
Abstract
Traumatic injury to the brain (TBI) results in a complex set of responses involving various symptoms and long-term consequences. TBI of any form can cause cognitive, behavioral and immunologic changes in later life, which underscores the problem of underdiagnosis of mild TBI that can cause long-term neurological deficits. TBI disrupts the blood–brain barrier (BBB) leading to infiltration of immune cells into the brain and subsequent inflammation and neurodegeneration. TBI-induced peripheral immune responses can also result in multiorgan damage. Despite worldwide research efforts, the methods of diagnosis, monitoring and treatment for TBI are still relatively ineffective. In this review, we delve into the mechanism of how TBI-induced central and peripheral immune responses affect the disease outcome and discuss recent developments in the continuing effort to combat the consequences of TBI and new ways to enhance repair of the damaged brain.
Similar content being viewed by others


Introduction
Traumatic brain injury (TBI) is a complex process involving a broad spectrum of symptoms and long-term consequences including disabilities. It is a serious health problem in the United States and around the world. Recent data show that approximately 1.7 million people sustain a TBI annually [1, 2] including U.S. soldiers involved in combat operations and public safety personnel surviving terrorist attacks. An estimated 150 to 300,000 military personnel from Operation Iraqi Freedom and Operation Enduring Freedom suffered from TBI [3–5]. It contributes to 30% of all injury-related deaths and costs about $60 billion annually. TBI of any form, mild to severe, can cause intellectual and cognitive deficits, mood and behavioral changes both short- and long-term [6–9]. In the long term, these can cause potentially permanent changes and may lead to post-traumatic stress disorder (PTSD) in the general population as well as those in the military. Besides psychological symptoms, immune suppression from TBI and subsequent infections are important consequences [10].
Although TBI can range from mild to severe, most TBI is mild and characterized by brief changes in mental status and cognitive ability [11]. Although the consequences of mild TBI are not readily appreciated, it can still cause infrastructural damage to the brain and secondary axonal injury [12] and shows symptoms like cognitive or intellectual deficits and behavioral and personality changes even six months after injury [10]. In most patients suffering from mild brain injury, the symptoms disappear within six months but many others suffer in a variety of ways that may be underappreciated and treated inadequately or improperly. Even under asymptomatic conditions, unhealed neurodegeneration may cause a spectrum of diseases with huge cost to society [10].
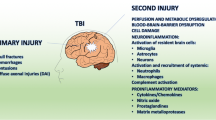
Once the brain suffers mechanical insult, the injury process evolves over time and includes (a) primary injury caused by direct or indirect contusion resulting in shearing or stretching of brain tissue, subdural hematoma and cerebral ischemia (b) secondary injury characterized by diffuse axonal injury and inflammatory reactions, and (c) regeneration. The secondary, that is, the nonmechanical injury phase, is progressive and lasts from hours to days [13, 14], significantly contributing to neurological disabilities [15]. Injury to the cerebral vasculature breaks the blood–brain barrier (BBB), allows entry of immune cells and stimulates inflammatory reactions. The molecular events result in apoptosis, inflammation, altered plasticity and neuronal regeneration. The complex nature of acute and chronic inflammatory reactions may aggravate the pathologic outcome or promote the repair process [16, 17]. Also, multiorgan damage in trauma patients can lead to elevated circulatory levels of inflammatory cytokines that may contribute to the post-TBI pathogenesis of the brain [18] and cause multiple organ dysfunction syndrome (MODS) and death [19]. In this review we discuss the mechanism of interaction between the systemic immune response and the brain after TBI and current novel treatment approaches to combat TBI-induced damage (Figure 1).

Possible mechanism and the interactions between brain and systemic immunity after traumatic brain injury (TBI). Blood–brain barrier (BBB) disruption allows peripheral immune cell infiltration into the brain. Interaction between brain and peripheral immune organs can cause either hyperinflammation or immune suppression. Anti-inflammatory cytokines may eventually lead to neuronal recovery.
Response of the central nervous system to TBI: neuroinflammation and pathobiology of the CNS
The BBB protects the brain and maintains the homeostasis. Following TBI, a massive release of excitatory amino acid neurotransmitters, particularly glutamate, takes place [20, 21]. These molecules interact with neurons and astrocytes and cause increased Ca2+, Na+, and K+ fluxes through overstimulation of glutamate receptors. As a consequence, catabolic processes are activated resulting in BBB breakdown [17]. The kinin system, excitotoxicity, activation of the innate immune system leading to neutrophil recruitment, mitochondrial alterations and microglial activation lead to generation of reactive oxygen species (ROS) which in turn trigger downstream pathways and cause oxidative damage, modifications in tight junctions and matrix metalloproteinase (MMP) activation. Thus ROS play an important role in mediating TBI-induced changes in BBB permeability [22]. ROS have also been implicated in fungal toxin T-2-mediated alteration in BBB permeability [23]. Recent animal studies have shown that BBB breakdown involves transcriptional changes in the neurovascular network and eventual neurodegeneration [24].
The leaky BBB allows the passage of inflammatory molecules and cells into and out of the injured brain initiating a cascade of responses in the brain and other organs. The most important events contributing toward the pathology of TBI are reactive astrogliosis, microglial activation, infiltration of immune cells in the CNS and neurodegeneration. Both the primary and secondary mechanisms of TBI cause neurodegeneration and contribute to post-traumatic neurological deficits [25, 26]. One of the major pathological outcomes of these mechanisms is diffuse axonal injury (DAI), the main clinical feature of human TBI, leading to diffuse degeneration of cerebral white matter [27, 28]. In a rodent model of diffuse TBI, Cernak et al. [26] have shown hypertension, brain edema, increased permeability of BBB, DAI and apoptosis of the cerebral cells following a high velocity impact. Alder et al. have characterized the pathological and behavioral changes in a lateral fluid percussion model (LFPI) of TBI in mice [29]. The process of TBI-induced neuronal cell death has multiple, overlapping and distinct molecular mechanisms [30]. Following TBI, neuronal cell death can be induced by caspase-dependent or -independent pathways [31], by cell cycle activation in which mature neuronal cells reenter the cell cycle and then die [32] or by autophagy [33]. In the caspase-dependent pathways, caspase 3 appears to play the major role in causing TBI-induced apoptosis, although caspase 6 and 7 have also been acknowledged as proapoptotic molecules [34]. The caspase-independent pathway is more complex and involves mitochondrial proapoptotic molecules including apoptosis-inducing factor (AIF) [35] and its regulators like PARP-1 [36, 37], cyclophilin [38, 39] and heat shock protein-70 (HSP-70) [40]. These mechanisms probably work together in stress-induced neuronal cell death and, therefore, inhibition of only one pathway may not be sufficient to protect neurons after TBI [41] (Table 1).
Role of neurocytokines and neurochemokines in the central response to TBI
In the 1980s, scientists observed that the brain, endocrine system and immune system function together to maintain homeostasis in health and prevent disease [55]. After Spangelo and co-workers identified cytokines and their role in inflammation and immunity [56], brain researchers began to study the actions of cytokines in the CNS. In 1992, Ban et al.[57] found that interleukin-1β (IL-1β) was synthesized in the brain under pathological conditions while others showed that peripherally synthesized cytokines were transported to the brain via the bloodstream or cerebrospinal fluid (CSF) and secreted into the brain parenchyma during breakdown of the BBB [58], thus linking the brain and immune system [59]. The chemokines are the chemotactic cytokines that play an important role in leukocytes migration [60]. Their role in signaling in the CNS was reported by investigators in the late 1990s [61–63]. Under inflammatory or neurodegenerative conditions in the CNS, chemokine molecules are synthesized by activated microglia or astrocytes which take part in the defense of the CNS by recruiting monocytes to the injury site [64–67]. Under normal physiological conditions the tight junctions of the BBB prevent infiltration of circulating leukocytes into the brain parenchyma [16, 68]. Pathological conditions like infections, mechanical trauma or toxicity may disrupt the BBB and allow immune cells to enter the brain parenchyma in response to chemokine signaling from resident immune cells.
