
Abstract
CX3CL1 (fractalkine) is the only member of the CX3C (delta) subfamily of chemokines which is unique and combines the properties of both chemoattractant and adhesion molecules. The two-form ligand can exist either in a soluble form, like all other chemokines, and as a membrane-anchored molecule. CX3CL1 discloses its biological properties through interaction with one dedicated CX3CR1 receptor which belongs to a family of G protein-coupled receptors (GPCR). The CX3CL1/CX3CR1 axis acts in many physiological phenomena including those occurring in the central nervous system (CNS), by regulating the interactions between neurons, microglia, and immune cells. Apart from the role under physiological conditions, the CX3CL1/CX3CR1 axis was implied to have a role in different neuropathologies such as traumatic brain injury (TBI) and spinal cord injury (SCI). CNS injuries represent a serious public health problem, despite improvements in therapeutic management. To date, no effective treatment has been determined, so they constitute a leading cause of death and severe disability. The course of TBI and SCI has two consecutive poorly demarcated phases: the initial, primary injury and secondary injury. Recent evidence has implicated the role of the CX3CL1/CX3CR1 axis in neuroinflammatory processes occurring after CNS injuries. The importance of the CX3CL1/CX3CR1 axis in the pathophysiology of TBI and SCI in the context of systemic and direct local immune response is still under investigation. This paper, based on a review of the literature, updates and summarizes the current knowledge about CX3CL1/CX3CR1 axis involvement in TBI and SCI pathogenesis, indicating possible molecular and cellular mechanisms with a potential target for therapeutic intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic central nervous system (CNS) injuries are the most heterogeneous as regards the individual response to injury and the results of treatment of the group of diseases. In spite of constant progress concerning the pathophysiology, diagnostic work-up, and treatment, they still pose a challenge for the modern society due to their complex and prolonged course, socioeconomic dimension, and also numerous existing controversies concerning the implementation of optimal operative and non-operative treatment [1–4]. CNS injuries may be divided into traumatic brain injury (TBI) and spinal cord injury (SCI), depending on the anatomical location. Neurotrauma is a name which comprises both these types [5, 6]. The analysis of currently available data concerning long-term epidemiological research revealed that CNS injuries are one of the leading causes of death and severe disability which drastically reduces health-related quality of life (HRQoL) in all age groups and all populations all over the world, especially in the developing countries [7, 8]. According to the Center for Disease Control and Prevention (CDC), the epidemiological data on TBI for the years 2002–2006 shows the average of 1.7 million new cases of TBI in the USA, resulting in over 52,000 deaths and 80,000 cases of long-term disability [9–11]. According to 23 independent studies, the yearly incidence of TBI is estimated at 235 per 100,000 people in Europe [1, 12]. World Health Organization (WHO) claims that globally, TBI will surpass many diseases as the major cause of death and disability by the year 2020 [13]. Epidemiological data on SCI, based on statistical studies provided by National Spinal Cord Injury Statistical Center (NSCISC), shows the annual incidence of SCI in the USA to be approximately 12,500–20,000 new cases and currently 240,000–337,000 living survivors [14, 15]. In Europe, the incidence of SCI is estimated at 15–16 per one million people [16, 17]. The global incidence rate is estimated at approximately 23 SCI cases per one million which equals to almost 180,000 new cases per annum [17, 18]. Apart from isolated TBI or SCI which occur quite rarely, it is also possible that these two conditions are concomitant and also accompanied by injuries to other body parts (extracranial or extraspinal injuries) which then means a multiple trauma in a patient [19–24]. CNS trauma and associated extracranial and extraspinal injuries give an image of a debilitating condition, which does not only create physical and emotional costs for individuals. It is also a significant financial burden for the whole society [25, 26]. With an increasing amount of knowledge about the pathogenesis of changes during TBI and SCI, it becomes clear that the natural course of these diseases does not only cover the direct moment of injury but, more importantly, long-term neurodegenerative processes [27, 28]. TBI- and SCI-related pathologies occur as a result of two subsequent complex mechanisms which are not clearly demarcated: primary injury and secondary (delayed) injury [1–3]. Brain and spinal cord injuries develop as a result of an external mechanical force, whose character, intensity, direction, and duration determine the severity of the injury. This mechanism is categorized as a primary injury [1–3]. Therefore, as regards primary injury, we can distinguish blunt TBI/SCI resulting from an external mechanical force and a rapid acceleration/deceleration, penetrating TBI/SCI which occurs by damaging the continuity of neural tissue by a ballistic object, and blast TBI/SCI resulting from different shock waves, e.g., acoustic, electromagnetic, light, and thermal waves or their combination, which are responsible for diffuse function disorders and neural tissue destruction [1–4, 29, 30]. The macrostructural image of primary injury includes contusion and edema of the neural tissue, discontinuation of meninges, concomitant fractures and dislocations of cranial and spinal bones, injuries and dislocations of ligamentous structures, and the development of intra- and extra-axial hemorrhages both in the brain and in the spinal cord [1, 31–33]. The effect of the primary injury is additionally strengthened by the dislocation and compression of edematous neural tissue by damaged osseous and ligamentous structures, hematomas, and also by possible compression of cerebrospinal fluid (CSF) cisterns which significantly influences the increase in intracranial pressure (ICP) and intraspinal pressure (ISP) [32–37]. Microscopically, the direct effects of primary injury include the immediate death of cells resulting from the direct mechanical force and secondary compression, the disruption of vascular regulation, hypoperfusion and hypoxia of injured tissues, the dysfunction of neurovascular units forming blood-brain and blood-spinal cord barriers (BBB and BSCB), microporation and the disorders of cell membrane permeability, changes in the ionic composition of intracellular and extracellular space, rapid release of neurotransmitters from damaged cells, and the possibility of diffuse axonal injury (DAI) [38, 39]. The essence of the multidimensional character of molecular and biochemical events occurring during secondary injury is the specific continuation of the processes initiated by primary injury, which contributes to the image of numerous synergistic neurodegenerative mechanisms [40–42]. The mechanisms of secondary injury include progressive disorders in the vascular regulation, progressive disorders of cell membrane permeability, disorders of energy homeostasis associated with the dysfunction of the synthesis of mitochondrial adenosine triphosphate (ATP), the production of free radicals and lipid peroxidation, glutamate (Glu) excitotoxicity, calcium (Ca2+)-mediated neurotoxicity, programmed cell death (apoptosis), and local and systemic inflammatory response related to the inflow of immune cells and secretion of inflammatory mediators [1, 3, 41, 42]. In case of an injury, all the abovementioned and other numerous mechanisms at the cellular and subcellular level lead to time-dependent neurochemical dysregulation, demyelination, loss of neurons (NeuN+), and the development of a glial scar resulting in the dysfunction of the injured neural tissue in the brain and the spinal cord [43–45]. Both the local and systemic body response constitute an essential element of CNS injuries [46, 47]. Immune system response influences all organs, not only the injured ones (the brain and the spinal cord), via marked changes encompassing gene expression changes, the recruitment of a broad spectrum of cells, and secretion of inflammatory mediators [46–48]. Neuroinflammation is considered to be one of the leading elements of secondary injury [49]. The role of immune response in the pathogenesis of TBI and SCI has been regarded controversial, due to its possible positive and negative consequences [49, 50]. In the past few years, independent authors who have been trying to fully elucidate the pathogenesis of TBI and SCI gave more attention to phenomena occurring during the secondary injury, particularly those connected with the immunological component [51]. Ongoing research most commonly concentrates on the mechanisms connected with the inflammatory mediators (e.g., cytokines, including chemokines) secreted by cells located residually in the brain and the spinal cord and also by infiltrating CNS structures via blood vessels [52, 53]. It seems that the most promising therapeutic direction is the modulation of the inflammatory response by limiting its neurotoxic effect, enhancing the neuroprotective properties and promoting the regeneration of injured neural tissue [54, 55]. As regards the wide range of inflammatory mediators connected with CNS functions, chemokines have been attributed an increasing role both in physiological and pathological conditions in the past few years [56]. The knowledge about the role of chemokines in CNS functioning is so wide, that researchers distinguished a new class of neurotransmitters and neuromodulators and named them neurochemokines [57]. The multidirectional actions of neurochemokines include the participation in the embryogenesis of the nervous system, modulation of synaptic conductance, plasticity, and also their function in the pathogenesis of neurodegenerative disorders [57]. Researchers have described almost 50 chemokine ligands, out of which chemokine CX3CL1 (fractalkine) and its receptor CX3CR1 deserve special attention. Apart from properties which are typical for the remaining compounds in this group, chemokine has a different molecular structure and may function not only as a chemoattractant but also as an adhesive molecule. The increasing role of the CX3CL1/CX3CR1 axis has been noted in the physiological communication between neurons and microglial cells. Moreover, there are numerous newly discovered phenomena as regards the influence of the CX3CL1/CX3CR1 axis on CNS physiology and pathology, such as the effect on the synaptic plasticity, maturation, and activity and a marked effect on the functioning of hippocampal formation [58]. Up to now, the role of CX3CL1/CX3CR1 axis has not been widely discussed in the context of its presence and possible functions in the pathophysiology of TBI- and SCI-related phenomena. Therefore, it seems justified to describe and summarize the participation of CX3CL1 and its CX3CR1 receptor in the course of CNS injuries basing on the available professional literature. In order to present a comprehensive overview of the problem, we will perform a thorough analysis of the structure and functions of the CX3CL1/CX3CR1 axis, present its role in the physiological processes related to CNS functioning, and then, we will discuss its possible role in the course of CNS injuries with particular attention paid to each stage of the pathology and its subsequent consequences and also possible clinical implications.
