
Abstract
Sarcopenia, broadly defined as the age-related decline in skeletal muscle mass, quality, and function, is associated with chronic low-grade inflammation and an increased likelihood of adverse health outcomes. The regulation of skeletal muscle mass with ageing is complex and necessitates a delicate balance between muscle protein synthesis and degradation. The secretion and transfer of cytokines, long non-coding RNAs (lncRNAs) and microRNAs (miRNAs), both discretely and within extracellular vesicles, have emerged as important communication channels between tissues. Some of these factors have been implicated in regulating skeletal muscle mass, function, and pathologies and may be perturbed by excessive adiposity. Indeed, adipose tissue participates in a broad spectrum of inter-organ communication and obesity promotes the accumulation of macrophages, cellular senescence, and the production and secretion of pro-inflammatory factors. Pertinently, age-related sarcopenia has been reported to be more prevalent in obesity; however, such effects are confounded by comorbidities and physical activity level. In this review, we provide evidence that adiposity may exacerbate age-related sarcopenia and outline some emerging concepts of adipose-skeletal muscle communication including the secretion and processing of novel myokines and adipokines and the role of extracellular vesicles in mediating inter-tissue cross talk via lncRNAs and miRNAs in the context of sarcopenia, ageing, and obesity. Further research using advances in proteomics, transcriptomics, and techniques to investigate extracellular vesicles, with an emphasis on translational, longitudinal human studies, is required to better understand the physiological significance of these factors, the impact of obesity upon them, and their potential as therapeutic targets in combating muscle wasting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Beyond middle-age, skeletal muscle mass, and strength decline by 1–2% and 1–5% per year, respectively [1,2,3]. Skeletal muscle tissue is the human body’s principle protein bank, and given the absence of a protein pool for storage, it is critical for maintaining protein status via the delicate regulation of its turnover [4, 5]. Indeed, the maintenance of skeletal muscle mass, quality, and function with ageing is multifactorial and necessitates a balance of regulatory processes in response to inherent biological ageing, physical in/activity, injury, or illness [6]. These processes include the generation of muscle via adult myogenesis (comprising the activation of muscle satellite cells, the proliferation of myoblasts; the withdrawal of myoblasts from the cell cycle, their subsequent differentiation and fusion into multinucleated myofibres) [7, 8], the repair and remodelling of muscle tissue [9] and its inter- and intracellular content [10], the interaction between skeletal muscle and the nervous system via motor neurones [11], and the interplay between synthesis and breakdown of muscle protein [12, 13].
Sarcopenia, broadly defined as the age-related decline in skeletal muscle mass, quality, and function, is the product of a negative balance of these muscle regulatory processes and is associated with an increased likelihood of adverse health outcomes [14]. Sarcopenia has been reported to be more prevalent in adults with obesity [15]. Adiposity exacerbates sarcopenia, increases fat infiltration in muscle, and reduces physical function [14, 16, 17]. The state of concurrent obesity and sarcopenia is considered by some to constitute a discrete condition, termed sarcopenic obesity; however, there is a lack of consensus surroundings its definition and diagnostic criteria [18,19,20]. Individuals presenting with sarcopenic obesity may have greater absolute muscle mass than is typical of individuals with sarcopenia, but the quality and function of that muscle (including muscle strength) are reduced, which may exacerbate muscle deterioration with ageing [21,22,23,24].
Critically, as our population becomes increasingly aged and obese, the ramifications of obesity on the mass and function of skeletal muscle with age warrant rigorous investigation [25, 26]. In this review, we outline some emerging conduits of adipose-skeletal muscle communication in the context of sarcopenia and obesity including the secretion and processing of novel myokines and adipokines, as well as the significance of cellular senescence on the adipose tissue secretome, and the role of extracellular vesicles and non-coding RNAs in mediating inter-tissue cross talk. We principally focus on sarcopenia and obesity as two distinct but often converging conditions. We also present evidence investigating the phenomenon of sarcopenic obesity and discuss the myriad ways that obesity may interact with sarcopenia to the detriment of muscle mass, quality, and function.
Adipose tissue as an endocrine organ
Once considered a passive energy reservoir, the discovery of the adipose-secreted factors adipsin and leptin confirmed adipose tissue as an endocrine organ [27,28,29]. Following the initial observation that obese adipose tissue has increased expression and secretion of tumour necrosis factor-alpha (TNF-α), it has been shown that obesity leads to the accumulation of macrophages within adipose tissue, which promote the production and secretion of several pro-inflammatory cytokines [30, 31]. Moreover, cellular senescence in adipose tissue, common to both ageing and obesity, is now understood to contribute to inflammation, aberrant cytokine production, and metabolic dysfunction [32]. Adipose tissue is now known to participate in a broad spectrum of inter-organ communication [33] and excessive adiposity conveys pleiotropic effects on its endocrine and metabolic function, contributing to pathophysiological consequences with ageing [34, 35]. Importantly, this has to be considered in the context of evidence from studies that demonstrated a capacity for healthy adipose tissue to exert favourable effects on skeletal muscle that further supports the physiological importance of cross talk between the two tissues [36].
Inter-tissue communication via multiple means
Historically, communication between cells was thought to be mediated by cell-cell contact or by the extracellular secretion of molecules, principally cytokines, a broad family of small secreted proteins, which can confer autocrine, paracrine, and endocrine effects on multiple tissues [37]. While cytokines can be secreted discretely, accumulating evidence has demonstrated that they may also be transported by the secretion and transfer of extracellular vesicles (EVs) and that certain cytokines may also serve to regulate the packaging and trafficking of these EVs [38]. EVs, principally divided into exosomes and microvesicles, are respectively released into the extracellular environment from endosomal and plasma membrane origins and are reviewed elsewhere [39, 40]. Alongside cytokines, EVs contain a broad array of cargo including proteins, lipids, organelle components, and myriad non-coding RNAs, which have emerged as an important communication channel between tissues [41]. Pertinently, the discrete and EV-mediated secretion and processing of novel cytokines, long non-coding RNAs (lncRNAs), and microRNAs (miRNAs), from adipose and skeletal muscle tissue, have been implicated in the regulation of skeletal muscle mass, function, and pathologies, in the context of obesity and ageing [42, 43] (Table 1).
Senescence-associated secretory phenotype
Cellular senescence is an evolutionarily conserved ageing mechanism characterised by upregulation of cyclin-dependent kinase inhibitor genes, which in turn activate retinoblastoma protein to block cell cycle progression [92,93,94]. Senescence arises in response to cellular stress and damage, including metabolic insults [95, 96]. While this serves to restrain harmful growth and replication of damaged cells, senescent cells also secrete an array of cytokines, chemokines, growth factors, and matrix-remodelling proteases, collectively termed the senescence-associated secretory phenotype (SASP) [97, 98]. Senescent cells are resistant to apoptosis and are ordinarily cleared by the immune system but accumulate with ageing [99, 100]. Conditioned media from primary human pre-adipocytes with induced senescence via serial passage or irradiation demonstrate a SASP that is rich in pro-inflammatory interleukins, interferon gamma (IFN-γ), and TNF-α, which could negatively impact upon muscle regulation [101].
Accumulation of senescent cells within metabolic tissues, such as adipose and skeletal muscle, is deleterious to metabolic homeostasis [95, 102,103,104], and obesity may promote the senescent state [105]. Indeed, the adipose tissue of individuals with obesity displays elevated oxidative stress and accelerated telomere shortening [106, 107], which are purported to induce and promote senescence [108, 109]. It has been proposed that obesity and ageing drive excessive turnover of adipose progenitors which may contribute to the onset of their senescence and subsequent SASP, conveying both local and systemic adipose tissue dysfunction and inflammation [110].
The potential benefit of modulating cellular senescence has been recently investigated. Administration of Dasatinib and Quercetin, which target and eliminate senescent cells, to omental adipose tissue explants from middle-aged adults with obesity reduced the abundance of senescent cells and decreased pro-inflammatory cytokine secretion [111]. No study to date has directly investigated whether adipose tissue senescence is implicated in sarcopenia and whether this is amplified by obesity. However, the accumulation of intramuscular adipose tissue that is common to both ageing and obesity [112, 113], the pro-inflammatory adipose SASP observed in those states, and the ability of senescent cells to confer senescence to their microenvironment suggest that this relationship is plausible [17, 114]. To this effect, long-term studies into the implications of adipose senescence and the viability of anti-senescent therapeutics in sarcopenia are needed. It must be considered, however, that the human lifespan is vastly different to that of rodents, from whom most existing data is derived. Thus, any deleterious effects of cytotoxic therapies to remove senescent cells may be amplified over the course of human ageing [92].
Cytokines as mediators of adipose-muscle cross talk
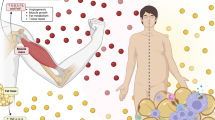
The decline of skeletal muscle mass in various inflammatory states is associated with elevated production of classical pro-inflammatory cytokines, including TNF-α, interleukin (IL)-1 beta (IL-1β), IL-6, and IFN-γ [115]. The low-grade inflammation associated with ageing, termed “inflammaging,” is influenced not only by overproduction of pro-inflammatory mediators but also by perturbation of anti-inflammatory factors, such as IL-10 [116,117,118,119]. While far from being fully understood, the roles of such classical cytokines in ageing muscle have been reviewed elsewhere [115, 120,121,122]. Accordingly, here we focus on cytokines which are implicated in adipose-muscle cross talk with emerging significance in the regulation of skeletal muscle mass, namely resistin, IL-15, adiponectin, leptin, lipocalin-2, and myostatin (Fig. 1; Table 1).

Cytokines secreted from skeletal muscle and adipose tissues with emerging significance in muscle-adipose cross talk and the regulation of skeletal muscle mass. The expression, secretion, extracellular-vesicle-mediated transport, and function of these cytokines may be perturbed by ageing and obesity, impairing normal muscle regulatory pathways. Processes regulating skeletal muscle mass have been condensed into four fundamental levels: activation of the muscle satellite cell pool; proliferation of myoblasts; differentiation of myoblasts into myotubes; and the ubiquitin (Ub) catabolic processes involved in the breakdown of muscle protein. Regardless of colour, arrows and block (inhibitory) lines indicate stimulatory and inhibitory effects, respectively, of a particular cytokine on these regulatory levels. Red lines (whether arrows or block lines) indicate pro-myogenic effects, which may act to preserve muscle mass, while black lines (whether arrows or block lines) indicate anti-myogenic effects which may confer adverse effects on muscle mass. The effects of ageing without obesity (silhouetted figure on the left) and ageing combined with obesity (silhouetted figure on the right) to increase or decrease the secretion and/or circulating abundance of these cytokines is indicated by thick black upward or downward-pointing arrows, respectively. Two upward arrows indicate a greater effect of obesity than the lean state on the relevant cytokine with ageing. A question mark indicates an unknown effect. An inhibitory line extending from a running person indicates that the proposed effect of ageing is offset when physical activity level is maintained with ageing. An inhibitory line extending to a cytokine receptor indicates that an increased abundance of that cytokine is associated with a reduction in expression of its receptor in skeletal muscle. Created with BioRender.com
Resistin
Resistin was first identified in rodents as an adipocyte-secreted factor that is upregulated in obesity and impairs glucose tolerance, insulin action, and fatty acid handling in skeletal muscle [123,124,125,126]. While the significance and even existence of resistin in human adipocytes has been contentious [127], by analysing conditioned medium from subcutaneous adipose tissue (SAT) from lean and obese humans, rather than isolated adipocytes, we recently found it to be secreted from SAT and increased with obesity [47]. This highlights the importance of stromal and infiltrating immune cells to the inflammatory in vivo phenotype of adipose tissue [128].
Notably, incubation of primary human myotubes from lean individuals with SAT-conditioned media obtained from obese subjects produced thinner, less multinucleated myotubes than those exposed to lean SAT secretome, which is concordant with findings using co-culture systems of human myotubes and adipocytes isolated from visceral adipose tissue (VAT) [47, 129]. Pertinently, the deleterious effects of the obese SAT secretome were more pronounced in myotubes derived from older than younger individuals, suggesting ageing impairs the ability of muscle to withstand inflammatory challenges. Since resistin was prolifically secreted from obese SAT, further investigation revealed that a physiologically relevant concentration of resistin (5 ng mL−1) impaired myoblast differentiation through activation of the classical nuclear factor-kappa beta (NF-κβ) pathway, similarly producing thinner myotubes with reduced nuclear fusion [47]. This adverse effect of resistin was confirmed in experiments showing that its depletion from obese SAT secretome restored myogenesis [47]. Consistent with these findings, overexpression of resistin impairs C2C12 myoblast differentiation, resulting in thinner myotubes with reduced expression of desmin and myoglobin [44].
In vivo research on the effects of resistin on regulating skeletal muscle mass and function is scant. However, human plasma resistin concentrations correlate with inflammatory markers [130], have been reported to correlate with age [45], and in the elderly are inversely associated with quadriceps torque [46] and computed tomography determinants of abdominal muscle density (a marker of muscle quality and composition) [131]. Collectively, the limited literature suggests an emerging role for resistin in suppressing myogenic differentiation, particularly in older skeletal muscle, which appears to be driven by inflammation and obesity.
