
Abstract
Southeast Asia experiences some of the highest deforestation in the world. Loss of tropical forest typically leads to widespread habitat fragmentation, with detrimental effects on dispersal ability and gene flow—particularly for large carnivores. We conducted mtDNA and microsatellite analysis to assess—for the first time—contemporary patterns of genetic diversity in the Malayan tiger. We collected 295 suspected carnivore samples in Peninsular Malaysia, from which we identified 26 as originating from tiger using 16 polymorphic microsatellite loci, comprising 22 individual tigers. Despite limitations of the study, our findings suggest tiger subpopulations in the north of the peninsula maintain some genetic connectivity and migration between two putative geographic subpopulations in the Main Range and Greater Taman Negara, with negligible population segregation due to dispersal barriers such as road infrastructure. We identified consistently lower levels of genetic diversity in tigers in the Greater Taman Negara region compared to tigers in the Main Range and small but emerging differences in nuclear and mitochondrial genetic diversity. Our mtDNA haplotype and nuclear DNA analyses suggest the levels of genetic diversity in Malayan tigers may be amongst some of the lowest of the surviving tiger subspecies, though the study is limited both in scale and genomic loci. Our findings are consistent with an expected lag between the rapid decline of tigers in Peninsular Malaysia by over 95% in the last 70 years and observed differences in their levels of genetic diversity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetic diversity is fundamental for species adaptation (Hoban et al. 2020), and is especially relevant to populations undergoing severe decline where genetic diversity can be rapidly lost and contribute to population extinction (Markert et al. 2010). A sudden population crash (or ‘bottleneck’) can cause a rapid loss of alleles during a species’ recent demographic history (Schmidt-Küntzel et al. 2018). The heightened extinction risk of endangered species is often caused by anthropogenic activities such as habitat loss, fragmentation, and excessive poaching for the illegal wildlife trade (Ten et al. 2021).
A reduction in population size and restricted gene flow typically leads to increased inbreeding compounded by loss of genetic diversity within small and fragmented populations (Frankham et al. 2002). This can result in a range of negative genetic consequences, including inbreeding depression (such as loss of population- and individual-level fitness), susceptibility to disease, and a reduced ability to adapt to changing environmental conditions (McManus et al. 2015; Kleinhans and Willows-Munro 2019; Labisko et al. 2019; Hardouin et al. 2021). Through these processes, biodiversity loss has been particularly severe in tropical forest ecosystems in the last few decades (Symes et al. 2018; Hansen et al. 2020).
The global population of tigers (Panthera tigris) has declined dramatically due to habitat loss and fragmentation, human persecution, and prey depletion (Ten et al. 2021). As a result, tigers now occupy less than 7% of their global historic range (Suttidate et al. 2021). Conservation scientists generally recognise six extant subspecies: the Bengal tiger (P. t. tigris, Endangered), Amur or Siberian tiger (P. t. altaica, Endangered), Sumatran tiger (P. t. sumatrae, Critically Endangered), Indochinese tiger (P. t. corbetti, Endangered), Malayan tiger (P. t. jacksoni, Critically Endangered), and South China tiger (P. t. amoyensis, Critically Endangered/possibly Extinct in the Wild) (Liu et al. 2018). Three subspecies recognised on the basis of morphology are now extinct: the Bali tiger (P. t. balica), Java tiger (P. t. sondaica), and Caspian tiger (P. t. virgata) (Goodrich et al. 2022). Although the taxonomic status of the nine subspecies has been disputed (Kitchener and Yamaguchi 2010; Wilting et al. 2015), the Malayan tiger is currently recognised by most global conservation bodies based on mitochondrial DNA (mtDNA) and microsatellite genetic evidence (Luo et al. 2004) and whole genome sequencing data (Liu et al. 2018), as well as the need to define each subspecies according to its unique set of strategic conservation requirements (Liu et al. 2018). The Malayan tiger’s recognition as a unique subspecies aligns with its biogeographic history of gradual segregation from Indochinese tigers in northern Indochina (northern Thailand, Myanmar, Laos PDR, Vietnam and Cambodia) by the Isthmus of Kra on the Malay peninsula (central Thailand) (Luo et al. 2004).
