
Abstract
Aims/hypothesis
Debate exists regarding the role of resistin in the pathophysiology of insulin resistance. The aim of this study was to directly assess the effects of resistin (0–24 h) on basal and insulin-stimulated glucose uptake and metabolism in skeletal muscle cells and to investigate the mechanisms responsible for the effects of resistin.
Methods
We used L6 rat skeletal muscle cells and examined [3H]2-deoxyglucose uptake, GLUT4 translocation and GLUT protein content. We assessed glucose metabolism by measuring the incorporation of D-[U-14C]glucose into glycogen, 14CO2 and lactate production, as well as the phosphorylation level and total protein content of insulin signalling proteins, including insulin receptor β-subunit (IRβ), insulin receptor substrate (IRS), Akt and glycogen synthase kinase-3β (GSK-3β).
Results
Treatment of L6 rat skeletal muscle cells with recombinant resistin (50 nmol/l, 0–24 h) reduced levels of basal and insulin-stimulated 2-deoxyglucose uptake and decreased insulin-stimulated GLUT4myc content at the cell surface, with no alteration in the production of GLUT4 or GLUT1. Resistin also decreased glycogen synthesis and GSK-3β phosphorylation. Insulin-stimulated oxidation of glucose via the Krebs cycle was reduced by resistin, whereas lactate production was unaltered. Although insulin receptor protein level and phosphorylation were unaltered by resistin, production of IRS-1, but not IRS-2, was downregulated and a decreased tyrosine phosphorylation of IRS-1 was detected. Reduced phosphorylation of Akt on T308 and S473 was observed, while total Akt and Akt1, but not Akt2 or Akt3, production was decreased.
Conclusions/interpretation
Our data show that resistin regulates the function of IRS-1 and Akt1 and decreases GLUT4 translocation and glucose uptake in response to insulin. Selective decreases in insulin-stimulated glucose metabolism via oxidation and conversion to glycogen were also induced by resistin. These observations highlight the potential role of resistin in the pathophysiology of type 2 diabetes in obesity.
Similar content being viewed by others


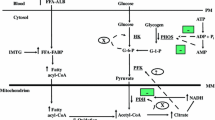
Introduction
Several years of capricious interest in the link between resistin and diabetes [1] have been enhanced by recent developments such as the generation of mice lacking or overexpressing resistin [2–5]. Resistin was discovered by virtue of its altered gene expression in mouse adipocytes in response to the peroxisome proliferator-activated receptor-γ ligands known as thiazolidinediones [6]. Addition of the recombinant protein to normal mice or cultured adipocytes impaired insulin action and neutralization of resistin function with anti-resistin antibody improved insulin action in mice with diet-induced obesity [6]. Plasma resistin levels are increased in ob/ob, db/db and diet-induced obese mice [6], although, concomitantly, resistin mRNA levels in obese rodents are often found to be decreased [7–9]. It is now appreciated that there is often a discrepancy between the circulating protein levels of resistin and the mRNA content in adipocytes [10, 11].
The gene encoding resistin produces a secreted protein consisting of 94 amino acids. The pathophysiological role of resistin in humans has been questioned because the human homologue of resistin is only 59% identical to mouse resistin at the amino acid level and the source of resistin appears to differ in humans [6, 12]. Resistin protein is almost undetectable in human adipocytes [13] and instead, macrophages appear to be the principal source [14]. This reinforces the need to view adipose tissue, rather than simply adipocytes, as a dynamic endocrine organ and to be aware of the importance of resistin (also called ‘found in inflammatory zone 3’, FIZZ3) in the low-grade inflammatory condition associated with obesity [15]. Nevertheless, despite the species differences outlined above, an increased plasma resistin concentration was observed in the serum of obese [16] or type 2 diabetics [17] and thiazolidinedione treatment resulted in decreased plasma resistin levels in patients with type 2 diabetes [18]. Furthermore, although some studies suggest a link between resistin and insulin sensitivity [19] others have failed to show an association with insulin resistance [16, 20, 21]. Similarly, genetic analyses have suggested that resistin plays a role in the aetiology of insulin resistance and diabetes [22, 23], whereas others contest this view [24].
The aim of this study was to assess the ability of recombinant resistin to directly alter glucose uptake and metabolism in skeletal muscle cells and to investigate the mechanisms responsible for the effect of resistin. This work, which involves various periods of resistin treatment (0–24 h), follows up our initial work examining the effects of acute resistin treatment [25].
