B genome of diploid O. punctata. In other two BBCC species (O. malampuzhaensis and O. minuta), the C-like copies diverged from their common paternal progenitor (O. officinalis alike) at ca. 0.6 Mya, later than the divergence time of their B-like copies from the B-genome of O. punctata (BB) at ca. 1.8 Mya. Similarly, the divergence time between C-like copies of CCDD tetraploids and their C-genome donor, was set at ca. 0.9 Mya, while the node to separate their D-genomes from O. australiensis (EE) was dated at ca. 2.8 Mya (Figure 4).

Divergence times of main lineages in O. officinalis complex. Calculated by Bayesian relaxed-clock methods (details see Materials and Methods). Estimated Mya and the standard deviation were noted above the branches. 2×: diploid; 4×: tetraploid (Gray Square: C and C-like copy; White Square: B and B-like copy; Black Square: E and E-like copy).
FISH analysis
Figure 5 shows multicolor fluorescent in situ hybridization images of O. officinalis complex, hybridized by C-genome-specific probes (red), together with B-genome probes (green) or E-genome probes (green), counterstained by DAPI (blue).

Multicolor fluorescent in situ hybridization images of O. officinalis complex. Hybridized by C genome-specific probes (red), together with B genome probes (green, e-g, n-s) or E genome probes (green, h-m, t-v), counterstained by DAPI (blue, a-d, k-m). Arrows indicated inter-genomic (B-C genomes) translocations, which were enlarged in the below box (e-g). (a) O. officinalis (CC), and its karyotype (b). (c) O. eichingeri (CC), and its karyotype (d). (e-f) O. punctata (BBCC). (g) O. malampuzhaensis (BBCC). (h, k) O. latifolia (CCDD). (i, l) O. grandiglumis (CCDD). (j, m) O. alta (CCDD). Parental genomes in the nuclei of allotetraploids, separated spatially in O. punctata (n, interphase; o, prophase; p, anaphase), in O. malampuzhaensis (q and r, interphase stages; s, anaphase), and in interphase nuclei of O. latifolia (t), O. grandiglumis (u), and O. alta (v), respectively. Bar, 5 μm.
Figures 5a-d show that the C-genome-specific probes were localized on all chromosomes of two diploid CC species, O. officinalis and O. eichingeri. The C- genome-specific sequences were scattered non-uniformly along each of the chromosome as well as among twelve homologous pairs, as the FISH patterns showed obviously (Figures 5a, c). The total 24 chromosomes were karyotypically arranged into twelve homologous pairs according to their FISH patterns, relative length, centromere position and heterochromatin, as showed in Figure 5b and Figure 5d.
Figures 5e and 5f show the FISH images of the tetraploid O. punctata using digoxigenin-labelled C-genome-specific probes and biotin-labelled total genomic DNA of diploid O. punctata (BB). B- and C-genomes were clearly discriminated in the same nucleus, where 24 chromosomes showed strong bright green signals of the B-genome probes, and the rest 24 chromosomes showed strong red signals of the C-genome-specific probes (Figures 5e, f). It is worth to notice that two pairs of B-genome chromosomes were clearly involved in inter-genomic translocations with the C-genome, one small and the other rather large.
Figure 5g shows that a prometaphase nucleus of O. malampuzhaensis was hybridized with C- genome-specific probes (red) together with B-genome probes (green). The 24 B-genome chromosomes exhibited strong green signals, and the rest 24 chromosomes belonging to C-genome showed bright red signals. Two B-genome signals were identified at short arm terminals of one pair of C-genome chromosomes. However, when O. malampuzhaensis was hybridized with B-genome probes (green) together with 45S rDNA probes (red), these two C-genome chromosomes with 45S rDNA signals were also painted by B-genome signals on same areas (Additional file 1). Therefore, in O. malampuzhaensis which was with different origin from tetraploid O. punctata, B-genome signals located on the two C-genome chromosomes may not be inter-genomic translocation but homologous sequences of 45S rDNAs.