In addition to macrophages and glial cells, neurons have also been found to express chemokines and chemokine receptors in the brain under physiological and pathological conditions [2, 62, 69, 70]. Fractalkine (CX3CL1) was the first chemokine seen to be constitutively expressed by the neuronal cells of the CNS [66]. Later, other chemokines like CXCL14/BRAK; [71, 72], CCL20 [45], CCL21 [47], CXCL12/SDF-1 and CCL2/MCP-1, were found in neuronal cells under various pathological conditions including TBI. Helmy et al.[43] have reviewed the temporal profile of 42 cytokines after TBI in human patients. Upregulation of CCL20 has been observed in human subjects one day after severe TBI [43]. Furthermore, a recent study identified CCL20 as a dual-acting chemokine with the potential for inhibiting immune reactions and more importantly in attracting inflammatory effectors and activators [44]. Studies in our laboratory showed cerebral as well as systemic expression of CCL20 after mild TBI in rats [45]. Recently, Biber and co-workers [46, 47, 73] showed that damaged neurons produce CCL21, which assumes a neuromodulatory function. In a spinal cord injury model, Zhao et al.[74] have shown that CCL21 expressed by the damaged neurons used the CXCR3 receptor instead of the usual CCR7 receptor to activate the local microglial cells [75–77] and initiate inflammatory reactions. These neurochemokines can also be involved in nonimmune-related functions like neuromodulation or neurotransmission, which could be important in TBI. As Rostene and colleagues have pointed out, this could be the complex communication network between the neurons and the cells in its microenvironment that informs them about the damage [2].
In addition to chemokines, various cytokines have also been reported to be expressed following TBI, including TNF-α associated with activated microglia and astrocytes that may initiate the inflammatory process [78]. IL-6 in the injured brain has been associated with reactive astrogliosis, neuronal injury, and infiltration of peripheral cells [78–81]. TGF-β expression in the astrocytes and microglia after injury has been implicated in the pathology and dysfunction of the CNS and IL-1, IL-6, IL-8, IL-10, granulocyte colony-stimulating factor, TNF-α, FAS ligand and monocyte chemo-attractant protein 1 [18, 82–84] are thought to account for the progressive injury. In a rat fluid percussion injury model a biphasic production of TGF-β, mainly of TGF-β 2, was detected in the ipsilateral cortex, with a first peak at 30 minutes and a second peak at 48 hours after the lesion. This response was accompanied by transient production of TNF-α and IL-6 occurring between five and eighteen hours after trauma. From this temporal pattern, Rimaniol et al. suggested an alternative pro- and anti-inflammatory role of TGF-β in the regulation of the brain cytokine network providing an endogenous mechanism for the control of the inflammatory reaction in traumatic brain injury [85].
Activation of resident immune cells of the CNS following TBI
Microglial activation is integral to the response of the brain and spinal cord to injury [86]. A number of factors including pro-inflammatory and anti-inflammatory cytokines, chemokines, growth factors, nitric oxide, prostaglandins, and superoxide and other reactive oxygen species are released by microglia and modulate secondary injury as well as recovery after injury. Microglial activation is regulated in part by poly(ADP-ribose) polymerase-1 (PARP-1) [87]. Using a PARP-knockout mouse model of TBI, Whalen et al.[54] showed improved motor and cognitive functions after TBI and thereby indicated a detrimental role of PARP in the pathogenesis of TBI. In 2006, Bernardo and colleagues [88] observed that inhibition of microglial activation by peroxisome proliferator-activated receptor (PPAR)-gamma and its synthetic agonists by expression of surface antigens, synthesis of nitric oxide, prostaglandins, inflammatory cytokines and chemokines by TBI-induced brain inflammation could be controlled [88]. Perivascular macrophages are reactive cells that produce IL-1β and TNFα after CNS injury. In the perivascular endothelium these cytokines induce the expression of adhesion molecules and promote leukocyte infiltration [89].
Response of the peripheral immune system to TBI: systemic immune activation and suppression after TBI
Multi-organ damage following TBI can lead to increased numbers of infiltrating inflammatory cells and levels of cytokines in the brain. Because of the compromised BBB, these cells and molecules gain access to the brain and aggravate the pathogenesis of TBI [18]. In spite of the importance of systemic inflammation and circulating inflammatory molecules in TBI, only limited investigations have been performed in this area. In a study on rats, Whalen et al.[54] observed systemic neutrophilia together with increased BBB permeability when granulocyte-colony stimulating factor (GCSF) was administered prior to cortical contusion injury (CCI). In another study Utagawa et al. demonstrated that systemically administered IL-1β markedly influenced the histopathological and behavioral outcome following fluid percussion injury. The leaking of pro-inflammatory molecules like cytokines, arachidonic acid metabolites, proteins of the contact-phase and coagulation systems, complement factors and acute-phase proteins, as well as hormonal mediators [90] through the compromised BBB into the circulation may generate a systemic immune response syndrome (SIRS) [90, 91] characterized by hyper-inflammation or may release anti-inflammatory molecules targeting IL-1β, IL-6 or TNFα resulting in compensatory anti-inflammatory response syndrome (CARS) to block development of SIRS [19].
The production of inflammatory mediators is regulated by the negative feedback provided by the hypothalamus-pituitary-adrenal (HPA) axis and sympathetic nervous system (SNS) efferent limbs in CARS [19]; but in TBI, an imbalance between these two can lead to immunological dysfunction like organ damage or susceptibility to infections [91]. Stress-mediated release of cortisol and catecholamines can enhance the immune suppression. Direct infection through a skull fracture in TBI or from the transmigration of enteric bacteria after a closed head injury may cause infection, pneumonia and sepsis which can be life threatening in TBI or immune-compromised patients [10]. Griffin [10] has also pointed out that immune suppression after TBI causes retardation of healing in the brain infrastructure. In a 2001 human study, severe immune suppression was observed following severe TBI. Eighteen to seventy-two hours after head trauma, the numbers of circulating T-cells, T-helper cells, T-suppressor [92, 93] and NK cells were reduced while the B-lymphocyte count remained normal [92]. There was also an increase in CD4+/CD45+ T cells [10, 93]. The immune regulatory functions within the CNS following TBI, for example, microglia and astrocyte activation lead to antigen presentation to T-cells that alters their cytokine response and this may contribute to TBI pathology. On the other hand, the ability of these neuroantigen-reactive T cells to specifically infiltrate the CNS can be used to deliver molecules to augment a recovery response in degenerating CNS tissues [94].