Interleukin-15
IL-15, a four-helix bundle family cytokine, is produced by a variety of tissues, including skeletal muscle and adipose. Its mRNA expression is upregulated during the differentiation of C2C12 myoblasts and it is also expressed in pre- and post-differentiated 3T3-L1 adipocytes, albeit at a comparatively much lower level [132]. IL-15 mRNA expression in mouse epididymal adipose tissue and adipose tissue macrophages is increased in response to a high-fat diet (HFD) [133]. IL-15 was originally observed to have pro-myogenic effects on murine C2 skeletal muscle cells by promoting the accumulation of contractile proteins [134, 135]. Such effects may be mediated by supressing muscle proteolytic pathways. Indeed, ex vivo incubation of rat muscle tissue with recombinant IL-15 decreased the rate of proteolysis, without a change in total protein synthesis or tissue amino acid uptake [136]. Similarly, in vivo studies showed that septic mice pre-treated with IL-15 had lower expression of the proteolytic E3 ubiquitin ligases muscle atrophy F-box gene/atrogin-1 (MAFbx) and muscle ring finger-1 (MuRF-1), and IL-15 treatment of cachexic rats decelerated protein degradation and suppressed expression of ubiquitin proteolysis pathway components [48, 137].
We also reported that exposing primary myogenic cultures from young and elderly adults to recombinant IL-15 during differentiation resulted in thicker myotubes that were protected against differentiation impairment by TNF-α [49]. However, subcutaneous administration of recombinant IL-15 in rats for 7 days did not impact upon net muscle mass or protein content, although it increased turnover by accelerating both protein synthesis and breakdown rates and decreased white adipose tissue (WAT) mass by 33% [138]. Taken together, the literature supports a muscle-sparing effect of IL-15, particularly in conditions of dysregulated protein turnover such as cancer cachexia and possibly sarcopenia, through the suppression of muscle protein catabolism. However, IL-15 appears to have limited effects on muscle anabolism, particularly in healthy states.
We recently demonstrated in healthy older adults that IL-15 muscle mRNA expression and plasma concentration are elevated 2-fold and 1.5-fold, respectively, relative to younger adults [49]. However, IL-15 receptor signalling subunit IL2RB expression was 80% lower in older muscle, suggesting that the intracellular pro-myogenic effects of IL-15 may be blunted in the elderly. In a separate study, plasma IL-15 concentrations were found to be lower in sarcopenic compared to non-sarcopenic older adults and inversely correlated with sarcopenic state and body mass index (BMI) [50]. With regard to adiposity and IL-15 expression, obese individuals present with lower serum IL-15 than normal-weight individuals and IL-15 knockout in mice results in weight gain [139]. Interestingly, the aforementioned study that examined the effect of recombinant IL-15 administration in rats reported a decline in WAT mass [138].
It is important to note that levels of tissue IL-15 mRNA are not necessarily concordant with IL-15 protein expression and secretion, on account of inefficient translation due to multiple AUG initiation sites in the 5′-untranslated region [140]. Furthermore, alternative splicing produces two mature isoforms, only one of which follows the secretory pathway [141]. The seemingly contradictory observations of increased IL-15 mRNA expression in ageing and reduced circulating IL-15 abundance in sarcopenia may therefore demonstrate a transcriptional attempt to offset some impaired translational or secretory capacity that may occur with inflammaging, in an attempt to mitigate the catabolic effects of declining circulating IL-15.
Collectively, the literature suggest that IL-15 expression and function is tightly regulated at multiple levels and plays a significant role in the preservation of skeletal muscle mass. Its significance appears greatest within an atrophic, inflammatory state, such as that commonly observed with ageing, where it may have therapeutic potential through amelioration of processes involved in protein degradation.
Adiponectin
Adiponectin is the most abundant peptide secreted by adipocytes and plays a key role in energy homeostasis [142,143,144]. Adiponectin functions through the binding of the adaptor protein APPL1 (phosphotyrosine interacting with PH domain and leucine zipper 1), to the AdipoR1 and AdipoR2 receptors, and promotes interaction between the insulin receptor (IR) and its substrates IRS1/2, consequently priming the phosphatidylinositol-3-kinase (PI3K) pathway [145,146,147]. In skeletal muscle, adiponectin enhances p38 mitogen-activated kinase (MAPK)/peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) signalling and mitochondrial biogenesis [148, 149], and stimulates activation of adenosine monophosphate-activated protein kinase (AMPK) and acetyl-CoA carboxylase [150,151,152].
Unlike many adipocyte-secreted factors, adiponectin expression and secretion are greatest from healthy adipocytes of lean individuals and are inhibited by pro-inflammatory agents [124, 153, 154]. Accordingly, adiponectin levels decrease with obesity and type 2 diabetes mellitus (T2D) and correlate positively with insulin sensitivity and negatively with visceral adiposity [155,156,157]. Pertinently, circulating adiponectin is lower in sarcopenic than non-sarcopenic older adults [51] but does not differ between physical activity-matched non-sarcopenic older and younger adults, suggesting that it is not ageing per se driving reduced adiponectin concentration, but rather an age-associated decline in habitual physical activity level [52]. Conversely, circulating adiponectin was found to be higher in cardiovascular disease (CVD) patients with sarcopenia than without. However, the sarcopenic CVD group were predominantly female (66% female compared to a 28% female non-sarcopenic group) who typically present with higher adiponectin concentrations [158,159,160]. Nevertheless, some studies have suggested that elevated adiponectin may predict reduced muscle strength and function in older adults [161, 162]. Therefore, the higher levels of adiponectin in sarcopenic individuals reported in some studies could reflect a compensatory adaptation. Such a compensatory induction could be an attempt to preserve the p38 MAPK translational initiation pathway, the activation of which is dysregulated in human ageing [163], thus protecting against age-related impairment of protein synthetic pathways.
Globular adiponectin activates muscle satellite cells, promotes their motility, induces muscle gene expression including myosin heavy chain (MHC), myogenin, and p21, and drives myoblasts to exit the cell cycle, promoting differentiation [53,54,55]. Adiponectin overexpression increased regenerating myotubes in a mouse model of ageing and heart failure [164]. Adiponectin was found to accumulate on the myofibre plasma membrane and intracellularly co-localised with endosomes positive for the multivesicular/exosome marker cluster of differentiation-63 in regenerating, but not intact, fibres. However, in T-cadherin-null mice myotube regeneration was not increased by adiponectin overexpression, suggesting an essential role of T-cadherin in mediating these effects [164]. The authors demonstrated that both globular adiponectin and a small molecule mimetic (GTDF) induced differentiation and supressed MURF1 and MAFbx in various atrophy models, preventing the loss of myotube area [165]. Crossing degenerative muscle-diseased mice with mice overexpressing adiponectin improved expression of the myogenic differentiation markers myogenic regulatory factor-4 and myogenin, while reducing muscle inflammation and oxidative stress, resulting in higher muscle force and endurance [166]. Although this appears to contradict findings from human studies [161, 162], it supports the aforementioned possibility of a protective role of adiponectin on skeletal muscle mass in dysregulated states.
Confoundedly, stimulation of C2C12 cells with an adiponectin receptor agonist (AdipoRon) reduced cellular protein content, myotube diameter, and myotube multinucleation in a dose-dependent fashion (5–20 μM). However, the relevance of the dose utilised to physiologically elevate adiponectin concentrations is not well understood [167]. In mice, the extensor digitorum longus (fast twitch), but not soleus (slow twitch) muscle, exhibited increased adiponectin expression with ageing, though adiponectin receptor expression was unchanged [167]. The authors proposed that excessively high circulating adiponectin may induce skeletal muscle atrophy within fast-type muscle. However, neither serum AdipoRon nor adiponectin were measured in this study, limiting extrapolation and comparison to humans. Given that adiponectin activates AMPK, and AMPK activity has an inhibitive effect on mammalian target of rapamycin (mTOR), which plays a central role in protein synthesis, it is plausible that highly elevated adiponectin could exert negative effects on muscle mass regulation by excessive activation of AMPK [168,169,170,171]. Thus, it has been speculated that there is a healthy circulating adiponectin range required to maintain normal adiponectin signalling, though this purported range has not yet been clearly defined [171].
Leptin
Leptin is a cytokine-like hormone that is abundantly secreted by adipocytes but is also expressed in skeletal muscle [172]. Leptin conveys satiety-promoting effects and plays a significant role in the regulation of energy balance and body mass, as well as lipolysis and insulin sensitivity, which has been reviewed elsewhere [173,174,175,176]. Circulating leptin concentration increases with obesity, correlates with BMI and adipose tissue mass [81, 177,178,179,180], and is reduced with weight loss [181,182,183,184]. Pertinent to the regulation of skeletal muscle mass, leptin administration increases hindlimb muscle mass and fibre size in aged mice [56], and prevents muscle atrophy in leptin-deficient mice [57]. Mechanistically, the binding of leptin to the long form of its receptor, activates Janus kinase 2 (JAK2), subsequently promoting PI3K activity and phosphorylation of protein kinase B (Akt) and p38 MAPK [58, 185,186,187,188]. Leptin promotes C2C12 myoblast proliferation but suppresses expression of myogenin and myoblast determination protein (MyoD), which is mediated by JAK-STAT (signal transducer and activator of transcription) and MAPK pathways [59]. Despite the propensity for leptin to promote pathways involved in muscle regulation, its obesity-mediated upregulation is associated with resistance to its action [176, 189,190,191]. Central leptin receptor expression may be reduced as a direct result of increased leptin abundance [192,193,194]. Concordantly, skeletal muscle protein expression of the long leptin receptor is lower in adults with obesity, suggesting one mechanism by which peripheral leptin resistance may occur [60].
While circulating leptin is elevated in animal models of ageing in the absence of obesity [195], it has been reported to decline in elderly adults with severe frailty, but not in community-dwellers [61]. This decline may reflect not only the low abundance of adipose tissue often seen in frail older adults but also the diminishing mass of skeletal muscle, which is also a significant source of leptin [196,197,198]. Conversely, in community dwelling older adults, appendicular skeletal muscle mass and thigh muscle cross-sectional area have been negatively associated with plasma leptin concentration, even after adjustments for bodyweight or body fat percentage [62, 63]. In these studies, circulating leptin abundance was greater in those presenting with either sarcopenia or obesity, and greater still in individuals presenting with sarcopenic obesity. Interestingly, physical inactivity in the form of bed rest, which is often characteristic of frail older adults, also increases leptin levels independent of changes in fat mass [199]. It could be argued that physical inactivity increases leptin secretion in an attempt to preserve muscle mass, which may be ineffective in an aged-obese environment due to existing leptin resistance. Collectively, the literature suggests that both sarcopenia and obesity may promote a hyperleptinaemic environment that drives peripheral leptin resistance via the downregulation of its receptor. Thus, obesity may amplify resistance to the muscle-preserving effects of leptin in ageing and contribute to the sarcopenic-obese state.
Lipocalin 2
Lipocalin 2 (Lcn2) is a secretory glycoprotein first identified in human neutrophils and later noted for its abundant expression in adipocytes [200,201,202]. Both Lcn2 and its cell surface receptor (24p3R) have also been found to be expressed in skeletal muscle [64, 203]. Lipocalins bind and transport small hydrophobic molecules such as fatty acids, steroids, and retinol [200]. Lcn2 is induced by factors that drive insulin resistance and inflammatory stimuli, with transactivation by the pro-inflammatory transcription factor NF-κβ [65, 204]. Circulating Lcn2 is elevated with obesity and correlates with adiposity, adipose distribution, and inflammatory markers, though the significance of this relationship is contestable and not necessarily causal, with Liu et al. observing circulating Lcn2 to neither correlate with nor predict the incidence of insulin resistance or cardiovascular risk factors [66, 202, 205, 206].
Adipose Lcn2 mRNA and protein expression is greater in humans and rodents that are obese, compared to those of normal-weight [67, 200, 202]. Catalán et al. found plasma Lcn2 concentration was not affected by obesity; however, circulating Lcn2/matrix metalloproteinase (MMP) complex abundance and VAT MMP-2 and MMP-9 activity were elevated [67]. Given the role of the MMP system in adipocyte differentiation and SAT and VAT remodelling, this may implicate Lcn2 in the development of obesity [207,208,209]. On the other hand, it has recently been argued that rather than being causal of metabolic dysfunction, elevated Lcn2 may instead provide a protective mechanism to mitigate obesity-induced dysregulation and preserve pancreatic β-cell function [210]. Indeed, global Lcn2 knockout in mice was recently shown to accelerate age-dependent weight gain and visceral fat deposition in female mice, although curiously this was not the case in male mice [211].