Multicolor FISH was also used for three CCDD species, where two probes were applied, one from O. australiensis (EE) genome (labelled in green), and one from the C-genome-specific probe (labelled in red). Figure 5h shows in O. latifolia, strong C-genome-specific signals (orange) painted 24 chromosomes, while green signals (from the E-genome probes) stained all chromosomes, in which 24 chromosomes with pure green signals should belong to the D-genome. The FISH patterns of O. latifolia differed remarkably from those of O. alta and O. grandiglumis. In O. latifolia all chromosomes were painted by E-genome signals (Figures 5h, K), whereas in O. alta and O. grandiglumis all chromosomes were painted by C-genome signals (red), in which merely some of the chromosomes showed the E-genome signals (green) faintly or strongly near centromere regions (Figures 5i-j; 5l-m). This difference could also be seen in the interphase cells, as showed in Figures 5t-v, where nuclei of O. latifolia were dominantly painted by E-genome probes while those of O. alta and O. grandiglumis were strongly painted by C-genome-specific probes with dot-like signals of E-genome probes.
Figure 5n-s shows each of the two parental genomes separated spatially in BBCC tetraploid species in interphase, prophase and anaphase nuclei. In O. malampuzhaensis, about 10 chromocenters of B-genome were found at late stage of interphase (Figure 5r); however, no similar chromocenters were found in O. punctata.
Discussion
The key to trace the complicated evolution process of polyploid complex lies on a universal criterion. C-genome in O. officinalis complex could play such a role. As the pivotal genome, C-genome participated each of the polyploid formation in the complex, and its evolution process in genomic differentiation and geographical patterning can therefore reflect the temporal and spatial history of polyploid evolution.
Genomic relationships in O. officinaliscomplex
In O. officinalis complex, four extant genomes, B, C, D or E, were identified [14, 21, 27]. The present study showed that each genome in the complex occurred only once when rooted by the outgroup, O. granulata, where E-genome sited at the basal position of the complex. The clade of O. officinalis complex was first divided into two clades, E-genome clade and the other clade involving B-and C-genomes. In E-genome clade, D-genome was located as E's sister group. These results were consistent with previous reports [14, 21–23]. In the other clade, C-genomes in different diploid species had differentiated apparently thereafter they partook in different polyploid formation (Figures 1, 2, 3), in agreement with other authors [58–61].
Although there is only one extant diploid with B-genome, O. punctata, the B-genomes in tetraploids were differentiated, as revealed by AFLP [21], RFLP [62], SSR [60] and GISH [29]. In this study, multicolor FISH (Figure 5g) revealed that the B-genome of O. punctata (BB) was clearly diverged from that of O. malampuzhaensis. Further evidence of molecular phylogeny and dating showed that the divergence happened even before polyploidization, which formed O. malampuzhaensis and O. minuta (Figures 1, 2, 3, 4). Therefore, a diploid B-genome species extinct nowadays in Asia was assumed to be the direct genomic donor of Asian distributed BBCC tetraploids.
Since no diploid DD species has ever been found, the D donor for the CCDD tetraploids has long been controversial [14, 27, 28, 32]. The Australian diploid, O. australiensis, as the unique E-genome holder, was assumed to be D-genome donor by several authors [14, 32]. Nevertheless, genomic comparison by GISH and retrotransposon analysis found obvious differences between D- and E-genomes, and thus suspected E as the direct donor [28, 63]. Based on a universal criterion of C-genome differentiation, our study in phylogeny and molecular dating (Figures 1, 2, 3, 4) showed that D- and E-genome were tied together as sister group, but they diverged much earlier than CCDD tetraploid formation. Multicolor FISH using E-genome probes for the CCDD tetraploids also revealed obvious differentiation between D- and E-genome, and this was even remarkable in D-genome itself, as showed in Figure 5, where the D-genomes of O. alta and O. grandiglumis exhibited sharply different from that of O. latifolia.
C-genome variation and polyploid evolution in O. officinaliscomplex
To date six tetraploid species, three BBCC and three CCDD, have been recorded in O. officinalis complex, and all are C-genome carriers [16, 64]. The relationship and origin of the tetraploids have long been in debate [14, 21, 22, 32]. In this study, C-genome of diploid O. eichingeri was localized at the basal of C-genomes, and it subsequently diverged, resulting two C-genome diploids, O. rhizomatis and O. officinalis. Later on, the three C diploids participated separately in hybridization and polyploidization, finally forming six tetraploids. For O. eichingeri, it merely joined formation of O. punctata (BBCC), while O. officinalis (CC) partook in formation of O. malampuzhaensis and O. minuta. On the other hand, a species closely related to present O. officinalis (or O. rhizomatis) offered its C-genome to the three CCDD tetraploids, O. alta, O. grandiglumis and O. latifolia (Figures 1, 2, 3).