Molecular Organization and Biological Functioning of CX3CL1 and Its Dedicated Receptor CX3CR1
CX3CL1—a Unique Two-Form Ligand Member of the CX3C (Delta) Subfamily
In 1997, Bazan et al. first identified and described the CX3C (delta) subfamily of chemokines [59]. The name “fractalkine” was then used for the first time. In the same year (at the interval of several weeks), Pan et al. confirmed the existence of the CX3C subfamily [60]. This group of researchers used the name “neurotactin” to emphasize its mode of action during experimental autoimmune encephalomyelitis (EAE) on a mouse model. Regarding the latest classification of chemokines including the arrangement of amino acids and cysteine (Cys) residues with disulfide bonds, CX3CL1 is the only representative of the CX3C subfamily which is also classified as a dual-function chemokine (category D) which has the properties of homeostatic chemokines (category H) and inflammatory chemokines (category I) [61, 62]. In human genome, the gene coding CX3CL1 is composed of three exons and is located within the long arm of chromosome 16 (16q13) [63]. It is highly conservative with the genes of a mouse (Mus musculus) or a rat (Rattus norvegicus). The respective genes are located on chromosome 8 (8qC5) in the mouse and chromosome 19 (19p12) in the rat [64, 65]. In the body, CX3CL1 occurs in two different isoforms: membrane-anchored CX3CL1 and soluble CX3CL1 (sCX3CL1). Therefore, it has chemotactic and adhesive properties [66]. Ultrastructurally, the precursor form of membrane-anchored CX3CL1 is a polypeptide chain composed of 397 amino acid residues containing signal peptide (SP) included in 24 amino acid residues, N-terminal chemokine domain (CD) with CX3C motif included in 76 amino acid residues, forming a globular structure which is approximately 3 nm long, mucin-like stalk included in 241 amino acid residues which are 26 nm long, also including 17 degenerated mucin-like repeats with 26 potential O-glycosylations at serine (Ser) and threonine (Thr) residues, transmembrane region (TM) included in 21 amino acid residues, and C-terminal intracellular cytoplasmic tail (CT) included in 35 amino acid residues [59, 60, 67, 68] (Fig. 1). The mature membrane-anchored form of CX3CL1 does not contain SP and is composed of 373 amino acid residues of the total molecular weight of approximately 17.5 kDa, and 95 kDa after glycosylation [59, 60]. The connection with cell membranes is a factor which, apart from the regulation at the level of transcription and translation observed in other chemokines, is associated with an additional phenomenon of functional regulation via undergoing constitutive internalization, producing a dynamic balance in membrane compartments between the plasma membrane and the intracellular endocytic compartment [69–71]. The structure of intracellular CT includes two adaptor protein 2 (AP2)-binding motifs, such as YQSL at positions 362–365 and YVLV at positions 392–395 (PSORT II analysis), predicted to bind clathrin-coated pit which allows constitutive clathrin-mediated endocytosis [70]. The distribution of CX3CL1 in distinct subcellular compartments is also associated with the properties of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE), such as syntaxin 13 (STX13) and vesicle-associated membrane protein 3 (VAMP3) [71]. This type of dynamic balance facilitates the protection of presynthesized CX3CL1 against premature degradation on the external side of cell membrane and, under appropriate conditions, accelerates the mobility of intracellular content [70, 71]. Another form to be described is sCX3CL1 included in 317 amino acid residues, approximately 29 nm long, whose structure encompasses a CD-containing segment and a mucin-like stalk of the total molecular weight of approximately 14.7 kDa, and 80 kDa after glycosylation [59, 60]. The separation of sCX3CL1 results from proteolytic cleavage process which occurs at the di-arginine (di-Arg) motif (RR) near the external surface of plasma membrane [59]. The process is based on ectodomain shedding in which sCX3CL1 acts as a soluble ectodomain and is mediated by two transmembrane proteases from a disintegrin and metalloproteinase (ADAM) family and by cathepsin S (CTSS) [72, 73]. The proteases from ADAM family, which take part in this process are ADAM metallopeptidase domain 10 (ADAM10) and ADAM metallopeptidase domain 17 (ADAM17) which is also called tumor necrosis factor-alpha-converting enzyme (TACE) [72]. ADAM10 was identified as a protease responsible for the constitutive and ionomycin-induced shedding of CX3CL1, and ADAM17 is involved in the 12-O-tetradecanoylphorbol-13-acetate (PMA)-induced shedding of CX3CL1 [74, 75]. It is worth noting that the described shedding events may be pharmacologically targeted by the use of a specific inhibitor for ADAM10 (GI254023X) and the dual-specific ADAM10 and ADAM17 inhibitor (GW280264X) [74, 76]. The remaining C-terminal cleavage fragment (CTF) undergoes degradation associated with the activity of membrane-associated secretase complexes and the proteasome [77].

The schematic structure of two-form chemokine ligand CX3CL1 (fractalkine). a Membrane-anchored form of CX3CL1 showing specific regions of the molecule and the site of the cleaving action of the metalloproteinase ADAM10 and ADAM17 (TACE). b The soluble form of CX3CL1 (sCX3CL1), produced by metalloproteinase cleaving. The N-terminal chemokine domain (CD) containing the CX3C motif is shown in greater detail including important parts of the secondary and tertiary protein structure. Adopted and modified with permission from Wojdasiewicz P, Poniatowski ŁA et al. (2014) The Chemokine CX3CL1 (Fractalkine) and its Receptor CX3CR1: Occurrence and Potential Role in Osteoarthritis. Arch Immunol Ther Exp (Warsz) 62(5):395-403. doi: 10.1007/s00005-014-0275-0
Characteristics of CX3CR1 Chemokine Receptor
Unlike most ligands belonging to the chemokine family with the affinity for numerous receptors, CX3CL1 reveals its biological activity via an interaction with only one dedicated receptor—CX3CR1 [78]. During a study on a rat model conducted by Harrison et al. in 1994, CX3CR1 was first described as RBS11 constituting the orphan G protein-coupled receptor (oGPCR) [79]. In further study by Raport et al., a homologous human gene was named V28, and its product was described as oGPCR containing a highly conserved 20 amino acid region typical for receptors from G protein-coupled receptor (GPCR) family and seven amino acid sequence Asp-Arg-Tyr-Leu-Ala-Ile-Val (DRYLAIV motif) in the second cytoplasmic loop (IL2) which is conserved among chemokine receptors [80]. The identification of the connection of CX3CL1 and CX3CR1 in a signaling axis was described by Imai et al. in 1997 [81]. In human genome, the gene coding CX3CR1 is composed of four exons and is located within the short arm of chromosome 3 (3p21.3) [82, 83]. It is conservative with mouse or rat genes, which, in turn, are located on chromosome 9 (9qF4) in the mouse and on chromosome 8 (8q32) in the rat [80, 82–84]. According to existing classifications, CX3CR1 belongs to the biggest A class (rhodopsin-like receptors) of GPCR proteins, which includes all chemokine receptors [85, 86]. Ultrastructurally, the polypeptide chain of CX3CR1 includes 355 amino acid residues of the total molecular weight of approximately 40 kDa [80] (Fig. 2). The polypeptide chain includes extracellular N-terminus, seven α-helical domains (TM1–TM7) penetrating the whole thickness of the cell membrane, three intracellular (IL1, IL2, IL3) and three extracellular loops (EL1, EL2, EL3), and an intracellular C-terminus [87]. Two conservative Cys residues located respectively at the top of the third transmembrane domain (TM3) and the second extracellular loop (EL2) are connected with a disulfide bond [88]. N-terminus and loops formed by the polypeptide chain extracellularly create the location of binding two functional ligands, which can be both CX3CL1 and CCL26 (eotaxin-3) and also antibodies and some pathogens such as bacteria and viruses [89–92]. It was also demonstrated that the appropriate level of tyrosine (Tyr) sulfation of N-terminus is necessary for maintaining normal activity of the majority of GPCR receptors for chemokines [93]. C-terminus and loops located on the cytoplasmatic side form the site where heterotrimeric Gαi protein is bound [81]. The phosphorylation of C-terminus, which occurs easily due to numerous Ser and Thr residues, facilitates the appropriate modulation of the process of transmitting a signal associated with Gαi protein [94]. Gαi protein, which has a function in the transmission of intracellular signals associated with CX3CR1 activation, may be inhibited by pertussis toxin (PTX), which was implemented in some experimental models [90]. The majority of chemokine GPCR receptors (including CX3CR1) are characterized by a marked polymorphism. This may be responsible for intraindividual and interindividual variability of chemokine effects and may account for changeable risk of the development and course of inflammatory, autoimmune, and hyperplastic diseases [95, 96]. It is also worth noting that the existence of only one receptor for CX3CL1 significantly facilitates the interpretation and possible clinical implications of reported biological effects concerning the CX3CL1/CX3CR1 axis.

The schematic structure of CX3CR1 chemokine receptor. The molecular structure of the receptor includes seven α-helical transmembrane domains (TM1–TM7), three extracellular (EL1, EL2, EL3) and three intracellular loops (IL1, IL2, IL3), an extracellular N-terminus, and an intracellular C-terminus. The disulphide bond is shown between two highly conserved cysteines (Cys) which are located respectively at the top of the third transmembrane domain (TM3) and the second extracellular loop (EL2). The second intracellular loop (IL2) contains a conserved seven amino acid sequence Asp-Arg-Tyr-Leu-Ala-Ile-Val (DRYLAIV motif) which serves as Gαi heterotrimeric protein docking site. a CX3CR1 receptor shown from the side perspective. b CX3CR1 receptor shown from the intracellular perspective
Systemic Expression and Distribution of the CX3CL1/CX3CR1 Axis
The main source of chemokines is white blood cells (WBC), but CX3CL1 is mainly produced in endothelial cells and neurons [97]. It is not surprising that CX3CL1 expression is elevated in highly vascularized and well-innervated organs and also in locations with an increased concentration of immune system cells, such as the CNS, lungs, cardiac muscle, liver, intestines, and placenta [98–104]. In spite of the wide distribution and expression throughout the body, CX3CL1 presents a high specificity for some cell types. CX3CR1+ cells present chemotactic properties toward sCX3CL1. They include a significant percentage of immune cell population, i.e., monocytes (CD14+), macrophages (MΦ), NK cells (CD16+), lymphocytes (CD4+ and CD8+), mast cells (MC), and dendritic cells (DC) [105–109].
CX3CL1/CX3CR1 Axis in the Regulation of Cellular Mechanism Involved in Cellular Adhesion and Migration
Leukocyte transendothelial migration (TEM) from vascular lumen into extravascular tissues triggers a dynamic cascade of molecular events connected with interactions between leukocytes and endothelial cells both under physiological and pathological conditions [110]. Apart from the conventional leukocyte extravasation based on integrin-mediated adhesion, it is also possible to employ the adhesive activity of membrane-anchored CX3CL1 [111]. It was noted that the CX3CL1/CX3CR1 axis is involved at all migration stages, but the adhesive ability of CX3CL1 is determined by the presence of mucin-like stalk [111, 112]. Leukocytes which have an appropriate level of CX3CR1 expression adheres CD to membrane-anchored CX3CL1 [112]. Following the adhesion associated with an interaction between CX3CL1 and CX3CR1, leukocytes are able to migrate through vessel walls in a directly selectin- and integrin-independent manner [113]. Moreover, after the adhesion of CX3CL1 to CX3CR1, there is an activation and synthesis of other adhesive molecules which strengthens the adhesion via synergism [114, 115]. It was also noted that integrins may act as receptors and co-receptors as they are able to adhere to CX3CL1 with no participation of CX3CR1 [114, 115]. Multilevel interactions of the CX3CL1/CX3CR1 axis with different types of cell adhesion molecules (CAM), such as selectins and integrins, are currently thought to be key phenomena in the pathogenesis of many inflammatory diseases, which may be useful as potential locations of modulating immune response associated with the recruitment of individual cell types [116].
CX3CL1/CX3CR1 Axis Interplay with Related Activators/Repressors and Intracellular Multiple Signaling Pathways
The regulation of the activation and functioning of the CX3CL1/CX3CR1 axis encompasses the control at the level of transcription and also regulation during post-translational modifications. Intracellular transmission of signals via CX3CR1 and then heterotrimeric Gαi protein is associated with the activation of numerous signaling molecules, such as several different secondary messengers and transcription factors, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), cAMP response element-binding protein (CREB), signal transducer and activator of transcription (STAT), and activator protein 1 (AP-1) [117–119]. The activation of the transcription factors associated with the CX3CL1/CX3CR1 axis may be connected with activating a potentially wide range of functions of a given cell, such as messenger RNA (mRNA) transcription for many proteins (including cytokines), cytoskeletal rearrangement and migration, apoptosis, and proliferation [117, 119]. Local production and membrane expression of CX3CL1 and also CX3CR1 are controlled by other cytokines with the most important ones including tumor necrosis factor alpha (TNFα), interleukin 1 (IL-1), interferon gamma (IFNγ), and soluble interleukin 6 receptor alpha (sIL-6Rα) [120, 121]. Similar reciprocal correlations were also observed as regards the presence of lipopolysaccharide (LPS), nitric oxide (NO), adenosine (ADO), 15-deoxy-delta(12,14)-prostaglandin J(2) (15d-PGJ(2)), 8-isoprostane, and hypoxia [122–126]. CX3CL1 production induced by various factors is also subjected to autoregulation via modulating the expression of their CX3CR1 receptor [117]. The activation and functioning of individual cascades of intracellular pathways associated with the CX3CL1/CX3CR1 axis under particular physiological and pathological conditions demonstrate differences, and a direct recapitulation of their activity in a specific clinical status within various cell and tissue types requires further research which would confirm these potential correlations.