Rebalka et al. investigated the role of Lcn2 in regenerating skeletal muscle and the effects of Lcn2 deletion [64]. While Lcn2 protein was lowly expressed in uninjured mouse skeletal muscle, its expression in Pax7+ muscle satellite cells was increased in response to cardiotoxin injury. In contrast, global Lcn2 knockout mice had reduced satellite cell activation and diminished muscle regeneration, with decreased embryonic MHC expression and smaller myofibre areas. However, the effects on adipose tissue were not studied. Finally, consistent with the role of Lcn2 in MMP regulation, Lcn2 knockout mice displayed greater fibrosis and lower MMP-9 activity during muscle regeneration.
The relationship between adipose-derived Lcn2 and skeletal muscle regulation is not yet fully understood, with reports of global Lnc2 knockout in C57BL/6 mice conferring both protection from, and potentiation of, diet- and ageing-induced metabolic dysregulation [212,213,214]. Initial investigations have shown that anti-diabetic drugs can counteract obesity-upregulated Lcn2 expression and circulating abundance in rats, T2D mice, and humans with T2D [200, 206]. However, reports of acute, but not chronic, exercise elevating circulating Lcn2 abundance in obese adults suggests that the relationship between Lcn2 and metabolic health is complex [215, 216]. Furthermore, it is not yet clear how modulation of circulating Lcn2 might impact skeletal muscle regulation. Therefore, studies employing muscle and adipose tissue-specific deletion as well as overexpression or administration of recombinant Lcn2 within rodent models of ageing and muscle wasting are warranted.
Myostatin
Myostatin is a transforming growth factor-beta (TGF-β) superfamily member that negatively regulates muscle mass and whose disruption produces hyper-muscularity [68]. Myostatin signals through activin-responsive type II receptors (ACTRIIA and ACTRIIB) and the type I receptors, activin-like kinases, triggering activation of small mothers against decapentaplegic (SMAD)2 and SMAD3 transcription factors, resulting in the inhibition of hypertrophic and activation of atrophic pathways [69, 217,218,219,220,221]. Recombinant myostatin impairs myogenesis through suppression of satellite cell activation and myoblast proliferation and differentiation, while genetic inactivation impairs myotube formation in vitro [70,71,72, 222, 223]. Myostatin expression and abundance is reportedly, though not universally, elevated with obesity, advancing age and muscle wasting, and decreased by weight loss [73, 224,225,226,227,228,229].
Whether the deleterious association of obesity and myostatin expression reflects an effect of excessive adipose tissue per se, or the product of an obesogenic muscle environment, remains unclear. Similarly, exercise is a potent suppressor of myostatin expression [230,231,232], and thus physical activity status adds a further confounding variable that is not always controlled for. Nevertheless, primary myotubes generated from morbidly obese individuals exhibit ~ 2-fold greater myostatin protein content and ~ 3-fold greater secretion than myotubes from lean donors, and mRNA expression is similarly elevated in morbidly obese muscle [73, 233].
Myostatin is also expressed in adipose tissue; however, data from ob/ob mice suggest mRNA expression is 50–100-fold lower than in skeletal muscle [234]. Importantly, myostatin expression was assessed in the tibialis anterior, a muscle comprised almost exclusively of fast fibres [235]. Since MHC IIb (a fast twitch myosin) expression correlates with myostatin mRNA expression, this may in part explain the magnitude of difference reported between muscle and adipose myostatin expression [236]. Nevertheless, both myostatin and ActRIIB mRNA expression were 50–100-fold higher in ob/ob than wild-type adipose tissue. Conversely, despite elevated serum abundance, myostatin expression in SAT and VAT was not different between age-matched severely obese, lean, and overweight humans [237]. It is plausible that the contradictory findings between obese human and murine adipose tissue myostatin expression reflect fundamental differences between human and murine adipose expression profiles and brown adipocyte abundance [238, 239].
Adipose tissue-specific deletion of myostatin in mice fed a HFD did not affect muscle weight nor body composition; however, whole-body knockout partially supressed HFD-induced fat accumulation [77, 240]. Conversely, muscle-specific inhibition of myostatin signalling increased lean mass and decreased fat mass in both chow and high-fat diets [77]. These studies suggest that secretion and signalling of myostatin in skeletal muscle influence the regulation of muscle and adipose mass. However, myostatin secretion and its signalling in adipose tissue have little impact on either adipose or muscle regulation, reflecting its vastly lower expression in adipose tissue.
Ageing appears to upregulate myostatin expression independent of adiposity, which if translated to increased myostatin protein secretion could contribute to sarcopenia by promoting atrophic and inhibiting hypertrophic pathways via canonical TGF-β signalling. Muscle myostatin mRNA and protein expression was 2.0-fold and 1.4-fold higher, respectively, in older than younger males [224] and myostatin mRNA expression was 56% greater in elderly than young women [74]. Older males matched with younger males for total- and lean-body mass and body fat percentage tended to have higher basal muscle myostatin mRNA expression and significantly greater muscle myostatin protein content [75]. Whether elevated myostatin expression with advancing age is a function of ageing per se or a product of age-associated physical inactivity, systemic inflammation, or nutritional status remains poorly investigated. Interestingly, muscle myostatin mRNA was found to be lower in both ambulatory and non-ambulatory elderly women than young women [76]. Myostatin protein content was greatest in the old ambulatory group, with no difference in plasma concentrations between groups. Similarly, higher concentrations of serum myostatin have been reported in non-frail compared to frail nursing home residents, and its abundance was increased in male residents, of both frail and non-frail status, following chronic exercise [241]. Evidently, further research is necessary to characterise and delineate between the effects of advancing age, adiposity, and physical inactivity on human myostatin expression and function in vivo.
Long non-coding RNAs: emerging roles in skeletal muscle regulation
Only ~ 1% of the human genome is translated into proteins; however, much is transcribed into non-protein-coding RNAs [242, 243]. Of these, lncRNAs, with transcripts of > 200 nucleotides, are the largest group and have been implicated in the regulation of RNA transcription, splicing, and trafficking, miRNA regulation, and RNA stability [84, 244,245,246,247,248]. lncRNAs have been implicated in obesity and in obesity-associated disorders including muscle wasting [249, 250]. Sequencing of SAT from females with and without obesity identified 86 lncRNAs that were differentially expressed [251]. Skeletal muscle from individuals with inclusion body and anti-Jo-1-associated myositis, a collection of diseases characterised by chronic muscle inflammation and weakening, revealed a similar number of differentially expressed lncRNAs, relative to healthy controls [78]. Of these, 16 lncRNAs were differentially expressed in both myositis groups, including H19, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), plasmacytoma variant translocation 1 (PVT1), and long non-coding myoblast determination protein (lncMyoD), which have been previously characterised and implicated in skeletal muscle regulation [84, 252, 253] (Table 1).
H19
H19 is upregulated during myoblast differentiation and its inhibition impairs skeletal muscle differentiation [254]. Pertinently, two miRNAs encoded by H19 exon1, miR-675-3p and miR-675-5p, which are induced during differentiation, can rescue the effects of H19 depletion through suppression of the TGF-β/bone morphogenetic protein pathway. H19 expression in SAT and VAT negatively correlates with BMI; however, whether an obesity-associated decline in adipose H19 affects the regulation of skeletal muscle mass remains to be investigated [255]. Given the aforementioned differential skeletal muscle expression of H19 in myositis patients, its potential involvement in age-related muscle wasting also warrants investigation.
MALAT1
MALAT1 expression increases during differentiation of primary human myoblasts and its knockdown is associated with impaired proliferation and differentiation [79, 256]. Treatment of mice with recombinant myostatin drastically reduced MALAT1 expression, which given the positive impact of myostatin deficiency on muscle function with ageing suggests MALAT1 may be an important downstream target of myostatin and regulator of myogenesis with ageing that deserves examination [256,257,258]. MALAT1 can competetively bind miR-133, de-repressing the transcription factor serum response factor (SRF) in myoblasts, facilitating upregulation of muscle-specific gene expression [79]. Conversely, it has been reported that MALAT1 is downregulated during early myogenesis and regulates differentiation through modulation of MyoD [80]. The impact of adiposity on MALAT1 expression in skeletal muscle and on myogenesis has not yet been explored.
PVT1
PVT1 is activated during muscle atrophy, affecting myofibre size, apoptosis, and mitochondrial function [252]. Downexpression of PVT1 in muscle induces resistance to atrophic processes in response to a denervation model of muscle wasting, by modulating apoptosis and autophagy. PVT1 is implicated in a variety of disease states characterised by inflammation, including obesity where its expression in murine adipose tissue is increased and promotes adipogenesis [82] and in osteoarthritis, where its suppression may ameliorate disease progression through the repression of catabolism and inflammation [83, 259]. However, PVT1 in ageing has not yet received much attention and its role in the cross talk between adipose tissue and skeletal muscle remains unknown.
lncMyoD
Encoded next to the MyoD gene, lncMyoD is activated by MyoD during myoblast differentiation and binds to insulin-like growth factor (IGF) mRNA-binding protein 2 (IMP2), blocking IMP-mediated shuttling of proliferation-promoting RNAs, inhibiting their translation and thus proliferation [84, 85]. The MyoD-lncMyoD-IMP2 pathway demonstrates a mechanism involved in the proliferation-inhibitive effects of MyoD, promoting a permissive state for differentiation. In mice, skeletal muscle lncMyoD expression is upregulated during models of disuse atrophy including denervation, casting, and tail suspension, but not in models of systemic muscle wasting including dexamethasone administration, cancer cachexia, and fasting [260]. Given the prevalence of physical inactivity and immobilisation amongst the elderly, such findings warrant investigations into the role and therapeutic potential of lncMyoD in human muscle ageing.
MicroRNAs: novel candidate factors in adipose-muscle cross talk
The non-coding miRNAs are well established as key mediators of gene expression through post-transcriptional downregulation, translational repression, and deadenylation-dependent decay and are abundant within EVs [261,262,263,264,265,266,267,268]. Most miRNAs are first transcribed into primary miRNAs (pri-miRNAs), processed by the RNase III enzymes Drosha, forming precursor miRNAs (pre-miRNAs), and Dicer, yielding mature miRNAs [269,270,271]. Recent reviews and systematic analyses of microarrays and next-generation sequencing have highlighted a plethora of miRNAs that are differentially expressed in skeletal muscle and/or plasma with ageing and muscle wasting, though the functional roles of many of these novel miRNAs remain to be elucidated [89, 272,273,274,275]. Similarly, within metabolic tissues and the circulation, numerous miRNAs have been implicated in the pathological development of obesity and may serve important functions in metabolic organ cross talk [276,277,278,279,280,281].
Whether obesity exacerbates sarcopenia through miRNA-mediated adipose-muscle cross talk is unclear. However, the capacity for adipose tissue to mediate metabolic cross talk with skeletal muscle via miRNAs is evident. Indeed, Wang et al. showed that miR-130b is secreted from adipocytes during adipogenesis; its WAT mRNA expression is increased with obesity; its circulating abundance is elevated in human and murine obesity, correlates with BMI, and predicts the metabolic syndrome; and it is able to target muscle cells, supressing expression of its target gene, PGC-1α [282].
While direct investigations of possible miRNA-mediated cross talk in the context of age-related muscle wasting and obesity remain to be undertaken, separate investigations have uncovered differentially expressed miRNAs common to both ageing and obesity, including miR-31, miR-223, and miR-33a [283, 284]. Such miRNAs may present candidate mediators of the adverse effects of obesity on muscle mass regulation (Table 1).
miR-31, miR-223, and miR-33a
Administration of leptin to aged mice increased muscle mass and expression of miR-31 and miR-223, which are known to be elevated during muscle regeneration and repair [56]. Given the aforementioned (“Leptin” section) potential role of leptin in the regulation of skeletal muscle mass, this suggests a channel by which miRNAs may be involved in mediating a beneficial effect of leptin in ageing muscle [56, 284].
miR-33a is expressed in both human skeletal muscle and adipose tissue [285] and encoded within sterol regulatory element-binding protein 2, a transcription factor involved in lipid biosynthesis and trafficking [286]. The circulating level of miR-33a is lower in older than younger adults in both the presence and absence of insulin resistance [283]. It has been shown that miR-33a knockout mice develop obesity with increased proliferation of pre-adipocytes, increased lipid uptake, and impaired lipolysis [287, 288]; however, the effect of obesity per se on miR-33a expression and abundance is not known. Administration of a miR-33a mimetic to primary duck myoblasts impaired proliferation, while a miR-33a inhibitor enhanced proliferation, and it was demonstrated that miR-33a may directly target IGF-1, follistatin, and cyclin-D1 to inhibit myoblast proliferation by suppressing the PI3K/Akt/mTOR signalling pathway [289]. Taken together, these data suggest that miR-33a is essential for normal adipocyte development and function; however, its excessive abundance may impair myogenesis. Thus, the declining levels of circulating miR-33a seen with ageing may act to offset the otherwise declining myogenic capacity of skeletal muscle. However, more research is needed to unravel the role of miR-33a in regulating muscle mass in the context of ageing and obesity.