C-genomes in different BBCC tetraploids confronted variable fates, such as changes by inter-genomic translocation. Multicolor FISH probing different genomes in an allopolyploid can be a powerful indicator for identifying such changes. As showed in multicolor FISH (Figure 5e-g), inter-genomic translocations between C-and B-genomes were visualized for the first time in two tetraploids of the complex, which was speculated as the result of diploidization impact [5, 8, 65, 66]. In O. punctata two fragments of C-chromosomes were translocated to different B-chromosomes, while in O. malampuzhaensis no obvious inter-genomic translocation was found. Although C-genomes experienced different history in various polyploid formations, few fragments of C-genome-specific were detectably lost after hybridization and polyploidization, as found in multicolor FISH with genome-specific probes (Figures 5e-g, n-s).
The fate of C-genomes in CCDD tetraploids was different even more. In O. alta and O. grandiglumis C-genome-specific probes apparently dominated the nuclei, most probably by inter-genomic invasion [5, 8, 67, 68], as showed in multicolor FISH images, while in O. latifolia C-genome kept almost unchanged (Figures 5 5h-m, t-v). Considering that O. alta and O. grandiglumis diverged from O. latifolia (Figure 4), the inter-genomic invasion would have happened during their speciation.
Temporal and spatial evolution of O. officinaliscomplex
Geographical pattern of intercontinental pantropics in O. officinalis complex, framed by its relatively recent history, makes its evolution process paradoxical for long time. Based on molecular clock of matK and GAP1 sequences, the origin of the complex was dated at late Miocene (ca.9 Mya) [69], and speciation of O. australiensis was set in ca. 8.5 Mya through Adh2 gene [63]. However, re-dating the origin and divergence times became necessary, because (a) previous dating dealt mainly with diploids while the polyploids evolution history remained unclear; (b) new molecular timescales based on non-parametric rate smoothing, penalized likelihood, and Bayesian-relaxed clock methods have been recently developed for the grasses [70].
In this study, the estimated divergence time between O. officinalis complex and its outgroup, O. granulata, was 13.6 ± 3.6 Mya, earlier than the previous suggestion, and the time of the first species divergence in the complex, was 7.9 ± 1.6 Mya (Figure 4). C-genome was separated with B-genome at about 4.8 Mya, and then C-genome itself was split into two clades in approximately 1.8-0.9 Mya, one including O. eichingeri and the other including the rest two CC diploid species. These times were earlier than previously suggested [58], but closed to recent research [24, 25]. The time of polyploidization to synthesize tetraploids was estimated to be ca. 0.3-0.9 Mya in Pleistocene, in which the CCDD species (ca. 0.7-0.9 Mya) were formed obviously earlier than BBCC species, also closed to that recently reported [24].
If all C-genome species separated no earlier than two Mya, the distribution of these species can be feasibly explained by long-distance dispersal rather than vicariance, given that the continental splitting was much earlier than the species radiation. As suggest by Vaughan et al [11, 12], animal migration may play a role for this complex in seed dispersal between Asia and Africa. Bird could be another carrier, which could account for the disjunctive distribution of some Oryza species, such as O. eichingeri [11, 58]. For the CCDD tetraploids, this and previous studies [32] both revealed that their putative parents were O. officinalis and O. australiensis. The problem was that the putative parents were confined to south Asia-Australia but the CCDD tetraploids were nowadays endemic to the tropics of Americas. Therefore, a new pathway to bridge these two continents for long-distance dispersal was put forward (Figure 6). The strong floristic affinities between the South America and the antipodes were also confirmed by biogeographical studies of other Poaceae species [70, 71]. However, how the species in the complex could transcontinentally spread across the oceans, remains mysterious.

Biogeographical scenario of species with C genome in O. officinalis complex. Above: divergence time calculated using Bayesian relaxed-clock methods (the cladegram narrowed from Figure 2). Below: Distribution areas (outline and noted) and inferred migration procedure among continents. CC diploids (eic = O. eichingeri, off = O. officinalis, rhi = O. rhizomatis), BBCC tetraploids (pun = O. punctata, mal = O. malampuzhaensis, min = O. minuta), and CCDD tetraploids (lat = O. latifolia, alt = O. alta, gra = O. grandiglumis).