Materials and methods
Materials
The cell culture medium (α-minimal essential medium [α-MEM]), foetal bovine serum (FBS) and antibiotics/antimycotic solution were purchased from Wisent (St Bruno, QC, Canada). Cytochalasin B, glycogen and O-phenylenediamine di-hydrochloride (OPD), triethanolamine were purchased from Sigma (St Louis, MO, USA). Human insulin (Humulin) was from Eli Lilly (Toronto, ON, Canada) and d-[U–14C]glucose and deoxy-d-[2-3H]glucose were from Amersham (QC, Canada). Recombinant murine resistin was used in this study and was purchased from Peprotech (Ottawa, ON, Canada). Anti-myc antibody (9E10) and insulin receptor-α subunit and insulin receptor substrate-1 and -2 (IRS-1 and IRS-2) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Lactacystin was obtained from Calbiochem (La Jolla, CA, USA). Insulin receptor-β subunit (Ab-6) and glycogen synthase kinase-3β (GSK-3β) antibody and phospho-specific antibodies for insulin receptor β-subunit (IRβ; Y972), IRS-1 (Y612 and S616) were purchased from Biosource (Montreal, QC, Canada). Antibodies against total Akt, Akt1, Akt2 and Akt3 were from Oncogene Research Products (La Jolla, CA, USA). Polyclonal phosphospecific antibodies to Akt (T308 and S473), IRS-1 (S307) and GSK-3β (S-9) were from Cell Signaling Technology (Beverly, MA, USA). Antibodies to glucose transporter 1 (GLUT1) and GLUT4 were obtained from Biogenesis (Poole, UK). Horseradish peroxidase-conjugated secondary antibodies were purchased as follows: anti-rabbit-IgG (Cell Signaling Technology), anti-goat IgG (Calbiochem) and anti-mouse IgG (Bio-Rad, Mississauga, ON, Canada). All other reagents used were of the highest purity available.
Cell culture
L6-GLUT4myc myoblasts (a kind gift from A. Klip, The Hospital for Sick Children, Toronto, ON, Canada) were cultured as described previously [26] in α-MEM supplemented with 10% (v/v) FBS and 1% antibiotic/antimycotic solution (100 units/ml penicillin, 100 μg/ml streptomycin, 250 ng/ml amphotericin B) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were passaged before reaching confluence. The standard growth medium was changed every 2 days.
Glucose uptake
Cells were cultivated in 24-well plates with or without resistin for periods of 0–24 h and were serum starved for 3–5 h then incubated, in the continued presence of resistin, for different times (0–20 min) with or without insulin as indicated in the figure legend. Subsequently, glucose transport was assayed for 5 min at room temperature as previously described [27]. The incubation medium was aspirated, the cells were washed with ice-cold saline, and 200 μl of KOH (1 mol/l) was added to each well. Aliquots of cell lysates were transferred to scintillation vials for radioactivity counting and the remainder were used for protein assay. Non-specific uptake was determined in the presence of cytochalasin B (10 μmol/l) and was subtracted from all values. Results are calculated as pmol of glucose uptake per min per mg protein.
Determination of cell surface GLUT4-myc
The amount of GLUT4-myc present at the cell surface was measured by an antibody-coupled colorimetric assay as described previously [27]. The L6GLUT4-myc cells are stably transfected to express GLUT4 tagged on its first exofacial loop with a myc epitope. The exofacial placement of the myc epitope on GLUT4 allows the analysis of GLUT4 localisation in intact cells. Briefly, L6 myoblasts were grown in 12-well plates in the presence or absence of resistin (50 nmol/l) for 24 h followed by the 3–5 h of serum starvation in the continued presence of resistin. Cells were then treated with or without insulin (100 nmol/l) for 20 min. Subsequently, cells were quickly washed in ice-cold PBS and incubated with anti-c-myc antibody (9E10, 1:200 dilution) for 60 min at 4°C. Cells were washed and fixed in 3% paraformaldehyde for 3 min on ice. The fixative was neutralised by incubation in 10 mmol/l glycine in ice-cold PBS for 10 min. Cells were incubated in 10% goat serum for 10 min and then with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000 dilution, 4°C) for 60 min. Cells were washed five times with ice-cold PBS and incubated for 30 min at room temperature with 1 ml of OPD reagent per well. The reaction was stopped by adding 0.25 ml of HCl (3 mol/l) and supernatant absorbance was measured at 492 nm. Absorbance associated with non-specific binding (primary antibody omitted) was used as a blank and was subtracted from all values.