MyomiRs
Numerous muscle-enriched miRNAs (myomiRs) have been implicated in myogenesis through their involvement with myogenic regulatory factors [290,291,292,293]. Using microarrays, miR-133a and miR-133b, which promote myoblast proliferation through repression of SRF [86], were found to be downregulated in muscle from healthy older men [89]. Conversely, elevated levels of pri-miRNA-1-1, pri-miRNA-1-2, pri-miRNA-133a-1, and pri-miRNA-133a-2 were found in the muscle from elderly men, relative to younger men [90]. However, these differences did not exist at the mature miRNA level, suggesting differential processing of pri- and pre-miRNAs with ageing, although dissimilar to prior findings, Drosha and Dicer protein or mRNA expression were not different between young and older men [90, 294].
Combined injection of miR-1, miR-133, and miR-206 accelerates muscle regeneration in vivo, induces myogenic expression, and promotes myoblast differentiation in vitro [87, 293]. miR-1 promotes myogenesis through targeting of the muscle gene transcriptional repressor histone deacetylase 4 (HDAC4), and its expression and circulating abundance are suppressed by HFD in mice [295, 296]; however, skeletal muscle miR-1 mRNA expression was elevated in a murine model of progeroid ageing [88]. miR-206 promotes myoblast differentiation and directly represses follistatin-like-1 and utrophin; however, its mRNA expression was also found to be elevated in aged mouse muscle [56, 91]. Pertinently, miR-206 expression in plasma was found to be downregulated 0.5-fold in children with overweight/obesity relative to normal-weight children, suggesting a potential mechanism by which obesity might adversely affect myogenesis independent of ageing [297]. Similarly, obesity is associated with downregulation of miR-133a-3p in human SAT [298] and in mice HFD-induced obesity downregulates miR-133b in WAT [299]. Conversely, HFD-induced obesity upregulates miR-133a in mouse muscle [295]; however, the functional significance of these observations in the context of regulating skeletal muscle mass remains unclear.
It is growing ever more apparent that miRNAs play vital roles in myogenesis and preliminary studies have identified both interacting and diverging effects of obesity and ageing on their expression and function. The lack of consensus between these studies highlights the need for further research, with an emphasis on translational studies to confirm findings from sequencing and microarray studies, to delineate the impact of ageing and obesity per se on the miRNA-mediated regulation of skeletal muscle mass. The subsequent identification and confirmation of differentially regulated adipose-muscle cross talking miRNAs will doubtless present novel potential therapeutic targets for muscle wasting conditions.
Conclusions and future directions
The regulation of skeletal muscle mass with ageing is complex and delicately balanced, involving many secreted molecules (including proteins and RNAs), which often display pleiotropic effects. Many of these factors appear perturbed by excessive adiposity, but effects are confounded by age, physical activity status, and comorbidities, frequently resulting in conflicting findings and limited reproducibility. Indeed, the potent beneficial effects of exercise on muscle-remodelling are a stark contrast to the rapid deterioration of muscle mass and function observed during periods of physical inactivity and immobilisation, which feature in the lives of some, but not all, elders. It is imperative therefore that every effort is taken to control for physical activity status when conducting human studies. Advances in proteomics and transcriptomics are enabling greater investigation of the secretomes of human muscle and adipose tissue in healthy, dysfunctional, and senescent states. There is a need, however, to extend the application of these techniques beyond the cross-sectional studies that have identified the differences in these secretomes, and to apply them to longitudinal studies of human ageing. Similarly, co-culture techniques using human primary cells from well-characterised donors are improving our understanding of these factors across the spectrums of health and ageing and offer greater relevance to the human condition. Finally, advances in techniques to investigate extracellular vesicles, to identify novel non-coding RNAs, and to establish their functional significance in regulating skeletal muscle mass may identify novel therapeutic targets for preserving muscle mass with ageing and increasing adiposity and may enhance our understanding of the sarcopenic-obese phenomenon.
Abbreviations
- ACTRIIB:
-
Activin receptor type IIB
- Akt:
-
Protein kinase B
- AMPK:
-
Adenosine monophosphate kinase
- APPL1:
-
Adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1
- BMI:
-
Body mass index
- CVD:
-
Cardiovascular disease
- HDAC4:
-
Histone deacetylase 4
- HFD:
-
High-fat diet
- IFN-γ:
-
Interferon gamma
- IGF:
-
Insulin-like growth factor
- IL:
-
Interleukin
- IMP2:
-
Insulin-like growth factor 2 mRNA-binding protein 2
- IRS:
-
Insulin receptor substrate
- JAK:
-
Janus kinase
- Lcn2:
-
Lipocalin-2
- lncMyoD:
-
Long non-coding myoblast determination protein
- lncRNA:
-
Long non-coding RNA
- MAFbx:
-
Muscle atrophy F-box gene/atrogin-1
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MHC:
-
Myosin heavy chain
- miRNA:
-
MicroRNA
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mammalian target of rapamycin
- MuRF-1:
-
Muscle ring finger-1
- MyoD:
-
Myoblast determination protein
- NF-κβ:
-
Nuclear factor-kappa beta
- P38 MAPK:
-
P38 mitogen-activated kinase
- Pax7:
-
Paired box 7
- PGC1-α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PI3K:
-
Phosphatidylinositol-3-kinase
- Pre-miRNA:
-
Precursor-microRNA
- Pri-miRNA:
-
Primary-microRNA
- PVT1:
-
Plasmacytoma variant translocation 1
- SASP:
-
Senescence-associated secretory phenotype
- SAT:
-
Subcutaneous adipose tissue
- SMAD:
-
Small mothers against decapentaplegic
- SRF:
-
Serum response factor
- STAT:
-
Signal transducer and activator of transcription
- T2D:
-
Type 2 diabetes mellitus
- TGF-β:
-
Transforming growth factor-beta
- TNF-α:
-
Tumour necrosis factor-alpha
- VAT:
-
Visceral adipose tissue
- WAT:
-
White adipose tissue
References
Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci. 2006;61(10):1059–64.
Frontera WR, Hughes VA, Fielding RA, Fiatarone MA, Evans WJ, Roubenoff R. Aging of skeletal muscle: a 12-yr longitudinal study. J Appl Physiol. 2000;88(4):1321–6. https://doi.org/10.1152/jappl.2000.88.4.1321.
Rantanen T, Masaki K, Foley D, Izmirlian G, White L, Guralnik JM. Grip strength changes over 27 yr in Japanese-American men. Journal of applied physiology (Bethesda, Md : 1985). 1998;85(6):2047–53. https://doi.org/10.1152/jappl.1998.85.6.2047.
Guillet C, Boirie Y, Walrand S. An integrative approach to in-vivo protein synthesis measurement: from whole tissue to specific proteins. Curr Opin Clin Nutr Metab Care. 2004;7(5):531–8.
Poortmans JR, Carpentier A, Pereira-Lancha LO, Lancha A. Protein turnover, amino acid requirements and recommendations for athletes and active populations. Braz J Med Biol Res. 2012;45(10):875–90. https://doi.org/10.1590/S0100-879X2012007500096.
McCormick R, Vasilaki A. Age-related changes in skeletal muscle: changes to life-style as a therapy. Biogerontology. 2018;19(6):519–36. https://doi.org/10.1007/s10522-018-9775-3.
Le Grand F, Rudnicki MA. Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol. 2007;19(6):628–33. https://doi.org/10.1016/j.ceb.2007.09.012.
Yamakawa H, Kusumoto D, Hashimoto H, Yuasa S. Stem cell aging in skeletal muscle regeneration and disease. Int J Mol Sci. 2020;21(5):1830. https://doi.org/10.3390/ijms21051830.
Carosio S, Berardinelli MG, Aucello M, Musarò A. Impact of ageing on muscle cell regeneration. Ageing research reviews. 2011;10(1):35–42. https://doi.org/10.1016/j.arr.2009.08.001.
Addison O, Drummond MJ, LaStayo PC, Dibble LE, Wende AR, McClain DA, et al. Intramuscular fat and inflammation differ in older adults: the impact of frailty and inactivity. J Nutr Health Aging. 2014;18(5):532–8. https://doi.org/10.1007/s12603-014-0019-1.
Wilkinson DJ, Piasecki M, Atherton PJ. The age-related loss of skeletal muscle mass and function: measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing research reviews. 2018;47:123–32. https://doi.org/10.1016/j.arr.2018.07.005.
Breen L, Phillips SM. Skeletal muscle protein metabolism in the elderly: interventions to counteract the 'anabolic resistance' of ageing. Nutr Metab (Lond). 2011;8(1):68. https://doi.org/10.1186/1743-7075-8-68.
Wall BT, Gorissen SH, Pennings B, Koopman R, Groen BB, Verdijk LB, et al. Aging is accompanied by a blunted muscle protein synthetic response to protein ingestion. PLoS One. 2015;10(11):e0140903. https://doi.org/10.1371/journal.pone.0140903.
Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. 2018:afy169-afy. https://doi.org/10.1093/ageing/afy169.
Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One. 2010;5(5):e10805. https://doi.org/10.1371/journal.pone.0010805.
Roh E, Choi KM. Health consequences of sarcopenic obesity: a narrative review. Front Endocrinol (Lausanne). 2020;11(332). https://doi.org/10.3389/fendo.2020.00332.
Kalinkovich A, Livshits G. Sarcopenic obesity or obese sarcopenia: a cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing research reviews. 2017;35:200–21. https://doi.org/10.1016/j.arr.2016.09.008.
Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med. 2001;137(4):231–43. https://doi.org/10.1067/mlc.2001.113504.
ROUBENOFF R. Sarcopenic obesity: does muscle loss cause fat gain?: lessons from rheumatoid arthritis and osteoarthritisa. Ann N Y Acad Sci. 2000;904(1):553–557. https://doi.org/10.1111/j.1749-6632.2000.tb06515.x.
Batsis JA, Villareal DT. Sarcopenic obesity in older adults: aetiology, epidemiology and treatment strategies. Nature reviews Endocrinology. 2018;14(9):513–37. https://doi.org/10.1038/s41574-018-0062-9.
Villareal DT, Banks M, Siener C, Sinacore DR, Klein S. Physical frailty and body composition in obese elderly men and women. Obes Res. 2004;12(6):913–20. https://doi.org/10.1038/oby.2004.111.
Roubenoff R. Sarcopenic obesity: the confluence of two epidemics. Obes Res. 2004;12(6):887–8. https://doi.org/10.1038/oby.2004.107.
Stephen WC, Janssen I. Sarcopenic-obesity and cardiovascular disease risk in the elderly. J Nutr Health Aging. 2009;13(5):460–6. https://doi.org/10.1007/s12603-009-0084-z.
Chen L, Nelson DR, Zhao Y, Cui Z, Johnston JA. Relationship between muscle mass and muscle strength, and the impact of comorbidities: a population-based, cross-sectional study of older adults in the United States. BMC Geriatr. 2013;13(1):74. https://doi.org/10.1186/1471-2318-13-74.
Han TS, Tajar A, Lean MEJ. Obesity and weight management in the elderly. Br Med Bull. 2011;97(1):169–96. https://doi.org/10.1093/bmb/ldr002.
Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet (London, England). 2009;374(9696):1196–208. https://doi.org/10.1016/S0140-6736(09)61460-4.
Siiteri PK. Adipose tissue as a source of hormones. Am J Clin Nutr. 1987;45(1 Suppl):277–82. https://doi.org/10.1093/ajcn/45.1.277.
Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237(4813):402–5. https://doi.org/10.1126/science.3299705.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32. https://doi.org/10.1038/372425a0.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. https://doi.org/10.1126/science.7678183.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. https://doi.org/10.1172/jci19246.
Liu Z, Wu KKL, Jiang X, Xu A, Cheng KKY. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin Sci. 2020;134(2):315–30. https://doi.org/10.1042/cs20190966.
Funcke JB, Scherer PE. Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res. 2019;60(10):1648–84. https://doi.org/10.1194/jlr.R094060.
Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. American Journal of Physiology-Endocrinology and Metabolism. 2001;280(6):E827–E47. https://doi.org/10.1152/ajpendo.2001.280.6.E827.
Mathus-Vliegen EMH, Basdevant A, Finer N, Hainer V, Hauner H, Micic D, et al. Prevalence, pathophysiology, health consequences and treatment options of obesity in the elderly: a guideline. Obesity Facts. 2012;5(3):460–83. https://doi.org/10.1159/000341193.
Stanford KI, Middelbeek RJW, Townsend KL, Lee M-Y, Takahashi H, So K, et al. A novel role for subcutaneous adipose tissue in exercise-induced improvements in glucose homeostasis. Diabetes. 2015;64(6):2002–14. https://doi.org/10.2337/db14-0704.
Zhang J-M, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45(2):27–37. https://doi.org/10.1097/AIA.0b013e318034194e.
Barnes BJ, Somerville CC. Modulating cytokine production via select packaging and secretion from extracellular vesicles. Front Immunol. 2020;11(1040). https://doi.org/10.3389/fimmu.2020.01040.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. The Journal of cell biology. 2013;200(4):373–83. https://doi.org/10.1083/jcb.201211138.