Response of peripheral immune organs to TBI
Despite ongoing research, the effect of TBI on other organs is largely unknown. In one study Mirzayan et al. [95] evaluated the histopathological changes in lung and liver. Following a single TBI event, they observed migration of immunocompetent cells to peripheral organs leading to various degrees of organ dysfunction. The spleen is a reservoir of peripheral macrophages and other immune cells in the body, and it is now well known that splenic signaling contributes to injury of various tissues after ischemic insult. For example, splenectomy prior to insult protects both the liver [96] and brain [84] from ischemic damage. They have also observed a reduction in spleen size following ischemic insult 84]. Li et al.[97] showed that splenectomy immediately after severe TBI induced by weight drop in rats decreased pro-inflammatory cytokine production, mortality rate and improved cognitive function. It was observed by Das et al.[45] that splenectomy immediately after the induction of mild TBI by lateral fluid percussion in rats attenuated neurodegeneration and CCL20 chemokine expression in the brain. Although the mechanism of spleen-brain interaction is not clear, it was found by Lee et al. [98] that the spleen participates in cerebral inflammation following intracerebral hemorrhage in a stroke model, as splenectomy reduced cerebral edema and inflammatory cell counts (probably by increased circulating catecholamines) [99]. Stewart and McKenzie [100] suggested that sympathetic stimulation can cause the release of immune cells from the spleen and subsequent infiltration into brain tissues. Regardless of the neural mechanism, removal of the spleen immediately after the insult would remove the largest pool of immune cells, which should decrease infiltration and consequent neuroinflammation. The thymus is the major source of maturing T-cells in the body. Although a great deal of investigation has been done to elucidate the relationship between brain trauma and the immune system, very little has been done to explore the function of the thymus after TBI. In a study of LFPI in rats, Das et al. found elevated CCL20 expression in the thymus following TBI [45]. Further investigation is needed to identify the specific function of thymus after TBI in adult rats. In a model of polytrauma combined with shock, Guan et al. observed apoptosis in the thymus, spleen, lung, liver and intestine which could cause the early organ injury and late organ failure seen in polytrauma patients [101]. In an effort to elucidate the hepatic response to acute brain injury, Campbell et al.[102] observed that clodronate-mediated Kupffer cell (KC) depletion reduced neutrophil- and ED-1-positive macrophage infiltration in IL-1β-injected brain or contusion-injured spinal cord by 70% and 50% respectively. Suppression of KC proliferation may, therefore, reduce secondary injury. Previously this group had pointed out that hepatic cytokines or chemokines produced as a result of acute injury may inhibit neutrophil recruitment to the CNS [102–105]. In recent studies, decreased liver weight and protein content, altered energy metabolism [106] and p450 dysfunction [107] have been observed following TBI.
Cytokines and chemokines secreted peripherally control TBI
Following TBI, the signaling pathways are activated, inflammatory cells are mobilized and there is enhanced secretion of multiple inflammatory mediators like cytokines, chemokines and damage-associated molecular patterns (DAMPs). DAMPs in turn reactivate the inflammatory mediators and aggravate the damage [108]. The exact role of cytokines in brain trauma is not fully known, although experimental evidences suggest that cytokines play a major role in the body’s response to TBI. The major cytokines produced after TBI include tumor necrosis factor–alpha (TNF-α), IL-1β, IL-2, IL-6, IL-8, [91, 109], IL-4 [110] and IL-18 [111]. Free radical nitric oxide (NO) is produced by the enzyme inducible NO synthase (iNOS) [112], which is an important inflammatory mediator after trauma in mice [113]. Among peripherally secreted chemokines in response to TBI the role of CCL20 has recently been described. This unique chemokine interacts specifically with the CC chemokine receptor 6 (CCR6) and induces chemotaxis of dendritic cells, T cells and B cells [114]. These cells are residents of the spleen and have the potential to promote neuroinflammation. CCL20 is expressed in inflamed epithelial cells [115] and in the synovial tissues of rheumatoid arthritis patients [116, 117]. It has also been shown to be upregulated under normothermic conditions in a rat middle cerebral artery occlusion (MCAO) model [118]. Upregulation of CCL20 along with other cytokines has been observed in human subjects one day after severe traumatic brain injury [43]. Furthermore, CCL20 has been identified as a dual-acting chemokine with the potential for inhibiting immune reactions and more importantly in attracting inflammatory effectors and activators [44]. In a recent study using the LFPI rat model of TBI, Das et al. showed the expression of CCL20 mRNA and protein in spleen and thymus 24 hours after TBI, which is 24 hours before its expression in the brain. Since the thymus is the major source of mature circulating T cells, CCL20 expression in the thymus in adult rats as observed in this study seems significant [45] and should be further investigated.
Is TBI associated with other neurodegenerative disorders?
There is increasing evidence showing that TBI is associated with neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), and amyotropic lateral sclerosis (ALS) [119, 120]. Epidemiological data indicates a single TBI event may trigger or accelerate the onset of Alzheimer’s disease (AD) in later life [121–124]. On the other hand, repetitive mild TBI has been associated with progressive neurodegeneration [125]. Since, Rudelli et al.[126] reported a case of classic AD pathology in a 38 year old severe head trauma patient, both tau pathologies and Aβ plaques were identified in survivors of single TBI [121, 123] Subsequently, cases of AD-like pathology including neurofibrillary tangles and Aβ deposition [124, 127–130] were reported in head trauma victims, including boxers, irrespective of age [131]. Although the Aβ plaques in AD and TBI are morphologically different, both contain primarily Aβ1- 42 with some occurrence of Aβ1-40 in TBI [129, 130, 132]. Aβ1-42 has also been observed in the CSF of severe TBI patients and is thought to be directly related to the increased level of cerebral Aβ [133] and neuronal amyloidogenic amyloid precursor protein (APP) levels after TBI [134]. Although results of animal studies on TBI induced AD pathologies are conflicting, it has been observed that post TBI activation of microglia and proinflammatory cytokine release exacerbates the AD like pathologies [135] in rats and is involved in APP processing that leads to generation of Aβ plaques [136, 137].
In contrast to AD, studies attempting to correlate TBI and MS, another neurodegenerative, demyelinating disease of the CNS, are limited. Goldacre and colleagues [138] and Kurland [139] found no evidence of association between TBI and the development of MS. However, risk analysis using Taiwan’s National Health Insurance Research Database, indicated higher risk of incidence of MS in patients with a history of TBI compared to non TBI control group [140]. Parkinson’s disease (PD) is a neurodegenerative disorder, which affects the dopaminergic neurons of the substantia nigra. PD-associated mitochondrial dysfunction and pathology was observed after mild to moderate TBI and trichloroethylene (TCE) exposure in rats [141]. Also, TBI was reported to cause the nigrostriatal dopaminergic neurodegeneration in a rat model of LFPI suggesting that TBI is a risk factor of PD development [142]. Thus, although TBI appears to be associated with the development of some neurodegenerative diseases, conflicting data exist and detailed human and animal studies are necessary in this field. The most studied association between TBI and AD appears to suggest that TBI activation of immune mechanisms and proinflammatory cytokine activation of microglia contribute to neurodegenerative processes.
Therapeutic approaches for TBI
A number of drugs for TBI have been tested in clinical trials but none has shown much promise. Most of the approaches to TBI therapy aim at treating the secondary neurodegeneration as a single component. Recently, a therapeutic regimen using multifunctional drugs has been proposed and tested in experimental neurotrauma models. The therapeutic agents included hormones like thyrotropin releasing hormone (TRH) and progesterone, heat shock proteins, neurotrophic factors, erythropoietin, statin drugs and antibiotics [143, 144], substance P antagonists, cyclosporine, and magnesium salts among others [145].
Anti-inflammatories for TBI
The inflammation following TBI causes tissue damage correlating with the secondary injury phase. Recently much attention has been drawn to the potential therapeutic benefits of inhibiting reactive oxygen species (ROS), reactive nitrogen species (RNS), and several types of tissue-digesting enzymes (matrix metalloproteinases), prostanoids, leukotrienes, and proinflammatory/inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α). Inhibition of TNF-α with cannabinoids like pentoxifylline and dexanabinol, and use of corticosteroids or NSAIDs like ibuprofen or minocycline to reduce inflammation in the brain have shown promise in animals but failed in clinical trials [146]. Corticosteroids are a family of anti-inflammatory drugs that are widely used in autoimmune and allergic conditions and to reduce tumor-induced cerebral edema; but they failed to show any benefit in human trials of TBI involving adults and children [147]. Reduction of oligodendrocyte death and axonal degeneration by minocycline, a tetracycline derivative was observed in a spinal cord injury model [42]. Cederberg et al. [148] suggested that timing is crucial in inflammatory intervention, as IL-1, IL-6, and TNF-α may also play an anti-inflammatory role in a later stage of TBI-induced brain inflammation. Also, the PPAR-gamma agonist 15d-prostaglandin J(2) was shown to control brain inflammation by inhibiting microglial activation after TBI [88].