Overview of the Role of CX3CL1 and CX3CR1 in the Physiological Functioning of the Brain and Spinal Cord
CX3CL1/CX3CR1 Axis—Neuroanatomical Ultrastructural Location and Regional Distribution
The analysis of in vitro experiments performed on cellular models and knowledge about transgenic mouse and rat models facilitates deep insight into the pattern of CX3CL1 and CX3CR1 expression within the CNS. In order to comprise the extensive neuroanatomical distribution of CX3CL1 and CX3CR1, it needs to be noted that high levels of CX3CL1 chemokine are constitutively produced by neurons within the telencephalon and diencephalon and particularly in the cerebral cortex, hippocampus, amygdala, basal ganglia, thalamus, and olfactory bulb [127–129]. A particularly high level of expression of CX3CL1 mRNA transcripts is present in the hippocampal formation demonstrating the highest concentration in CA1, CA2, and CA3 regions and in the cerebral cortex—layers II, III, V, and VI [127–129]. The hypothalamus and mesencephalon are the areas where the levels of mRNA expression for CX3CL1 are very low or even below detectable limits [127–129]. A similarly low expression level was noted within the metencephalon and myelencephalon encompassing the pons, cerebellum, and medulla oblongata [127–129]. Interestingly, mRNA expression for CX3CL1 is mainly associated with expression within the gray matter and the lack of its expression within the white matter, e.g., within the corpus callosum and fimbria/fornix (FF) structures [129, 130]. CX3CL1 expression within the spinal cord is limited to neurons in the dorsal horn and dorsal root ganglia (DRG) [131, 132]. Interestingly, it remains controversial that endothelial cells in the brain and the spinal cord, as opposed to those in other locations, do not present constitutive CX3CL1 expression on the surface, which suggests that it is rather dependent on the activation occurring as a result of an inflammation in certain CNS pathologies [127, 129, 133, 134]. Constitutive mRNA expression for CX3CL1 and CX3CR1 is also described by astrocyte cells (GFAP+) [135, 136]. In the ventricular system, CX3CL1 and CX3CR1 expression is connected with the choroid plexus (CP) [137, 138]. CX3CL1 occurs physiologically in the CSF at the concentration which is approximately 500-fold below serum concentration [139]. It is widely accepted that CX3CR1 demonstrates a relatively homogeneous expression via microglial cells in the brain and the spinal cord which, at the same time, do not demonstrate mRNA expression for CX3CL1 [127–129, 135]. Apart from the abundant presence of CX3CR1 receptor within microglial cells it is also important to note its expression via neurons [140–142]. When analysing the presence of CX3CR1 both on neurons and on microglial cells, it is important to note the possibility of distinguishing between the direct modulating effect of CX3CL1 on neurons and its indirect effect via previously activated microglial cells [142, 143]. Making such a distinction is possible by the analysis of the effect of CX3CR1 activation on glutamatergic transmission which results in the inhibition of phosphorylation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and, more precisely, its glutamate receptor 1 (GluR1) subunit associated with calcium oscillator, increased Ca2+ entry, and the reduction of excitatory postsynaptic current (EPSC) amplitude [144, 145]. It was also noted that CX3CL1 inhibits hippocampal long-term potentiation (LTP) in CA1 region through adenosine A3 receptor (A3R) activity [146]. Differences in mRNA expression for CX3CL1 and CX3CR1 are also correlated with age, where the reduction in CX3CL1 expression and level and the increase in CX3CR1 level within the hippocampus are accompanied by an increase in microglial activity through the phosphorylation of protein kinase B (Akt) and activation of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3-K) pathway [147].
CX3CL1/CX3CR1 Axis in the Context of Homeostatic Bidirectional Cross Talk Between Neurons and Microglia
Regarding both the neuroanatomical CX3CL1 and CX3CR1 distribution in the context of bidirectional interaction between neurons and microglial cells, it can be noted that the CX3CL1/CX3CR1 signaling axis probably has a crucial role in modulating the homeostasis of these multilevel interactions throughout the ontogenetic development of mammals [148]. Microglia, which seem to be constantly active, undergo tonic signaling through the CX3CL1/CX3CR1 axis under physiological conditions, which facilitate the maintenance of its cells in a quiescent state and maintain homeostasis in the neuronal network [148, 149]. The phenomenon seems to corroborate the fact that structural connectivity reflects functional connectivity, particularly influencing synaptic transmission in specific neuroanatomical areas, in processes like learning, memory, and behavior [58, 150]. However, the unambiguous determination of the role of the CX3CL1/CX3CR1 axis as inflammatory or anti-inflammatory seems controversial in the context of functioning within the CNS [148]. Microglia, which is the non-neuronal element of the CNS, includes specific residual macrophages performing the function of a specific sensor sensitive to injuries and developing pathologies in the neural tissue, e.g., an injury or a neoplastic, autoimmune, or infectious process [151, 152]. Under physiological conditions, CX3CL1 seems to inhibit microglial activation, while in particular pathologies a paradoxical promotion of inflammatory response may occur [151, 153, 154]. The factors which determine the specific type of neuroprotective or neurotoxic response are most probably dependent on the type of the primary destructive factor, CNS area, and the local concentration of CX3CL1 and CX3CR1 [148]. The disruption of homeostatic paracrine and autocrine interactions of the CX3CL1/CX3CR1 axis in the context of neuron-microglia communication may be seen as one of fundamental elements in the pathogenesis of CNS-related diseases [155] (Fig. 3). The understanding and determination of the precise location of the CX3CL1/CX3CR1 signaling axis as a physiological element of cell properties within the CNS still require further detailed research.

The schematic representation of the physiological and pathological role of CX3CL1/CX3CR1 axis in the context of bidirectional cross talk between neurons and microglia. a Under physiological conditions, microglia undergo tonic signaling through the CX3CL1/CX3CR1 axis, which facilitates the maintenance of its cells in a quiescent state. b Under pathological conditions, the homeostasis cross talk between neurons and microglia through the CX3CL1/CX3CR1 axis is disrupted. Upregulated levels of several cytokines, chemokines, and other mediators create a specific inflammatory environment, which results in paradoxical promotion through the CX3CL1/CX3CR1 axis, activation and proliferation of microglia, and the infiltration of peripheral immune cells
Role of CX3CL1 and CX3CR1 Axis in the Pathophysiology of Traumatic Brain and Spinal Cord Injury
Expression Pattern of the CX3CL1 and CX3CR1
The properties of CX3CL1 and CX3CR1 and also of the remaining chemokines and their receptors are still the focus of research on humans and various animal models, to present their potential role and possibility of implementation in the clinical therapy of TBI and SCI. The analysis of data available in the professional literature demonstrates the scarcity of studies conducted in patients and the domination of animal model and cell culture approach.
Traumatic Brain Injury
Available studies concerning the level of CX3CL1 and CX3CR1 expression in the course of TBI present data including both clinical trials with patients suffering from severe head injuries and also data obtained from animal model and cell culture studies (Table 1). Rancan et al., who conducted a study in patients with head trauma and in mice after standardized cortical experimental contusion, were the first to analyze the posttraumatic profile of CX3CL1/CX3CR1 axis expression in TBI [156]. The study group included 12 patients (n = 12) with Glasgow Coma Scale (GCS) of ≤8 on admission and with visible changes in computed tomography scans according to Marshall classification [157]. The patients had intraventricular catheters (IVCs) for the direct measurement of ICP. CSF drainage was performed when the pressure was over 15 mm Hg. The control group for CSF analysis included five patients (n = 5) without neuropathologies, and as regards serum analysis, there were eight (n = 8) healthy volunteers. The levels of CX3CL1 in CSF obtained in the control group were 12.6 to 57.3 pg/mL, while in the study group, they were 29.92 to 535.33 pg/mL demonstrating the most abundant values on the day of admission, with a gradual decreasing tendency during 14 days of the study. CX3CL1 levels in the serum of the control group patients were 21.288 to 74.548 pg/mL, while in the study group, they amounted to 3.1 to 59.159 pg/mL. Therefore, the authors reported that physiologically, the concentration of CX3CL1 in humans is higher in the serum than in the CSF and the values are reversed as a result of TBI, correlating with BBB dysfunction and suggesting the possibility of shifting CX3CL1 from the serum to the CSF. The second part of the study was performed on a mouse model (n = 40). No differences were observed as regards CX3CL1 levels and the expression of its mRNA in the study group and sham-operated control group. However, the authors noted an elevated level of mRNA for CX3CR1 in the study group which gradually increased between the trauma and the seventh day of the experiment. Helmy et al. conducted a study which consisted in the determination of cytokine profile in patients with TBI [158]. They performed cerebral microdialysis (CM) with two types of perfusates, such as crystalloid perfusate and 3.5 % human albumin solution (HAS) and the serum coming from sampled arterial and venous blood. The qualified patients (n = 12) had the GCS of ≤8 on admission and typical changes in the computed tomography scan. With crystalloid perfusate, the median CX3CL1 concentration was 9.21 pg/mL, and with 3.5 % HAS, it was 20.39 pg/mL. The median microdialysate concentration/arterial plasma ratio was 0.96. Gaetani et al. performed an analysis of human brain samples harvested during decompressive craniotomy for TBI and after spontaneous intracranial hemorrhage (ICH) [159]. They were studied in terms of CX3CL1 and CX3CR1 expression. The level of CX3CL1 expression demonstrated upregulation in the neural compartment compared to samples from the control group harvested during gyrectomy, which was a part of a surgery for unruptured intracranial aneurysyms (UIAs). It was demonstrated that the level of CX3CL1 expression correlated with lower ICP within the glial compartment. CX3CR1 expression was observed in lower concentrations both in neurons and glia, while its higher concentrations in neurons were analyzed with regard to the GCS of patients on admission. Fahlenkamp et al. analyzed organotypic hippocampal slice cultures in a mouse model [160]. The cultures were subjected to in vitro mechanical dropweight trauma of moderate severity in the CA1 region to assess local cytokine and chemokine reaction. The results demonstrated the downregulation of the expression of transcript mRNA for CX3CL1 with simultaneous upregulation of the expression of transcript mRNA for CX3CR1.
Spinal Cord Injury
The analysis of CX3CL1 and CX3CR1 expression levels in the course of SCI has only been possible to perform with studies conducted on animal models (Table 2). Detloff et al. conducted a series of experiments concerning behavioral and cellular responses after thoracic (Th8) SCI with the use of electromagnetic impactor in rats, occurring below the level of injury in the lumbar spinal cord (L5) [161]. It was demonstrated that within L5 dorsal horn, the level of CX3CL1 presented no changes after 7, 21, and 35 days following the SCI, while the level of mRNA expression for CX3CL1 decreased after 35 days following the SCI. The level of mRNA expression for CX3CR1 was elevated compared to control groups. Donnelly et al. conducted a study on a mouse model with SCI in the thoracic spine (Th9–Th10) [162]. It was demonstrated that the local mRNA expression for CX3CL1 was decreased between days 1 and 7 after SCI, and it reached its normal level after 14 days. On day 28, it had a markedly elevated level. However, mRNA expression for CX3CR1 was reduced between days 1 and 3 after SCI. Then, the expression gradually increased between days 3 and 28 in comparison with control groups. The expression of CX3CL1 remained unchanged for 6 weeks after SCI, while CX3CR1 demonstrated an increased expression starting on the third day after SCI which corresponded with the level of expression of its mRNA. Cizkova et al. reported spatial changes in the expression of CX3CR1 within rostro-caudal axis after SCI on the rat model after 3 days, with the spinal cord damaged in the thoracic region (Th8–Th9) with the use of modified balloon compression technique (2-French Fogarty catheter) [163]. It was noted that the CX3CR1 expression significantly increased after 3 days in the study group (n = 4) in comparison with sham-operated control group (n = 4) where CX3CR1 expression was homogeneous and poorly pronounced. The expression was particularly intensified within the gray and white matter of the rostral segment (Th2–Th6) compared to the caudal segment (Th12–L3). Blomster et al. (2013) analyzed CX3CL1 concentrations in the serum of the blood sampled with cardiac puncture from the left ventricle (LV) in mice subjected to SCI in the thoracic spine (Th9) [164]. It was demonstrated that 7 days after, SCI mice (n = 6) presented a slightly elevated CX3CL1 concentration in comparison with a group of non-injured (n = 5) and sham-operated (n = 5) controls.