Rome S, Forterre A, Mizgier ML, Bouzakri K. Skeletal muscle-released extracellular vesicles: state of the art. Front Physiol. 2019;10(929). https://doi.org/10.3389/fphys.2019.00929.
Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–32. https://doi.org/10.1016/j.cell.2016.01.043.
Wang H, Wang B. Extracellular vesicle microRNAs mediate skeletal muscle myogenesis and disease. Biomed Rep. 2016;5(3):296–300. https://doi.org/10.3892/br.2016.725.
Bittel DC, Jaiswal JK. Contribution of extracellular vesicles in rebuilding injured muscles. Front Physiol. 2019;10:828. https://doi.org/10.3389/fphys.2019.00828.
Sheng CH, Du ZW, Song Y, Wu XD, Zhang YC, Wu M, et al. Human resistin inhibits myogenic differentiation and induces insulin resistance in myocytes. BioMed Research International. 2013;2013:804632. https://doi.org/10.1155/2013/804632.
Won JC, Park C-Y, Lee WY, Lee ES, Oh SW, Park SW. Association of plasma levels of resistin with subcutaneous fat mass and markers of inflammation but not with metabolic determinants or insulin resistance. J Korean Med Sci. 2009;24(4):695–700. https://doi.org/10.3346/jkms.2009.24.4.695.
Bucci L, Yani SL, Fabbri C, Bijlsma AY, Maier AB, Meskers CG, et al. Circulating levels of adipokines and IGF-1 are associated with skeletal muscle strength of young and old healthy subjects. Biogerontology. 2013;14(3):261–72. https://doi.org/10.1007/s10522-013-9428-5.
O’Leary MF, Wallace GR, Davis ET, Murphy DP, Nicholson T, Bennett AJ, et al. Obese subcutaneous adipose tissue impairs human myogenesis, particularly in old skeletal muscle, via resistin-mediated activation of NFκB. Sci Rep. 2018;8(1):15360. https://doi.org/10.1038/s41598-018-33840-x.
Kim HC, Cho H-Y, Hah Y-S. Role of IL-15 in sepsis-induced skeletal muscle atrophy and proteolysis. Tuberc Respir Dis. 2012;73(6):312–9. https://doi.org/10.4046/trd.2012.73.6.312.
O’Leary MF, Wallace GR, Bennett AJ, Tsintzas K, Jones SW. IL-15 promotes human myogenesis and mitigates the detrimental effects of TNFα on myotube development. Sci Rep. 2017;7(1):12997. https://doi.org/10.1038/s41598-017-13479-w.
Yalcin A, Silay K, Balik AR, Avcioğlu G, Aydin AS. The relationship between plasma interleukin-15 levels and sarcopenia in outpatient older people. Aging Clin Exp Res. 2018;30(7):783–90. https://doi.org/10.1007/s40520-017-0848-y.
Can B, Kara O, Kizilarslanoglu MC, Arik G, Aycicek GS, Sumer F, et al. Serum markers of inflammation and oxidative stress in sarcopenia. Aging Clin Exp Res. 2017;29(4):745–52. https://doi.org/10.1007/s40520-016-0626-2.
Hioki M, Kanehira N, Koike T, Saito A, Takahashi H, Shimaoka K, et al. Associations of intramyocellular lipid in vastus lateralis and biceps femoris with blood free fatty acid and muscle strength differ between young and elderly adults. Clin Physiol Funct Imaging. 2016;36(6):457–63. https://doi.org/10.1111/cpf.12250.
Fiaschi T, Giannoni E, Taddei ML, Chiarugi P. Globular adiponectin activates motility and regenerative traits of muscle satellite cells. PLoS One. 2012;7(5):e34782. https://doi.org/10.1371/journal.pone.0034782.
Fiaschi T, Cirelli D, Comito G, Gelmini S, Ramponi G, Serio M, et al. Globular adiponectin induces differentiation and fusion of skeletal muscle cells. Cell Res. 2009;19(5):584–97. https://doi.org/10.1038/cr.2009.39.
Fiaschi T, Tedesco FS, Giannoni E, Diaz-Manera J, Parri M, Cossu G, et al. Globular adiponectin as a complete mesoangioblast regulator: role in proliferation, survival, motility, and skeletal muscle differentiation. Mol Biol Cell. 2010;21(6):848–59. https://doi.org/10.1091/mbc.e09-04-0310.
Hamrick MW, Herberg S, Arounleut P, He HZ, Shiver A, Qi RQ, et al. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem Biophys Res Commun. 2010;400(3):379–83. https://doi.org/10.1016/j.bbrc.2010.08.079.
Sáinz N, Rodríguez A, Catalán V, Becerril S, Ramírez B, Gómez-Ambrosi J, et al. Leptin administration favors muscle mass accretion by decreasing FoxO3a and increasing PGC-1α in ob/ob mice. PLoS One. 2009;4(9):e6808. https://doi.org/10.1371/journal.pone.0006808.
Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Häring HU. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia. 1997;40(11):1358–62. https://doi.org/10.1007/s001250050832.
Pijet M, Pijet B, Litwiniuk A, Pajak B, Gajkowska B, Orzechowski A. Leptin impairs myogenesis in C2C12 cells through JAK/STAT and MEK signaling pathways. Cytokine. 2013;61(2):445–54. https://doi.org/10.1016/j.cyto.2012.11.002.
Fuentes T, Ara I, Guadalupe-Grau A, Larsen S, Stallknecht B, Olmedillas H, et al. Leptin receptor 170 kDa (OB-R170) protein expression is reduced in obese human skeletal muscle: a potential mechanism of leptin resistance. Exp Physiol. 2010;95(1):160–71. https://doi.org/10.1113/expphysiol.2009.049270.
Hubbard RE, O'Mahony MS, Calver BL, Woodhouse KW. Nutrition, inflammation, and leptin levels in aging and frailty. J Am Geriatr Soc. 2008;56(2):279–84. https://doi.org/10.1111/j.1532-5415.2007.01548.x.
Kohara K, Ochi M, Tabara Y, Nagai T, Igase M, Miki T. Leptin in sarcopenic visceral obesity: possible link between adipocytes and myocytes. PLoS One. 2011;6(9):e24633. https://doi.org/10.1371/journal.pone.0024633.
Waters DL, Qualls CR, Dorin RI, Veldhuis JD, Baumgartner RN. Altered growth hormone, cortisol, and leptin secretion in healthy elderly persons with sarcopenia and mixed body composition phenotypes. The Journals of Gerontology: Series A. 2008;63(5):536–41. https://doi.org/10.1093/gerona/63.5.536.
Rebalka IA, Monaco CMF, Varah NE, Berger T, D’souza DM, Zhou S, et al. Loss of the adipokine lipocalin-2 impairs satellite cell activation and skeletal muscle regeneration. American Journal of Physiology-Cell Physiology. 2018;315(5):C714–C21. https://doi.org/10.1152/ajpcell.00195.2017.
Cowland JB, Muta T, Borregaard N. IL-1beta-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IkappaB-zeta. J Immunol. 2006;176(9):5559–66. https://doi.org/10.4049/jimmunol.176.9.5559.
Auguet T, Quintero Y, Terra X, Martínez S, Lucas A, Pellitero S, et al. Upregulation of lipocalin 2 in adipose tissues of severely obese women: positive relationship with proinflammatory cytokines. Obesity (Silver Spring). 2011;19(12):2295–300. https://doi.org/10.1038/oby.2011.61.
Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, et al. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med. 2009;87(8):803. https://doi.org/10.1007/s00109-009-0486-8.
McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. https://doi.org/10.1038/387083a0.
Lee S-J, McPherron AC. Regulation of myostatin activity and muscle growth. Proceedings of the National Academy of Sciences. 2001;98(16):9306–11. https://doi.org/10.1073/pnas.151270098.
McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. The Journal of cell biology. 2003;162(6):1135–47. https://doi.org/10.1083/jcb.200207056.
Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275(51):40235–43. https://doi.org/10.1074/jbc.M004356200.
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831–40. https://doi.org/10.1074/jbc.M204291200.
Hittel DS, Berggren JR, Shearer J, Boyle K, Houmard JA. Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes. 2009;58(1):30–8. https://doi.org/10.2337/db08-0943.
Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Myogenic gene expression at rest and after a bout of resistance exercise in young (18–30 yr) and old (80–89 yr) women. Journal of applied physiology (Bethesda, Md : 1985). 2006;101(1):53–9. https://doi.org/10.1152/japplphysiol.01616.2005.
McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J. 2012;26(6):2509–21. https://doi.org/10.1096/fj.11-198663.
Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology. 2013;14(3):303–23. https://doi.org/10.1007/s10522-013-9432-9.
Guo T, Jou W, Chanturiya T, Portas J, Gavrilova O, McPherron AC. myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One. 2009;4(3):e4937. https://doi.org/10.1371/journal.pone.0004937.
Hamann PD, Roux BT, Heward JA, Love S, McHugh NJ, Jones SW, et al. Transcriptional profiling identifies differential expression of long non-coding RNAs in Jo-1 associated and inclusion body myositis. Sci Rep. 2017;7(1):8024. https://doi.org/10.1038/s41598-017-08603-9.
Han X, Yang F, Cao H, Liang Z. Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. The FASEB Journal. 2015;29(7):3054–64. https://doi.org/10.1096/fj.14-259952.
Chen X, He L, Zhao Y, Li Y, Zhang S, Sun K, et al. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov. 2017;3:17002. https://doi.org/10.1038/celldisc.2017.2.
van Dielen FMH, van't Veer C, Schols AM, Soeters PB, Buurman WA, Greve JWM. Increased leptin concentrations correlate with increased concentrations of inflammatory markers in morbidly obese individuals. Int J Obes. 2001;25(12):1759–66. https://doi.org/10.1038/sj.ijo.0801825.
Zhang L, Zhang D, Qin Z-Y, Li J, Shen Z-Y. The role and possible mechanism of long noncoding RNA PVT1 in modulating 3T3-L1 preadipocyte proliferation and differentiation. IUBMB life. 2020;72(7):1460–7. https://doi.org/10.1002/iub.2269.
Zhao Y, Zhao J, Guo X, She J, Liu Y. Long non-coding RNA PVT1, a molecular sponge for miR-149, contributes aberrant metabolic dysfunction and inflammation in IL-1β-simulated osteoarthritic chondrocytes. Biosci Rep. 2018;38(5):BSR20180576. https://doi.org/10.1042/BSR20180576.
Gong C, Li Z, Ramanujan K, Clay I, Zhang Y, Lemire-Brachat S, et al. A long non-coding RNA, LncMyoD, regulates skeletal muscle differentiation by blocking IMP2-mediated mRNA translation. Dev Cell. 2015;34(2):181–91. https://doi.org/10.1016/j.devcel.2015.05.009.
Boudoukha S, Cuvellier S, Polesskaya A. Role of the RNA-binding protein IMP-2 in muscle cell motility. Mol Cell Biol. 2010;30(24):5710–25. https://doi.org/10.1128/MCB.00665-10.
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38(2):228–33. https://doi.org/10.1038/ng1725.
Nakasa T, Ishikawa M, Shi M, Shibuya H, Adachi N, Ochi M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J Cell Mol Med. 2010;14(10):2495–505. https://doi.org/10.1111/j.1582-4934.2009.00898.x.
Mariño G, Ugalde AP, Fernández ÁF, Osorio FG, Fueyo A, Freije JMP, et al. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proceedings of the National Academy of Sciences. 2010;107(37):16268–73. https://doi.org/10.1073/pnas.1002696107.
Drummond MJ, McCarthy JJ, Sinha M, Spratt HM, Volpi E, Esser KA, et al. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol Genomics. 2011;43(10):595–603. https://doi.org/10.1152/physiolgenomics.00148.2010.
Drummond MJ, McCarthy JJ, Fry CS, Esser KA, Rasmussen BB. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am J Physiol Endocrinol Metab. 2008;295(6):E1333–40. https://doi.org/10.1152/ajpendo.90562.2008.
Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tapscott SJ. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol. 2006;175(1):77–85. https://doi.org/10.1083/jcb.200603039.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35. https://doi.org/10.1038/nm.4000.
Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8(11):1291–7. https://doi.org/10.1038/ncb1491.
Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A. 1996;93(24):13742–7. https://doi.org/10.1073/pnas.93.24.13742.
Schafer MJ, Miller JD, LeBrasseur NK. Cellular senescence: implications for metabolic disease. Mol Cell Endocrinol. 2017;455:93–102. https://doi.org/10.1016/j.mce.2016.08.047.
Baar MP, Perdiguero E, Muñoz-Cánoves P, de Keizer PLJ. Musculoskeletal senescence: a moving target ready to be eliminated. Curr Opin Pharmacol. 2018;40:147–55. https://doi.org/10.1016/j.coph.2018.05.007.
Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):e301. https://doi.org/10.1371/journal.pbio.0060301.
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144.