Glycogen synthesis
Glycogen synthesis was measured by the incorporation of d-[U–14C]glucose to glycogen as we described previously [28] with a few modifications. Briefly, cells were grown in six-well plates and pre-incubated with or without resistin (50 nmol/l) for 0–24 h and deprived of serum for 3–5 h before incubation with 5.55 kBq/ml d-[U–14C]glucose in the presence or absence of insulin (100 nmol/l) for 2 h. The cells were washed three times with cold PBS and lysed in 1 mol/l KOH. To measure insulin-stimulated incorporation of glucose into glycogen, cell lysates were used for overnight glycogen precipitation with ethanol. Precipitated glycogen was then dissolved in water and transferred to scintillation vials for radioactivity counting.
Glucose oxidation
Glucose oxidation was measured by the production of 14CO2 from d-[U–14C]glucose as previously described [28]. Briefly, cells were grown in 60 × 15 mm Petri dishes and pretreated with or without resistin (50 nmol/l) for 0–24 h followed by serum starvation. Cells were then incubated with medium containing 5.55 kBq/ml d-[U–14C]glucose with or without insulin for 2 h. Each Petri dish was sealed with parafilm containing a piece of Whatman paper attached to the inside. The Whatman paper was wet with 100 μl of phenylethylamine-methanol (1:1) to trap CO2 produced during the incubation period. After 2 h of incubation, 200 μl of H2SO4 (4 mol/l) was added, followed by further incubation for 1 h at 37°C. Finally, the pieces of Whatman paper were removed and transferred to scintillation vials for radioactivity counting.
Lactate assay
Lactate content was determined by the lactate oxidase method using a lactate assay kit (Sigma). Cells were incubated with resistin (50 nmol/l) for 24 h then serum starved for 3–5 h in the continued presence of resistin, before treatment with or without insulin for 2 h, after which the media was collected and used for analysis.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide assay
Cell viability was determined using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich, St Louis, MO). Briefly, cells were seeded at a density of 1 × 106 cells/ml in 96-well plates and incubated in the presence or absence of resistin (50 nmol/l) for 24 h. MTT was then added and the ability of cells to reduce this substrate to the blue formazan product was determined colorimetrically (550 nm) as an indicator of metabolically active cells.
Western blotting
To prepare lysates for immunoblotting, cells were grown in six-well plates and treated with resistin (50 nmol/l, 0–24 h) or after 24 h in the presence or absence of resistin (50 nmol/l), they were incubated for the times indicated (0–30 min) with or without insulin. We also treated cells with lactacystin (10 μmol/l, 1 h pretreatment) to prepare lysates for examination of IRS-1 or Akt1 protein content. Lysates were prepared exactly as we previously described [27]. Prior to loading onto SDS-PAGE gels, the samples were diluted 1:1 (v/v) with 2 × Laemmli sample buffer (62.5 mmol/l Tris–HCl [pH 6.8], 2% [w/v] SDS, 50 mmol/l dithiothreitol, 0.01% [w/v] bromophenol blue). Equal amounts of muscle proteins (30 μg) were resolved by SDS-PAGE (8–10%), and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Burlington, ON, Canada). Membranes were probed with primary antibodies against proteins of interest (phospho GSK-3β at 1:3,000, all Akt antibodies, IRS-1, IRS-2 and phospho IRS-1 at 1:1,000 dilution; GLUT4, phospho IRS Y612 and S616 1:2,000; GLUT1, insulin receptor α-subunit [IRα] and phospho IRβ 1:500; and IRβ and GSK-3β 1:200) and appropriate horseradish peroxidase-conjugated secondary antibodies (anti-rabbit at 1:10,000 dilution, goat 1:3,000 and mouse 1:3,000) were used in each case and detected by the enhanced chemiluminescence method.
Statistical analysis
Data are expressed as means±SEM. Statistical analysis was undertaken using one-way ANOVA or the paired Student's t-test where appropriate. Differences between groups were considered statistically significant when p<0.05.