Implication on Systemic and Local Mechanisms Involved in the Course of Traumatic Brain and Spinal Cord Injury
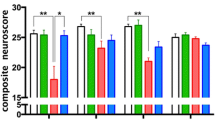
Regarding the kinetics of the changes of CX3CL1 and CX3CR1 expression occurring as a result of TBI and SCI, it becomes obvious that the regulation within this signaling axis should be regarded as an endogenous mechanism of the control of an inflammation resulting from this type of injuries (Table 3). CNS trauma triggers the local activation and migration of microglia, enhances the secretion of inflammatory mediators like cytokines and chemokines, and creates a specific inflammatory environment, which also results in the infiltration of peripheral immune cells [165, 166]. As regards the possible destruction and regeneration of CNS tissues, the presence of activated macrophages seems to be of key importance. In spite of the fact that the macrophages are of different origin and, partially, phenotype, they are similar in numerous aspects [167–169]. The sources of the activated macrophages include both microglia and the population of monocytes mobilized from circulation originating from bone marrow and splenic reservoir [164, 169]. Both cell types demonstrate a similar morphology and the expression of similar superficial markers [170]. The analysis of a mouse model in terms of the superficial expression of receptor markers demonstrated that microglia are immunophenotypically described as Ly6Clow/CX3CR1high/CD45low/Iba-1+, while circulating monocytes are described as two subpopulations, such as inflammatory monocytes (classically activated, M1) defined as Ly6Chigh/CX3CR1low/CCR2high and anti-inflammatory monocytes (alternatively activated, M2) defined as Ly6Clow/CX3CR1high/CCR2low [171–175]. Human counterparts of Ly6Chigh and Ly6Clow monocytes are defined as CD14++/CD16− and CD14+/CD16++ [175, 176]. The main source of knowledge regarding the direct functioning of the CX3CL1/CX3CR1 axis in TBI and SCI is studies on animal models which were subjected to substitution on one or both CX3CR1-coding alleles using gene for the green fluorescent protein (GFP) in this way obtaining heterozygotes (CX3CR1+/GFP) or knockout (KO) homozygotes (CX3CR1GFP/GFP). If we analyze studies by Donnelly et al. and Blomster et al. (2013) at a different angle, we may note that the modulation of monocyte activation via changes in the CX3CL1/CX3CR1 signaling axis on chimeric mouse models with SCI may affect their course [162, 164]. In the study of Donnelly et al., it was observed that the bone marrow of CX3CR1GFP/GFP chimeric mouse, contrary to wild-type (WT) mouse, presents a negative effect on the recruitment and maturation of macrophages defined as Ly6Clow/iNOS+/MHCII+/CD11c− at the site of injury along with their capacity of the production of inflammatory cytokines and oxidative metabolites, with a positive effect on the recruitment of macrophages defined as Ly6Chigh/CCR2+/MHCII−/CD11c+ [162]. Microglia activation was also altered, which was expressed as the reduced production of mRNA for interleukin 6 (IL-6) and inducible nitric oxide synthase (iNOS) with the absence of changes in the production of mRNA for interleukin 1 beta (IL-1β) and TNFα. Moreover, when using the standardized Basso Mouse Scale (BMS), CX3CR1GFP/GFP mice demonstrated a faster and sustained resumption of mobility, unlike WT mice [177]. Blomster et al. (2013) performed a study on the bone marrow of CX3CR1GFP/GFP mouse chimeras [164]. They noted that CX3CR1 deficiency contributed to an increased recruitment of monocytes with no effect on their distribution and content at the site of injury. The BMS of CX3CR1GFP/GFP mice indicated the deteriorated recovery of locomotor function in comparison with WT mice. Another study conducted by Blomster et al. (2011) concerning olfactory bulbectomy (OBX) in CX3CR1GFP/GFP mice showed some properties of the CX3CL1/CX3CR1 axis in the course of TBI, because a model injury of olfactory system revealed some aspects if the injury did not result from a direct impact, but from surgical intervention [178]. An increased recruitment of monocytes was observed in the olfactory neuroepithelium of CX3CR1GFP/GFP mice in comparison with WT mice. The level of mRNA expression for ADAM10 was slightly increased, while no changes were noted in mRNA expression for ADAM17 which were independent from the genotypes of the mice in the study. Significant changes were noted as regards the expression of inflammatory cytokines, which was demonstrated as an increased expression of mRNA for IL-1β, IL-6, and TNFα in CX3CR1GFP/GFP mice in comparison with WT mice. Morganti et al. studied TBI on a mouse model using CX3CR1GFP/GFP chimeras [179]. They observed neurotoxic responses at acute (24 h) and chronic (3 months) stages after controlled cortical impact (CCI). It was noted that 24 h after an injury, the inflammatory response in CX3CR1GFP/GFP mice was decreased which manifested as reduced IL-1β, IL-6, TNFα, and iNOS expression in comparison with WT mice. The analysis of hippocampal-dependent cognitive function 3 months following an injury revealed that CX3CR1GFP/GFP mice made fewer mistakes in the radial arm water maze (RAWM) test than WT mice. As regards changes in synaptic conductance, it was demonstrated that a quantitative change of a NR2B subunit of N-methyl-D-aspartate (NMDA) receptor occurred within the hippocampus of a WT mouse, while it was not observed in CX3CR1GFP/GFP mice. Other investigated differences in both types of mice include the effect on the activity of Src kinase, p44/42 MAP kinase, postsynaptic density protein 95 (PSD-95), and tau protein phosphorylation. Zanier et al. conducted a similar study on CX3CR1−/− mouse model [180]. The mice were subjected to CCI, and a number of parameters were compared after 4 days and 5 weeks following the TBI. It was noted that 4 days after the injury, there were no changes in IL-1β and TNFα expression in CX3CR1−/− mice in comparison with WT mice. However, there was a reduction in iNOS expression with simultaneous unchanged level of CD11b+ marker. During the fifth week, the expression of IL-1β and TNFα still remained unchanged, but the expression of iNOS and CD11b+ marker increased. Moreover, the levels of expression of interleukin 10 (IL-10) and insulin-like growth factor 1 (IGF-1) were also assessed. After 4 days and during the fifth week, their expression was lower in CX3CR1−/− mice in comparison with WT mice, and their elevated levels were noted only 1 day after TBI. As regards the neurological outcome, CX3CR1−/− mice demonstrated superior results on day 4 after TBI in comparison with WT mice in neuroscore test [181]. However, during week 5, their results were poorer. Regarding numerous parameters assessed in this study, it was stated that blocking CX3CL1/CX3CR1 axis conduction leads to positive effects soon after TBI, but the long-term outcome is poorer. Another valuable study with a similar methodology is one conducted by Febinger et al. on the mouse model of KO CX3CR1GFP/GFP homozygotes which were subjected to CCI [182]. The mice were compared as regards the assessed parameters in two time intervals since the injury, both in the acute phase (24 h to 15 days) and in the chronic phase (15 to 30 days) with particular emphasis put on the assessment of locomotor activity, motor learning, anxiety behavior, and cognitive function with the following tests: neuroscore, open field, elevated plus maze (EPM), rota-rod, and Morris water maze (MWM). It was noted that in the acute phase, the CX3CR1GFP/GFP chimeric mice demonstrated superior results in comparison with WT mice in neuroscore test, while in the chronic phase, there was a reverse trend in which WT mice accomplished superior results. No differences between individual genotypes were demonstrated in mice undergoing open field and EPM test after 30 days since the injury. At the same time, the results of rota-rod and MWM tests were poorer in CX3CR1GFP/GFP mice compared to WT mice. The observation of the degree of neuronal loss with Fluoro-Jade B+ and NeuN+ staining demonstrated that CX3CR1GFP/GFP homozygotes lost fewer neurons in the acute phase in comparison with WT mice. However, in the chronic phase, the trend was reversed. The analysis of cytokine level showed the reduced expression on days 7 and 15 after CCI for IL-1β, reduced IL-4 and IL-6 on day 15, and reduced iNOS on day 7. No changes were observed as regards the expression of transforming growth factor beta (TGF-β) on day 7 following the injury. The authors, just like Zanier et al., suggested that the inhibition of CX3CL1/CX3CR1 axis conduction may be a potential therapeutic target in the acute phase after an injury basing on the analysis of the studied parameters and the activity and proliferation of microglia, observing the CD11b+ cell count on days 15 and 30 after the injury and the kinetics of the expression of superficial markers for specific subtypes of activated microglia (YM1, CD206, CD68, and MARCO) within the studied tissues. Regarding the immune response occurring in case of ICH which is a common consequence of TBI, we need to mention the findings of Taylor et al. [183]. They used chimeric CX3CR1GFP/GFP mice in which ICH was modeled via the injection of whole blood into the striatum. It was demonstrated that in this model, CX3CR1 deficiency did not affect monocyte recruitment, functional recovery, or immune response manifested as the changes of cytokine expression levels.
