
Abstract
Many species with currently continuously distributed populations have histories of geographic range shifts and successive shifts between decline or fragmentation, growth and spatial expansion. The moose (Alces alces) colonised Scandinavia after the last ice age. Historic records document a high abundance and a wide distribution across Norway in the middle ages, but major decline and fragmentation in the eighteenth and nineteenth centuries. After growth and expansion during the twentieth century, the Norwegian population is currently abundant and continuously distributed. We examined the distribution of genetic variation, differentiation and admixture in Norwegian moose, using 15 microsatellites. We assessed whether admixture has homogenised the population or if there are any genetic structures or discontinuities that can be related to recent or ancient shifts in demography or distribution. The Bayesian clustering algorithm STRUCTURE without any spatial information showed that there is currently a genetic dichotomy dividing the population into one southern and one northern subpopulation. Including spatial information, the Bayesian clustering algorithm TESS, which considers gradients of genetic variation and spatial autocorrelation, suggests that the population is divided into three subpopulations along a latitudinal axis, the southern one identical to the one identified with STRUCTURE. Present convergence zones of high admixture separate the identified subpopulations, which are delimited by genetic discontinuities corresponding to geographic barriers against dispersal, e.g. wide fiords and mountain ranges. The distribution of the subpopulations is supported by spatial autocorrelation analysis. However, some loci are not in Hardy–Weinberg equilibrium and the STRUCTURE analysis suggests that a lower hierarchical structure may exist within the southernmost subpopulation. No bottlenecks or founder events are indicated by the levels of genetic variation, rather a high degree of private alleles in the northern subpopulations indicates introgression. Coalescent-based Approximate Bayesian Computation estimates unambiguously suggest that the genetic structure is a result of an ancient divergence event and a more recent admixture event a few centuries ago. This indicates that the central Scandinavian subpopulation constitutes a relatively recent convergence zone of secondary contact.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Knowledge of genetic structure is important for estimation of effective population size, and for wildlife management and conservation (Wang and Caballero 1999; Nunney 2000). In species with continuous distributions, discontinuities in genetic variation are commonly addressed through spatial models and landscape genetics. Such analyses identify breaks in gene flow, secondary contact zones, geographic barriers for dispersal and interactions between landscape features and micro-evolutionary processes, such as gene flow, genetic drift and selection (Manel et al. 2003). Many species with currently continuously distributed populations have histories of geographic range shifts and/or successive shifts between decline, fragmentation, growth, spatial expansion and continuous distribution. The effects of such shifts are of great concern (Pertoldi et al. 2007), given the major range shifts expected and found in many species following the significant climatic changes since pre-industrial times (IPCC 2001, 2007; Parmesan 2006). During the ice ages’ extensive oscillations in climate and distribution of continental ice sheets, such shifts resulted in vast diversification of genetic variation and adaptations in many species, and many secondary contact zones (Taberlet et al. 1998; Hewitt 1999, 2000, 2004). Historic records also tell of extreme decline and fragmentation during the last centuries in several species, which today are numerous and continuously distributed (Gill 1990). Investigations thus often focus on determining whether genetic structure and signals of population reduction derive from ancient natural events or from events of the last centuries that may be related to anthropogenic factors (e.g. Storz and Beaumont 2002; Goossens et al. 2006; Okello et al. 2008; Scandura et al. 2008).
In expanding populations, new demes may become bottlenecked or genetically differentiated because of founder events and subsequent genetic drift, depending on the number of founders and migration rates (Austerlitz et al. 1997; Excoffier 2004), especially when dispersers move long distances and become isolated (Nichols and Hewitt 1994; Ibrahim et al. 1996). Strong genetic drift and reduced genetic variation may therefore characterise both new demes and bottlenecked indigenous populations. However, increased gene flow opposes genetic drift and structure (Slatkin 1987), and when genetically different subpopulations merge, the level of genetic variation can increase as a result of the isolate breaking (Hartl and Clark 1997). Present genetic structure may thus result from founder events after local extinction or from still remaining bottlenecked indigenous populations, or it may be broken down by high levels of migration and dispersal. Remaining indigenous populations may possess local adaptations but may also be subject to introgression from non-native populations.
Many deer species have histories of both glacial and recent shifts between fragmentation, spatial expansion and new ranges (Groves and Grubb 1987; Gill 1990; Geist 1998). Among these, the moose (Alces alces) is a potentially far-ranging ungulate species, and a highly valued game species. In Scandinavia, moose often conduct seasonal migrations extending up to 200 km (Sæther et al. 1992, Bunnefeld et al. 2011). The high mobility and frequent, long dispersal distances facilitate fast range expansion given appropriate conditions for population growth. Moose are long-lived and females have a high reproductive rate after maturing at 1.5–2.5 years old (Garel et al. 2009). There is large geographic variation in life history traits among Norwegian moose populations (Grøtan et al. 2009; Herfindal et al. 2006a; Garel et al. 2009), which mainly has been linked to environmental variation (Grøtan et al. 2009; Herfindal et al. 2006a). In Norway, about 35,000–39,000 moose are harvested each year, based on an estimated winter population size of about 100,000–120,000 individuals (i.e. about 1 moose per km2 moose land, Solberg et al. 2005, 2006). However, previous investigations have indicated a generally low level of genetic variation (Baccus et al. 1983; Røed 1998; Røed and Midthjell 1998). Archaeological finds suggest that moose have existed in Norway at least since the Atlantic period of the stone age (8000 bp), with a wide distribution and abundant population until the end of the Middle Ages (Collett 1912; Hufthammer 2006; Rosvold 2006). Historical records indicate major population reductions and fragmentation in the seventeenth and eighteenth century, leading to only a few remaining subpopulations at low density in Norway, Sweden and Finland in the early nineteenth century (Collett 1912; Ryman et al. 1980; Nygrén 1987; Thörnqvist 1997). Since then, following the introduction of restrictive hunting regulations, these subpopulations have been growing and expanding their ranges, resulting in the present high abundance and continuous distributions within and across all three countries (Gill 1990; Lavsund et al. 2003).