The Malayan tiger (P. t. jacksoni) is listed on the IUCN (International Union for Conservation of Nature) Red List as Critically Endangered (Kawanishi 2015), and currently represents 5% of the remaining global tiger population (Biswas et al. 2020). This globally important but dwindling tiger population comprises fewer than 150 individuals, with the wild population now heavily fragmented in isolated pockets of rainforest after seven decades of intensive hunting and habitat loss (Ten et al. 2021; Abdullah 2022). To date, an absence of a population genetic study for the wild population of Malayan tiger has meant the extent of population connectivity and gene flow between fragmented subpopulations are unknown, at a crucial time when such information could inform future habitat management and conservation strategy.
It is important for conservation managers tasked with recovering the Malayan tiger to know what levels of genetic diversity exist within wild populations and understand how that genetic diversity is distributed across their fragmented landscape. This knowledge can help inform conservation actions that seek to maximise the tiger’s ability to adapt to complex and changing landscapes (McManus et al. 2015), and prioritise the management of key populations and habitats so that threats towards tigers can be effectively mitigated. Information on patterns of gene flow and population differentiation can aid in guiding decisions on how best to improve the connectivity of natural habitats, reunite fragmented subpopulations, slow the rate of loss of genetic diversity, and encourage species’ long-term population viability (Thapa et al. 2018; Mueller et al. 2020; Kopatz et al. 2021). Conservation actions to inform these decisions include enhancing our understanding of the effects of human and environmental barriers to population connectivity (Sharma et al. 2013), understanding patterns of tiger movement, translocation and gene flow across heterogeneous landscapes (Sarkar et al. 2016; Mardiastuti 2018), and the strategic targeting of resources towards creating or reinforcing habitat corridors to reconnect isolated subpopulations (Dutta et al. 2018).
Here, we use non-invasive methods to quantify levels of genetic diversity in subpopulations of the Malayan tiger. We applied a suite of microsatellite DNA markers used in previous tiger studies and mtDNA sequencing of the cytochrome-b gene to: (i) quantify nuclear and mitochondrial DNA genetic diversity across remaining strongholds in the Malayan tiger’s northern range, (ii) compare levels of genetic diversity to those documented for other tiger subspecies, and (iii) interpret observed patterns of genetic diversity and structure in relation to the current backdrop of habitat loss and fragmentation in Peninsular Malaysia.
Methods
Study area
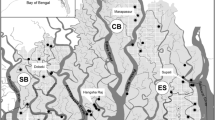
We conducted intensive field surveys in the Belum Temengor Forest Complex (2900 km2) in Perak State, Peninsular Malaysia, between May 2019 and March 2020. This landscape comprises a 1175 km2 protected primary forest (Royal Belum State Park) and a 1440 km2 timber production forest (Temengor Forest Reserve), both comprising the Belum Temengor Forest Complex (BTFC). The forest complex is bisected by a public highway, with the remaining strip of forest either side of the highway recognised as a habitat corridor known as Primary Linkage 2 (PL2/A-PL1) (Fig. 1). Additional old and contemporary tiger scat, spray and blood samples were sourced from the Main Range (MR), Taman Negara National Park and Kenyir.
Malaysia’s wildlife corridors (Fig. 1) were originally identified in areas where it was deemed crucial to re-establish forest connectivity, under the Central Forest Spine (CFS) Masterplan for Ecological Linkages (DTCP 2009). Linkages tend to take the form of linear corridors (i.e. unbroken stretches of forested habitats connecting forest islands which, due to their locations, are critically important for large mammals to move from one forest patch to another). The Belum Temengor Forest Complex is a priority area for wildlife conservation and lies within a Class 1 Tiger Conservation Landscape (TCL 16), which is a national and global priority for tiger conservation (Sanderson et al. 2010).