Gene therapy for TBI
Gene therapy is a promising approach for the treatment of several diseases and conditions including TBI. With the advent of improved experimental techniques like microarrays for gene expression analysis, new targets are emerging for the treatment of diseases, drug development, immunotherapeutics and gene therapy. Colak et al. have identified several gene networks potentially involved in TBI that includes the C1ql2, Cbnl, Sdc1, Bdnf, MMP9, and Cd47 genes [149]. Redell et al. observed changes in hippocampal miRNA expression corresponding to the pathophysiological changes following injury and identified these as potential targets for gene therapy [150]. Degeorge and coworkers demonstrated that administration of viral-mediated glial cell-line derived neurotrophic factor (AdGDNF) one week prior to cortical contusion injury in rats resulted in neuroprotection but not functional recovery [151]. Attempts have been made to target mRNA translational regulation to combat neurodegeneration. Aberrant RNA oxidation, RNA degradation, altered RNA splicing and ribosomal changes – all leading to mRNA translational abnormalities have been described by many authors in different neurodegenerative conditions [152, 153]. The mRNA translational regulation is affected by small non-coding microRNAs. The miRNA-argonaute complex suppresses the translation of target mRNA and each miRNA can regulate the translation of hundreds of mRNA targets and control the expression of many genes. Under cellular stress, a subset of microRNAs increases while expression of other miRNAs is decreased [154]. High throughput sequencing has shown that the human brain expresses over 1000 miRNAs, the functions of only approximately 500 of which have been determined [155]. MiRNAs have been implicated in various neurodegenerative conditions including TBI. Using microarray analysis, Redell and coworkers observed changes in the hippocampal expression levels of 444 miRNAs at 3 and 24 hours after controlled cortical impact injury in rats. In this study, 50 miRNAs were overexpressed including targets for proteins known to be initiated after injury [150]. Lei et al. also observed up- and down-regulation of rat cerebral miRNA up to 72 hours after TBI [156] while Liu et al. reported altered miRNA profiles after traumatic spinal cord injury in mice [157]. The potential exists for using miRNAs and small interfering RNAs (siRNAs) as therapeutic agents, but much work needs to be done before they will become a regular part of the physician’s tool kit. The si/miRNAs can be delivered using various transfection agents including liposomes, polyethylenimine (PEI), chitosan nanoparticles or by electroporation. Apart from the potential disadvantage of off-target effects, RNA knockdown can be useful in treating TBI.
Transplantation-based approaches for treating TBI
In the past two decades, restorative therapeutic approaches focusing on repair or replacement of damaged or dead cells following TBI have gained importance [158]. Cellular transplantation is the method of choice because the brain itself has a limited capacity for self-repair. Early experiments with transplantation of fetal neural tissues with or without nerve growth factor (NGF) were effective [159], but raised issues of practicality and ethics. NT2N cells showed promise in graft survival [160, 161]. It was found that ex vivo NGF gene therapy improved cognitive deficits following CCI in rodents [162, 163]. Both rodent and human embryonic stem cells have shown encouraging results in survival, integration and attenuation of post-traumatic sequellae. Stem cells have the ability to self- renew and differentiate depending on specific cues. Neural stem cells in particular can divide unlimitedly and differentiate into neurons or glial cells. It was observed that E14.5 mouse embryonic stem cells transplanted with or without a fibronectin scaffold following CCI improved behavioral symptoms [164]. Xenotransplanted human neural stem cells have been found to survive in injured rodent brains and to express astrocytic and neuronal antigens [165, 166]. They migrated to the hippocampus, corpus callosum and ipsilateral sub-ependymal zone [167] and decreased the number of degenerating neurons [168]. Bone marrow-derived stem cells (BMSCs), either hematopoietic or mesenchymal, are advantageous in that they can be harvested from the same animal and thereby avoid the problems of cell availability and immune rejection. These cells have successfully been transplanted into injured rats by different routes where they express neural and glial cell markers (35, 36) and migrate to the subventricular zone, hippocampus and pericontusional areas [169] indicating neurogenesis and improved neurobehavioral outcome [170]. Ma et al.[171] transplanted neural stem cells (NSCs) modified to encode brain derived neurotrophic factor (BDNF) in rats after TBI and found significant improvement in graft survival, neurogenesis and behavioral outcome. In another study in Wistar rats, functional improvement and colonization of BMSCs were observed after TBI and the recovery was found to be facilitated by granulocyte colony stimulating factor (G-CSF) [172]. Human fetal neural stem cells (hfNPCs) transplanted after CCI in SD rats increased angiogenesis and reduced astrogliosis [173]. As a long term effect they observed functional improvement, reduced lesion volume and increased neuronal survival surrounding the lesion [173].
The potential of therapeutic transplantation of immortalized progenitor cell lines after TBI, has also been tested by various authors. HiB5 cells derived from embryonic rat hippocampus [174, 175], MHP36, the fibroblast growth factor 2 (FGF-2)-responsive Maudsley hippocampal cell line clone 36 [176] and C17.2, which is a clonal multipotent progenitor cell from murine cerebellum [177], have been tested for their efficacy in improving repair of the contusion site, migration, neurogenesis and neurobehavioral outcome. Hunang et al.[178] reviewed successful preclinical studies and clinical trials of cell-based therapeutics for different neurodegenerative conditions including TBI. They mentioned the use of restorative transplantation involving fetal/embryonic brain and spinal cord tissue, stem cells including embryonic, neural, hematopoietic, adipose-derived adult stem/precursor cells, skin-derived precursor and induced pluripotent stem cells, glial cells (Schwann cells, oligodendrocyte, olfactory ensheathing cells, astrocytes, microglia, tanycytes), neuronal cells (various phenotypic neurons and Purkinje cells), mesenchymal stromal cells originating from bone marrow, umbilical cord, and umbilical cord blood, epithelial cells derived from the layer of retina and amnion, menstrual blood-derived stem cells, Sertoli cells, and active macrophages. Functional recovery and angiogenesis were observed following transplantation of endothelial progenitor cells derived from adipose tissues in the injured rat brain [179] showing promise. Some of these approaches have also gone to clinical trials for SCI/TBI [180, 181], and the clinical and scientific communities are paying more attention to the restorative treatment options for TBI.
Conclusion
Traumatic brain injury is a complex process evoking systemic immune responses as well as direct local responses in the brain tissues. The primary or direct damage disrupts the BBB and injures the neurons. This initiates a cascade of inflammatory reactions including chemokine production and activation of resident immune cells. The leakage of the inflammatory molecules through the compromised BBB attracts peripheral immune cells to the site of injury. The effect of TBI is not restricted to the brain; it can cause multi-organ damage and evoke systemic immune response including cytokine and chemokine production. This facilitates the recruitment of immune cells to the site of injury and progression of the inflammatory reaction and subsequent repair processes. In spite of the socioeconomic burden of TBI and worldwide research efforts, an effective treatment is still not available. Translational regulation of mRNA by si/mi RNA shows promise as a safe and specific treatment to combat neurodegeneration. Transplantation-based therapies also have the potential to repair and restore brain structure and function but continued in-depth investigations are needed before they become successful therapeutics.