Here, we examine the present distribution of genetic variation in the Norwegian moose to assess whether there are any genetic discontinuities in this continuously distributed population. We test the null hypothesis that demographic and spatial expansion during the last decades has homogenised the distribution of genetic variation and that no genetic structure will be observed in spite of the previous population reductions. We also aim to estimate the time of divergence between any identified genetically differentiated subpopulations to assess whether any genetic structure may be of recent or ancient origin.
Methods and materials
Sampling and laboratory procedures
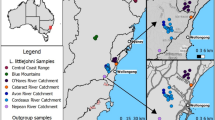
From 2005 to 2007 we sampled tissue from 585 wild Norwegian moose in 154 municipalities across Norway (Fig. 1), mostly calves (n = 340), yearlings (n = 125) and sub-adults (n = 64). To deal with biases that may be caused by discontinuous sampling on non-spatial clustering algorithms (Serre and Pääbo 2004; Chen et al. 2007; Schwartz and McKelvey 2009), we used a random sampling design with samples spatially spread as much as possible (Fig. 1), but according to local population densities (Solberg et al. 2009). Genomic DNA was isolated from ear and muscle tissue (Qiagen, DNeasy Blood and Tissue kit). We selected 15 dinucleotide microsatellite loci: CSSM03 (Moore et al. 1994), RT1, RT5, RT6, RT9, RT27 and RT30 (Wilson et al. 1997), NVHRT01 and NVHRT21 (Røed and Midthjell 1998), MAF46 (Swarbrick et al. 1992), McM58 (Hulme et al. 1994), OarFCB193 (Buchanan and Crawford 1993), BM888, BM4107 and BM4513 (Bishop et al. 1994). The microsatellites were amplified with a GeneAmp PCR System 9700 (Applied Biosystems) in 10 μl reaction mixtures of 2 μl of genomic template DNA, 2 pmol of each primer, Ammonium buffer with 1.5 mM MgCl2, 0.2 mM dNTP, and 0.25 U of TaqDNA polymerase (Ampliqon). Thermocycling parameters after denaturation at 95°C for 2 min were 28 cycles with 95°C for 30 s, 55°C for 30 s and 72°C for 45 s, followed by an additional 10 min at 72°C. The PCR products were then separated by size with capillary electrophoresis (3100 Genetic Analyzer, Applied Biosystems) and electromorphs were genotyped with GeneMapper 3.7 (Applied Biosystems).

The distribution of samples (n = 585), topography and forest cover in Norway (a), and the geographic distribution of admixture estimates for Norwegian moose with K = 2 and no prior information in STRUCTURE (b, c), and with K-max = 3 in TESS (d–f), which consider geographical clines of genetic variation and spatial autocorrelation. For both analyses, admixture models were applied and admixture estimates were averaged for the 10 of 50 runs with highest posterior probability. The spatial distribution is extrapolated using triangulation from the admixture estimates of the unique moose observations
Population genetics analysis
We used Bayesian assignment as implemented in STRUCTURE 2.3 (Pritchard et al. 2000) without any prior spatial information to assess whether discontinuities existed in the distribution of genetic variation within the data set. For each of a different number of genetic clusters (K ∈ [1,13]), a model with uniform priors, admixture (α = 1, αmax = 50), 100,000 burnins and 500,000 MCMC iterations was run 50 times. The model was run with both correlated and independent gene frequencies (Falush et al. 2003), because the age of any detected genetic structure was unknown. To assess the effect of any present null alleles, it was also run with missing values interpreted as null alleles (RECESSIVE ALLELES = 1). For each K-value, average posterior probability among runs and standard error was calculated. The main genetic structure of the data set was interpreted from ΔK. Larger values of K often involves higher posterior probabilities and an increasing variance among runs, and since ΔK is negatively related to the variance among runs, it can be used to identify major breakpoints (Evanno et al. 2005). For the identified K-value, the individuals’ assignment coefficients (q) for each genetic cluster were averaged over the 10 runs with lowest posterior probability, using the CLUMPP software (Jakobsson and Rosenberg 2007). The geographic distribution of genetic clusters was assessed by comparing the individuals’ assignment to each genetic cluster (q) with their geographic sampling locations, identifying subpopulations from areas with a high cluster membership and convergence zones from areas with a high degree of admixture.
To address the extent of spatial genetic structure within subpopulations identified with STRUCTURE, we analysed the spatial genetic autocorrelation among individuals using GenAlEx 6 (Peakall and Smouse 2006). From pairwise Euclidean, linear geographic and squared genetic distances between individuals, a spatial genetic autocorrelation coefficient (r) is calculated. Assuming statistical independence among loci, genetic distances are calculated as the number of different alleles per locus among pairs of individuals, treating all loci simultaneously (Smouse and Peakall 1999). The geographic distance range is divided into a set of distance classes of a specified size (km), r is calculated within each and can be evaluated as a function of geographic distance. Positive significant spatial autocorrelation reflects a higher genetic similarity than expected at random, but autocorrelation is not independent among distance classes (Smouse and Peakall 1999). Spatial genetic autocorrelation was therefore assessed for distance classes of an increasing size (100–1700 km, increment 100 km), and the largest significant distance class was interpreted as the true extent of positive spatial genetic structure (Peakall et al. 2003). Non-parametric tests for statistical significance include (1) bootstrap estimates (n = 999) of the 95% confidence interval (CI) around the r value of each distance class size, and (2) random permutation estimates of the 95% CI around the null hypothesis of no positive genetic structure, r = 0.