Conclusion
The genomic relationships, polyploid formations and divergence times in Oryza officinalis complex of the genus Oryza, were revealed based on DNA sequences and FISH evidence. Focused on C-genome, the "pivotal genome" of the polyploids, we found that the polyploids were formed by stepwise polyploidizations in ca. 0.3-0.9 Mya, followed by a series of inter-genomic translocations and invasions. The pantropical distribution of the complex was suggested to be formed by long-distance dispersal transcontinentally, instead of vicariance. This study offers a typical example in tracing tempo-spatial process of polyploidization, and for the first time it gives new stands for the complex in dating the detailed times of polyploid formation, visualizing inter-genomic changes, and viewing the spatial evolution history of the polyploids.
References
Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng CF, Sankoff D, dePamphilis CW, Wall PK, Soltis PS: Polyploidy and angiosperm diversification. American Journal of Botany. 2009, 96 (1): 336-348. 10.3732/ajb.0800079.
Cui LY, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A, et al: Widespread genome duplications throughout the history of flowering plants. Genome Research. 2006, 16 (6): 738-749. 10.1101/gr.4825606.
Comai L: The advantages and disadvantages of being polyploid. Nature Reviews Genetics. 2005, 6 (11): 836-846. 10.1038/nrg1711.
Hegarty M, Hiscock S: Polyploidy: Doubling up for evolutionary success. Current Biology. 2007, 17: R927-R929. 10.1016/j.cub.2007.08.060.
Wendel JF: Genome evolution in polyploids. Plant Molecular Biology. 2000, 42 (1): 225-249. 10.1023/A:1006392424384.
Adams KL, Wendel JF: Polyploidy and genome evolution in plants. Current Opinion in Plant Biology. 2005, 8 (2): 135-141. 10.1016/j.pbi.2005.01.001.
Chen ZJ, Ha M, Soltis D: Polyploidy: genome obesity and its consequences. New Phytologist. 2007, 174 (4): 717-720. 10.1111/j.1469-8137.2007.02084.x.
Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, Wendel JF: Evolutionary Genetics of Genome Merger and Doubling in Plants. Annual Review of Genetics. 2008, 42: 443-461. 10.1146/annurev.genet.42.110807.091524.
Chen ZJ, Ni ZF: Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays. 2006, 28 (3): 240-252. 10.1002/bies.20374.
Zohary D, Feldman M: Hybridization between amphidiploids and evolution of polyploids in wheat (Aegilops-Triticum) group. Evolution. 1962, 16 (1): 44-61. 10.2307/2406265.
Vaughan DA, Ge S, Kaga A, Tomooka N: Phylogeny and Biogeography of the Genus Oryza. Biotechnology in Agriculture and Forestry. Edited by: Hirano HHA, Sano Y, Sasaki T. 2008, 219-234. full_text.
Vaughan DA, Kadowaki K, Kaga A, Tomooka N: On the phylogeny and biogeography of the genus Oryza. Breeding Science. 2005, 55 (2): 113-122. 10.1270/jsbbs.55.113.
Vaughan DA, Morishima H, Kadowaki K: Diversity in the Oryza genus. Current Opinion in Plant Biology. 2003, 6 (2): 139-146. 10.1016/S1369-5266(03)00009-8.
Ge S, Sang T, Lu BR, Hong DY: Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proceedings of the National Academy of Sciences of the United States of America. 1999, 96 (25): 14400-14405. 10.1073/pnas.96.25.14400.
Nishikawa T, Vaughan DA, Kadowaki K: Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theoretical and Applied Genetics. 2005, 110 (4): 696-705. 10.1007/s00122-004-1895-2.
Vaughan DA: The genus Oryza L. Current status of taxonomy. IRRI Res Paper Series. 1989, 138: 1-21.
Lu BR, Khush GS, Kinoshita T: Taxonomy of the genus Oryza (Poaceae): Historical perspective and current status. International Rice Research Notes. 1999, 24 (3): 4-8.
Tateoka T: Taxonomic studies of Oryza. III. Key to the species and their enumeration. Bot Mag Tokoy. 1963, 76: 165-173.
Tateoka T: Taxonomic studies of Oryza. II. Several species complexes. Bot Mag Tokyo. 1962, 75: 455-461.
Joshi SP, Gupta VS, Aggarwal RK, Ranjekar PK, Brar DS: Genetic diversity and phylogenetic relationship as revealed by inter simple sequence repeat (ISSR) polymorphism in the genus Oryza. Theoretical and Applied Genetics. 2000, 100 (8): 1311-1320. 10.1007/s001220051440.