Potential Therapeutic Options
The analysis of the literature shows that the possibility of linking CX3CL1 and CX3CR1 concentrations with such prognostic factors as BBB dysfunction, ICP, and GCS seems particularly promising in the clinical aspect [156, 159]. Regrettably, the scarcity of studies conducted so far make it impossible to determine the details of the functional role of CX3CL1 and CX3CR1 in the pathomechanism of TBI and SCI and unambiguous qualification of this signaling axis as neuroprotective or neurotoxic. According to the literature describing the mouse model of KO CX3CR1GFP/GFP homozygotes, the inhibition of CX3CL1/CX3CR1 axis conduction contributes to an intensified recruitment of monocytes at the injury site in the majority of cases, which leads to varied effects as regards the expression of other cytokines (IL-1β, IL-6, TNFα) and iNOS. Therefore, scoring and testing bring heterogeneous results of the cognitive and motor neurological functions of the evaluated animals. The discrepancies in the interpretation of study results and a paucity of research on patients result from differences in the modeling of animal experiments and the difficulty obtaining uniform patient groups qualified for the study, because CNS injuries occur unexpectedly and are a highly heterogeneous group of injuries. The present authors believe that it is necessary to conduct multicenter research comprising larger patient groups and to analyze other parameters, such as concomitant inflammatory diseases or patients’ drug history, which may significantly contribute to overcoming the difficulty obtaining more reliable results. According to the current state of knowledge about CX3CL1 and CX3CR1 both as regards physiological and pathological CNS conditions, it is still fundamental and valid to determine the role of this signaling axis precisely in individual medical conditions and to search for a proper therapeutic approach. It implies a distinct necessity to seek new types of immunomodulatory therapies. It seems that inhibiting more than one chemokine signaling axis in TBI, SCI, and other neuropathologies might have a positive effect on the specificity of obtained results and provide researchers with new data concerning the role of chemokines in the CNS and cells of various immunophenotypes which infiltrate injured neural tissue. Seemingly, a factor which plays a particularly significant role may be the effect on the inflammatory environment developing at the time of injury via modulating the microglia cell phenotype in individual phases following the injury in order to direct the polarization of its activity toward the neuroprotective aspect [180, 182]. The use of a selective CX3CR1 antagonist (AZD8797) may be a promising treatment of TBI and SCI in this case [184, 185]. Moreover, if we bear in mind the fact that the migration of mesenchymal stem cells (MSCs) is also dependent on CX3CL1/CX3CR1 axis, then their potential administration, either intravenously or directly to the cerebral ventricles, with or without simultaneous inhibition of the CX3CL1/CX3CR1 axis in various periods since the injury, might contribute to the success of this type of therapy. However, additional research is necessary [186–191]. Glucocorticoid (GC) administration is another pharmacological modality in TBI and SCI, which has been very well-known and commonly used for many years. However, it is still disputable as regards the effectiveness and long-term consequences [192–195]. Regarding the influence of GC on CX3CL1/CX3CR1 axis functioning, it needs to be emphasized that GC administration may contribute to the reduction of mRNA expression both for CX3CL1 and also CX3CR1 [196]. It may be due to the effect of GC on IL-1β expression level or the activity of NF-κB transcription factor [196]. In the light of this analysis, it is difficult to state whether GC modulation of the CX3CL1/CX3CR1 axis is a neuroprotective or neurotoxic therapeutic option in case of CNS trauma. Regrettably, the effect of GC and other pharmacological agents on the CX3CL1/CX3CR1 axis in the course of TBI and SCI has not been widely discussed. A thorough analysis of the role of the CX3CL1/CX3CR1 axis, particularly in the context of TBI and SCI, and the possibility of its modulation with drugs and immunomodulatory treatment may bring tangible benefits associated with the understanding of this type of injury and the possibility of comprehensive influence exerted on the immune response in order to obtain superior clinical results (Fig. 4).

Overview and summary of potential CX3CL1/CX3CR1 axis-associated therapeutic options for management of traumatic brain and spinal cord injury. GC glucocorticoids, MSC mesenchymal stem cells; (+) refer to activation and (−) to inhibition of CX3CL1/CX3CR1 axis by acting on protein expression and CX3CL1/CX3CR1 interaction
Conclusions and Future Perspectives
Traumatic CNS injuries pose a significant problem and a challenge for the modern society due to their multidimensional character, necessary interdisciplinary approach, and the absence of efficient treatment options. More and more attention has been paid to phenomena occurring during the secondary injury, with a very important element being the local and systemic immune response associated with the migration of immune system cells and the secretion of cytokines and chemokines. The role of conduction related to the CX3CL1/CX3CR1 axis, which is analyzed in this paper, has been well-known in the context of physiological regulation between neurons and microglia, which confirms its position as one of the most important neurochemokines. However, its role has not been widely discussed as regards TBI and SCI. According to available professional literature, the expression of CX3CL1 and CX3CR1 undergoes dynamic regulation both in the course of TBI and SCI. The regulation occurs both at the transcriptional and post-translational levels which results from the changes in the levels of CX3CL1 and CX3CR1 proteins and dedicated mRNA transcripts. The general tendency as regards mRNA expression indicates that CX3CR1 undergoes regulation to a larger extent than CX3CL1 which seems to be more dependent on the regulation at the post-translational level. The post-translational expression of CX3CL1 may be conditioned by the following factors: BBB and BSCB dysfunction, proteolytic cleavage by ADAM10 and ADAM17 proteases, rapid binding to CX3CR1, and penetrating between three compartments including the extracellular space, CSF, and circulating blood [156, 197, 198]. According to the majority of studies, the significant increase in the expression of CX3CR1 and its mRNA after an injury may be justified by the increased monocyte infiltration and increased activity and migration of microglia (CX3CR1+) and also in the local changes of the immunophenotype of these cells [162, 164, 178, 199, 200]. Moreover, it needs to be mentioned that there are potential limitations of fluorescent mouse model which was most commonly used in studies. The limitations are associated with the insertion of GFP protein in the CX3CR1 promotor which leads to haploinsufficiency and the reduced superficial expression of CX3CR1 in heterozygotes which are commonly used as control groups [201]. A significant aspect which impedes the understanding whether the influence of the CX3CL1/CX3CR1 axis is the same as regards the injuries of the whole CNS is the fact that there are substantial morphological and associated neuroinflammatory response differences between the brain and the spinal cord [46, 202]. It seems that in this case, the most important factors are the differences in the quantity, distribution and phenotype of microglia cells, permeability of BBB and BSCB after injury, and the quantity of recruited immune cells [46, 202]. Until now, the analysis of CX3CL1/CX3CR1 axis properties in other neuropathologies has not brought an unambiguous answer concerning its positive or negative consequences. The positive effects may include CX3CR1 deficiency in the course of ischemic stroke [203–205]. However, in conditions like Parkinson’s disease or amyotrophic lateral sclerosis (ALS), CX3CL1 deficiency is associated with the worst outcome [206, 207]. The analysis of CX3CR1 deficiency models in Alzheimer’s disease still leaves some ambiguity [208–210]. The analysis of professional literature demonstrates that CX3CL1/CX3CR1 axis is an attractive potential therapeutic target due to its properties of an inflammatory response regulator, which may facilitate the introduction of immunomodulatory therapies that, depending on the needs, may enable selective intensification or inhibition of an inflammatory response in the course of various neuropathologies with the final aim of obtaining superior clinical results. Thus, after discussing the structure and function of CX3CL1/CX3CR1 axis, the physiological function within the CNS, and its role in the course of TBI and SCI, it may be stated that in spite of a variety of existing and available studies, it is still necessary to conduct further multicenter-detailed research to determine the accurate location of this signaling axis in the pathogenesis of secondary injury development and, consequently, to develop new possible therapeutic methods.
References
Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7(8):728–41. doi:10.1016/S1474-4422(08)70164-9
Kolias AG, Guilfoyle MR, Helmy A, Allanson J, Hutchinson PJ (2013) Traumatic brain injury in adults. Pract Neurol 13(4):228–35. doi:10.1136/practneurol-2012-000268
Silva NA, Sousa N, Reis RL, Salgado AJ (2014) From basics to clinical: a comprehensive review on spinal cord injury. Prog Neurobiol 114:25–57. doi:10.1016/j.pneurobio.2013.11.002
Varma AK, Das A, Wallace G 4th, Barry J, Vertegel AA, Ray SK, Banik NL (2013) Spinal cord injury: a review of current therapy, future treatments, and basic science frontiers. Neurochem Res 38(5):895–905. doi:10.1007/s11064-013-0991-6
Chen AY, Colantonio A (2011) Defining neurotrauma in administrative data using the International Classification of Diseases Tenth Revision. Emerg Themes Epidemiol 8(1):4. doi:10.1186/1742-7622-8-4
Chan V, Thurairajah P, Colantonio A (2013) Defining traumatic brain injury in children and youth using international classification of diseases version 10 codes: a systematic review protocol. Syst Rev 2:102. doi:10.1186/2046-4053-2-102
Bulstrode H, Nicoll JA, Hudson G, Chinnery PF, Di Pietro V, Belli A (2014) Mitochondrial DNA and traumatic brain injury. Ann Neurol 75(2):186–95. doi:10.1002/ana.24116
Bener A, Omar AO, Ahmad AE, Al-Mulla FH, Abdul Rahman YS (2010) The pattern of traumatic brain injuries: a country undergoing rapid development. Brain Inj 24(2):74–80. doi:10.3109/02699050903508192
Faul M, Xu L, Wald MM, Coronado VG (2010) Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, Atlanta (GA), http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf. Accessed 3 August 2015
Jacobsson L, Lexell J (2013) Life satisfaction 6-15 years after a traumatic brain injury. J Rehabil Med 45(10):1010–5. doi:10.2340/16501977-1204
Andelic N, Hammergren N, Bautz-Holter E, Sveen U, Brunborg C, Røe C (2009) Functional outcome and health-related quality of life 10 years after moderate-to-severe traumatic brain injury. Acta Neurol Scand 120(1):16–23. doi:10.1111/j.1600-0404.2008.01116.x
Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J (2006) A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 148(3):255–68. doi:10.1007/s00701-005-0651-y, discussion 268
Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC (2007) The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22(5):341–53
National Spinal Cord Injury Statistical Center (2015) Spinal cord injury facts and figures at a glance National Spinal Cord Injury Statistical Center. University of Alabama at Birmingham, Birmingham, https://www.nscisc.uab.edu/Public/Facts%202015.pdf. Accessed 3 August 2015