Results
The results shown in Fig. 1a demonstrate that resistin decreased both basal and insulin-stimulated glucose uptake. An approximate 20% decrease in basal glucose uptake values was observed in cells treated for 24 h with resistin. Although insulin did increase glucose uptake above control in the presence of resistin the magnitude of the response was significantly attenuated (e.g. 1.75-fold in the absence and 1.44-fold in the presence of resistin upon 10 min stimulation with insulin). It is important to point out that cell viability was unaffected by resistin over this time period as assessed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay (control: 1.0±0.02; resistin (50 nmol/l, 24 h): 1.03±0.03). We also tested the effect of a range of resistin preincubation times (0–24 h) on basal and insulin-stimulated glucose uptake and found that a significant decrease was elicited by resistin after as little as 2 h (Fig. 1b). A trend towards partial recovery of the insulin response at later time-points was observed. Further analysis of the mechanism underlying the effect of resistin (24 h) on glucose uptake revealed that the ability of insulin to increase the amount of glucose transporter GLUT4 at the cell surface was prevented by resistin (Fig. 2a). Resistin treatment for 24 h did not alter the total content of either GLUT1 (0.92±0.1-fold relative to control) or GLUT4 (1.06±0.04-fold relative to control) in these cells (Fig. 2b).

Effect of resistin on glucose uptake. a Uptake of 2-deoxyglucose was measured under basal conditions (open circles) and in response to insulin (100 nmol/l, 0–20 min, closed squares) in the presence or absence of resistin (50 nmol/l, 24 h). Values are the means±SEM of n≥3. *p<0.05 vs control (in the absence of resistin). b The effect of resistin over various times (0–24 h) on basal (black bars) and insulin-stimulated (100 nmol/l, 20 min, white bars) glucose uptake. Values are the means±SEM of n≥3 experiments. *p<0.05 vs control (in the absence of resistin) and # p<0.05 vs insulin (in absence of resistin)

Effect of resistin on glucose transporter protein production and regulation. The amount of GLUT4 detected at the cell surface was measured in L6-GLUT4myc cells in response to insulin (100 nmol/l, 20 min) in the presence or absence of resistin (50 nmol/l, 24 h). Values shown in a are the means±SEM of n≥3 experiments. *p<0.05 vs control. b Representative immunoblots (n≥3 experiments) from analysis of total GLUT1 and GLUT4 protein content in cells grown under control conditions or in the presence of resistin (50 nmol/l, 24 h)
We also investigated the metabolism of glucose and found that both basal and insulin-stimulated glycogen synthesis were decreased in cells treated with resistin for between 0 and 24 h (Fig. 3a). There was a decrease in both the basal (0.79-fold relative to control) and insulin-stimulated (e.g. at 5 min insulin stimulation 1.56-fold and 1.29-fold above control in the absence and presence of resistin, respectively) level of GSK-3β phosphorylation in response to resistin (24 h, Fig. 3b) but there was no significant change in the total protein level of GSK-3β protein (0.99±0.09-fold relative to control) in these cells (Fig. 3c). Resistin reduced both basal and insulin-stimulated glucose oxidation after as little as 2 h and up to 24 h pretreatment (Fig. 4a). Insulin normally elicits a small but significant increase in lactate production and neither this effect of insulin nor basal levels of lactate production were affected by resistin (24 h, Fig. 4b).

Effect of resistin on regulation of glycogen synthesis. a The effect of resistin (50 nmol/l, 0–24 h) on basal (black bars) and insulin-stimulated (100 nmol/l, 2 h, white bars) incorporation of d-[U–14C]glucose into glycogen. Values are the means±SEM of n≥3 experiments. *p<0.05 vs control (in the absence of resistin); # p<0.05 vs insulin (in the absence of resistin). b, c Representative immunoblots (n≥3 experiments) from analysis of GSK-3β phosphorylation in response to insulin (100 nmol/l, 0–30 min) b and total GSK-3β protein content c in cells grown under control conditions (Con) or in the presence of resistin (Res; 50 nmol/l, 24 h)

Effect of resistin on glucose oxidation and lactate production. a The effect of resistin (50 nmol/l, 0–24 h) on basal (black bars) and insulin-stimulated (100 nmol/l, 2 h, white bars) 14CO2 production from d-[U–14C]glucose. b Lactate production under the different conditions. Values are the means±SEM of n≥3 experiments. *p<0.05 vs control