Aggarwal RK, Brar DS, Nandi S, Huang N, Khush GS: Phylogenetic relationships among Oryza species revealed by AFLP markers. Theoretical and Applied Genetics. 1999, 98 (8): 1320-1328. 10.1007/s001220051198.
Wang ZY, Second G, Tanksley SD: Polymorphism and phylogenetic-relationships among species in the genus Oryza as determined by analysis of nuclear RFLP. Theoretical and Applied Genetics. 1992, 83 (5): 565-581. 10.1007/BF00226900.
Zou XH, Zhang FM, Zhang JG, Zang LL, Tang L, Wang J, Sang T, Ge S: Analysis of 142 genes resolves the rapid diversification of the rice genus. Genome Biology. 2008, 9 (3): R49-10.1186/gb-2008-9-3-r49.
Lu F, Ammiraju JSS, Sanyal A, Zhang SL, Song RT, Chen JF, Li GS, Sui Y, Song X, Cheng ZK, et al: Comparative sequence analysis of MONOCULM1-orthologous regions in 14 Oryza genomes. Proceedings of the National Academy of Sciences of the United States of America. 2009, 106 (6): 2071-2076. 10.1073/pnas.0812798106.
Ammiraju JSS, Lu F, Sanyal A, Yu Y, Song X, Jiang N, Pontaroli AC, Rambo T, Currie J, Collura K, et al: Dynamic Evolution of Oryza Genomes Is Revealed by Comparative Genomic Analysis of a Genus-Wide Vertical Data Set. Plant Cell. 2008, 20 (12): 3191-3209. 10.1105/tpc.108.063727.
Zuccolo A, Sebastian A, Talag J, Yu Y, Kim H, Collura K, Kudrna D, Wing RA: Transposable element distribution, abundance and role in genome size variation in the genus Oryza. BMC Evolutionary Biology. 2007, 7: 152-10.1186/1471-2148-7-152.
Fukui K, Shishido R, Kinoshita T: Identification of the rice D-genome chromosomes by genomic in situ hybridisation. Theoretical and Applied Genetics. 1997, 95 (8): 1239-1245. 10.1007/s001220050687.
Li CB, Zhang DM, Ge S, Lu BR, Hong DY: Differentiation and inter-genomic relationships among C, E and D genomes in the Oryza officinalis complex (Poaceae) as revealed by multicolor genomic in situ hybridization. Theoretical and Applied Genetics. 2001, 103 (2-3): 197-203. 10.1007/s001220100562.
Li CB, Zhang DM, Ge S, Lu BR, Hong DY: Identification of genome constitution of Oryza malampuzhaensis, O. minuta, and O. punctata by multicolor genomic in situ hybridization. Theoretical and Applied Genetics. 2001, 103 (2-3): 204-211. 10.1007/s001220100563.
Dally AM, Second G: Chloroplast DNA diversity in wild and cultivated species of rice (Genus Oryza, section Oryza). Cladistic-mutation and genetic-distance analysis. TAG Theoretical and Applied Genetics. 1990, 80 (2): 209-222.
Kanno A, Hirai A: Comparative studies of the structure of chloroplast DNA from four species of Oryza: cloning and physical maps. TAG Theoretical and Applied Genetics. 1992, 83 (6): 791-798.
Bao Y, Ge S: Origin and phylogeny of Oryza species with the CD genome based on multiple-gene sequence data. Plant Systematics and Evolution. 2004, 249 (1-2): 55-66. 10.1007/s00606-004-0173-8.
Jena KK, Kochert G: Restriction fragment length polymorphism analysis of CCDD genome species of the genus Oryza L. Plant Molecular Biology. 1991, 16 (5): 831-839. 10.1007/BF00015075.
Doyle JJ, Doyle JL: A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987, 19: 11-15.
Nakamura Y, Umemoto T, Ogata N, Kuboki Y, Yano M, Sasaki T: Starch debranching enzyme (R-enzyme or pullulanase) from developing rice endosperm: purification, cDNA and chromosomal localization of the gene. Planta. 1996, 199 (2): 209-218. 10.1007/BF00196561.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997, 25 (24): 4876-4882. 10.1093/nar/25.24.4876.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Swofford DL: PAUP*. Phylogenetic analysis using parsimony (* and other methods), Version 4.0b10. 2002, Sunderland, MA: Sinauer and Associates
Felsenstein J: Confidence limits on phylogeny: an approach using the bootstrap. Evolution. 1985, 39: 783-791. 10.2307/2408678.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818. 10.1093/bioinformatics/14.9.817.