Ma VY, Chan L, Carruthers KJ (2014) Incidence, prevalence, costs, and impact on disability of common conditions requiring rehabilitation in the United States: stroke, spinal cord injury, traumatic brain injury, multiple sclerosis, osteoarthritis, rheumatoid arthritis, limb loss, and back pain. Arch Phys Med Rehabil 95(5):986–995.e1. doi:10.1016/j.apmr.2013.10.032
Cripps RA, Lee BB, Wing P, Weerts E, Mackay J, Brown D (2011) A global map for traumatic spinal cord injury epidemiology: towards a living data repository for injury prevention. Spinal Cord 49(4):493–501. doi:10.1038/sc.2010.146
Lee BB, Cripps RA, Fitzharris M, Wing PC (2014) The global map for traumatic spinal cord injury epidemiology: update 2011, global incidence rate. Spinal Cord 52(2):110–6. doi:10.1038/sc.2012.158
Fitzharris M, Cripps RA, Lee BB (2014) Estimating the global incidence of traumatic spinal cord injury. Spinal Cord 52(2):117–22. doi:10.1038/sc.2013.135
Sharma B, Bradbury C, Mikulis D, Green R (2014) Missed diagnosis of traumatic brain injury in patients with traumatic spinal cord injury. J Rehabil Med 46(4):370–3. doi:10.2340/16501977-1261
Macciocchi S, Seel RT, Thompson N, Byams R, Bowman B (2008) Spinal cord injury and co-occurring traumatic brain injury: assessment and incidence. Arch Phys Med Rehabil 89(7):1350–7. doi:10.1016/j.apmr.2007.11.055
van Leeuwen N, Lingsma HF, Perel P, Lecky F, Roozenbeek B, Lu J, Shakur H, Weir J et al (2012) Prognostic value of major extracranial injury in traumatic brain injury: an individual patient data meta-analysis in 39,274 patients. Neurosurgery 70(4):811–8. doi:10.1227/NEU.0b013e318235d640, discussion 818
Wang CM, Chen Y, DeVivo MJ, Huang CT (2001) Epidemiology of extraspinal fractures associated with acute spinal cord injury. Spinal Cord 39(11):589–94
Hofbauer M, Jaindl M, Höchtl LL, Ostermann RC, Kdolsky R, Aldrian S (2012) Spine injuries in polytraumatized pediatric patients: characteristics and experience from a Level I trauma center over two decades. J Trauma Acute Care Surg 73(1):156–61. doi:10.1097/TA.0b013e31824e32b5
Pape HC, Lefering R, Butcher N, Peitzman A, Leenen L, Marzi I, Lichte P, Josten C et al (2014) The definition of polytrauma revisited: an international consensus process and proposal of the new ‘Berlin definition’. J Trauma Acute Care Surg 77(5):780–786
Scholten AC, Haagsma JA, Panneman MJ, van Beeck EF, Polinder S (2014) Traumatic brain injury in the Netherlands: incidence, costs and disability-adjusted life years. PLoS One 9(10):e110905. doi:10.1371/journal.pone.0110905
García-Altés A, Pérez K, Novoa A, Suelves JM, Bernabeu M, Vidal J, Arrufat V, Santamariña-Rubio E et al (2012) Spinal cord injury and traumatic brain injury: a cost-of-illness study. Neuroepidemiology 39(2):103–8. doi:10.1159/000338297
Washington PM, Villapol S, Burns MP (2016) Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol 275(Pt 3):381–8. doi:10.1016/j.expneurol.2015.06.015
Hausmann ON (2003) Post-traumatic inflammation following spinal cord injury. Spinal Cord 41(7):369–78
Rosenfeld JV, Bell RS, Armonda R (2015) Current Concepts in Penetrating and Blast Injury to the Central Nervous System. World J Surg 39(6):1352–62. doi:10.1007/s00268-014-2874-7
Magnuson J, Leonessa F, Ling GS (2012) Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep 12(5):570–9. doi:10.1007/s11910-012-0303-6
Tseng WC, Shih HM, Su YC, Chen HW, Hsiao KY, Chen IC (2011) The association between skull bone fractures and outcomes in patients with severe traumatic brain injury. J Trauma 71(6):1611–4. doi:10.1097/TA.0b013e31823a8a60, discussion 1614
Oertel M, Kelly DF, McArthur D, Boscardin WJ, Glenn TC, Lee JH, Gravori T, Obukhov D et al (2002) Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg 96(1):109–16
Chen H, Guo Y, Chen SW, Wang G, Cao HL, Chen J, Gu Y, Tian HL (2012) Progressive epidural hematoma in patients with head trauma: incidence, outcome, and risk factors. Emerg Med Int 2012:134905. doi:10.1155/2012/134905
Cuenca PJ, Tulley EB, Devita D, Stone A (2004) Delayed traumatic spinal epidural hematoma with spontaneous resolution of symptoms. J Emerg Med 27(1):37–41
Khuyagbaatar B, Kim K, Hyuk Kim Y (2014) Effect of bone fragment impact velocity on biomechanical parameters related to spinal cord injury: a finite element study. J Biomech 47(11):2820–5. doi:10.1016/j.jbiomech.2014.04.042
Persson C, McLure SW, Summers J, Hall RM (2009) The effect of bone fragment size and cerebrospinal fluid on spinal cord deformation during trauma: an ex vivo study. J Neurosurg Spine 10(4):315–23. doi:10.3171/2009.1.SPINE08286
Wilcox RK, Boerger TO, Allen DJ, Barton DC, Limb D, Dickson RA, Hall RM (2003) A dynamic study of thoracolumbar burst fractures. J Bone Joint Surg Am 85-A(11):2184–9
McAllister TW (2011) Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci 13(3):287–300
Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. doi:10.1016/j.expneurol.2012.01.013
Guha A (2004) Management of traumatic brain injury: some current evidence and applications. Postgrad Med J 80(949):650–3
Borgens RB, Liu-Snyder P (2012) Understanding secondary injury. Q Rev Biol 87(2):89–127
Oyinbo CA (2011) Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars) 71(2):281–99
Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N (2009) Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil 24(6):439–51. doi:10.1097/HTR.0b013e3181c15600
Huang YH, Yang TM, Lin WC, Ho JT, Lee TC, Chen WF, Rau CS, Wang HC (2009) The prognosis of acute blunt cervical spinal cord injury. J Trauma 66(5):1441–5. doi:10.1097/TA.0b013e318184ba88
Kawano H, Kimura-Kuroda J, Komuta Y, Yoshioka N, Li HP, Kawamura K, Li Y, Raisman G (2012) Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res 349(1):169–80. doi:10.1007/s00441-012-1336-5
Anthony DC, Couch Y (2014) The systemic response to CNS injury. Exp Neurol 258:105–11. doi:10.1016/j.expneurol.2014.03.013
Das M, Mohapatra S, Mohapatra SS (2012) New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation 9:236. doi:10.1186/1742-2094-9-236
White TE, Ford GD, Surles-Zeigler MC, Gates AS, Laplaca MC, Ford BD (2013) Gene expression patterns following unilateral traumatic brain injury reveals a local pro-inflammatory and remote anti-inflammatory response. BMC Genomics 14:282. doi:10.1186/1471-2164-14-282
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26(8):1191–201. doi:10.1016/j.bbi.2012.06.008
Finnie JW (2013) Neuroinflammation: beneficial and detrimental effects after traumatic brain injury. Inflammopharmacology 21(4):309–20. doi:10.1007/s10787-012-0164-2
Estes ML, McAllister AK (2014) Alterations in immune cells and mediators in the brain: it’s not always neuroinflammation! Brain Pathol 24(6):623–30. doi:10.1111/bpa.12198
Ziebell JM, Morganti-Kossmann MC (2010) Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7(1):22–30. doi:10.1016/j.nurt.2009.10.016
Sandhir R, Puri V, Klein RM, Berman NE (2004) Differential expression of cytokines and chemokines during secondary neuron death following brain injury in old and young mice. Neurosci Lett 369(1):28–32
Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T (2007) Modulation of immune response by head injury. Injury 38(12):1392–400
Bowes AL, Yip PK (2014) Modulating inflammatory cell responses to spinal cord injury: all in good time. J Neurotrauma 31(21):1753–66. doi:10.1089/neu.2014.3429
de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP (2007) Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol 36(2):137–51
Rostène W, Dansereau MA, Godefroy D, Van Steenwinckel J, Reaux-Le Goazigo A, Mélik-Parsadaniantz S, Apartis E, Hunot S et al (2011) Neurochemokines: a menage a trois providing new insights on the functions of chemokines in the central nervous system. J Neurochem 118(5):680–94. doi:10.1111/j.1471-4159.2011.07371.x
Paolicelli RC, Bisht K, Tremblay MÈ (2014) Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci 8:129. doi:10.3389/fncel.2014.00129
Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A et al (1997) A new class of membrane-bound chemokine with a CX3C motif. Nature 385(6617):640–4
Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, Vath J, Gosselin M et al (1997) Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature 387(6633):611–7
Zlotnik A, Yoshie O (2012) The chemokine superfamily revisited. Immunity 36(5):705–16. doi:10.1016/j.immuni.2012.05.008
Zlotnik A, Yoshie O, Nomiyama H (2006) The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol 7(12):243
Nomiyama H, Imai T, Kusuda J, Miura R, Callen DF, Yoshie O (1998) Human chemokines fractalkine (SCYD1), MDC (SCYA22) and TARC (SCYA17) are clustered on chromosome 16q13. Cytogenet Cell Genet 81(1):10–1
Rossi DL, Hardiman G, Copeland NG, Gilbert DJ, Jenkins N, Zlotnik A, Bazan JF (1998) Cloning and characterization of a new type of mouse chemokine. Genomics 47(2):163–70
Xiao J, Dong H, Wu Y, Tian W, Liu L (2009) Gene expression profiling of Cx3cl1 in bone marrow mesenchymal stem cells by osteogenic induction. OMICS 13(4):337–43. doi:10.1089/omi.2009.0018
Chapman GA, Moores KE, Gohil J, Berkhout TA, Patel L, Green P, Macphee CH, Stewart BR (2000) The role of fractalkine in the recruitment of monocytes to the endothelium. Eur J Pharmacol 392(3):189–95
Fong AM, Erickson HP, Zachariah JP, Poon S, Schamberg NJ, Imai T, Patel DD (2000) Ultrastructure and function of the fractalkine mucin domain in CX(3)C chemokine domain presentation. J Biol Chem 275(6):3781–6
Hermand P, Pincet F, Carvalho S, Ansanay H, Trinquet E, Daoudi M, Combadière C, Deterre P (2008) Functional adhesiveness of the CX3CL1 chemokine requires its aggregation. Role of the transmembrane domain. J Biol Chem 283(44):30225–34. doi:10.1074/jbc.M802638200
Patel S, Mukovozov I, Robinson LA (2011) Assessment of the recycling of the membrane-bound chemokine, CX3CL1. Methods Mol Biol 748:143–53. doi:10.1007/978-1-61779-139-0_10
Huang YW, Su P, Liu GY, Crow MR, Chaukos D, Yan H, Robinson LA (2009) Constitutive endocytosis of the chemokine CX3CL1 prevents its degradation by cell surface metalloproteases. J Biol Chem 284(43):29644–53. doi:10.1074/jbc.M109.045682
Liu GY, Kulasingam V, Alexander RT, Touret N, Fong AM, Patel DD, Robinson LA (2005) Recycling of the membrane-anchored chemokine, CX3CL1. J Biol Chem 280(20):19858–66
Dreymueller D, Pruessmeyer J, Groth E, Ludwig A (2012) The role of ADAM-mediated shedding in vascular biology. Eur J Cell Biol 91(6-7):472–85. doi:10.1016/j.ejcb.2011.09.003
Clark AK, Malcangio M (2012) Microglial signalling mechanisms: Cathepsin S and Fractalkine. Exp Neurol 234(2):283–92. doi:10.1016/j.expneurol.2011.09.012
Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F et al (2003) The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102(4):1186–95
Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW (2001) Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem 276(41):37993–8001
Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, Leesnitzer MA, Becherer JD (2005) Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen 8(2):161–71
Schulte A, Schulz B, Andrzejewski MG, Hundhausen C, Mletzko S, Achilles J, Reiss K, Paliga K et al (2007) Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by alpha- and gamma-secretases. Biochem Biophys Res Commun 358(1):233–40
Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, Horuk R, Sparre-Ulrich AH et al (2013) International Union of Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol Rev 66(1):1–79. doi:10.1124/pr.113.007724
Harrison JK, Barber CM, Lynch KR (1994) cDNA cloning of a G-protein-coupled receptor expressed in rat spinal cord and brain related to chemokine receptors. Neurosci Lett 169(1-2):85–9
Raport CJ, Schweickart VL, Eddy RL Jr, Shows TB, Gray PW (1995) The orphan G-protein-coupled receptor-encoding gene V28 is closely related to genes for chemokine receptors and is expressed in lymphoid and neural tissues. Gene 163(2):295–9
Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S et al (1997) Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91(4):521–30
Maho A, Bensimon A, Vassart G, Parmentier M (1999) Mapping of the CCXCR1, CX3CR1, CCBP2 and CCR9 genes to the CCR cluster within the 3p21.3 region of the human genome. Cytogenet Cell Genet 87(3-4):265–8