Having determined the effect of resistin on glucose uptake and metabolism, we next examined the effect of resistin on signalling pathways normally employed by insulin in the regulation of glucose uptake and metabolism. First, we found that resistin did not appear to mediate effects at the level of insulin receptor because neither the level of α and β subunit proteins (1.01±0.03-fold and 1.01±0.02-fold, respectively, relative to control) were altered by resistin (Fig. 5a). Similarly, basal and time-dependent insulin-stimulated tyrosine phosphorylation of the IRβ (Fig. 5b) were not affected by resistin (e.g. at 5 min insulin stimulation 2.01-fold and 2.05-fold above control in the absence and presence of resistin, respectively). However, we found that resistin selectively decreased production of total IRS-1 protein (0.52±0.09-fold relative to control), without altering levels of IRS-2 (0.98±0.08-fold relative to control) as shown in Fig. 6a. Further analysis of the temporal nature of the resistin-induced decrease in IRS-1 protein content (Fig. 6a) demonstrated a time-course similar to that observed for the metabolic effects of resistin in previous figures. The insulin-stimulated tyrosine phosphorylation of IRS-1 on amino acid 612 was reduced in the presence of resistin (e.g. at 5 min insulin stimulation 2.61-fold and 1.78-fold above control in the absence and presence of resistin, respectively, Fig. 6b). There was no significant change upon resistin treatment in the level of insulin-stimulated IRS-1 S616 phosphorylation (e.g. at 15 min insulin stimulation 1.59-fold and 1.68-fold above control in the absence and presence of resistin, respectively) or IRS-1 S307 phosphorylation (e.g. at 15-min insulin stimulation 3.23-fold and 3.47-fold above control in the absence and presence of resistin, respectively) as shown in Fig. 6c,d. However, because of the reduced level of IRS-1 protein found in resistin-treated cells it is likely that the stoichiometry of tyrosine phosphorylation is not altered by resistin whereas the relative amount of serine phosphorylation is increased.

Effect of resistin on insulin receptor protein production and phosphorylation. a Representative immunoblots (n≥3 experiments) from analysis of production of both IRα and IRβ subunits in control cells and cells grown in the presence of resistin (50 nmol/l, 24 h). b Representative immunoblots from analysis of phosphorylation of IRβ on Y972 by insulin (100 nmol/l, 0–30 min) in the presence or absence of resistin (50 nmol/l, 24 h). Con, control; Res, resistin

Effect of resistin on IRS protein production and regulation by phosphorylation. a IRS-1 and IRS-2 content of control cells and cells grown in the presence of resistin (50 nmol/l, 24 h), with quantitative analysis for a representative time course (0–24 h) of resistin-mediated decreases in IRS-1 protein content. b–d Phosphorylation of IRS-1 on Y612, S616 and S307, respectively, by insulin (100 nmol/l, 0–30 min) in the presence or absence of resistin (50 nmol/l, 24 h). Representative immunoblots (n≥3 experiments) are shown in all cases. Con, control, Res, resistin
Resistin also mediated isoform-specific effects at the level of Akt. We found a decrease in the total Akt content upon treatment of L6 cells with resistin (0.69±0.06-fold relative to control) and further examination demonstrated that this was accounted for by a selective decrease in the level of Akt1 (0.42±0.07) with no change in Akt2 (0.96±0.04) or Akt3 (1.02±0.03) production (Fig. 7a). Having shown that both IRS-1 and Akt1 production were reduced by resistin we used lactacystin to examine the potential role of proteasome-mediated degradation. Surprisingly, we found that lactacystin caused a decrease in basal IRS-1 and Akt-1 protein content while others (e.g. β-actin) were unaffected under these conditions. Resistin caused no significant decrease in IRS-1 or Akt-1 production in the presence of this inhibitor (data not shown). There was a decrease in the basal level of Akt phosphorylation in response to resistin and analysis of the time-course of Akt activation in response to insulin, assessed by its phosphorylation on T308 and S473, demonstrated that this was attenuated by resistin as shown in Fig. 7b,c (e.g. at 5 min insulin stimulation T308 phosphorylation was 1.73- and 1.22-fold above control in the absence and presence of resistin, respectively).