Farris JS, Kallersjo M, Kluge AG, Bult C: Testing significance of incongruence. Cladistics-the International Journal of the Willi Hennig Society. 1994, 10 (3): 315-319. 10.1111/j.1096-0031.1994.tb00181.x.
Cunningham CW: Can three incongruence tests predict when data should be combined?. Molecular Biology and Evolution. 1997, 14 (7): 733-740.
Thorne JL, Kishino H, Painter IS: Estimating the rate of evolution of the rate of molecular evolution. Molecular Biology and Evolution. 1998, 15 (12): 1647-1657.
Thorne JL, Kishino H: Divergence time and evolutionary rate estimation with multilocus data. Systematic Biology. 2002, 51 (5): 689-702. 10.1080/10635150290102456.
Kishino H, Thorne JL, Bruno WJ: Performance of a divergence time estimation method under a probabilistic model of rate evolution. Molecular Biology and Evolution. 2001, 18 (3): 352-361.
Yang ZH: PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences. 1997, 13 (5): 555-556.
Vicentini A, Barber JC, Aliscioni SS, Giussani LM, Kellogg EA: The age of the grasses and clusters of origins of C-4 photosynthesis. Global Change Biology. 2008, 14 (12): 2963-2977. 10.1111/j.1365-2486.2008.01688.x.
Jacobs BF, Kingston JD, Jacobs LL: The origin of grassdominated ecosystems. Annals of the Missouri Botanical Garden. 1999, 86: 590-643. 10.2307/2666186.
Kellogg EA: Evolutionary history of the grasses. Plant Physiology. 2001, 125: 1198-1205. 10.1104/pp.125.3.1198.
Prasad V, Stromberg CAE, Alimohammadian H, Sahni A: Dinosaur coprolites and the early evolution of grasses and grazers. Science. 2005, 310 (5751): 1177-1180. 10.1126/science.1118806.
Kishino H, Hasegawa M: Evaluation of the maximum-likelihood estimate of the evolutionary tree topologies from DNA-sequence date, and the branching order in hominoidea. Journal of Molecular Evolution. 1989, 29 (2): 170-179. 10.1007/BF02100115.
Gurskaya NG, Diatchenko L, Chenchik A, Siebert PD, Khaspekov GL, Lukyanov KA, Vagner LL, Ermolaeva OD, Lukyanov SA, Sverdlov ED: Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: Cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Analytical Biochemistry. 1996, 240 (1): 90-97. 10.1006/abio.1996.0334.
Fukui K, Iijima K: Somatic chromosome map of rice by imaging methods. Theoretical and Applied Genetics. 1991, 81 (5): 589-596. 10.1007/BF00226723.
Fukui K, Ohmido N, Khush GS: Variability in rDNA loci in the genus Oryza detected through fluorescence in-situ hybridization. Theoretical and Applied Genetics. 1994, 87 (8): 893-899. 10.1007/BF00225782.
Kohany O, Gentles AJ, Hankus L, Jurka J: Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. Bmc Bioinformatics. 2006, 7: 10.1186/1471-2105-7-474.
Okada N, Shedlock AM, Nikaido M: Retroposon Mapping in Molecular Systematics. Mobile genetic elements: protocols and genomic applications. Edited by: Capy WJMaP. 2004, Totowa (NJ): Humana Press, 189-226.
Zhang LB, Ge S: Multilocus analysis of nucleotide variation and speciation in Oryza officinalis and its close relatives. Molecular Biology and Evolution. 2007, 24 (3): 769-783. 10.1093/molbev/msl204.
Bao Y, Ge S: Phylogenetic relationships among diploid species of Oryza officinalis complex revealed by multiple gene sequences. Acta Phytotaxonomica Sinica. 2003, 41 (6): 497-508.
Bao Y, Zhou HF, Hong DY, Ge S: Genetic diversity and evolutionary relationships of Oryza species with the B- and C-genomes as revealed by SSR markers. Journal of Plant Biology. 2006, 49 (5): 339-347.