References
Faul M, Xu L, Wald MM, Coronado VG: Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, Atlanta (GA); 2010.
Rostene W, Dansereau MA, Godefroy D, Van Steenwinckel J, Reaux-Le Goazigo A, Melik-Parsadaniantz S, Apartis E, Hunot S, Beaudet N, Sarret P: Neurochemokines: a menage a trois providing new insights on the functions of chemokines in the central nervous system. J Neurochem 2011, 118:680–694.
Fabrizio KS, Keltner NL: Traumatic brain injury in operation enduring freedom/operation iraqi freedom: a primer. Nurs Clin North Am 2010, 45:569–580. vi
Galarneau MR, Woodruff SI, Dye JL, Mohrle CR, Wade AL: Traumatic brain injury during operation Iraqi freedom: findings from the United States navy-marine corps combat trauma registry. J Neurosurg 2008, 108:950–957.
Macgregor AJ, Dougherty AL, Galarneau MR: Injury-specific correlates of combat-related traumatic brain injury in Operation Iraqi Freedom. J Head Trauma Rehabil 2011, 26:312–318.
Ettenhofer ML, Abeles N: The significance of mild traumatic brain injury to cognition and self-reported symptoms in long-term recovery from injury. J Clin Exp Neuropsychol 2009, 31:363–372.
Ettenhofer ML, Barry DM: A comparison of long-term postconcussive symptoms between university students with and without a history of mild traumatic brain injury or orthopedic injury. J Int Neuropsychol Soc 2012, 18:451–460.
Halldorsson JG, Flekkoy KM, Arnkelsson GB, Tomasson K, Magnadottir HB, Arnarson EO: The scope of early traumatic brain injury as a long-term health concern in two nationwide samples: prevalence and prognostic factors. Brain Inj 2012, 26:1–13.
Ozen LJ, Fernandes MA: Slowing down after a mild traumatic brain injury: a strategy to improve cognitive task performance? Arch Clin Neuropsychol 2012, 27:85–100.
Griffin GD: The injured brain: TBI, mTBI, the immune system, and infection: connecting the dots. Mil Med 2011, 176:364–368.
Centers for Disease Control and Prevention: Concussion and Mild TBI. http://www.cdc.gov/concussion
Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM: White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain 2007, 130:2508–2519.
McIntosh TK, Smith DH, Meaney DF, Kotapka MJ, Gennarelli TA, Graham DI: Neuropathological sequelae of traumatic brain injury: relationship to neurochemical and biomechanical mechanisms. Lab Invest 1996, 74:315–342.
Cernak I: Animal models of head trauma. NeuroRx 2005, 2:410–422.
DeKosky ST, Kochanek PM, Clark RS, Ciallella JR, Dixon CE: Secondary injury after head trauma: subacute and long-term mechanisms. Semin Clin Neuropsychiatry 1998, 3:176–185.
Lucas SM, Rothwell NJ, Gibson RM: The role of inflammation in CNS injury and disease. Br J Pharmacol 2006,147(Suppl 1):S232-S240.
Werner C, Engelhard K: Pathophysiology of traumatic brain injury. Br J Anaesth 2007, 99:4–9.
Utagawa A, Truettner JS, Dietrich WD, Bramlett HM: Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol 2008, 211:283–291.
Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S: Systemic inflammatory response following acute traumatic brain injury. Front Biosci 2009, 14:3795–3813.
Bramlett HM, Dietrich WD: Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 2004, 24:133–150.
Robertson CL, Bell MJ, Kochanek PM, Adelson PD, Ruppel RA, Carcillo JA, Wisniewski SR, Mi Z, Janesko KL, Clark RS, Marion DW, Graham SH, Jackson EK: Increased adenosine in cerebrospinal fluid after severe traumatic brain injury in infants and children: association with severity of injury and excitotoxicity. Crit Care Med 2001, 29:2287–2293.
Pun PB, Lu J, Moochhala S: Involvement of ROS in BBB dysfunction. Free Radic Res 2009, 43:348–364.
Ravindran J, Agrawal M, Gupta N, Lakshmana Rao PV: Alteration of blood brain barrier permeability by T-2 toxin: role of MMP-9 and inflammatory cytokines. Toxicology 2011, 280:44–52.
Shlosberg D, Benifla M, Kaufer D, Friedman A: Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 2010, 6:393–403.
Faden AI: Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Curr Opin Neurol 2002, 15:707–712.
Cernak I, Vink R, Zapple DN, Cruz MI, Ahmed F, Chang T, Fricke ST, Faden AI: The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol Dis 2004, 17:29–43.
Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR: Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 1989, 15:49–59.
Gennarelli TA, Thibault LE, Tipperman R, Tomei G, Sergot R, Brown M, Maxwell WL, Graham DI, Adams JH, Irvine A: Axonal injury in the optic nerve: a model simulating diffuse axonal injury in the brain. J Neurosurg 1989, 71:244–253.
Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S: Lateral fluid percussion: model of traumatic brain injury in mice. J Vis Exp 2011., 54:
Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark R: Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem 2002, 82:181–91.
Ravagnan L, Roumier T, Kroemer G: Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002, 192:131–7.
Stoica BA, Byrnes KR, Faden AI: Cell cycle activation and CNS injury. Neurotox Res 2009, 16:221–37.
Bredesen DE: Programmed cell death mechanisms in neurological disease. Curr Mol Med 2008, 8:173–86.
Yakovlev AG, Faden AI: Caspase-dependent apoptotic pathways in CNS injury. Mol Neurobiol 2001, 24:131–44.
Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS: Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 2002, 158:507–17.
Hong SJ, Dawson TM, Dawson VL: Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 2004, 25:259–64.
Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham SH, Virág L, Haskó G, Stachlewitz R, Szabó C, Kochanek PM: Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab 1999, 19:835–42.
Galat A: Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity–targets–functions. Curr Top Med Chem 2003, 3:1315–47.
Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G: AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 2004, 23:1514–21.
Yasuda H, Shichinohe H, Kuroda S, Ishikawa T, Iwasaki Y: Neuroprotective effect of a heat shock protein inducer, geranylgeranylacetone in permanent focal cerebral ischemia. Brain Res 2005, 1032:176–82.
Stoica BA, Faden AI: Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7:3–12.
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W: Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 2004, 24:2182–90.
Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ: The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab 2010, 68:126–35.
Comerford I, Bunting M, Fenix K, Haylock-Jacobs S, Litchfield W, Harata-Lee Y, Turvey M, Brazzatti J, Gregor C, Nguyen P, Kara E, McColl SR: An immune paradox: how can the same chemokine axis regulate both immune tolerance and activation? CCR6/CCL20: a chemokine axis balancing immunological tolerance and inflammation in autoimmune disease. Bioessays 2010, 32:1067–76.
Das M, Leonardo CC, Rangooni S, Pennypacker K, Mohapatra S, Mohapatra SS: Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflammation 2011, 8:148.
Biber K, Rappert A, Kettenmann H, Brouwer N, Copray SC, Boddeke HW: Neuronal SLC (CCL21) expression: implications for the neuron-microglial signaling system. Ernst Schering Res Found Workshop 2002, 39:45–60.
de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K: Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci 2005, 25:7548–57.
Rothwell NJ: Annual review prize lecture cytokines - killers in the brain? J Physiol 1999,514(Pt 1):3–17.
Hans VH, Imhof HG, Stocker R, Grob P, Trentz O, Morganti-Kossmann C: Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 1995, 4:311–7.