Wildlife corridors, major roads, and tiger sample locations. Sample locations include old samples donated to this study and new samples collected during field surveys
Sample collection
We implemented a programme of intensive sample collection in the BTFC only, as surveys were limited to this site due to COVID-19 restrictions. Surveys yielded 113 scat and one environmental DNA (eDNA, soil) sample. An additional 182 DNA samples were contributed by the following organisations from locations across northern Peninsular Malaysia: the National Wildlife Forensics Laboratory of Department of Wildlife and National Parks Peninsular Malaysia (n = 4, blood and n = 1, scat), WWF-Malaysia (n = 138, scat), Perak State Parks Corporation Rangers (n = 5, scat), Rimba (n = 1, scat), Panthera Malaysia (n = 12, eDNA, spray) and Malayan Rainforest Station (n = 21, scat).
The home range size of the Malayan tiger has yet to be determined through wildlife telemetry techniques, but is expected to be similar to that of the Indochinese tiger (average 77 km2 for females and 280 km2 for males; Simcharoen et al. 2014). Therefore, surveys were conducted across accessible sites spanning the north and south regions of the Belum Temengor Forest Complex to ensure sufficient home range coverage. Approximately 300 km2 of forest to the west of Royal Belum State Park with prohibited or ill-advised access due to army activity or poaching was avoided. To maximise the probability of locating tiger scat in each survey location, and to ensure broad, landscape-scale coverage (Oyler-McCance et al. 2013), we adopted a cluster sampling regime at key tiger sites across the forest complex. This approach involved intensively surveying each site at least once over 1–11 days. We conducted scat searches along 1 km line-transect surveys, targeting landscape features typically used by tigers, including known tiger trails, ridges and riverbeds, based on the higher likelihood of tiger detection indicated by previous surveys (Rayan and Linkie 2015).
DNA extraction
All DNA extraction and sequencing took place at the National Wildlife Forensic Laboratory in Cheras, Kuala Lumpur, with all samples remaining in Malaysia as property of the Department of Wildlife and National Parks Peninsular Malaysia. Samples were prepared in separate dedicated areas for faecal and blood sample preparation to avoid cross-contamination. All surfaces were cleaned prior to extraction with 10% bleach, and all extraction equipment was sterilised under a UV hood and thereafter with 10% bleach before and throughout the extraction process, to limit the possibility of contamination.
We used the Qiagen QIAamp DNeasy Blood and Tissue Kit (Qiagen, Inc.) for faecal and blood sample extraction using the manufacturer’s protocol with the following modifications: (i) an extended initial incubation step (from three hours to overnight homogenisation in an incubator shaker at 56 °C); (ii) an additional 30 µl Proteinese K added during the initial incubation step; (iii) 4 µl of carrier RNA (cRNA) added to 330 µl of AL buffer during the second incubation step; (iv) warming the elution buffer AE at 70 °C; (v) extending the final elution step: to increase DNA yield, 50 µl of warmed elution buffer was added to the spin column containing the sample; (vi) standing eluted DNA at room temperature for 10 min, then spinning for two minutes; (vii) steps (v) and (vi) were repeated to produce a final elution of 100 µl. We found the Qiagen QIAamp DNeasy Blood and Tissue Kit with AL buffer more effective for DNA extraction compared to the Qiagen Stool kit with ASL buffer during initial lab testing, and so continued with the DNeasy Kit for all subsequent extractions. Samples were processed in batches of eight, with extractions repeated between three to six times to maximize the likelihood of obtaining sufficient quantity and quality of DNA for PCR amplification. A negative control was included with each batch to monitor for contamination during both the extraction and PCR process.
Species identification
In addition to tiger, there are six other wild cat species known to currently inhabit the forests of Peninsular Malaysia: Indochinese leopard (Panthera pardus delacouri), Clouded leopard (Neofelis nebulosa), Asian golden cat (Catopuma temminckii), Flat-headed cat (Prionailurus planiceps), Marbled cat (Pardofelis marmorata), and Leopard cat (Prionailurus bengalensis) (Ratnayeke et al. 2018). Therefore, genetic authentication was required to identify those faecal samples originating from tiger and those from non-target felid species. All subsequent analyses used samples that were only from tiger.