Discrete sampling along genetic clines may confound non-spatial Bayesian clustering (Serre and Pääbo 2004; Rosenberg et al. 2005; Schwartz and McKelvey 2009). Therefore, a good solution is to combine STRUCTURE with a spatial model like implemented in the program TESS (Chen et al. 2007). To assess whether clusters identified with STRUCTURE were robust to hypotheses of continuous geographic variation in gene frequencies (Francois et al. 2006) and local spatial autocorrelation, we included spatial information and analysed the data set with the Bayesian clustering in TESS 2.3 (Chen et al. 2007; Durand et al. 2009). In the admixture model of TESS, trend surfaces account for geographic clines and spatial autocorrelation residuals account for isolation by distance (Francois and Durand 2010). TESS uses hidden Markov random fields to model geographic clines of gene frequencies (Francois et al. 2006) and more efficiently infers the correct K, especially when differentiation is low (Chen et al. 2007) and when including spatial autocorrelation (Durand et al. 2009). The spatial information considered is a neighbourhood network of the samples, obtained from a Dirichlet tessellation of their coordinates. The network distances are by default weighted by one, and because our data set has a skewed sampling density, the analyses were also performed with these weights scaled by the geographic distances between sample coordinates. The admixture model of TESS was run 50 times for different maximal numbers of genetic clusters (K-max ∈ [2,10]), using a linear trend surface, a conditional auto-regressive (CAR) variance of 1.0 and a spatial interaction strength of 0.6 for spatial autocorrelation, and 50,000 MCMC iteration sweeps with a burnin period of 40,000. For each run, a measure of the models’ predictive ability is calculated, the Deviance Information Criterion (DIC), which expresses model fit (posterior mean deviance) penalized by model complexity (Spiegelhalter et al. 2002). For each K-max value the 10 runs with the lowest DIC values were selected. Genetic structure was interpreted from the effective number of clusters identified by the K-max model where the decreasing DIC averages reach a plateau (Durand et al. 2009), and from the delta statistic of Evanno et al. (2005) calculated for DIC. Admixture estimates were averaged (CLUMPP), and the distribution of subpopulations and their admixture was assessed in the same way as with STRUCTURE.
We described the level and distribution of genetic variation within and among subpopulations identified with STRUCTURE and TESS, as well as in the whole population. Exact tests of Hardy–Weinberg (H–W) equilibrium across the 15 loci were performed using GENEPOP 3.4 with default settings (Raymond and Rousset 1995). To assess the level of genetic variation, we counted the number of private alleles and used FSTAT 2.9.3 (Goudet 2001) to calculate means of allelic richness (rarefaction, El Mousadik and Petit 1996), F IS, observed and expected heterozygosity (unbiased H, Nei 1987, 2000) for each subpopulation across loci. Among identified subpopulations each pair of loci were tested for genotypic linkage disequilibrium (GENEPOP 3.4). We used F-statistics (e.g., Weir 1996), as implemented in FSTAT to assess genetic structure among subpopulations (F ST).
To assess for previous occurrences of recent subpopulation reductions, we used BOTTLENECK 1.2 (Cornuet and Luikart 1996) to perform one-tailed Wilcoxon tests of heterozygosity excess (10,000 iterations). Most microsatellites fit a two-phase model of mutation (TPM) with 80–95% of the stepwise mutation model (SMM; Ohta and Kimura 1973) better than a strict one-step model (Di Rienzo et al. 1994). Since the infinite allele model (IAM; Kimura and Crow 1964) more easily exhibits heterozygosity as a consequence of bottlenecks (Piry et al. 1999), we assumed a TPM model with 20% variation (IAM) and 80% SMM, to be certain to detect signals of recent bottlenecks. To examine for older and more severe bottlenecks, we calculated the M-ratio (Garza and Williamson 2001) between the observed number of alleles and the number of repeats in the whole allele size range of each locus, across loci for each subpopulation.
Divergence times and effective sizes were assessed both for the subpopulations identified with STRUCTURE and for the subpopulations identified through TESS, applying the coalescent-based Approximate Bayesian Computation (ABC) algorithm of DIY ABC (Cornuet et al. 2008). This algorithm simulates data sets for each of a specified set of scenarios of historic and/or demographic events, and compares summary statistics from these with the summary statistics of the observed data. The posterior probability and distribution of parameters of each scenario are estimated and alternative scenarios can be compared. We applied different scenarios with various splitting and admixture events to explain today’s observed subpopulations, some involving un-sampled populations (outside the study area). With a present dichotomy (STRUCTURE results), one scenario with one divergence event and two scenarios with additional admixture events were explored. With a present trichotomy (TESS results), three scenarios with different divergence orders and three scenarios with both divergence and admixture events were explored. Secondarily we explored the same scenarios with demographic events like population reductions and expansions, but compared these only with the most probable scenario without demographic events. For all simulations, wide priors were used for all parameters (e.g. 10, 10,000), and conditions set for the sequence order of historic events. Similarly, for the demographic models, previous population reduction was imposed through conditions: present size > previous size < older size. The generalised stepwise model of mutation was applied with default values (GSM; Fu and Chakraborty 1998; Estoup et al. 2002). For each simulated data set, all 4 within- and 5 of the 7 default among—populations summary statistics were calculated. For each explored scenario, 500,000 data sets were simulated. The 500 and 5,000 sets with summary statistics most similar to the observed data were identified through the direct and logistic regression rejections steps of the algorithm, respectively, and used for ABC estimation of parameters. The program assumes that populations evolve independently without migration between the historic and demographic events.
Results
Within the whole data set, we found 112 alleles, an allelic richness of 7.4 (SD = 2.5) and an expected heterozygosity of 0.66 (SD = 0.13). Only four of 15 loci were in H–W equilibrium (CSMM03, OarFCB193, NVHRT21 and RT09).