Salminen A, Ojala J, Kaarniranta K. Apoptosis and aging: increased resistance to apoptosis enhances the aging process. Cell Mol Life Sci. 2011;68(6):1021–31. https://doi.org/10.1007/s00018-010-0597-y.
Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing. 2020;17(1):16. https://doi.org/10.1186/s12979-020-00187-9.
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proceedings of the National Academy of Sciences. 2015;112(46):E6301–E10. https://doi.org/10.1073/pnas.1515386112.
Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes. 2015;64(7):2289–98. https://doi.org/10.2337/db14-1820.
Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15(9):1082–7. https://doi.org/10.1038/nm.2014.
Farah H, Young SP, Mauro C, Jones SW. Metabolic dysfunction and inflammatory disease: the role of stromal fibroblasts. The FEBS journal. n/a(n/a). https://doi.org/10.1111/febs.15644.
Burton DGA, Faragher RGA. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology. 2018;19(6):447–59. https://doi.org/10.1007/s10522-018-9763-7.
Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract. 2013;7(5):e330–e41. https://doi.org/10.1016/j.orcp.2013.05.004.
Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, et al. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366(9486):662–4. https://doi.org/10.1016/s0140-6736(05)66630-5.
Passos JF, Simillion C, Hallinan J, Wipat A, von Zglinicki T. Cellular senescence: unravelling complexity. Age (Dordr). 2009;31(4):353–63. https://doi.org/10.1007/s11357-009-9108-1.
Tzanetakou IP, Katsilambros NL, Benetos A, Mikhailidis DP, Perrea DN. "Is obesity linked to aging?": adipose tissue and the role of telomeres. Ageing research reviews. 2012;11(2):220–9. https://doi.org/10.1016/j.arr.2011.12.003.
Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat tissue, aging, and cellular senescence. Aging cell. 2010;9(5):667–84. https://doi.org/10.1111/j.1474-9726.2010.00608.x.
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–56. https://doi.org/10.1038/s41591-018-0092-9.
Study HAC. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. The American Journal of Clinical Nutrition. 2009;90(6):1579–85. https://doi.org/10.3945/ajcn.2009.28047.
Goodpaster BH, Thaete FL, Kelley DE. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. The American Journal of Clinical Nutrition. 2000;71(4):885–92. https://doi.org/10.1093/ajcn/71.4.885.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–90. https://doi.org/10.1038/ncb2784.
Costamagna D, Costelli P, Sampaolesi M, Penna F. Role of inflammation in muscle homeostasis and myogenesis. Mediators Inflamm. 2015;2015:805172. https://doi.org/10.1155/2015/805172.
Dagdeviren S, Jung DY, Lee E, Friedline RH, Noh HL, Kim JH, et al. Altered interleukin-10 signaling in skeletal muscle regulates obesity-mediated inflammation and insulin resistance. Mol Cell Biol. 2016;36(23):2956–66. https://doi.org/10.1128/MCB.00181-16.
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology. 2018;14(10):576–90. https://doi.org/10.1038/s41574-018-0059-4.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. https://doi.org/10.1093/gerona/glu057.
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105. https://doi.org/10.1016/j.mad.2006.11.016.
Peake J, Gatta PD, Cameron-Smith D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2010;298(6):R1485–R95. https://doi.org/10.1152/ajpregu.00467.2009.
Degens H. The role of systemic inflammation in age-related muscle weakness and wasting. Scand J Med Sci Sports. 2010;20(1):28–38. https://doi.org/10.1111/j.1600-0838.2009.01018.x.
Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 2013;14(12):877–82. https://doi.org/10.1016/j.jamda.2013.05.009.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307–12. https://doi.org/10.1038/35053000.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nature Reviews Immunology. 2011;11(2):85–97. https://doi.org/10.1038/nri2921.
Palanivel R, Sweeney G. Regulation of fatty acid uptake and metabolism in L6 skeletal muscle cells by resistin. FEBS Lett. 2005;579(22):5049–54. https://doi.org/10.1016/j.febslet.2005.08.011.
Palanivel R, Maida A, Liu Y, Sweeney G. Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia. 2006;49(1):183–90. https://doi.org/10.1007/s00125-005-0060-z.
Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, et al. Resistin / Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50(10):2199–202. https://doi.org/10.2337/diabetes.50.10.2199.
Blaber SP, Webster RA, Hill CJ, Breen EJ, Kuah D, Vesey G, et al. Analysis of in vitro secretion profiles from adipose-derived cell populations. J Transl Med. 2012;10:172. https://doi.org/10.1186/1479-5876-10-172.
Pellegrinelli V, Rouault C, Rodriguez-Cuenca S, Albert V, Edom-Vovard F, Vidal-Puig A, et al. Human adipocytes induce inflammation and atrophy in muscle cells during obesity. Diabetes. 2015;64(9):3121–34. https://doi.org/10.2337/db14-0796.
Qi Q, Wang J, Li H, Yu Z, Ye X, Hu FB, et al. Associations of resistin with inflammatory and fibrinolytic markers, insulin resistance, and metabolic syndrome in middle-aged and older Chinese. Eur J Endocrinol. 2008;159(5):585–93. https://doi.org/10.1530/eje-08-0427.
VAN HOLLEBEKE RB, CUSHMAN M, SCHLUETER EF, ALLISON MA. Abdominal muscle density is inversely related to adiposity inflammatory mediators. Med Sci Sports Exerc. 2018;50(7):1495–501. https://doi.org/10.1249/mss.0000000000001570.
Quinn LS, Strait-Bodey L, Anderson BG, Argilés JM, Havel PJ. Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: evidence for a skeletal muscle-to-fat signaling pathway. Cell Biol Int. 2005;29(6):449–57. https://doi.org/10.1016/j.cellbi.2005.02.005.
Lee B-C, Kim M-S, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. 2016;23(4):685–98. https://doi.org/10.1016/j.cmet.2016.03.002.
Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science. 1994;264(5161):965–8. https://doi.org/10.1126/science.8178155.
Quinn LS, Haugk KL, Grabstein KH. Interleukin-15: a novel anabolic cytokine for skeletal muscle. Endocrinology. 1995;136(8):3669–72. https://doi.org/10.1210/endo.136.8.7628408.
Busquets S, Figueras MT, Meijsing S, Carbó N, Quinn LS, Almendro V, et al. Interleukin-15 decreases proteolysis in skeletal muscle: a direct effect. Int J Mol Med. 2005;16(3):471–6.
Carbó N, López-Soriano J, Costelli P, Busquets S, Alvarez B, Baccino FM, et al. Interleukin-15 antagonizes muscle protein waste in tumour-bearing rats. Br J Cancer. 2000;83(4):526–31. https://doi.org/10.1054/bjoc.2000.1299.
Carbó N, López-Soriano J, Costelli P, Alvarez B, Busquets S, Baccino FM, et al. Interleukin-15 mediates reciprocal regulation of adipose and muscle mass: a potential role in body weight control. Biochim Biophys Acta. 2001;1526(1):17–24. https://doi.org/10.1016/s0304-4165(00)00188-4.
Barra NG, Reid S, MacKenzie R, Werstuck G, Trigatti BL, Richards C, et al. Interleukin-15 contributes to the regulation of murine adipose tissue and human adipocytes. Obesity. 2010;18(8):1601–7. https://doi.org/10.1038/oby.2009.445.
Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol. 1999;17:19–49. https://doi.org/10.1146/annurev.immunol.17.1.19.
Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17(4):259–80. https://doi.org/10.1016/j.cytogfr.2006.05.001.
Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care. 2003;26(8):2442–50. https://doi.org/10.2337/diacare.26.8.2442.
Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun. 1996;221(2):286–9. https://doi.org/10.1006/bbrc.1996.0587.
Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. 2017;18(6):1321. https://doi.org/10.3390/ijms18061321.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762–9. https://doi.org/10.1038/nature01705.
Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, et al. Crystal structures of the human adiponectin receptors. Nature. 2015;520(7547):312–6. https://doi.org/10.1038/nature14301.
Ryu J, Galan AK, Xin X, Dong F, Abdul-Ghani MA, Zhou L, et al. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep. 2014;7(4):1227–38. https://doi.org/10.1016/j.celrep.2014.04.006.
Xin X, Zhou L, Reyes CM, Liu F, Dong LQ. APPL1 mediates adiponectin-stimulated p38 MAPK activation by scaffolding the TAK1-MKK3-p38 MAPK pathway. American Journal of Physiology-Endocrinology and Metabolism. 2011;300(1):E103–E10. https://doi.org/10.1152/ajpendo.00427.2010.
Qiao L, Kinney B. Yoo Hs, Lee B, Schaack J, Shao J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes. 2012;61(6):1463–70. https://doi.org/10.2337/db11-1475.
Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang Cc C, Itani SI, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99(25):16309–13. https://doi.org/10.1073/pnas.222657499.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–95. https://doi.org/10.1038/nm788.
Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98(4):2005–10. https://doi.org/10.1073/pnas.041591798.
Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol. 2003;14(6):561–6. https://doi.org/10.1097/00041433-200312000-00003.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–49. https://doi.org/10.1161/01.Res.0000163635.62927.34.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83. https://doi.org/10.1006/bbrc.1999.0255.
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20(6):1595–9. https://doi.org/10.1161/01.atv.20.6.1595.
Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M, et al. Adiponectin as a biomarker of the metabolic syndrome. Circ J. 2004;68(11):975–81. https://doi.org/10.1253/circj.68.975.
Harada H, Kai H, Shibata R, Niiyama H, Nishiyama Y, Murohara T, et al. New diagnostic index for sarcopenia in patients with cardiovascular diseases. PLoS One. 2017;12(5):e0178123. https://doi.org/10.1371/journal.pone.0178123.
Song HJ, Oh S, Quan S, Ryu O-H, Jeong J-Y, Hong K-S, et al. Gender differences in adiponectin levels and body composition in older adults: Hallym aging study. BMC Geriatr. 2014;14(1):8. https://doi.org/10.1186/1471-2318-14-8.
Eglit T, Ringmets I, Lember M. Obesity, high-molecular-weight (HMW) adiponectin, and metabolic risk factors: prevalence and gender-specific associations in Estonia. PLoS One. 2013;8(9):e73273. https://doi.org/10.1371/journal.pone.0073273.
Huang C, Tomata Y, Kakizaki M, Sugawara Y, Hozawa A, Momma H, et al. High circulating adiponectin levels predict decreased muscle strength among older adults aged 70 years and over: a prospective cohort study. Nutr Metab Cardiovasc Dis. 2015;25(6):594–601. https://doi.org/10.1016/j.numecd.2015.03.010.
Karvonen-Gutierrez CA, Zheng H, Mancuso P, Harlow SD. Higher leptin and adiponectin concentrations predict poorer performance-based physical functioning in midlife women: the Michigan Study of Women’s Health Across the Nation. J Gerontol A Biol Sci Med Sci. 2016;71(4):508–14. https://doi.org/10.1093/gerona/glv123.
Williamson D, Gallagher P, Harber M, Hollon C, Trappe S. Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. The Journal of physiology. 2003;547(Pt 3):977–87. https://doi.org/10.1113/jphysiol.2002.036673.
Tanaka Y, Kita S, Nishizawa H, Fukuda S, Fujishima Y, Obata Y, et al. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci Rep. 2019;9(1):16. https://doi.org/10.1038/s41598-018-37115-3.
Singh AK, Shree S, Chattopadhyay S, Kumar S, Gurjar A, Kushwaha S, et al. Small molecule adiponectin receptor agonist GTDF protects against skeletal muscle atrophy. Mol Cell Endocrinol. 2017;439:273–85. https://doi.org/10.1016/j.mce.2016.09.013.
Abou-Samra M, Lecompte S, Schakman O, Noel L, Many MC, Gailly P, et al. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skeletal Muscle. 2015;5(1):25. https://doi.org/10.1186/s13395-015-0051-9.
Ito R, Higa M, Goto A, Aoshima M, Ikuta A, Ohashi K, et al. Activation of adiponectin receptors has negative impact on muscle mass in C2C12 myotubes and fast-type mouse skeletal muscle. PLoS One. 2018;13(10):e0205645. https://doi.org/10.1371/journal.pone.0205645.
Rennie MJ, Wackerhage H, Spangenburg EE, Booth FW. Control of the size of the human muscle mass. Annu Rev Physiol. 2004;66(1):799–828. https://doi.org/10.1146/annurev.physiol.66.052102.134444.
Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr. 2012;52(5):373–81. https://doi.org/10.1080/10408398.2010.500245.
Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology. 2006;21(5):362–9. https://doi.org/10.1152/physiol.00024.2006.
Krause MP, Milne KJ, Hawke TJ. Adiponectin-consideration for its role in skeletal muscle health. Int J Mol Sci. 2019;20(7):1528. https://doi.org/10.3390/ijms20071528.
Guerra B, Santana A, Fuentes T, Delgado-Guerra S, Cabrera-Socorro A, Dorado C et al. Leptin receptors in human skeletal muscle. Journal of applied physiology (Bethesda, Md : 1985). 2007;102(5):1786–92. https://doi.org/10.1152/japplphysiol.01313.2006.