Effect of resistin on Akt production and regulation by phosphorylation. The content of total Akt as well as each of the three individual isoforms was examined in control cells and cells grown in the presence of resistin (50 nmol/l, 24 h) with quantitative analysis also provided for a representative time course (0–24 h) of resistin-mediated decreases in IRS-1 production. b, c Phosphorylation of Akt on T308 and S473, respectively, by insulin (100 nmol/l, 0–30 min) in the presence or absence of resistin (50 nmol/l, 24 h). Representative immunoblots (n≥3 experiments) are shown in all cases. Con, control; Res, resistin
Discussion
In this study we examined the direct effect of resistin on glucose uptake and metabolism in rat skeletal muscle cells. Considerable debate exists regarding the role of resistin in the pathophysiology of insulin resistance in humans and animals and as to whether resistin acts primarily in muscle, liver or fat [2, 3, 5, 29–32]. However, several studies provide convincing evidence for an effect of resistin in skeletal muscle. For example, use of adenovirus to overexpress murine resistin at supraphysiological concentrations for 7 days in male Wistar rats caused glucose intolerance, hyperinsulinaemia and an impaired ability of insulin to lower blood glucose [31]. Resistin caused insulin resistance at the level of both skeletal muscle (decreased insulin-stimulated glucose infusion during clamp) and liver (attenuation of suppression of hepatic glucose output) [31]. Furthermore, preventing resistin action in transgenic mice expressing a dominant inhibitory version of the protein improved insulin sensitivity and glucose tolerance in mice [5]. A recent elegant study where adipocytes transfected in vitro to express resistin were surgically inserted into mice showed that these mice then exhibited high plasma resistin levels [32]. Importantly, this was associated with whole body insulin resistance and decreased IRS-1 phosphorylation in muscle [32], although it was suggested that these in vivo effects of resistin may be mediated via increased TNF-α production. We have also recently demonstrated that resistin acutely decreased glucose uptake in skeletal muscle cells [25].
Although results from both ourselves [25] and others [6] have established the ability of acute resistin treatment to reduce glucose uptake in muscle and fat, few studies have examined the direct effect of chronic resistin on glucose uptake. We believe that because chronic hyperresistinaemia is typically found in obese animals and humans that chronic effects are more pertinent to the pathophysiology of diabetes. One recent study has shown that chronic resistin treatment of adipocytes caused a small but significant decrease in glucose uptake [17]. Notably, the effect of resistin on glucose metabolism remains to be determined. Here we further investigated the role of resistin in regulating basal and insulin-stimulated glucose uptake and metabolism in rat skeletal muscle cells by examining the effect of resistin treatment times up to 24 h. We have shown that resistin caused a decrease in both basal and insulin-stimulated glucose uptake. This expands upon our previous work [25] using shorter resistin pretreatment times and is similar to studies performed in adipocytes [6, 17]. Furthermore, we measured the cell surface content of myc-tagged GLUT4 in intact cells to identify that the mechanism underlying the effect of resistin on insulin-stimulated glucose uptake involved a decrease in the extent of translocation of GLUT4 to the cell surface. In attempting to explain the decreased basal glucose uptake we found no overall change in GLUT1 or GLUT4 protein content.
To further investigate the mechanism whereby resistin decreased GLUT4 translocation and glucose uptake we examined components of the insulin signalling pathway known to regulate these phenomena [33]. Resistin had no effect on insulin signalling at the level of the receptor because total protein content was unaltered and insulin-stimulated tyrosine phosphorylation remained intact. Interestingly, we found effects on specific isoforms of both IRS and Akt. Production of IRS-1 but not IRS-2 was decreased by resistin and this correlated with a reduced level of IRS-1 tyrosine phosphorylation (Y612). Thus, the decrease in the observed level of IRS-1 Y612 phosphorylation may be the result of a reduced level of protein production rather than a change in the stoichiometry of phosphorylation. Based upon previous work documenting the inhibitory effect of phosphorylation of this residue on insulin signalling [34], we speculated that this may represent another way in which resistin leads to insulin resistance or degradation of IRS-1 [34]. However, we observed no change in the basal or insulin-stimulated total levels of IRS-1 phosphorylation on S616 after 24 h of resistin treatment. However, as alluded to above, because the IRS-1 protein production is decreased in resistin-treated cells this implies an increased stoichiometry of S616 phosphorylation in the presence of resistin which may contribute to depression of insulin signalling. We also investigated the regulation of IRS-1 S307 phosphorylation because this residue has been implicated in degradation of the protein via a proteasome pathway, yet we did not detect a significant change in the level of insulin-induced phosphorylation of this residue in the presence of resistin. We found that resistin reduced Akt1 isoform production but did not alter Akt2 or Akt3 isoform production. We confirmed the functional consequence of resistin's effect on Akt by demonstrating reduced phosphorylation of the kinase on both T308 and S473. Importantly, a recent study demonstrated that phosphorylation of IRS-1 and Akt was reduced in rats overexpressing resistin [31]. It was recently demonstrated in L6 cells that IRS-1 regulates the activation of Akt1, GLUT4 translocation, and glucose uptake whereas IRS-2 regulates ERK signalling [35]. Thus, a specific effect of resistin here on IRS-1 and Akt1 function is in keeping with decreased glucose uptake and metabolism.