The STRUCTURE algorithm showed that with both independent (not shown) and correlated allele frequencies, the most likely main partitioning of the genetic variation in the data set was a dichotomy. A pronounced higher delta K demonstrated a major break in the data set with K = 2, while an increasing posterior probability up to K = 5 indicated the presence of lower hierarchical structure (Supplementary Fig. S1). The same results were achieved when incorporating the effect of null alleles (RECESSIVE ALLELE MODEL). Ordered by latitude, the probabilities of individual assignment to each of two clusters (K = 2) suggest the presence of two subpopulations of relatively low admixture and one area of admixture (Supplementary Fig. S2). In the two suggested subpopulations a large proportion of the individuals have a high cluster membership, while many individuals in the convergence zone between the subpopulations have a divided cluster membership, suggesting mixed origin and a convergence zone (Table 1). Among individuals with a high cluster assignment (q > 0.8), 422 were assigned to the subpopulation that corresponded to their geographic sampling location, whereas only four individuals sampled within the southern and one sampled within the northern subpopulation were assigned to the opposite cluster. Only eight individuals with a high cluster assignment were found within the approximately 150 km wide convergence zone. Contour plots of the admixture estimates (q) support the geographic distribution of the two subpopulations (Fig. 1b, c). The discontinuities between the subpopulations and the convergence zone fall together with mountain ranges and fiords, indicating a lower degree of admixture across these. The northern cluster is delimited to the south by a very long and wide fiord (Trondheimsfjorden) and along most its length to the east by mountain ranges. The southern cluster is delimited to the north by high, alpine and continuous mountain ranges (Dovre, Rondane, Forelhogna and Sylan).
The spatial autocorrelation analyses showed that the two subpopulations suggested by STRUCTURE both contained positive genetic structure significantly different from the null hypothesis of no genetic structure, but to different spatial extents (Fig. 2). Within the southern subpopulation the positive genetic structure extends to up to 300 km. In the northern subpopulation it extends much further, i.e. up to 650 km, indicating the presence of additional genetic structure. By comparison, spatial autocorrelation was significant for distance class sizes up to 1,300 kilometres within the whole data set (not shown).

Spatial autocorrelation (r) for distance classes of increasing size, for the northern (n = 374, grey) and southern (n = 164, black) subpopulation of Norwegian moose, identified with STRUCTURE, and their 95% confidence intervals (CI). The 95% CI around the null hypothesis of no genetic structure are shown with dashed lines
The TESS algorithm, incorporating spatial information and the occurrence of geographical clines and spatial autocorrelation, suggested that an additional partitioning of the northern subpopulation was the most likely division of the data set. A plateau in the decreasing DIC averages and a much higher delta DIC at K-max = 3 both suggested a partition of the genetic variation in three clusters (Supplementary Fig. S3). Plotting of the cluster assignment of individuals against their latitudinal order of sampling showed that all higher K-max values had the same effective number and distribution of clusters (Fig. 3). Scaling of the weights used in the neighbouring network by geographic distances gave the same results. The distribution of the suggested southern subpopulation and its convergence zone to the north corresponded well to the pattern derived by STRUCTURE. In addition TESS suggested a third subpopulation in the high north and a convergence zone toward a middle (the northern) subpopulation (Fig. 1d, e, f). The latitudinal distribution and proportionate cluster memberships of these areas are given in Table 1. Totally 430 individuals were assigned with a high stringency (q > 0.8) to the subpopulation that corresponded to their geographic sampling location. By comparison, only one individual sampled in the middle subpopulation was assigned to the southern subpopulation, and only eight individuals from the southern and four from the middle subpopulation were assigned to inside the southern convergence zone, with high stringency. The genetic discontinuities fall well together with fiords and mountain ranges. Two long fiords (Saltfjorden and Foldafjorden) and two high mountain ranges (Sulitjelma and Rago) are situated to the north of the middle cluster and a very long and wide fiord (Ofotfjorden) and steep mountains (Istind and Bjørnfjell) are situated south of the high northern subpopulation. The spatial genetic autocorrelation analyses verified that the extent of genetic structure now was similar between the southern, middle and high northern subpopulations.

By latitudinal order (not a continuous geographic scale), individual cluster assignment (q ∈ [0, 1]) for 585 Norwegian moose, averaged for the 10 of 50 runs with lowest DIC for each of a different number of clusters (K-max ∈ [2, 5]) in an admixture model of TESS considering geographical clines of genetic variation and spatial autocorrelation
With the data set divided according to STRUCTURE, the number of loci not in H–W equilibrium within subpopulations included two loci in the northern (RT01 and RT27) and five loci in the southern subpopulation (BM888, MAF46, RT6, NVHRT21 and RT30). Significant deviations from linkage equilibrium were found in one pair of loci in the northern subpopulation (RT27-CSMM03) and three pair of loci in the southern population (RT1-RT5, RT5-RT6, MAF46-RT6). With the data divided according to TESS only the five loci in the southern subpopulation were out of H–W equilibrium and significant deviations from linkage equilibrium were observed in the same three pairs of loci. Average expected heterozygosity was similar between subpopulations for both divisions, but allelic richness was higher in the two northernmost subpopulations (Table 2). With the dichotomy in STRUCTURE, both the number of private alleles and allelic richness were higher in the northern subpopulation. With the trichotomy in TESS, two private alleles were “lost” to the northernmost convergence zone and the rest were divided between the middle and the high northern subpopulations. Compared to their small sample sizes the numbers of private alleles and the allelic richness in these subpopulations were still high. A highly significant genetic structure (P < 0.005) was found between the subpopulations identified with STRUCTURE (F ST = 0.07), as well as between the subpopulations identified with TESS (F ST; south–north = 0.06, south–high north = 0.11, north–high north = 0.05). Neither the Bottleneck analysis nor the M-ratios suggested any previous subpopulation reductions (Table 2).