Harris RBS. Leptin—much more than a satiety signal. Annu Rev Nutr. 2000;20(1):45–75. https://doi.org/10.1146/annurev.nutr.20.1.45.
Park HK, Ahima RS. Physiology of leptin: energy homeostasis, neuroendocrine function and metabolism. Metabolism. 2015;64(1):24–34. https://doi.org/10.1016/j.metabol.2014.08.004.
Cui H, López M, Rahmouni K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nature Reviews Endocrinology. 2017;13(6):338–51. https://doi.org/10.1038/nrendo.2016.222.
Izquierdo AG, Crujeiras AB, Casanueva FF, Carreira MC. Leptin, obesity, and leptin resistance: where are we 25 years later? Nutrients. 2019;11(11):2704. https://doi.org/10.3390/nu11112704.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334(5):292–5. https://doi.org/10.1056/nejm199602013340503.
Liuzzi A, Savia G, Tagliaferri M, Lucantoni R, Berselli ME, Petroni ML, et al. Serum leptin concentration in moderate and severe obesity: relationship with clinical, anthropometric and metabolic factors. Int J Obes Relat Metab Disord. 1999;23(10):1066–73. https://doi.org/10.1038/sj.ijo.0801036.
Schwartz MW, Prigeon RL, Kahn SE, Nicolson M, Moore J, Morawiecki A, et al. Evidence that plasma leptin and insulin levels are associated with body adiposity via different mechanisms. Diabetes Care. 1997;20(9):1476–81. https://doi.org/10.2337/diacare.20.9.1476.
Perry HM, Morley JE, Horowitz M, Kaiser FE, Miller DK, Wittert G. Body composition and age in african-american and caucasian women: relationship to plasma leptin levels. Metabolism. 1997;46(12):1399–405. https://doi.org/10.1016/S0026-0495(97)90138-4.
Rissanen P, Mäkimattila S, Vehmas T, Taavitsainen M, Rissanen A. Effect of weight loss and regional fat distribution on plasma leptin concentration in obese women. Int J Obes. 1999;23(6):645–9. https://doi.org/10.1038/sj.ijo.0800896.
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1(11):1155–61. https://doi.org/10.1038/nm1195-1155.
Havel PJ, Kasim-Karakas S, Mueller W, Johnson PR, Gingerich RL, Stern JS. Relationship of plasma leptin to plasma insulin and adiposity in normal weight and overweight women: effects of dietary fat content and sustained weight loss. J Clin Endocrinol Metab. 1996;81(12):4406–13. https://doi.org/10.1210/jcem.81.12.8954050.
Thong FSL, Hudson R, Ross R, Janssen I, Graham TE. Plasma leptin in moderately obese men: independent effects of weight loss and aerobic exercise. American Journal of Physiology-Endocrinology and Metabolism. 2000;279(2):E307–E13. https://doi.org/10.1152/ajpendo.2000.279.2.E307.
Kamikubo Y, Dellas C, Loskutoff DJ, Quigley JP, Ruggeri ZM. Contribution of leptin receptor N-linked glycans to leptin binding. Biochem J. 2008;410(3):595–604. https://doi.org/10.1042/bj20071137.
Bjørbaek C, Kahn BB. Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res. 2004;59:305–31. https://doi.org/10.1210/rp.59.1.305.
Maroni P, Bendinelli P, Piccoletti R. Early intracellular events induced by in vivo leptin treatment in mouse skeletal muscle. Mol Cell Endocrinol. 2003;201(1–2):109–21. https://doi.org/10.1016/s0303-7207(02)00427-6.
Maroni P, Bendinelli P, Piccoletti R. Intracellular signal transduction pathways induced by leptin in C2C12 cells. Cell Biol Int. 2005;29(7):542–50. https://doi.org/10.1016/j.cellbi.2005.03.008.
Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1(12):1311–4. https://doi.org/10.1038/nm1295-1311.
Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20(11):1341–5. https://doi.org/10.1016/s0196-9781(99)00139-4.
El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105(12):1827–32. https://doi.org/10.1172/jci9842.
Wilsey J, Scarpace PJ. Caloric restriction reverses the deficits in leptin receptor protein and leptin signaling capacity associated with diet-induced obesity: role of leptin in the regulation of hypothalamic long-form leptin receptor expression. J Endocrinol. 2004;181(2):297–306. https://doi.org/10.1677/joe.0.1810297.
Scarpace PJ, Matheny M, Tümer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104(4):1111–7. https://doi.org/10.1016/s0306-4522(01)00142-7.
Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52(15):1201–10. https://doi.org/10.1016/j.jacc.2008.05.060.
Guadalupe-Grau A, Larsen S, Guerra B, Calbet JAL, Dela F, Helge JW. Influence of age on leptin induced skeletal muscle signalling. Acta Physiologica. 2014;211(1):214–28. https://doi.org/10.1111/apha.12273.
Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature. 1998;393(6686):684–8. https://doi.org/10.1038/31474.
Fernández-Real JM, Vayreda M, Casamitjana R, Gonzalez-Huix F, Ricart W. The fat-free mass compartment influences serum leptin in men. Eur J Endocrinol. 2000;142(1):25–9. https://doi.org/10.1530/eje.0.1420025.
Wolsk E, Mygind H, Grøndahl TS, Pedersen BK, van Hall G. Human skeletal muscle releases leptin in vivo. Cytokine. 2012;60(3):667–73. https://doi.org/10.1016/j.cyto.2012.08.021.
Guerra B, Ponce-González JG, Morales-Alamo D, Guadalupe-Grau A, Kiilerich K, Fuentes T, et al. Leptin signaling in skeletal muscle after bed rest in healthy humans. Eur J Appl Physiol. 2014;114(2):345–57. https://doi.org/10.1007/s00421-013-2779-4.
Zhang J, Wu Y, Zhang Y, LeRoith D, Bernlohr DA, Chen X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol. 2008;22(6):1416–26. https://doi.org/10.1210/me.2007-0420.
Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268(14):10425–32.
Yan QW, Yang Q, Mody N, Graham TE, Hsu CH, Xu Z, et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56(10):2533–40. https://doi.org/10.2337/db07-0007.
Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123(7):1293–305. https://doi.org/10.1016/j.cell.2005.10.027.
Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281(34):24138–48. https://doi.org/10.1074/jbc.M604597200.
Liu X, Hamnvik OP, Petrou M, Gong H, Chamberland JP, Christophi CA, et al. Circulating lipocalin 2 is associated with body fat distribution at baseline but is not an independent predictor of insulin resistance: the prospective Cyprus Metabolism Study. Eur J Endocrinol. 2011;165(5):805–12. https://doi.org/10.1530/eje-11-0660.
Wang Y, Lam KSL, Kraegen EW, Sweeney G, Zhang J, Tso AW, et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53(1):34–41. https://doi.org/10.1373/clinchem.2006.075614.
Bouloumié A, Sengenès C, Portolan G, Galitzky J, Lafontan M. Adipocyte produces matrix metalloproteinases 2 and 9: involvement in adipose differentiation. Diabetes. 2001;50(9):2080–6. https://doi.org/10.2337/diabetes.50.9.2080.
Christiaens V, Scroyen I, Lijnen HR. Role of proteolysis in development of murine adipose tissue. Thromb Haemost. 2008;99(2):290–4. https://doi.org/10.1160/th07-10-0589.
Berg G, Barchuk M, Miksztowicz V. Behavior of metalloproteinases in adipose tissue, liver and arterial wall: an update of extracellular matrix remodeling. Cells. 2019;8(2):158. https://doi.org/10.3390/cells8020158.
Mosialou I, Shikhel S, Luo N, Petropoulou PI, Panitsas K, Bisikirska B, et al. Lipocalin-2 counteracts metabolic dysregulation in obesity and diabetes. J Exp Med. 2020;217(10). https://doi.org/10.1084/jem.20191261.
Meyers K, López M, Ho J, Wills S, Rayalam S, Taval S. Lipocalin-2 deficiency may predispose to the progression of spontaneous age-related adiposity in mice. Sci Rep. 2020;10(1):14589. https://doi.org/10.1038/s41598-020-71249-7.
Ishii A, Katsuura G, Imamaki H, Kimura H, Mori KP, Kuwabara T, et al. Obesity-promoting and anti-thermogenic effects of neutrophil gelatinase-associated lipocalin in mice. Sci Rep. 2017;7(1):15501. https://doi.org/10.1038/s41598-017-15825-4.
Guo H, Jin D, Zhang Y, Wright W, Bazuine M, Brockman DA, et al. Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes. 2010;59(6):1376–85. https://doi.org/10.2337/db09-1735.
Law IK, Xu A, Lam KS, Berger T, Mak TW, Vanhoutte PM, et al. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes. 2010;59(4):872–82. https://doi.org/10.2337/db09-1541.
Choi KM, Kim TN, Yoo HJ, Lee KW, Cho GJ, Hwang TG, et al. Effect of exercise training on A-FABP, lipocalin-2 and RBP4 levels in obese women. Clin Endocrinol (Oxf). 2009;70(4):569–74. https://doi.org/10.1111/j.1365-2265.2008.03374.x.
Damirchi A, Rahmani-Nia F, Mehrabani J. Lipocalin-2: response to a progressive treadmill protocol in obese and normal-weight men. Asian J Sports Med. 2011;2(1):44–50. https://doi.org/10.5812/asjsm.34821.
de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15(1):1–11.
Breitbart A, Auger-Messier M, Molkentin JD, Heineke J. Myostatin from the heart: local and systemic actions in cardiac failure and muscle wasting. Am J Physiol Heart Circ Physiol. 2011;300(6):H1973–82. https://doi.org/10.1152/ajpheart.00200.2011.
Chen Y, Mironova E, Whitaker LL, Edwards L, Yost HJ, Ramsdell AF. ALK4 functions as a receptor for multiple TGF beta-related ligands to regulate left-right axis determination and mesoderm induction in Xenopus. Dev Biol. 2004;268(2):280–94. https://doi.org/10.1016/j.ydbio.2003.12.035.
Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, et al. Activin signaling as an emerging target for therapeutic interventions. Cell communication and signaling : CCS. 2009;7:15. https://doi.org/10.1186/1478-811x-7-15.
Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol. 2011;12(6):349–61. https://doi.org/10.1038/nrm3118.
Tsao J, Vernet DA, Gelfand R, Kovanecz I, Nolazco G, Bruhn KW, et al. Myostatin genetic inactivation inhibits myogenesis by muscle-derived stem cells in vitro but not when implanted in the mdx mouse muscle. Stem Cell Res Ther. 2013;4(1):4. https://doi.org/10.1186/scrt152.
Artaza JN, Bhasin S, Magee TR, Reisz-Porszasz S, Shen R, Groome NP, et al. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multipotent cells. Endocrinology. 2005;146(8):3547–57. https://doi.org/10.1210/en.2005-0362.
Leger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation research. 2008;11(1):163-75b. https://doi.org/10.1089/rej.2007.0588.
Allen DL, Hittel DS, McPherron AC. Expression and function of myostatin in obesity, diabetes, and exercise adaptation. Med Sci Sports Exerc. 2011;43(10):1828–35. https://doi.org/10.1249/MSS.0b013e3182178bb4.
Milan G, Dalla Nora E, Pilon C, Pagano C, Granzotto M, Manco M, et al. Changes in muscle myostatin expression in obese subjects after weight loss. J Clin Endocrinol Metab. 2004;89(6):2724–7. https://doi.org/10.1210/jc.2003-032047.
Ratkevicius A, Joyson A, Selmer I, Dhanani T, Grierson C, Tommasi AM, et al. Serum concentrations of myostatin and myostatin-interacting proteins do not differ between young and sarcopenic elderly men. J Gerontol A Biol Sci Med Sci. 2011;66(6):620–6. https://doi.org/10.1093/gerona/glr025.
Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998;95(25):14938–43.
Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. Journal of cachexia, sarcopenia and muscle. 2011;2(3):143–51. https://doi.org/10.1007/s13539-011-0035-5.
Louis E, Raue U, Yang Y, Jemiolo B, Trappe S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J Appl Physiol. 2007;103(5):1744–51. https://doi.org/10.1152/japplphysiol.00679.2007.
Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc. 2010;42(11):2023–9. https://doi.org/10.1249/MSS.0b013e3181e0b9a8.
MacKenzie MG, Hamilton DL, Pepin M, Patton A, Baar K. Inhibition of myostatin signaling through Notch activation following acute resistance exercise. PLoS One. 2013;8(7):e68743-e. https://doi.org/10.1371/journal.pone.0068743.
Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, et al. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284(4):E741–7. https://doi.org/10.1152/ajpendo.00514.2002.
Allen DL, Cleary AS, Speaker KJ, Lindsay SF, Uyenishi J, Reed JM, et al. Myostatin, activin receptor IIb, and follistatin-like-3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am J Physiol Endocrinol Metab. 2008;294(5):E918–27. https://doi.org/10.1152/ajpendo.00798.2007.