In this study we also examined the effect of resistin on glucose metabolism in rat skeletal muscle cells. We found a decrease in both glycogen synthesis and glucose oxidation. This is in agreement with one previous study which demonstrated that treatment of skeletal muscle from hypertensive rats with 600 ng/ml of recombinant resistin for 2 h inhibited both insulin-stimulated glycogen synthesis and glucose oxidation [4]. The reduction in glycogen synthesis is particularly important and we went on to show that this effect correlated with decreased GSK-3β phosphorylation, whereas total protein content was unaltered. The ability of insulin to stimulate hepatic Akt and GSK-3 phosphorylation was previously shown to be enhanced by antisense-mediated knockdown of circulating resistin levels [30]. Here, our results are consistent with a mechanism involving defects in IRS-1, Akt-1 and GSK-3β leading to decreased glycogen synthesis in skeletal muscle cells. The reduced glucose oxidation also observed in response to resistin will probably also place a higher demand on fatty acid oxidation to maintain the energetic needs of muscle.
In summary, we have shown that resistin reduced basal and insulin-stimulated glucose uptake, oxidation and glycogen synthesis. The mechanism of resistin action may involve alterations in IRS-1 and Akt1 function or additional parameters not investigated here. Our results agree with prior studies suggesting that chronic elevation in resistin levels can cause whole body insulin resistance and defects in insulin signalling in skeletal muscle [31]. These effects would be expected to contribute to the development of hyperglycaemia and our observations confirm resistin as a potential direct regulator of glucose homeostasis.
Abbreviations
- GLUT:
-
glucose transporter
- GSK-3β:
-
glycogen synthase kinase-3β
- IRα:
-
insulin receptor α-subunit
- IRβ:
-
insulin receptor β-subunit
- IRS:
-
insulin receptor substrate
- MTT:
-
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide
References
Steppan CM, Lazar MA (2004) The current biology of resistin. J Intern Med 255:439–447
Banerjee RR, Rangwala SM, Shapiro JS et al. (2004) Regulation of fasted blood glucose by resistin. Science 303:1195–1198
Rangwala SM, Rich AS, Rhoades B et al. (2004) Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes 53:1937–1941
Pravenec M, Kazdova L, Landa V et al. (2003) Transgenic and recombinant resistin impair skeletal muscle glucose metabolism in the spontaneously hypertensive rat. J Biol Chem 278:45209–45215
Kim KH, Zhao L, Moon Y, Kang C, Sul HS (2004) Dominant inhibitory adipocyte-specific secretory factor (ADSF)/resistin enhances adipogenesis and improves insulin sensitivity. Proc Natl Acad Sci USA 101:6780–6785
Steppan CM, Bailey ST, Bhat S et al. (2001) The hormone resistin links obesity to diabetes. Nature 409:307–312
Rajala MW, Lin Y, Ranalletta M et al. (2002) Cell type-specific expression and coregulation of murine resistin and resistin-like molecule-alpha in adipose tissue. Mol Endocrinol 16:1920–1930
Steppan CM, Lazar MA (2002) Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab 13:18–23
Juan CC, Au LC, Fang VS et al. (2001) Suppressed gene expression of adipocyte resistin in an insulin-resistant rat model probably by elevated free fatty acids. Biochem Biophys Res Commun 289:1328–1333
Rajala MW, Qi Y, Patel HR et al. (2004) Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes 53:1671–1679
Lee JH, Bullen JW Jr, Stoyneva VL, Mantzoros CS (2004) Circulating resistin in lean, obese and insulin-resistant mouse models: lack of association with insulinemia and glycemia. Am J Physiol Endocrinol Metab 288(3):E625–632
Ghosh S, Singh AK, Aruna B, Mukhopadhyay S, Ehtesham NZ (2003) The genomic organization of mouse resistin reveals major differences from the human resistin: functional implications. Gene 305:27–34
Nagaev I, Smith U (2001) Insulin resistance and type 2 diabetes are not related to resistin expression in human fat cells or skeletal muscle. Biochem Biophys Res Commun 285:561–564
Fain JN, Cheema PS, Bahouth SW, Lloyd Hiler M (2003) Resistin release by human adipose tissue explants in primary culture. Biochem Biophys Res Commun 300:674–678
Lehrke M, Reilly MP, Millington SC, Iqbal N, Rader DJ, Lazar MA (2004) An inflammatory cascade leading to hyperresistinemia in humans. Plos Med 1:e45