The DIY ABC estimates involved wide posterior distributions but unanimously suggested that ancient divergence and recent admixture explain the observed genetic structure, both as a dichotomy and as a trichotomy (Figs. 4, 5). With the data set divided according to STRUCTURE (dichotomy), all the explored coalescent scenarios involved estimates of an old to ancient divergence (Fig. 4). In the most likely scenario (posterior probability = 0.77), the southern subpopulation and an un-sampled population diverged 1,533 generations ago (t) and much more recent admixture of these resulted in the northern subpopulation (t = 186). In the other scenario with admixture, the estimated divergence was even more ancient, while in the single-divergence scenario the estimated time was lower (t = 318). Estimates of effective sizes of present subpopulations were similar among the scenarios (Supplementary Table S1). Inclusion of demographic events did not involve any scenarios with higher posterior probability or any large differences in estimates (data not presented). With a present trichotomy and the data set divided according to TESS, all explored scenarios involved an old to ancient divergence (Fig. 5). The most likely scenario involved an old divergence between the southern and high northern subpopulation (t = 830) and a recent admixture event (t = 90) explaining the middle subpopulation (Fig. 5). The second most probable scenario involved an ancient divergence between the southern subpopulation and an un-sampled population (t = 1984), an old divergence between the southern and the middle subpopulation (t = 275) and a more recent admixture event between the un-sampled population and the middle subpopulation explaining the high northern subpopulation (t = 117). The scenarios where the southern subpopulation first diverged from either of the northern subpopulations were the least probable. Estimates of present effective sizes were similar among scenarios (Supplementary Table S2), and inclusion of demographic events involved only low posterior probabilities (data not presented).

Historic scenarios (A2–C2) explored with DIYABC to explain a present dichotomy in the Norwegian moose population. Pop 1 is the southern subpopulation (n = 373) and pop 2 the northern (n = 164). Posterior probabilities (PP (95% CI)) after logistic regression on the 1% simulated data most similar to the observed data, and median (95% CI) of estimated time since divergence and admixture events (numbers of generations, t) below each scenario. N1–N4 are the effective population sizes, r and r − 1 are admixture proportions (Supplementary Table S1)

Historic scenarios (A3–F3) explored with DIY ABC to explain a present trichotomy in the Norwegian moose population. Pop 1 is the southern subpopulation (n = 373), pop 2 the middle (n = 117) and pop 3 the high northern subpopulation (n = 13). Posterior probabilities (PP (95% CI)) after logistic regression on the 1% simulated data most similar to the observed data, and median (95% CI) of estimated time since divergence and admixture events (numbers of generations, t) below each scenario. N1–N4 are the effective population sizes, r and r − 1 are admixture proportions (Supplementary Table S2)
Discussion
Our results clearly show that the Norwegian moose population is not a panmictic population. Treated as one population, only four of the investigated microsatellite loci are in H–W equilibrium, while Bayesian clustering methods both without and with spatial information suggest genetic structure. The STRUCTURE analysis shows one main latitudinal division of the genetic variation (63ºN), into two distinct subpopulations separated by an area of high admixture, a convergence zone. With spatial information, TESS indicates an additional latitudinal division of the northern subpopulation (69ºN), and a third distinct subpopulation in the high north delimited to the south by a second convergence zone. With both the dichotomous and the trichotomous division, the degree of admixture has relatively sharp boundaries at each side of the convergence zones. We therefore reject our null hypothesis that there are no genetic discontinuities in this continuously distributed population. Both divisions imply moderate genetic structure (Wright 1978; Hartl and Clark 1997) and involve H–W equilibrium in most loci, as would be expected when dividing according to genetic structure. However, both STRUCTURE and the five loci not in H–W equilibrium indicate additional lower hierarchical structure within the southernmost subpopulation. The estimates of time since divergence between the subpopulations unambiguously suggest that the main genetic structure is old and comparison of alternative historic scenarios imply that the two northern subpopulations are the result of admixture between the southern subpopulation and an un-sampled population (Figs. 4, 5).
Using the ΔK ad hoc statistic, STRUCTURE may accurately detect the main or true genetic structure (Evanno et al. 2005), and in our study showed that the most probable main partitioning of genetic variation was a dichotomy. The increasing posterior probabilities up to K = 5 indicate also a lower hierarchical genetic structure, which involves the same partitioning of the northern subpopulation as in TESS and a division of the southern subpopulation into three admixed units. The loci not in H–W equilibrium in the southern subpopulation support that this is a real substructure, but for three reasons this was not pursued any further: First, when genetic variation has a continuous distribution, it has been debated whether Bayesian clusters identified without the use of spatial information represent actual genetic structure or sampling artefacts (Serre and Pääbo 2004; Rosenberg et al. 2005). This represents an acute problem in the presence of family groups (Anderson and Dunham 2008) and local spatial autocorrelation (Schwartz and McKelvey 2009). It is therefore recommended to check whether clusters obtained without use of spatial information are robust to the hypothesis of discontinuous geographic variation (Francois et al. 2006; Chen et al. 2007; Durand et al. 2009). When spatial information was considered for our data set, and genetic clines and local spatial autocorrelation taken into account, the CAR model in the TESS algorithm did not detect the lower hierarchical structure in the southern subpopulation. Second, the substructure in the southern subpopulation involved a high degree of admixture and it would be difficult to define and treat these units separately in a divergence analysis, which would also become far too complex. Finally, with the dichotomy according to STRUCTURE, the southern subpopulation showed a much lower geographic extent of genetic structure in the spatial autocorrelation analysis, compared to the northern subpopulation. With the trichotomous division of TESS, the geographic extent of genetic structure was similar among the three subpopulations. We therefore assessed both a present dichotomy and a present trichotomy.