Camats J, Hadberg H, Quintana A, Rojas S, Giralt M, Molinero A, Campbell IL, Hidalgo J: Astrocyte-targeted expression of interleukin-6 protects the central nervous system during neuroglial degeneration induced by 6-aminonicotinamide. J Neurosci Res 2003, 73:481–96.
Kremlev SG, Palmer C: Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J Neuroimmunol 2005, 162:71–80.
Kim KS, Wass CA, Cross AS, Opal SM: Modulation of blood–brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res 1992, 11:293–8.
Ramilo O, Saez-Llorens X, Mertsola J, Jafari H, Olsen KD, Hansen EJ, Yoshinaga M, Ohkawara S, Nariuchi H, McCracken GH Jr: Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J Exp Med 1990, 172:497–507.
Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham S, Virag L, Hasko G, Stachlewitz R, Szabo C, Kochanek PM: Traumatic brain injury in mice deficient in poly-ADP(ribose) polymerase: a preliminary report. Acta Neurochir Suppl 2000, 76:61–4.
Kordon C, Bihoreau C: Integrated communication between the nervous, endocrine and immune systems. Horm Res 1989, 31:100–4.
Spangelo BL, Gorospe WC: Role of the cytokines in the neuroendocrine-immune system axis. Front Neuroendocrinol 1995, 16:1–22.
Ban E, Haour F, Lenstra R: Brain interleukin 1 gene expression induced by peripheral lipopolysaccharide administration. Cytokine 1992, 4:48–54.
Ban E, Marquette C, Sarrieu A, Fitzpatrick F, Fillion G, Milon G, Rostene W, Haour F: Regulation of interleukin-1 receptor expression in mouse brain and pituitary by lipopolysaccharide and glucocorticoids. Neuroendocrinology 1993, 58:581–7.
Breder CD, Dinarello CA, Saper CB: Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 1988, 240:321–4.
Tran PB, Miller RJ: Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci 2003, 4:444–55.
Bajetto A, Bonavia R, Barbero S, Piccioli P, Costa A, Florio T, Schettini G: Glial and neuronal cells express functional chemokine receptor CXCR4 and its natural ligand stromal cell-derived factor 1. J Neurochem 1999, 73:2348–57.
Rostene W, Kitabgi P, Parsadaniantz SM: Chemokines: a new class of neuromodulator? Nat Rev Neurosci 2007, 8:895–903.
Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR: Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393:595–9.
Biber K, Neumann H, Inoue K, Boddeke HW: Neuronal 'On' and 'Off' signals control microglia. Trends Neurosci 2007, 30:596–602.
Harrison JKL: Lukacs NW (eds): The chemokine receptors. Humana Press, New York; 2007.
Ransohoff RM: Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 2009, 31:711–21.
Semple BD, Kossmann T, Morganti-Kossmann MC: Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 2010, 30:459–73.
Wilson EH, Weninger W, Hunter CA: Trafficking of immune cells in the central nervous system. J Clin Invest 2010, 120:1368–79.
Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C: The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol 2010, 224:93–100.
Coughlan CM, McManus CM, Sharron M, Gao Z, Murphy D, Jaffer S, Choe W, Chen W, Hesselgesser J, Gaylord H, Kalyuzhny A, Lee VM, Wolf B, Doms RW, Kolson DL: Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience 2000, 97:591–600.
Yamamoto T, Yamashita A, Yamada K, Hata R: Immunohistochemical localization of chemokine CXCL14 in rat hypothalamic neurons. Neurosci Lett 2011, 487:335–40.
Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD, Sutcliffe JG, Carson MJ: Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. J Neurochem 2009, Suppl 1:117–25.
de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP: Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol 2007, 36:137–51.
Zhao P, Waxman SG, Hains BC: Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci 2007, 27:8893–902.
Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW: Ischemia-induced neuronal expression of the microglia attracting chemokine Secondary Lymphoid-tissue Chemokine (SLC). Glia 2001, 34:121–33.
Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H: Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol 2002, 168:3221–6.
Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H: CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci 2004, 24:8500–9.
Ghirnikar RS, Lee YL, Eng LF: Inflammation in traumatic brain injury: role of cytokines and chemokines. Neurochem Res 1998, 23:329–40.
Norris JG, Tang LP, Sparacio SM, Benveniste EN: Signal transduction pathways mediating astrocyte IL-6 induction by IL-1 beta and tumor necrosis factor-alpha. J Immunol 1994, 152:841–50.
Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML: Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol 1991, 33:227–36.
Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F: Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol 1993, 42:177–85.
Walker PA, Jimenez F, Cox CS Jr: Progenitor cell therapy for traumatic brain injury: effect of serum osmolarity on cell viability and cytokine production. Regen Med 2010, 5:65–71.
Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T: Modulation of immune response by head injury. Injury 2007, 38:1392–400.
Ajmo CT Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR: The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res 2008, 86:2227–34.
Rimaniol AC, Lekieffre D, Serrano A, Masson A, Benavides J, Zavala F: Biphasic transforming growth factor-beta production flanking the pro-inflammatory cytokine response in cerebral trauma. Neuroreport 1995, 7:133–6.
Adibhatla RM, Hatcher JF, Dempsey RJ: Lipids and lipidomics in brain injury and diseases. AAPS J 2006, 8:E314–21.
D'Avila JC, Lam TI, Bingham D, Shi J, Won SJ, Kauppinen TM, Massa S, Liu J, Swanson RA: Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation 1:31.
Bernardo A, Minghetti L: PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des 2006, 12:93–109.
Konsman JP, Drukarch B, Van Dam AM: (Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clin Sci (Lond) 2007, 112:1–25.
Keel M, Trentz O: Pathophysiology of polytrauma. Injury 2005, 36:691–709.
Smrcka M, Mrlian A, Karlsson-Valik J, Klabusay M: The effect of head injury upon the immune system. Bratisl Lek Listy 2007, 108:144–8.
Wolach B, Sazbon L, Gavrieli R, Broda A, Schlesinger M: Early immunological defects in comatose patients after acute brain injury. J Neurosurg 2001, 94:706–11.
Dziedzic T, Slowik A, Szczudlik A: Nosocomial infections and immunity: lesson from brain-injured patients. Crit Care 2004, 8:266–70.
Becher B, Prat A, Antel JP: Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia 2000, 29:293–304.
Mirzayan MJ, Probst C, Krettek C, Samii M, Pape HC, van Griensven M, Samii A: Systemic effects of isolated brain injury: an experimental animal study. Neurol Res 2008, 30:457–60.
Okuaki Y, Miyazaki H, Zeniya M, Ishikawa T, Ohkawa Y, Tsuno S, Sakaguchi M, Hara M, Takahashi H, Toda G: Splenectomy-reduced hepatic injury induced by ischemia/reperfusion in the rat. Liver 1996, 16:188–94.
Li M, Li F, Luo C, Shan Y, Zhang L, Qian Z, Zhu G, Lin J, Feng H: Immediate splenectomy decreases mortality and improves cognitive function of rats after severe traumatic brain injury. J Trauma 2011, 71:141–7.
Lee ST, Chu K, Jung KH, Kim SJ, Kim DH, Kang KM, Hong NH, Kim JH, Ban JJ, Park HK, Kim SU, Park CG, Lee SK, Kim M, Roh JK: Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain 2008, 131:616–29.
Ajmo CT Jr, Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, Cuevas J, Willing AE, Pennypacker KR: Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol 2009, 218:47–55.
Stewart IB, McKenzie DC: The human spleen during physiological stress. Sports Med 2002, 32:361–9.
Guan J, Jin D, Jin L: Apoptosis in multiple organs of rats in early stage of polytrauma combined with shock. Zhonghua Yi Xue Za Zhi 1998, 78:741–5.
Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, van Rooijen N, Morsali D, Davis AE, Anthony DC: Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology 2008, 55:780–7.