Degawa-Yamauchi M, Bovenkerk JE, Juliar BE et al. (2003) Serum resistin (FIZZ3) protein is increased in obese humans. J Clin Endocrinol Metab 88:5452–5455
McTernan PG, Fisher FM, Valsamakis G et al. (2003) Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes. J Clin Endocrinol Metab 88:6098–6106
Bajaj M, Suraamornkul S, Hardies LJ, Pratipanawatr T, DeFronzo RA (2004) Plasma resistin concentration, hepatic fat content, and hepatic and peripheral insulin resistance in pioglitazone-treated type II diabetic patients. Int J Obes Relat Metab Disord 28:783–789
Smith SR, Bai F, Charbonneau C, Janderova L, Argyropoulos G (2003) A promoter genotype and oxidative stress potentially link resistin to human insulin resistance. Diabetes 52:1611–1618
Lee JH, Chan JL, Yiannakouris N et al. (2003) Circulating resistin levels are not associated with obesity or insulin resistance in humans and are not regulated by fasting or leptin administration: cross-sectional and interventional studies in normal, insulin-resistant, and diabetic subjects. J Clin Endocrinol Metab 88:4848–4856
Heilbronn LK, Rood J, Janderova L et al. (2004) Relationship between serum resistin concentrations and insulin resistance in nonobese, obese, and obese diabetic subjects. J Clin Endocrinol Metab 89:1844–1848
Conneely KN, Silander K, Scott LJ et al. (2004) Variation in the resistin gene is associated with obesity and insulin-related phenotypes in Finnish subjects. Diabetologia 47:1782–1788
Wang H, Chu WS, Hemphill C, Elbein SC (2002) Human resistin gene: molecular scanning and evaluation of association with insulin sensitivity and type 2 diabetes in Caucasians. J Clin Endocrinol Metab 87:2520–2524
Sentinelli F, Romeo S, Arca M et al. (2002) Human resistin gene, obesity, and type 2 diabetes: mutation analysis and population study. Diabetes 51:860–862
Moon B, Kwan JJ, Duddy N, Sweeney G, Begum N (2003) Resistin inhibits glucose uptake in L6 cells independently of changes in insulin signaling and GLUT4 translocation. Am J Physiol Endocrinol Metab 285:E106–115
Tajmir P, Kwan JJ, Kessas M, Mozammel S, Sweeney G (2003) Acute and chronic leptin treatment mediate contrasting effects on signaling, glucose uptake, and GLUT4 translocation in L6-GLUT4myc myotubes. J Cell Physiol 197:122–130
Ceddia RB, Somwar R, Maida A, Fang X, Bikopoulos G, Sweeney G (2005) Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 48:132–139
Ceddia RB, Sweeney G (2004) Creatine supplementation increases glucose oxidation and AMPK phosphorylation and reduces lactate production in L6 rat skeletal muscle cells. J Physiol 555:409–421
Rajala MW, Obici S, Scherer PE, Rossetti L (2003) Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 111:225–230
Muse ED, Obici S, Bhanot S et al. (2004) Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest 114:232–239
Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM (2004) Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest 114:224–231
Kitagawa Y, Bujo H, Takahashi K et al. (2004) Impaired glucose tolerance is accompanied by decreased insulin sensitivity in tissues of mice implanted with cells that overexpress resistin. Diabetologia 47:1847–1853
Rudich A, Klip A (2003) Push/pull mechanisms of GLUT4 traffic in muscle cells. Acta Physiol Scand 178:297–308
Gual P, Le Marchand-Brustel Y, Tanti JF (2005) Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87:99–109
Huang C, Thirone AC, Huang X, Klip A (2005) Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem 280:19426–19435
Acknowledgements
Funding was provided by the Natural Science and Engineering Research Council (NSERC) via a Discovery grant to G. Sweeney and by the Canadian Diabetes Association via a scholarship award to G. Sweeney in honour of the late M. A. Bodington. G. Sweeney was also supported by the Ontario Ministry of Economic Development and Trade via a Premiers Research Excellence Award. Funding from the Canada Foundation for Innovation and Ontario Innovation Trust is also acknowledged. A. Maida was supported by an NSERC summer studentship award.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Palanivel, R., Maida, A., Liu, Y. et al. Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia 49, 183–190 (2006). https://doi.org/10.1007/s00125-005-0060-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0060-z