With both the dichotomous and the trichotomous division, few individuals were miss-assigned, suggesting a low degree of first-generation long-distance dispersal. This was expected given our samples, which consisted mostly of calves and 1.5-year-olds. However, the identified genetic discontinuities bordering the convergence zones fall very well together with long fiords and high mountain ranges, which probably in general act as geographic barriers for dispersal, indicating that the observed genetic structure may not be of transient nature. There are no indications that the main genetic structure is the result of recent events. Genetic structure in a spatially expanding population may result from both long distance dispersal and limited migration among demes (Nichols and Hewitt 1994; Ibrahim et al. 1996; Austerlitz et al. 1997; Excoffier 2004). However, neither the level of genetic variation in the population as a whole, nor the even heterozygosity among the suggested subpopulations, indicates any recent reductions of genetic variation from bottlenecks or founder events. Indeed, the BOTTLENECK analysis did not indicate any recent bottlenecks in any subpopulation, and because only M-ratios smaller than 0.68 can be assumed to represent previous population reductions (Garza and Williamson 2001), the observed values (0.66–0.80, Table 2) do not support the historic records of recent nor any older bottlenecks. The slightly lower M-ratio in the northernmost subpopulation is caused by private alleles outside the ordinary microsatellite mutation ranges, which probably are introduced by introgression. The lower hierarchical structure within the southern subpopulation may however be a trace of recent fragmentation during the last centuries, but to ascertain this and exclude immigration, sampling should also include the neighbouring Swedish moose. The Swedish population has, like the Norwegian, increased and expanded much during the twentieth century and might, considering (1) the fragmentation in the eighteenth and nineteenth centuries (Collett 1912; Nygrén 1987; Thörnqvist 1997) and (2) the currently continuous Scandinavian distribution (Gill 1990; Lavsund et al. 2003), represent genetically differentiated subpopulations that have expanded into the Norwegian population. Comparably, the higher allelic richness in the two identified Norwegian northern subpopulations indicates introgression from an un-sampled population. This is supported by the relatively high degree of admixture in the high northern subpopulation identified with TESS and may provide an explanation for the origin of the observed genetic structure.
The DIY ABC estimation of the genealogical history of the main genetic structure suggested an ancient rather than recent divergence, which in the most likely scenarios was between north and south. The most likely scenario with a present dichotomy according to STRUCTURE involved admixture with an un-sampled population, giving support to the trichotomous division of the data set suggested by the spatial model of TESS. However, the lower hierarchical genetic structure within the southern subpopulation was not included, which can stem from more recent fragmentation, and estimates may involve some bias. The consistency between analyses based on a present dichotomy and a present trichotomy, in both the estimated divergence times and the most likely scenario, however suggests robustness. Our interpretations are therefore: (1) that the population contains an old to ancient genetic structure that divides the population by latitude, that (2) subsequent admixture has resulted in the middle subpopulation, and/or that (3) both the northern subpopulations are the result of admixture with an un-sampled population. In both cases, the observed genetic structure is the result of ancient differentiation and a subsequent relatively recent admixture into the middle (northern) subpopulation, suggesting this is an area of secondary contact, probably from the period before population reduction in the last centuries. The remaining question is whether the high northern subpopulation represents an original part of the ancient divergence or whether it is the result of subsequent and more recent admixture with an un-sampled population.
In Scandinavia, many species followed two colonisation routes after the last glaciation, i.e. from the south across a land bridge and from the north-east, and today, their populations typically meet in a convergence zone or an area of admixture in central Scandinavia (Taberlet et al. 1998; Jaarola et al. 1999; Hewitt 1999, 2000, 2004). This pattern is seen in other highly mobile mammals, like the brown bear (Ursus arctos; Taberlet and Bouvet 1994; Taberlet et al. 1995), as well as in smaller and less mobile rodents (Jaarola et al. 1999). In the Swedish moose genetic variation is differentiated between areas in the south and north, which have levels of genetic variation and differentiation (Charlier et al. 2008) that are similar to the ones detected by the study at hand. This coincides well with the Norwegian genetic structure, supporting the existence of two anciently differentiated moose populations in the south and north of Scandinavia. It therefore seems most probable that the middle subpopulation represents an old convergence zone resulting from admixture of these two populations. This suggests two routes of colonisation by the moose in Scandinavia. Secondary immigration also seems very likely considering: (1) the higher allelic richness and lower M-ratio in the high northern subpopulation, indicating introgression, and (2) the existence of large moose populations in northern Sweden, Finland and Russia. The high northern subpopulation in Norway thus probably results from recent admixture from one or more of these un-sampled populations. The historical records of absence of moose in the high north during the nineteenth century (Collett 1912) and a more pure origin seem less likely with no signatures of founder events and given the admixture suggested by both the TESS and DIY ABC analyses. Considering the estimated old to ancient divergence time, populations located far away in Finland or Russia seem to be the most likely source of origin, but additional sampling is required to determine which. Further, the DIY ABC algorithm assumes no migration between scenario events (Cornuet et al. 2008), and immigration from un-sampled populations may have been more continuous, possibly earlier into the middle and more recently into the high northern subpopulation.
The ancient estimated divergence time, in combination with the relatively high level of genetic variation, suggest that the Norwegian moose was not as fragmented or reduced in size as indicated by historic records. Rather we suggest the existence of two anciently differentiated moose populations in the north and south of Scandinavia, which may be indigenous and possibly possess local adaptations. We did not investigate for differentiation and structuring of phenotypic traits within the population, but given the relatively sharp boundaries of the subpopulations we recommend further studies to clarify whether different adaptive traits exist in these anciently genetically differentiated subpopulations. If expansion continues, further studies should be conducted of the admixture and transience of the present convergence zones. Dispersal and admixture into the southern subpopulation, which probably involves hybridisation between anciently differentiated populations, should especially be of interest to management. This is particularly true in face of the predicted climatic changes (IPCC 2001, 2007). Variation in moose life history traits is closely linked to environmental variation (Sæther et al. 1996; Solberg et al. 2004; Herfindal et al. 2006a), and such traits are thus likely to respond to changes in the environment. However, populations differ in their response to environmental variation (Herfindal et al. 2006b, Grøtan et al. 2009). Management strategies should also account for such heterogeneity in environmental responses, which could be linked to genetic variation and structure (Sæther et al. 2009). Further studies should also include samples from all of Scandinavia to improve the models of divergence times and to be able to determine whether the lower hierarchical structure in the south results from different colonisation, immigration, recent fragmentation, or sampling artefacts.