Kammoun M, Cassar-Malek I, Meunier B, Picard B. A simplified immunohistochemical classification of skeletal muscle fibres in mouse. European journal of histochemistry : EJH. 2014;58(2):2254. https://doi.org/10.4081/ejh.2014.2254.
Carlson CJ, Booth FW, Gordon SE. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. Am J Physiol. 1999;277(2 Pt 2):R601–6. https://doi.org/10.1152/ajpregu.1999.277.2.r601.
Amor M, Itariu BK, Moreno-Viedma V, Keindl M, Jurets A, Prager G, et al. Serum myostatin is upregulated in obesity and correlates with insulin resistance in humans. Exp Clin Endocrinol Diabetes. 2019;127(8):550–6. https://doi.org/10.1055/a-0641-5546.
Baboota RK, Sarma SM, Boparai RK, Kondepudi KK, Mantri S, Bishnoi M. Microarray based gene expression analysis of murine brown and subcutaneous adipose tissue: significance with human. PLoS One. 2015;10(5):e0127701. https://doi.org/10.1371/journal.pone.0127701.
Zuriaga MA, Fuster JJ, Gokce N, Walsh K. Humans and mice display opposing patterns of “browning” gene expression in visceral and subcutaneous white adipose tissue depots. Frontiers in Cardiovascular Medicine. 2017;4(27). https://doi.org/10.3389/fcvm.2017.00027.
McPherron AC, Lee SJ. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest. 2002;109(5):595–601. https://doi.org/10.1172/jci13562.
Arrieta H, Hervás G, Rezola-Pardo C, Ruiz-Litago F, Iturburu M, Yanguas JJ, et al. Serum myostatin levels are higher in fitter, more active, and non-frail long-term nursing home residents and increase after a physical exercise intervention. Gerontology. 2019;65(3):229–39. https://doi.org/10.1159/000494137.
Mattick JS. The central role of RNA in human development and cognition. FEBS Lett. 2011;585(11):1600–16. https://doi.org/10.1016/j.febslet.2011.05.001.
Pertea M. The human transcriptome: an unfinished story. Genes (Basel). 2012;3(3):344–60. https://doi.org/10.3390/genes3030344.
Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–69. https://doi.org/10.1534/genetics.112.146704.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10(3):155–9. https://doi.org/10.1038/nrg2521.
Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14(2):103–5. https://doi.org/10.1038/nsmb0207-103.
Wang Kevin C, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–14. https://doi.org/10.1016/j.molcel.2011.08.018.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(suppl_1):R17–29. https://doi.org/10.1093/hmg/ddl046.
Nanus DE, Wijesinghe SN, Pearson MJ, Hadjicharalambous MR, Rosser A, Davis ET, et al. Regulation of the inflammatory synovial fibroblast phenotype by metastasis-associated lung adenocarcinoma transcript 1 long noncoding RNA in obese patients with osteoarthritis. Arthritis & Rheumatology. 2020;72(4):609–19. https://doi.org/10.1002/art.41158.
Wijesinghe SN, Nicholson T, Tsintzas K, Jones SW. Involvements of long noncoding RNAs in obesity-associated inflammatory diseases. Obes Rev.n/a(n/a). https://doi.org/10.1111/obr.13156.
Gao H, Kerr A, Jiao H, Hon C-C, Rydén M, Dahlman I, et al. Long non-coding RNAs associated with metabolic traits in human white adipose tissue. EBioMedicine. 2018;30:248–60. https://doi.org/10.1016/j.ebiom.2018.03.010.
Alessio E, Buson L, Chemello F, Peggion C, Grespi F, Martini P, et al. Single cell analysis reveals the involvement of the long non-coding RNA Pvt1 in the modulation of muscle atrophy and mitochondrial network. Nucleic Acids Res. 2019;47(4):1653–70. https://doi.org/10.1093/nar/gkz007.
Liu M, Li B, Peng W, Ma Y, Huang Y, Lan X, et al. LncRNA-MEG3 promotes bovine myoblast differentiation by sponging miR-135. J Cell Physiol. 2019;234(10):18361–70. https://doi.org/10.1002/jcp.28469.
Dey BK, Pfeifer K, Dutta A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014;28(5):491–501. https://doi.org/10.1101/gad.234419.113.
Schmidt E, Dhaouadi I, Gaziano I, Oliverio M, Klemm P, Awazawa M, et al. LincRNA H19 protects from dietary obesity by constraining expression of monoallelic genes in brown fat. Nature Communications. 2018;9(1):3622. https://doi.org/10.1038/s41467-018-05933-8.
Watts R, Johnsen VL, Shearer J, Hittel DS. Myostatin-induced inhibition of the long noncoding RNA Malat1 is associated with decreased myogenesis. Am J Physiol Cell Physiol. 2013;304(10):C995–1001. https://doi.org/10.1152/ajpcell.00392.2012.
Schirwis E, Agbulut O, Vadrot N, Mouisel E, Hourdé C, Bonnieu A, et al. The beneficial effect of myostatin deficiency on maximal muscle force and power is attenuated with age. Exp Gerontol. 2013;48(2):183–90. https://doi.org/10.1016/j.exger.2012.11.008.
Kim J, Kim KM, Noh JH, Yoon J-H, Abdelmohsen K, Gorospe M. Long noncoding RNAs in diseases of aging. Biochim Biophys Acta. 2016;1859(1):209–21. https://doi.org/10.1016/j.bbagrm.2015.06.013.
Xing D, Liang JQ, Li Y, Lu J, Jia HB, Xu LY, et al. Identification of long noncoding RNA associated with osteoarthritis in humans. Orthop Surg. 2014;6(4):288–93. https://doi.org/10.1111/os.12147.
Hitachi K, Nakatani M, Funasaki S, Hijikata I, Maekawa M, Honda M, et al. Expression levels of long non-coding RNAs change in models of altered muscle activity and muscle mass. Int J Mol Sci. 2020;21(5):1628. https://doi.org/10.3390/ijms21051628.
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–9. https://doi.org/10.1261/rna.2183803.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. https://doi.org/10.1016/0092-8674(93)90529-y.
Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–62. https://doi.org/10.1126/science.1065062.
Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294(5543):862–4. https://doi.org/10.1126/science.1065329.
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. https://doi.org/10.1038/cr.2008.282.
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proceedings of the National Academy of Sciences. 2011;108(12):5003–8. https://doi.org/10.1073/pnas.1019055108.
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9. https://doi.org/10.1038/ncb1596.
Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119(3):756–66. https://doi.org/10.1182/blood-2011-02-338004.
O'Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9(402). https://doi.org/10.3389/fendo.2018.00402.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nature Reviews Molecular Cell Biology. 2014;15(8):509–24. https://doi.org/10.1038/nrm3838.
Yang JS, Lai EC. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell. 2011;43(6):892–903. https://doi.org/10.1016/j.molcel.2011.07.024.
Yanai K, Kaneko S, Ishii H, Aomatsu A, Ito K, Hirai K, et al. MicroRNAs in sarcopenia: a systematic review. Frontiers in Medicine. 2020;7(180). https://doi.org/10.3389/fmed.2020.00180.
Brown DM, Goljanek-Whysall K. microRNAs: Modulators of the underlying pathophysiology of sarcopenia? Ageing research reviews. 2015;24:263–73. https://doi.org/10.1016/j.arr.2015.08.007.
He N, Zhang YL, Zhang Y, Feng B, Zheng Z, Wang D, et al. Circulating microRNAs in plasma decrease in response to sarcopenia in the elderly. Frontiers in Genetics. 2020;11(167). https://doi.org/10.3389/fgene.2020.00167.
Kim JY, Park Y-K, Lee K-P, Lee S-M, Kang T-W, Kim H-J, et al. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging. 2014;6(7):524–44. https://doi.org/10.18632/aging.100677.
Heneghan HM, Miller N, Kerin MJ. Role of microRNAs in obesity and the metabolic syndrome. Obes Rev. 2010;11(5):354–61. https://doi.org/10.1111/j.1467-789X.2009.00659.x.
Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. The FEBS journal. 2009;276(8):2348–58. https://doi.org/10.1111/j.1742-4658.2009.06967.x.
Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, et al. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279(50):52361–5. https://doi.org/10.1074/jbc.C400438200.
Xie H, Lim B, Lodish HF. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes. 2009;58(5):1050–7. https://doi.org/10.2337/db08-1299.
Iacomino G, Siani A. Role of microRNAs in obesity and obesity-related diseases. Genes Nutr. 2017;12(1):23. https://doi.org/10.1186/s12263-017-0577-z.
Ji C, Guo X. The clinical potential of circulating microRNAs in obesity. Nature Reviews Endocrinology. 2019;15(12):731–43. https://doi.org/10.1038/s41574-019-0260-0.
Wang Y-c, Li Y, Wang X-y, Zhang D, Zhang H, Wu Q, et al. Circulating miR-130b mediates metabolic crosstalk between fat and muscle in overweight/obesity. Diabetologia. 2013;56(10):2275–85. https://doi.org/10.1007/s00125-013-2996-8.
Corona-Meraz F-I, Vázquez-Del Mercado M, Ortega FJ, Ruiz-Quezada S-L, Guzmán-Ornelas M-O, Navarro-Hernández R-E. Ageing influences the relationship of circulating miR-33a and miR-33b levels with insulin resistance and adiposity. Diabetes and Vascular Disease Research. 2019;16(3):244–53. https://doi.org/10.1177/1479164118816659.
Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23(10):3335–46. https://doi.org/10.1096/fj.08-128579.
Goedeke L, Vales-Lara FM, Fenstermaker M, Cirera-Salinas D, Chamorro-Jorganes A, Ramírez CM, et al. A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol Cell Biol. 2013;33(11):2339–52. https://doi.org/10.1128/MCB.01714-12.
Näär AM. miR-33: a metabolic conundrum. Trends in endocrinology and metabolism: TEM. 2018;29(10):667–8. https://doi.org/10.1016/j.tem.2018.04.004.
Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. 2013;4:2883. https://doi.org/10.1038/ncomms3883.
Price NL, Singh AK, Rotllan N, Goedeke L, Wing A, Canfrán-Duque A, et al. Genetic ablation of mir-33 increases food intake, enhances adipose tissue expansion, and promotes obesity and insulin resistance. Cell Rep. 2018;22(8):2133–45. https://doi.org/10.1016/j.celrep.2018.01.074.
Li X, Qiu J, Liu H, Deng Y, Hu S, Hu J, et al. MicroRNA-33a negatively regulates myoblast proliferation by targeting IGF1, follistatin and cyclin D1. Biosci Rep. 2020;40(6):BSR20191327. https://doi.org/10.1042/BSR20191327.
Güller I, Russell AP. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. The Journal of physiology. 2010;588(Pt 21):4075–87. https://doi.org/10.1113/jphysiol.2010.194175.
Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem Sci. 2007;32(4):189–97. https://doi.org/10.1016/j.tibs.2007.02.006.
Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436(7048):214–20. https://doi.org/10.1038/nature03817.
Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci U S A. 2006;103(23):8721–6. https://doi.org/10.1073/pnas.0602831103.
Mori Marcelo A, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012;16(3):336–47. https://doi.org/10.1016/j.cmet.2012.07.017.
Frias FT, de Mendonça M, Martins AR, Gindro AF, Cogliati B, Curi R, et al. MyomiRs as markers of insulin resistance and decreased myogenesis in skeletal muscle of diet-induced obese mice. Front Endocrinol (Lausanne). 2016;7:76. https://doi.org/10.3389/fendo.2016.00076.
de Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JWA, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. 2017;7(1):47. https://doi.org/10.1038/s41598-017-00070-6.
Iacomino G, Russo P, Stillitano I, Lauria F, Marena P, Ahrens W, et al. Circulating microRNAs are deregulated in overweight/obese children: preliminary results of the I.Family study. Genes Nutr. 2016;11(1):7. https://doi.org/10.1186/s12263-016-0525-3.
Oliveira MS, Rheinheimer J, Moehlecke M, Rodrigues M, Assmann TS, Leitão CB, et al. UCP2, IL18, and miR-133a-3p are dysregulated in subcutaneous adipose tissue of patients with obesity. Mol Cell Endocrinol. 2020;509:110805. https://doi.org/10.1016/j.mce.2020.110805.
Chartoumpekis DV, Zaravinos A, Ziros PG, Iskrenova RP, Psyrogiannis AI, Kyriazopoulou VE, et al. Differential expression of microRNAs in adipose tissue after long-term high-fat diet-induced obesity in mice. PLoS One. 2012;7(4):e34872. https://doi.org/10.1371/journal.pone.0034872.
Funding
PhD Studentship (AW) funded by the MRC Versus Arthritis Centre for Musculoskeletal Ageing Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wilhelmsen, A., Tsintzas, K. & Jones, S.W. Recent advances and future avenues in understanding the role of adipose tissue cross talk in mediating skeletal muscle mass and function with ageing. GeroScience 43, 85–110 (2021). https://doi.org/10.1007/s11357-021-00322-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00322-4