Campbell SJ, Rob MJ D, Yanyan J, Carina F, Pitossi FJ, Anthony DC: Overexpression of IL-1beta by adenoviral-mediated gene transfer in the rat brain causes a prolonged hepatic chemokine response, axonal injury and the suppression of spontaneous behaviour. Neurobiol Dis 2007, 27:151–63.
Campbell SJ, Hughes PM, Iredale JP, Wilcockson DC, Waters S, Docagne F, Perry VH, Anthony DC: CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J 2003, 17:1168–70.
Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, Dempster R, Anthony DC: Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol 2005, 166:1487–97.
Moinard C, Severine G, Valerie B, Beatrice M, Catherine M-L, Jean-Claude C, Luc C, Christine C: Evidence for impairment of hepatic energy homeostasis in head-injured rat. J Neurotrauma 2008, 25:124–9.
Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT: The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J Trauma 2006, 60:152–7.
Namas R, Ghuma A, Hermus L, Zamora R, Okonkwo D, Billiar TR, Vodovotz Y: The acute inflammatory response in trauma /hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J Med 2009, 4:97–103.
Smith RM, Giannoudis PV: Trauma and the immune response. J R Soc Med 1998, 91:417–20.
DiPiro JT, Howdieshell TR, Goddard JK, Callaway DB, Hamilton RG, Mansberger AR Jr: Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg 1995, 130:1159–62. Discussion, 1162–3
Marcu AC, Paccione KE, Barbee RW, Diegelmann RF, Ivatury RR, Ward KR, Loria RM: Androstenetriol immunomodulation improves survival in a severe trauma hemorrhage shock model. J Trauma 2007, 63:662–9.
Zamora R, Vodovotz Y, Billiar TR: Inducible nitric oxide synthase and inflammatory diseases. Mol Med 2000, 6:347–73.
Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, Peitzman AB, Billiar TR, Tweardy DJ: Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med 1998, 187:917–28.
Schutyser E, Struyf S, Van Damme J: The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev 2003, 14:409–26.
Dieu-Nosjean MC, Massacrier C, Homey B, Vanbervliet B, Pin JJ, Vicari A, Lebecque S, Dezutter-Dambuyant C, Schmitt D, Zlotnik A, Caux C: Macrophage inflammatory protein 3-alpha is expressed at inflamed epithelial surfaces and is the most potent chemokine known in attracting Langerhans cell precursors. J Exp Med 2000, 192:705–18.
Chabaud M, Page G, Miossec P: Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein 3-alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol 2001, 167:6015–20.
Matsui T, Akahoshi T, Namai R, Hashimoto A, Kurihara Y, Rana M, Nishimura A, Endo H, Kitasato H, Kawai S, Takagishi K, Kondo H: Selective recruitment of CCR6-expressing cells by increased production of MIP-3 alpha in rheumatoid arthritis. Clin Exp Immunol 2001, 125:155–61.
Adams CW, Bruton CJ: The cerebral vasculature in dementia pugilistica. J Neurol Neurosurg Psychiatry 1989, 52:600–4.
Masel BE, DeWitt DS: Traumatic brain injury: a disease process, not an event. J Neurotrauma 2010, 27:1529–40.
Kiraly M, Kiraly SJ: Traumatic brain injury and delayed sequelae: a review–traumatic brain injury and mild traumatic brain injury (concussion) are precursors to later-onset brain disorders, including early-onset dementia. Scientific World Journal 2007, 7:1768–76.
Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA: Traumatic brain injury elevates the Alzheimer's amyloid peptide A beta 42 in human CSF. A possible role for nerve cell injury. Ann N Y Acad Sci 2000, 903:118–22.
Van Den Heuvel C, Thornton E, Vink R: Traumatic brain injury and Alzheimer's disease: a review. Prog Brain Res 2007, 161:303–16.
Johnson VE, Stewart W, Smith DH: Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22:142–9.
Clinton J, Ambler MW, Roberts GW: Post-traumatic Alzheimer's disease: preponderance of a single plaque type. Neuropathol Appl Neurobiol 1991, 17:69–74.
Fleminger S, Oliver D, Lovestone S, Rabe-Hesketh S, Giora A: Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 2003, 74:857–62.
Rudelli R, Strom JO, Welsh PT, Ambler MW: Posttraumatic premature Alzheimer's disease. Neuropathologic findings and pathogenetic considerations. Arch Neurol 1982, 39:570–5.
Roberts GW, Gentleman SM, Lynch A, Graham DI: Beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991, 338:1422–3.
Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI: Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 1994, 57:419–25.
Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin WS, Graham DI: A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport 1997, 8:1519–22.
Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST: Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 2004, 190:192–203.
Roberts GW, Allsop D, Bruton C: The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 1990, 53:373–8.
Horsburgh K, Cole GM, Yang F, Savage MJ, Greenberg BD, Gentleman SM, Graham DI, Nicoll : Beta-amyloid (Abeta)42(43), abeta42, abeta40 and apoE immunostaining of plaques in fatal head injury. Neuropathol Appl Neurobiol 2000, 26:124–32.
Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR: Traumatic brain injury increases beta-amyloid peptide 1–42 in cerebrospinal fluid. J Neurochem 1998, 71:2505–9.
McKenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC: Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport 1994, 6:161–4.
Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM, Kozikowski AP, Lyeth BG: HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res 2008, 1226:181–91.
Marx F, Blasko I, Grubeck-Loebenstein B: Mechanisms of immune regulation in Alzheimer's disease: a viewpoint. Arch Immunol Ther Exp (Warsz) 1999, 47:205–9.
Marx F, Blasko I, Zisterer K, Grubeck-Loebenstein B: Transfected human B cells: a new model to study the functional and immunostimulatory consequences of APP production. Exp Gerontol 1999, 34:783–95.
Goldacre MJ, Abisgold JD, Yeates DGR, Seagroatt V: Risk of multiple sclerosis after head injury: record linkage study. J Neurol Neurosurg Psychiatry 2006, 77:351–53.
Kurland LT: Trauma and multiple sclerosis. Ann Neurol 1994,36(Suppl):S33–37.
Kang JH, Lin HC: Increased risk of multiple sclerosis after traumatic brain injury: a nationwide population-based study. J Neurotrauma 2012, 29:90–95.
Sauerbeck A, Hunter R, Bing G, Sullivan PG: Traumatic brain injury and trichloroethylene exposure interact and produce functional, histological, and mitochondrial deficits. Exp Neurol 2012, 234:85–94.
Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF: Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma 2011, 28:1783–801.
Faden AI, Stoica B: Neuroprotection: challenges and opportunities. Arch Neurol 2007, 64:794–800.
Stoica B, Byrnes K, Faden AI: Multifunctional drug treatment in neurotrauma. Neurotherapeutics 2009, 6:14–27.
Vink R, Nimmo AJ, Cernak I: An overview of new and novel pharmacotherapies for use in traumatic brain injury. Clin Exp Pharmacol Physiol 2001, 28:919–21.
Ziebell JM, Morganti-Kossmann MC: Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7:22–30.
Dearden NM, Gibson JS, McDowell DG: Effect of high-dose dexamethasone on outcome from severe head injury. J Neurosurg 1986, 64:81–8.
Cederberg D, Siesjo P: What has inflammation to do with traumatic brain injury? Childs Nerv Syst 2010, 26:221–6.
Colak T, Cine N, Bamac B, Kurtas O, Ozbek A, Bicer U, Sunnetci D, Savlı H: Microarray-based gene expression analysis of an animal model for closed head injury. Injury 2012, 43:1264–70.