References
Anderson EC, Dunham KK (2008) The influence of family groups on inferences made with the program STRUCTURE. Mol Ecol Res 8:1219–1229
Austerlitz F, JungMuller B, Godelle B, Gouyon PH (1997) Evolution of coalescence times, genetic diversity and structure during colonization. Theor Popul Biol 51:148–164
Baccus R, Ryman N, Smith MH, Reuterwall C, Cameron D (1983) Genetic variability and differentiation of large grazing mammals. J Mammal 64:109–120
Bishop MD, Kappes SM, Keele JW et al (1994) A genetic linkage map for cattle. Genetics 136:619–639
Buchanan FC, Crawford AM (1993) Ovine microsatellites at the OarFCB11, OarFCB128, OarFCB193, OarFCB226 and OarFCB304 loci. Anim Genet 24:145
Bunnefeld N, Börger L, Van Moorter B et al (2011) A model-driven approach to quantify migration patterns: individual, regional and yearly differences. J Anim Ecol 80: 466–476
Charlier J, Laikre L, Ryman N (2008) Genetic structure and evidence of a local bottleneck in moose in Sweden. J Wild Manag 72:411–415
Chen C, Durand E, Forbes F, Francois O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Collett R (1912) Norges patterdyr (in Norwegian). H.Aschehoug & Co (W. Nygaard), Kristiania, p 744
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cornuet JM, Santos F, Beaumont MA et al (2008) Inferring population history with DIY ABC: a user-friendly approach to Approximate Bayesian Computation. Bioinformatics 24:2713–2719
Di Rienzo A, Peterson AC, Garza JC et al (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Durand E, Jay F, Gaggiotti OE, Francois O (2009) Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26:1963–1973
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Gen 92:832–839
Estoup A, Jarne PJ, Cornuet JM (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetic analysis. Mol Ecol 11:1591–1604
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13:853–864
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Francois O, Ancelet S, Guillot G (2006) Bayesian clustering using hidden markov random fields in spatial population genetics. Genetics 174:805–816
Francois O, Durand E (2010) Spatially explicit Bayesian clustering models in population genetics. Mol Ecol 10:773–784
Fu YX, Chakraborty R (1998) Simultaneous estimation of all the parameters of a stepwise mutation model. Genetics 150:487–497
Garel M, Solberg EJ, Sæther BE, Grøtan V, Tufto J, Heim M (2009) Age, size, and spatiotemporal variation in ovulation patterns of a seasonal breeder, the Norwegian moose (Alces alces). Am Nat 173:89–104
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Geist V (1998) Deer of the World: Their Evolution Behaviour and Ecology. Swan Hill Press, UK
Gill R (1990) Monitoring the status of European and North American Cervids. United Nations environment programme, Nairobi, KE
Goossens B, Chikhi L, Ancrenaz M et al (2006) Genetic signature of anthropogenic population collapse in orang-utans. PLoS Biol 4:285–291
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices. Release 2.9.3.2. Available from http://www.unil.ch/izea/softwares/fstat.html
Grøtan V, Sæther BE, Lillegård M, Solberg EJ, Engen S (2009) Geographical variation in the influence of density dependence and climate on the recruitment of Norwegian moose. Oecologia 161:685–695
Groves CP, Grubb P (1987) Biology and management of the Cervidae. In: Wemmer CM (ed) Relationships of living deer. Smithsonian Institution Press, Washington DC, pp 21–59
Hartl DL, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer, Sunderland
Herfindal I, Solberg EJ, Sæther BE, Høgda KA, Andersen R (2006a) Environmental phenology and geographical gradients in moose body mass. Oecologia 150:213–224
Herfindal I, Sæther BE, Solberg EJ, Andersen R, Høgda KA (2006b) Population characteristics predict responses in moose body mass to temporal variation in the environment. J Anim Ecol 75:1110–1118
Hewitt G (1999) Post-glacial re-colonisation of European biota. Biol J Linn Soc 68:87–112
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hewitt G (2004) Genetic consequences of climate oscillations in the Quaternary. Phil Trans R Soc Lond B 359:183–195
Hufthammer AK (2006) The vertebrate fauna of eastern Norway—form the ice age to the middle ages. Skrifter (Kulturhistorisk Museum, Oslo) 4:191–202
Hulme DJ, Silk JP, Redwin JM, Barendse W, Beh KJ (1994) Ten polymorphic ovine microsatellites. Anim Genet 25:434–435
Ibrahim KM, Nichols RA, Hewitt GM (1996) Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77:282–291
IPCC (2001) Third assessment report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge
IPCC (2007) Fourth assessment report of the Intergovernmental Panel on Climate Change. http://www.ipcc.ch
Jaarola M, Tegelström H, Fredga K (1999) Colonisation history in Fennoscandian rodents. Biol J Linn Soc 68:113–127
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Lavsund S, Nygrèn T, Solberg ES (2003) Status of moose populations and challenges to moose management in Fennoscandia. Alces 39:109–130
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Moore SS, Byrne K, Berger KT et al (1994) Characterization of 65 bovine microsatellites. Mamm genome 5:84–90
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Nichols RA, Hewitt GM (1994) The genetic consequences of long-distance dispersal during colonization. Heredity 72:312–317
Nunney L (2000) The limits to knowledge in conservation genetics; The value of effective population size. In: Clegg MT, Hecht MK, MacIntyre RJ (eds) The limits to knowledge in conservation genetics. Kluwer, New York, pp 179–194
Nygrén T (1987) The history of moose in Finland. Swedish Wildl Res Suppl 1:49–54
Ohta T, Kimura M (1973) A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet Res 22:201–204
Okello JBA, Wittemyer G, Rasmussen HB et al (2008) Effective population size dynamics reveal impacts of historic climatic events and recent anthropogenic pressure in African elephants. Mol Ecol 17:3788–3799