Garin A, Pellet P, Deterre P, Debré P, Combadière C (2002) Cloning and functional characterization of the human fractalkine receptor promoter regions. Biochem J 368(Pt 3):753–60
Combadiere C, Gao J, Tiffany HL, Murphy PM (1998) Gene cloning, RNA distribution, and functional expression of mCX3CR1, a mouse chemotactic receptor for the CX3C chemokine fractalkine. Biochem Biophys Res Commun 253(3):728–32
Davenport AP, Alexander SP, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, Liew WC, Mpamhanga CP et al (2013) International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev 65(3):967–86. doi:10.1124/pr.112.007179
Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB (2006) Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 88(3):263–73
Raucci R, Costantini S, Castello G, Colonna G (2014) An overview of the sequence features of N- and C-terminal segments of the human chemokine receptors. Cytokine 70(2):141–50. doi:10.1016/j.cyto.2014.07.257
Szpakowska M, Perez Bercoff D, Chevigné A (2014) Closing the ring: a fourth extracellular loop in chemokine receptors. Sci Signal 7(341):pe21. doi:10.1126/scisignal.2005664
Mizoue LS, Bazan JF, Johnson EC, Handel TM (1999) Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry 38(5):1402–14
Nakayama T, Watanabe Y, Oiso N, Higuchi T, Shigeta A, Mizuguchi N, Katou F, Hashimoto K et al (2010) Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J Immunol 185(11):6472–9. doi:10.4049/jimmunol.0904126
Faure S, Meyer L, Costagliola D, Vaneensberghe C, Genin E, Autran B, Delfraissy JF, McDermott DH et al (2000) Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science 287(5461):2274–7
Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M et al (2005) CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307(5707):254–8
Ludeman JP, Stone MJ (2014) The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol 171(5):1167–79. doi:10.1111/bph.12455
Tobin AB (2008) G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol 153(Suppl 1):S167–76. doi:10.1038/sj.bjp.0707662
Zhao R, Wang Y, Shen R, Sun Y (2010) Relationship between CX3CR1 genetic polymorphism and carotid atherosclerosis. Artif Cells Blood Substit Immobil Biotechnol 38(1):19–23. doi:10.3109/10731190903495728
Arli B, Irkec C, Menevse S, Yilmaz A, Alp E (2013) Fractalkine gene receptor polymorphism in patients with multiple sclerosis. Int J Neurosci 123(1):31–7. doi:10.3109/00207454.2012.723079
Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, Vassileva G, Lira SA (2001) Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol 21(9):3159–65
Cardona AE, Sasse ME, Liu L, Cardona SM, Mizutani M, Savarin C, Hu T, Ransohoff RM (2008) Scavenging roles of chemokine receptors: chemokine receptor deficiency is associated with increased levels of ligand in circulation and tissues. Blood 112(2):256–63. doi:10.1182/blood-2007-10-118497
Zhu W, Acosta C, MacNeil B, Cortes C, Intrater H, Gong Y, Namaka M (2013) Elevated expression of fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) in the dorsal root ganglia and spinal cord in experimental autoimmune encephalomyelitis: implications in multiple sclerosis-induced neuropathic pain. Biomed Res Int 2013:480702. doi:10.1155/2013/480702
Zhang J, Patel JM (2010) Role of the CX3CL1-CX3CR1 axis in chronic inflammatory lung diseases. Int J Clin Exp Med 3(3):233–44
Harrison JK, Jiang Y, Wees EA, Salafranca MN, Liang HX, Feng L, Belardinelli L (1999) Inflammatory agents regulate in vivo expression of fractalkine in endothelial cells of the rat heart. J Leukoc Biol 66(6):937–44
Aoyama T, Inokuchi S, Brenner DA, Seki E (2010) CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 52(4):1390–400. doi:10.1002/hep.23795
Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR (2001) The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am J Pathol 158(3):855–66
Hannan NJ, Jones RL, White CA, Salamonsen LA (2006) The chemokines, CX3CL1, CCL14, and CCL4, promote human trophoblast migration at the feto-maternal interface. Biol Reprod 74(5):896–904
Hamann I, Unterwalder N, Cardona AE, Meisel C, Zipp F, Ransohoff RM, Infante-Duarte C (2011) Analyses of phenotypic and functional characteristics of CX3CR1-expressing natural killer cells. Immunology 133(1):62–73. doi:10.1111/j.1365-2567.2011.03409.x
Kobayashi T, Okamoto S, Iwakami Y, Nakazawa A, Hisamatsu T, Chinen H, Kamada N, Imai T et al (2007) Exclusive increase of CX3CR1 + CD28-CD4+ T cells in inflammatory bowel disease and their recruitment as intraepithelial lymphocytes. Inflamm Bowel Dis 13(7):837–46
Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D (2003) Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 197(12):1701–7
El-Shazly A, Berger P, Girodet PO, Ousova O, Fayon M, Vernejoux JM, Marthan R, Tunon-de-Lara JM (2006) Fraktalkine produced by airway smooth muscle cells contributes to mast cell recruitment in asthma. J Immunol 176(3):1860–8
Łyszkiewicz M, Witzlau K, Pommerencke J, Krueger A (2011) Chemokine receptor CX3CR1 promotes dendritic cell development under steady-state conditions. Eur J Immunol 41(5):1256–65. doi:10.1002/eji.201040977
Nourshargh S, Alon R (2014) Leukocyte migration into inflamed tissues. Immunity 41(5):694–707. doi:10.1016/j.immuni.2014.10.008
Ransohoff RM (2009) Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31(5):711–21. doi:10.1016/j.immuni.2009.09.010
Ostuni MA, Guellec J, Hermand P, Durand P, Combadière C, Pincet F, Deterre P (2014) CX3CL1, a chemokine finely tuned to adhesion: critical roles of the stalk glycosylation and the membrane domain. Biol Open 3(12):1173–82. doi:10.1242/bio.20149845
Haskell CA, Cleary MD, Charo IF (2000) Unique role of the chemokine domain of fractalkine in cell capture. Kinetics of receptor dissociation correlate with cell adhesion. J Biol Chem 275(44):34183–9
Fujita M, Takada YK, Takada Y (2014) The chemokine fractalkine can activate integrins without CX3CR1 through direct binding to a ligand-binding site distinct from the classical RGD-binding site. PLoS One 9(5):e96372. doi:10.1371/journal.pone.0096372
Fujita M, Takada YK, Takada Y (2012) Integrins αvβ3 and α4β1 act as coreceptors for fractalkine, and the integrin-binding defective mutant of fractalkine is an antagonist of CX3CR1. J Immunol 189(12):5809–19. doi:10.4049/jimmunol.1200889
Blauth K, Zhang X, Chopra M, Rogan S, Markovic-Plese S (2015) The role of fractalkine (CX3CL1) in regulation of CD4+ cell migration to the central nervous system in patients with relapsing-remitting multiple sclerosis. Clin Immunol 157(2):121–32. doi:10.1016/j.clim.2015.01.001
Chandrasekar B, Mummidi S, Perla RP, Bysani S, Dulin NO, Liu F, Melby PC (2003) Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J 373(Pt 2):547–58
Lauro C, Catalano M, Di Paolo E, Chece G, de Costanzo I, Trettel F, Limatola C (2015) Fractalkine/CX3CL1 engages different neuroprotective responses upon selective glutamate receptor overactivation. Front Cell Neurosci 8:472. doi:10.3389/fncel.2014.00472
Gan AM, Butoi ED, Manea A, Simion V, Stan D, Parvulescu MM, Calin M, Manduteanu I et al (2013) Inflammatory effects of resistin on human smooth muscle cells: up-regulation of fractalkine and its receptor, CX3CR1 expression by TLR4 and Gi-protein pathways. Cell Tissue Res 351(1):161–74. doi:10.1007/s00441-012-1510-9
Matsumiya T, Ota K, Imaizumi T, Yoshida H, Kimura H, Satoh K (2010) Characterization of synergistic induction of CX3CL1/fractalkine by TNF-alpha and IFN-gamma in vascular endothelial cells: an essential role for TNF-alpha in post-transcriptional regulation of CX3CL1. J Immunol 184(8):4205–14. doi:10.4049/jimmunol.0903212
Matsumiya T, Imaizumi T, Fujimoto K, Cui X, Shibata T, Tamo W, Kumagai M, Tanji K et al (2001) Soluble interleukin-6 receptor alpha inhibits the cytokine-Induced fractalkine/CX3CL1 expression in human vascular endothelial cells in culture. Exp Cell Res 269(1):35–41
Imaizumi T, Yoshida H, Satoh K (2004) Regulation of CX3CL1/fractalkine expression in endothelial cells. J Atheroscler Thromb 11(1):15–21
Zhang J, Hu H, Palma NL, Harrison JK, Mubarak KK, Carrie RD, Alnuaimat H, Shen X et al (2012) Hypoxia-induced endothelial CX3CL1 triggers lung smooth muscle cell phenotypic switching and proliferative expansion. Am J Physiol Lung Cell Mol Physiol 303(10):L912–22. doi:10.1152/ajplung.00014.2012
Kostadinova FI, Baba T, Ishida Y, Kondo T, Popivanova BK, Mukaida N (2010) Crucial involvement of the CX3CR1-CX3CL1 axis in dextran sulfate sodium-mediated acute colitis in mice. J Leukoc Biol 88(1):133–43. doi:10.1189/jlb.1109768
Zujovic V, Schussler N, Jourdain D, Duverger D, Taupin V (2001) In vivo neutralization of endogenous brain fractalkine increases hippocampal TNFalpha and 8-isoprostane production induced by intracerebroventricular injection of LPS. J Neuroimmunol 115(1-2):135–43
Lauro C, Cipriani R, Catalano M, Trettel F, Chece G, Brusadin V, Antonilli L, van Rooijen N et al (2010) Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacology 35(7):1550–9. doi:10.1038/npp.2010.26
Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, Ludwig A, Lira SA et al (2011) In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118(22):e156–67. doi:10.1182/blood-2011-04-348946
Tarozzo G, Bortolazzi S, Crochemore C, Chen SC, Lira AS, Abrams JS, Beltramo M (2003) Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res 73(1):81–8
Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A et al (1998) Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett 429(2):167–72
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN et al (1998) Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 95(18):10896–901
Yang JL, Xu B, Li SS, Zhang WS, Xu H, Deng XM, Zhang YQ (2012) Gabapentin reduces CX3CL1 signaling and blocks spinal microglial activation in monoarthritic rats. Mol Brain 5:18. doi:10.1186/1756-6606-5-18
Lindia JA, McGowan E, Jochnowitz N, Abbadie C (2005) Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain 6(7):434–8
Sunnemark D, Eltayeb S, Nilsson M, Wallström E, Lassmann H, Olsson T, Berg AL, Ericsson-Dahlstrand A (2005) CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation 2:17
Schwaeble WJ, Stover CM, Schall TJ, Dairaghi DJ, Trinder PK, Linington C, Iglesias A, Schubart A et al (1998) Neuronal expression of fractalkine in the presence and absence of inflammation. FEBS Lett 439(3):203–7
Hulshof S, van Haastert ES, Kuipers HF, van den Elsen PJ, De Groot CJ, van der Valk P, Ravid R, Biber K (2003) CX3CL1 and CX3CR1 expression in human brain tissue: noninflammatory control versus multiple sclerosis. J Neuropathol Exp Neurol 62(9):899–907
Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB (1999) Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol 163(3):1628–35
Mills JH, Alabanza LM, Mahamed DA, Bynoe MS (2012) Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J Neuroinflammation 9:193
Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH (2002) Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia 37(4):314–27
Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW (2003) CSF and serum levels of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system. J Neuroimmunol 137(1-2):210–7
Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU (2002) Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res 69(3):418–26
Meucci O, Fatatis A, Simen AA, Miller RJ (2000) Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A 97(14):8075–80
Heinisch S, Kirby LG (2009) Fractalkine/CX3CL1 enhances GABA synaptic activity at serotonin neurons in the rat dorsal raphe nucleus. Neuroscience 164(3):1210–23. doi:10.1016/j.neuroscience.2009.08.075
Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ (1998) Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A 95(24):14500–5
Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F (2005) Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol 166(1-2):19–28
Ragozzino D, Di Angelantonio S, Trettel F, Bertollini C, Maggi L, Gross C, Charo IF, Limatola C et al (2006) Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J Neurosci 26(41):10488–98