Redell JB, Liu Y, Dash PK: Traumatic brain injury alters expression of hippocampal microRNAs: potential regulators of multiple pathophysiological processes. J Neurosci Res 2009, 87:1435–48.
Degeorge ML, Marlowe D, Werner E, Soderstrom KE, Stock M, Mueller A, Bohn MC, Kozlowski DA: Combining glial cell line-derived neurotrophic factor gene delivery (AdGDNF) with L-arginine decreases contusion size but not behavioral deficits after traumatic brain injury. Brain Res 2011, 1403:45–56.
Markesbery WR, Lovell MA: Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol 2007, 64:954–6.
Nelson PT, Keller JN: RNA in brain disease: no longer just "the messenger in the middle". J Neuropathol Exp Neurol 2007, 66:461–8.
Nelson PT, Wang WX, Rajeev BW: MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol 2008, 18:130–8.
Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH: Diversity of microRNAs in human and chimpanzee brain. Nat Genet 2006, 38:1375–7.
Lei P, Li Y, Chen X, Yang S, Zhang J: Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain Res 2009, 1284:191–201.
Liu NK, Wang XF, Lu QB, Xu XM: Altered microRNA expression following traumatic spinal cord injury. Exp Neurol 2009, 219:424–9.
Royo NC, Shimizu S, Schouten JW, Stover JF, McIntosh TK: Pharmacology of traumatic brain injury. Curr Opin Pharmacol 2003, 3:27–32.
Sinson G, Voddi M, McIntosh TK: Combined fetal neural transplantation and nerve growth factor infusion: effects on neurological outcome following fluid-percussion brain injury in the rat. J Neurosurg 1996, 84:655–62.
Philips MF, Muir JK, Saatman KE, Raghupathi R, Lee VM, Trojanowski JQ, McIntosh TK: Survival and integration of transplanted postmitotic human neurons following experimental brain injury in immunocompetent rats. J Neurosurg 1999, 90:116–24.
Zhang C, Saatman K, Royo NC, Soltesz KM, Millard M, Schouten JW, Motta M, Hoover RC, McMillan A, Watson DJ, Lee VM, Trojanowski JQ, McIntosh TK: Delayed transplantation of human neurons following brain injury in rats: a long-term graft survival and behavior study. J Neurotrauma 2005, 22:1456–74.
Longhi L, Watson DJ, Saatman KE, Thompson HJ, Zhang C, Fujimoto S, Royo N, Castelbuono D, Raghupathi R, Trojanowski JQ, Lee VM, Wolfe JH, Stocchetti N, McIntosh TK: Ex vivo gene therapy using targeted engraftment of NGF-expressing human NT2N neurons attenuates cognitive deficits following traumatic brain injury in mice. J Neurotrauma 2004, 21:1723–36.
Longhi L, Zanier ER, Royo N, Stocchetti N, McIntosh TK: Stem cell transplantation as a therapeutic strategy for traumatic brain injury. Transpl Immunol 2005, 15:143–8.
Tate MC, et al.: Fibronectin promotes survival and migration of primary neural stem cells transplanted into the traumatically injured mouse brain. Cell Transplant 2002,11(3):283–95.
Englund U, Bjorklund A, Wictorin K: Migration patterns and phenotypic differentiation of long-term expanded human neural progenitor cells after transplantation into the adult rat brain. Brain Res Dev Brain Res 2002, 134:123–41.
Vescovi AL, Gritti A, Galli R, Parati EA: Isolation and intracerebral grafting of nontransformed multipotential embryonic human CNS stem cells. J Neurotrauma 1999, 16:689–93.
Wennersten A, Meier X, Holmin S, Wahlberg L, Mathiesen T: Proliferation, migration, and differentiation of human neural stem/progenitor cells after transplantation into a rat model of traumatic brain injury. J Neurosurg 2004, 100:88–96.
Hagan M, Wennersten A, Meijer X, Holmin S, Wahlberg L, Mathiesen T: Neuroprotection by human neural progenitor cells after experimental contusion in rats. Neurosci Lett 2003, 351:149–52.
Mahmood A, Lu D, Chopp M: Marrow stromal cell transplantation after traumatic brain injury promotes cellular proliferation within the brain. Neurosurgery 2004, 55:1185–93.
Mahmood A, Lu D, Yi L, Chen JL, Chopp M: Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg 2001, 94:589–95.
Walker PA, Harting MT, Shah SK, Day MC, El Khoury R, Savitz SI, Baumgartner J, Cox CS Jr: Progenitor cell therapy for the treatment of central nervous system injury: a review of the state of current clinical trials. Stem Cells Int 2010, 2010:369578.
Bakhtiary M, Marzban M, Mehdizadeh M, Joghataei MT, Khoei S, Pirhajati Mahabadi V, Laribi B, Tondar M, Moshkforoush A: Comparison of transplantation of bone marrow stromal cells (BMSC) and stem cell mobilization by granulocyte colony stimulating factor after traumatic brain injury in rat. Iran Biomed J 2010, 14:142–9.
Skardelly M, Gaber K, Burdack S, Scheidt F, Hilbig H, Boltze J, Förschler A, Schwarz S, Schwarz J, Meixensberger J, Schuhmann MU: Long-term benefit of human fetal neuronal progenitor cell transplantation in a clinically adapted model after traumatic brain injury. J Neurotrauma 2011, 28:401–14.
Lundberg C, Field PM, Ajayi YO, Raisman G, Bjorklund A: Conditionally immortalized neural progenitor cell lines integrate and differentiate after grafting to the adult rat striatum. A combined autoradiographic and electron microscopic study. Brain Res 1996, 737:295–300.
Renfranz PJ, Cunningham MG, McKay RD: Region-specific differentiation of the hippocampal stem cell line HiB5 upon implantation into the developing mammalian brain. Cell 1991, 66:713–29.
Sinden JD, Rashid-Doubell F, Kershaw TL, Nelson A, Chadwick A, Jat PS, Noble MD, Hodges H, Gray JA: Recovery of spatial learning by grafts of a conditionally immortalized hippocampal neuroepithelial cell line into the ischaemia-lesioned hippocampus. Neuroscience 1997, 81:599–608.
Ryder EF, Snyder EY, Cepko CL: Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. J Neurobiol 1990, 21:356–75.
Huang H, Chen L, Sanberg P: Cell therapy from bench to bedside translation in CNS neurorestoratology era. Cell Med 2010, 1:15–46.
Xue S, Zhang HT, Zhang P, Luo J, Chen ZZ, Jang XD, Xu RX: Functional endothelial progenitor cells derived from adipose tissue show beneficial effect on cell therapy of traumatic brain injury. Neurosci Lett 2010, 473:186–91.
Geffner LF, Santacruz P, Izurieta M, Flor L, Maldonado B, Auad AH, Montenegro X, Gonzalez R, Silva F: Administration of autologous bone marrow stem cells into spinal cord injury patients via multiple routes is safe and improves their quality of life: comprehensive case studies. Cell Transplant 2008, 17:1277–93.
Zhang ZX, Guan LX, Zhang K, Zhang Q, Dai LJ: A combined procedure to deliver autologous mesenchymal stromal cells to patients with traumatic brain injury. Cytotherapy 2008, 10:134–9.
Acknowledgements
This work is supported by the Office of Naval Research grant (N000140810914) to SSM and the Veterans Reintegration grant from University of South Florida to SM. We acknowledge Dr. Gary Hellermann for critical review and editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MD has researched and prepared the manuscript; SM and SSM have made critical suggestions on the content and reviewed the manuscript. All authors have read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Das, M., Mohapatra, S. & Mohapatra, S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation 9, 236 (2012). https://doi.org/10.1186/1742-2094-9-236
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-9-236