Parmesan C (2006) Responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 6:288–295
Peakall R, Ruibal M, Lindenmayer DB (2003) Spatial autocorrelation analysis offers new insight into gene flow in the Australian bush rat, Rattus fuscipes. Evolution 57:1182–1195
Pertoldi C, Bijlsma R, Loeschcke V (2007) Conservation genetics in a globally changing environment: present problems, paradoxes and future challenges. Biodivers Conserv 16:4147–4163
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (Version-1.2)—Population-Genetics Software for Exact Tests and Ecumenicism. J Hered 86:248–249
Røed KH (1998) Microsatellite variation in Scandinavian Cervidae using primers derived from Bovidae. Hereditas 129:19–25
Røed KH, Midthjell L (1998) Microsatellites in reindeer, Rangifer tarandus, and their use in other cervids. Mol Ecol 7:1773–1778
Rosenberg N, Mahajan S, Ramachadran S et al (2005) Clines, clusters, and the effect of study design on the inference of human population structure. PLOS 1:660–671
Rosvold J (2006) Do single prehistoric human dvelling sites reveal changes in the relative abundance of moose and red deer in Western Norway? Master thesis, Norwegian University of Science and Technology
Ryman N, Reuterwall C, Nygren K, Nygren T (1980) Genetic variation and differentiation in Scandinavian moose (Alces alces): are large mammals monomorphic? Evolution 34:1037–1049
Sæther BE, Solbraa K, Sødal DP, Hjeljord O (1992) Sluttrapport Elg-Skog-Samfunn. Norwegian Institute for Nature Research, Forskningsrapport 28 (in Norwegian)
Sæther BE, Andersen R, Hjeljord O, Heim M (1996) Ecological correlates of regional variation in life history of the moose Alces alces. Ecology 77:1493–1500
Sæther BE, Engen S, Solberg EJ (2009) Effective size of harvested ungulate populations. Anim Conserv 12:488–495
Scandura M, Iacolina L, Crestanello B et al (2008) Ancient vs. recent processes as factors shaping the genetic variation of the European wild boar: are the effects of the last glaciation still detectable? Mol Ecol 17:1745–1762
Schwartz MK, McKelvey KS (2009) Why sampling regime matters: the effect of sampling scheme on landscape genetics. Conserv Genet 10:441–452
Serre D, Pääbo S (2004) Evidence of gradients of human genetic diversity within and among continents. Genome Res 14:1679–1685
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Smouse PE, Peakall R (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetics structure. Heredity 82:561–573
Solberg EJ, Loison A, Gaillard JM, Heim M (2004) Lasting effects of conditions at birth on moose body mass. Ecography 27:677–687
Solberg EJ, Grøtan V, Rolandsen CM, Brøseth H, Brainerd S (2005) Change-in-sex-ratio as an estimator of population size for Norwegian moose. Wildl Biol 11:91–100
Solberg EJ, Rolandsen CM, Heim M et al (2006) Elgen i Norge sett med jegerøyne. En analyse av jaktmaterialet fra overvåkningsprogrammet for elg og det samlede sett elg-materialet for perioden 1966–2004. NINA Rapport 125 (In Norwegian), available at http://www.nina.no/archive/nina/PppBasePdf/rapport/2006/125.pdf
Solberg EJ, Røed KH, Flagstad Ø et al (2009) Elgens genetiske struktur i Norge (in Norwegian). Nina rapport 467
Spiegelhalter SD, Best NG, Carlin BP, Linde AVD (2002) Bayesian measures of model complexity and fit. J R Statist Soc B 64:583–639
Storz JF, Beaumont MA (2002) Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution 56:154–166
Swarbrick PA, Dietz AB, Womack JE, Crawford AM (1992) Ovine and bovine dinucleotide repeat morphism at the MAF46 locus. Anim Genet 23:182
Taberlet P, Bouvet J (1994) Mitochondrial DNA polymorphism, phylogeography, and conservation genetics of brown bear (Ursus arctos) in Europe. Proc R Soc Lond B Biol Sci 255:195–200
Taberlet P, Swenson JE, Sandegren F, Bjärvall A (1995) Localization of a contact zone between two highly divergent mitochondrial DNA lineages of the brown bear (Ursus arctos) in Scandinavia. Conserv Biol 9:1255–1261
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonisation routes in Europe. Mol Ecol 7:453–464
Thörnqvist M (1997) Historien bakom 100 års älgstatistikk. Svensk Jakt 1:4–12 (in Swedish)
Wang JL, Caballero A (1999) Developments in predicting the effective size of subdivided populations. Heredity 82:212–226
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer, Sunderland
Wilson GA, Strobeck C, Wu L, Coffin J (1997) Characterization of microsatellite loci in caribou Rangifer tarandus, and their use in other artiodactyls. Mol Ecol 6:697–699
Wright S (1978) Evolution and the genetics of populations. Volume 4 Variability within and among natural populations. University of Chicago Press, Chicago
Acknowledgments
We acknowledge the Directorate for Nature Management, the Norwegian Research Council and a large number of municipality wildlife boards for funding of the project, and thank R. Andersen, Ø. Flagstad, M. Heim and C. M. Rolandsen for professional input during the project. For help handling samples in the laboratory we are in debt to Liv Midthjell at the Norwegian School of Veterinary Science, as well as E. Bjørkvoll, T. Moe and M. G. Hansen at NINA. For help with clarifying questions about the applied statistical models we thank Eric Durand (TESS). We are in debt to all the moose hunters and local moose managers that collected tissue samples from their moose kills and organised the sampling in the field. Finally, we thank Associate Professor David Griffiths at the Norwegian School of Veterinary Sciences for linguistic proof reading.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Haanes, H., Røed, K.H., Solberg, E.J. et al. Genetic discontinuities in a continuously distributed and highly mobile ungulate, the Norwegian moose. Conserv Genet 12, 1131–1143 (2011). https://doi.org/10.1007/s10592-011-0214-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0214-0