Bao WD, Zhang WL, Yang QY, Zhang Y, Han B, Gu MH, Xue YB, Cheng ZK: Diversity of centromeric repeats in two closely related wild rice species, Oryza officinalis and Oryza rhizomatis. Molecular Genetics and Genomics. 2006, 275 (5): 421-430. 10.1007/s00438-006-0103-2.
Federici MT, Shcherban AB, Capdevielle F, Francis M, Vaughan D: Analysis of genetic diversity in the Oryza officinalis complex. Electronic Journal of Biotechnology. 2002, 5 (2): 173-181.
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA, et al: Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Research. 2006, 16 (10): 1262-1269. 10.1101/gr.5290206.
Vaughan DA: The wild relatives of rice: a genetic resources handbook. 1994, Manila, The Philippines: International Rice Research Institution
Lim KY, Kovarik A, Matyasek R, Chase MW, Clarkson JJ, Grandbastien MA, Leitch AR: Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytologist. 2007, 175 (4): 756-763. 10.1111/j.1469-8137.2007.02121.x.
Weiss-Schneeweiss H, Schneeweiss GM, Stuessy TF, Mabuchi T, Park JM, Jang CG, Sun BY: Chromosomal stasis in diploids contrasts with genome restructuring in auto- and allopolyploid taxa of Hepatica (Ranunculaceae). New Phytologist. 2007, 174 (3): 669-682. 10.1111/j.1469-8137.2007.02019.x.
Belyayev A, Raskina O, Korol A, Nevo E: Coevolution of A and B genomes in allotetraploid Triticum dicoccoides. Genome. 2000, 43 (6): 1021-1026. 10.1139/gen-43-6-1021.
Clarkson JJ, Lim KY, Kovarik A, Chase MW, Knapp S, Leitch AR: Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytologist. 2005, 168 (1): 241-252. 10.1111/j.1469-8137.2005.01480.x.
Guo YL, Ge S: Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. American Journal of Botany. 2005, 92 (9): 1548-1558. 10.3732/ajb.92.9.1548.
Inda LA, Segarra-Moragues JG, Muller J, Peterson PM, Catalan P: Dated historical biogeography of the temperate Loliinae (Poaceae, Pooideae) grasses in the northern and southern hemispheres. Molecular Phylogenetics and Evolution. 2008, 46 (3): 932-957. 10.1016/j.ympev.2007.11.022.
Soreng RJ, Davis JI: Phylogenetic structure in Poaceae subfamily Pooideae as inferred from molecular and morphological characters: Misclassification versus reticulation. Grasses: Systematics and Evolution. Edited by: Jacobs SWL, Everett J. 2000, CSIRO, Collingwood, 61-74.
Acknowledgements
We thank Drs. Yuzhu Dong, Junxia Yuan and Zhukuan Cheng for their technical assistance and helpful suggestions on the manuscript. We are also grateful to the International Rice Research Institute (Los Banos, Philippines) for providing seed samples. This work was supported by the National Natural Science Foundation of China (30430030) and Program for Key International S & T Cooperation project of P. R. China (2009CB119102).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
BW carried out the molecular and cytogenetic studies, wrote the manuscript and participated in the design of the study. ZD, WL and JP made equal contributions in chromosome preparation, data analyses, and phylogenetic inference. CL provided partial DNA sequences for phylogenetic analysis. SG identified all the Oryza materials and modified the manuscript. DZ contributed to the design of the study, supervised the experiment steps, and prepared the manuscript. All authors read and approved the final manuscript.
Zhuoya Ding, Wei Liu, Jin Pan contributed equally to this work.
Electronic supplementary material
12862_2009_1164_MOESM1_ESM.TIFF
Additional file 1: Multicolour fluorescent in situ hybridization images of O. malampuzhaensis. Prometaphase chromosomes of O. malampuzhaensis were hybridized by 45S rDNA probes (red) together with B-genome probes (green), and counterstained by DAPI (blue). Arrow indicated one pair of C-genome chromosomes which painted by both 45S rDNA and B-genome signals on same areas. (a) and (d) Two pairs of 45S rDNA loci (red). (b) and (e) The B-genome chromosomes showing blue-green signals. (c) and (f) The chromosomes counterstained by DAPI after hybridized with both 45S rDNA and B-genome probes. Bar, 5 μm. (TIFF 8 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, B., Ding, Z., Liu, W. et al. Polyploid evolution in Oryza officinalis complex of the genus Oryza. BMC Evol Biol 9, 250 (2009). https://doi.org/10.1186/1471-2148-9-250
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-250