Maggi L, Trettel F, Scianni M, Bertollini C, Eusebi F, Fredholm BB, Limatola C (2009) LTP impairment by fractalkine/CX3CL1 in mouse hippocampus is mediated through the activity of adenosine receptor type 3 (A3R). J Neuroimmunol 215(1-2):36–42. doi:10.1016/j.jneuroim.2009.07.016
Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA (2009) Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem 110(5):1547–56. doi:10.1111/j.1471-4159.2009.06253.x
Sheridan GK, Murphy KJ (2013) Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol 3(12):130181. doi:10.1098/rsob.130181
Wolf Y, Yona S, Kim KW, Jung S (2013) Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 7:26. doi:10.3389/fncel.2013.00026
Sheridan GK, Wdowicz A, Pickering M, Watters O, Halley P, O’Sullivan NC, Mooney C, O’Connell DJ et al (2014) CX3CL1 is up-regulated in the rat hippocampus during memory-associated synaptic plasticity. Front Cell Neurosci 8:233. doi:10.3389/fncel.2014.00233
Zujovic V, Taupin V (2003) Use of cocultured cell systems to elucidate chemokine-dependent neuronal/microglial interactions: control of microglial activation. Methods 29(4):345–50
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14(11):1189–97
Zujovic V, Benavides J, Vigé X, Carter C, Taupin V (2000) Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 29(4):305–15
Mattison HA, Nie H, Gao H, Zhou H, Hong JS, Zhang J (2013) Suppressed pro-inflammatory response of microglia in CX3CR1 knockout mice. J Neuroimmunol 257(1-2):110–5. doi:10.1016/j.jneuroim.2013.02.008
Lee S, Xu G, Jay TR, Bhatta S, Kim KW, Jung S, Landreth GE, Ransohoff RM et al (2014) Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J Neurosci 34(37):12538–46. doi:10.1523/JNEUROSCI.0853-14.2014
Rancan M, Bye N, Otto VI, Trentz O, Kossmann T, Frentzel S, Morganti-Kossmann MC (2004) The chemokine fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J Cereb Blood Flow Metab 24(10):1110–8
Marshall LF, Marshall SB, Klauber MR, Clark MVB, Eisenberg HM, Jane JA, Luerssen TG, Marmarou A et al (1991) A new classification of head injury based on computerized tomography. J Neurosurg 75(suppl):S14–S20
Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ (2011) The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab 31(2):658–70. doi:10.1038/jcbfm.2010.142
Gaetani P, Pisano P, Solinas G, Colombo P, Destro A, Levi D, Aimar E, Rodriguez R et al (2013) Immunohistohemical expression of the chemokine fractalkine and its receptor in the human brain cortex after severe traumatic brain injury and brain hemorrhage. J Neurosurg Sci 57(1):55–62
Fahlenkamp AV, Coburn M, Czaplik M, Ryang YM, Kipp M, Rossaint R, Beyer C (2011) Expression analysis of the early chemokine response 4 h after in vitro traumatic brain injury. Inflamm Res 60(4):379–87. doi:10.1007/s00011-010-0281-6
Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM (2008) Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol 212(2):337–47. doi:10.1016/j.expneurol.2008.04.009
Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG (2011) Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci 31(27):9910–22. doi:10.1523/JNEUROSCI.2114-11.2011
Cizkova D, Le Marrec-Croq F, Franck J, Slovinska L, Grulova I, Devaux S, Lefebvre C, Fournier I et al (2014) Alterations of protein composition along the rostro-caudal axis after spinal cord injury: proteomic, in vitro and in vivo analyses. Front Cell Neurosci 8:105. doi:10.3389/fncel.2014.00105
Blomster LV, Brennan FH, Lao HW, Harle DW, Harvey AR, Ruitenberg MJ (2013) Mobilisation of the splenic monocyte reservoir and peripheral CX3CR1 deficiency adversely affects recovery from spinal cord injury. Exp Neurol 247:226–40. doi:10.1016/j.expneurol.2013.05.002
Loane DJ, Byrnes KR (2010) Role of microglia in neurotrauma. Neurotherapeutics 7(4):366–77. doi:10.1016/j.nurt.2010.07.002
Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ (2010) Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133(Pt 2):433–47. doi:10.1093/brain/awp322
Jung S, Schwartz M (2012) Non-identical twins - microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation. Front Immunol 3:89. doi:10.3389/fimmu.2012.00089
Donnelly DJ, Popovich PG (2008) Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol 209(2):378–88
David S, Kroner A (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 12(7):388–99. doi:10.1038/nrn3053
Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14(10):1227–35. doi:10.1038/nn.2923
Guillemin GJ, Brew BJ (2004) Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol 75(3):388–97
Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF (2010) Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5(10):e13693. doi:10.1371/journal.pone.0013693
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–8
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29(43):13435–44. doi:10.1523/JNEUROSCI.3257-09.2009
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19(1):71–82
Urra X, Villamor N, Amaro S, Gómez-Choco M, Obach V, Oleaga L, Planas AM, Chamorro A (2009) Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab 29(5):994–1002. doi:10.1038/jcbfm.2009.25
Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG (2006) Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23(5):635–59
Blomster LV, Vukovic J, Hendrickx DA, Jung S, Harvey AR, Filgueira L, Ruitenberg MJ (2011) CX3CR1 deficiency exacerbates neuronal loss and impairs early regenerative responses in the target-ablated olfactory epithelium. Mol Cell Neurosci 48(3):236–45. doi:10.1016/j.mcn.2011.08.004
Morganti JM, Jopson TD, Riparip LK, Rosi S (2014) CX3CR1 deficiency ameliorates TBI-induced inflammatory response and cognitive dysfunction. J Neurotrauma 31(12): A-1-A-126. doi:10.1089/neu.2014.9935.abstracts
Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, Sammali E, Pischiutta F, De Simoni MG (2015) Fractalkine receptor deficiency is associated with early protection, but late worsening of outcome following brain trauma in mice. J Neurotrauma. doi:10.1089/neu.2015.4041
Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK (2004) Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev 28(4):365–78
Febinger HY, Thomasy HE, Pavlova MN, Ringgold KM, Barf PR, George AM, Grillo JN, Bachstetter AD et al (2015) Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J Neuroinflammation 2(12):154. doi:10.1186/s12974-015-0386-5
Taylor RA, Hammond MD, Ai Y, Sansing LH (2014) CX3CR1 signaling on monocytes is dispensable after intracerebral hemorrhage. PLoS One 9(12):e114472. doi:10.1371/journal.pone.0114472
Karlström S, Nordvall G, Sohn D, Hettman A, Turek D, Åhlin K, Kers A, Claesson M et al (2013) Substituted 7-amino-5-thio-thiazolo[4,5-d]pyrimidines as potent and selective antagonists of the fractalkine receptor (CX3CR1). J Med Chem 56(8):3177–90. doi:10.1021/jm3012273
Zanier ER, Pischiutta F, Riganti L, Marchesi F, Turola E, Fumagalli S, Perego C, Parotto E et al (2014) Bone marrow mesenchymal stromal cells drive protective M2 microglia polarization after brain trauma. Neurotherapeutics 11(3):679–95. doi:10.1007/s13311-014-0277-y
Ridderstad Wollberg A, Ericsson-Dahlstrand A, Juréus A, Ekerot P, Simon S, Nilsson M, Wiklund SJ, Berg AL et al (2014) Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc Natl Acad Sci U S A 111(14):5409–14. doi:10.1073/pnas.1316510111
Zhu J, Zhou Z, Liu Y, Zheng J (2009) Fractalkine and CX3CR1 are involved in the migration of intravenously grafted human bone marrow stromal cells toward ischemic brain lesion in rats. Brain Res 1287:173–83. doi:10.1016/j.brainres.2009.06.068
Wang S, Cheng H, Dai G, Wang X, Hua R, Liu X, Wang P, Chen G et al (2013) Umbilical cord mesenchymal stem cell transplantation significantly improves neurological function in patients with sequelae of traumatic brain injury. Brain Res 1532:76–84. doi:10.1016/j.brainres.2013.08.001
Neirinckx V, Cantinieaux D, Coste C, Rogister B, Franzen R, Wislet-Gendebien S (2014) Concise review: Spinal cord injuries: how could adult mesenchymal and neural crest stem cells take up the challenge? Stem Cells 32(4):829–43. doi:10.1002/stem.1579
Giunti D, Parodi B, Usai C, Vergani L, Casazza S, Bruzzone S, Mancardi G, Uccelli A (2012) Mesenchymal stem cells shape microglia effector functions through the release of CX3CL1. Stem Cells 30(9):2044–53. doi:10.1002/stem.1174
Sheikh AM, Nagai A, Wakabayashi K, Narantuya D, Kobayashi S, Yamaguchi S, Kim SU (2011) Mesenchymal stem cell transplantation modulates neuroinflammation in focal cerebral ischemia: contribution of fractalkine and IL-5. Neurobiol Dis 41(3):717–24. doi:10.1016/j.nbd.2010.12.009
Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF (2008) Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med 14(11-12):731–40. doi:10.2119/2008-00050.Beauchamp
Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P et al (2004) Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 364(9442):1321–8
Bydon M, Lin J, Macki M, Gokaslan ZL, Bydon A (2014) The current role of steroids in acute spinal cord injury. World Neurosurg 82(5):848–54. doi:10.1016/j.wneu.2013.02.062
Bracken MB (2012) Steroids for acute spinal cord injury. Cochrane Database Syst Rev 1:CD001046. doi:10.1002/14651858.CD001046.pub2
Sorrells SF, Munhoz CD, Manley NC, Yen S, Sapolsky RM (2014) Glucocorticoids increase excitotoxic injury and inflammation in the hippocampus of adult male rats. Neuroendocrinology 100(2-3):129–40. doi:10.1159/000367849
Warren KM, Reeves TM, Phillips LL (2012) MT5-MMP, ADAM-10, and N-cadherin act in concert to facilitate synapse reorganization after traumatic brain injury. Neurotrauma 29(10):1922–40. doi:10.1089/neu.2012.2383
Vidal PM, Lemmens E, Avila A, Vangansewinkel T, Chalaris A, Rose-John S, Hendrix S (2013) ADAM17 is a survival factor for microglial cells in vitro and in vivo after spinal cord injury in mice. Cell Death Dis 4:e954. doi:10.1038/cddis.2013.466
Wang X, Cao K, Sun X, Chen Y, Duan Z, Sun L, Guo L, Bai P et al (2015) Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris. Glia 63(4):635–51. doi:10.1002/glia.22774
Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, Huang AY, Silver J (2014) High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol 254:109–20. doi:10.1016/j.expneurol.2014.01.013
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20(11):4106–14
Zhang B, Gensel JC (2014) Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp Neurol 258:112–20. doi:10.1016/j.expneurol.2014.04.007
Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q et al (2002) Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol 125(1-2):59–65
Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ (2008) Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab 28(10):1707–21. doi:10.1038/jcbfm.2008.64
Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, Perego C, De Simoni MG et al (2011) CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci 31(45):16327–35. doi:10.1523/JNEUROSCI.3611-11.2011
Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC (2011) CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation 8:9. doi:10.1186/1742-2094-8-9
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9(7):917–24
Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L (2011) CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 286(37):32713–22. doi:10.1074/jbc.M111.254268
Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol 177(5):2549–62. doi:10.2353/ajpath.2010.100265
Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM et al (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci 13(4):411–3. doi:10.1038/nn.2511
Acknowledgments
The authors would like to thank Prof. Włodzimierz Maśliński (Department of Pathophysiology and Immunology, Eleonora Reicher National Institute of Geriatrics, Rheumatology and Rehabilitation, Warsaw, Poland) for major effort of critically reading this manuscript and for his valuable editorial suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Poniatowski, Ł.A., Wojdasiewicz, P., Krawczyk, M. et al. Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents. Mol Neurobiol 54, 2167–2188 (2017). https://doi.org/10.1007/s12035-016-9787-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9787-4