To discriminate tiger scats from samples produced by non-target felids, we amplified a mitochondrial cytochrome-b fragment using primer set TIGCYTB2 (5’-CGTCTGTCTATACATGCA-3’, 5’-TACTCTACTAGGTCGGTC-3’) developed by Mondol et al. (2009a). Primers were tested and optimised with positive samples of blood, hair/skin, and extracted faecal DNA from captive Malayan tiger, clouded leopard and leopard. The TIGCYTB2 primer set produced a ~ 200 base-pair fragment of the cytochrome-b gene from faecal samples from captive tiger, clouded leopard, leopard and leopard cat, and was thus included in the initial screening process. To identify and discriminate lesser (small and medium) cat species, we designed a shorter 165 base-pair cytochrome-b primer set GCCYTB (5’-CTTTGGATCCCTGCTAGGAGTC-3’, 5’- GCTCCGTTGGCATGTATGTATC) to amplify in wild faecal samples belonging to Asian golden cat, using the Primer Design function in Geneious Prime v2020.2.4 (Biomatters, Auckland, New Zealand). At least two PCRs were performed for each sample to confirm a positive result, indicated by a single, clear PCR band of the expected size, whilst negative samples produced no band or multiple stutter bands indicating non-specific primer binding. PCR cycling conditions for the initial screening process comprised the following: initial hot start of 95 ºC for 1 min; 45 cycles of 95 °C for 30 s; 52 °C for 15 s; 72 °C for 30 s; final incubation period of 10 min at 72 °C. PCRs were performed in reaction volumes of 20 µl, containing 2 µl of template DNA, 10 µl of MyTaq Hot start Red mix (Matrioux, Malaysia), 0.2 µl of each forward and reverse primer, and 7.6 µl of ddH2O. Positive PCR products were purified using ExoProStar 1-Step PCR Clean up (Illustra, USA), and outsourced to Apical Scientific Malaysia for direct Sanger sequencing. We visually checked, edited and aligned resulting forward and reverse sequences using Geneious Prime v2020.2.4, then compared the aligned sequences with reference sequences for each species from GenBank (National Center for Biotechnology Information, NCBI) for further checking and edits of any ambiguous nucleotides.
A common problem when discriminating between species in the genus Panthera (e.g. tiger and leopard) arises from the presence of pseudogenes (‘numts’): regions of mtDNA which integrate into the nuclear genome, creating nuclear gene ‘copies’ which, when amplified, can cause misidentification of a species (Morgan et al. 2021). To identify stop codons present in the mitochondrial data, we pairwise aligned sequences, and translated consensus sequences into six reading frames and checked for stop codons (represented as an ‘asterix’ in a mitochondrial sequence) which indicates the presence of a pseudogene (presence of two or more stop codons was taken to confirm the presence of a pseudogene). We investigated the presence of a single stop codon in a sequence further by screening the original sequence quality at positions of the three known stop codon motifs: TAG TAA TGA. Unambiguous sequence at these nucleotide positions confirmed the presence of a stop codon. Once amplified pseudogenes were removed, we used the remaining true mtDNA sequences as a search query in GenBank using the Nucleotide Basic Local Alignment Search Tool (BLAST) to confirm species identity.
Mitochondrial gene fragments are often used to assess genetic diversity in tiger populations (Mondol et al. 2009b. To generate mtDNA data from verified tiger DNA samples, three primer sets optimised from Mondol et al. (2009a) were used to amplify fragments from the cytochrome-b gene region: TIGCYTB2, TIGCYTB3, and TIGCYTB4 (Supplementary Information, Table 1). We initially amplified three mtDNA gene regions: control region (CR), cytochrome-b and NADH dehydrogenase subunits 5 (ND5) but due to incomplete amplification of fragments for each gene region, we were unable to concatenate sequences to form complete datasets for each gene region. PCR conditions and reaction volumes were the same as those given above for primer set TIGCYTB2. Sequences were edited and aligned with Geneious Prime v2020.2.4. Samples with multiple fragments of sequence missing were removed from the dataset (n = 4) leaving a final dataset of n = 22 sequences of a single 174 bp cytochrome-b fragment present across all samples.
Microsatellite amplification and genotyping
We tested and optimised a panel of 16 polymorphic microsatellite markers from an initial set of 40 drawn from previous studies on tiger, clouded leopard and leopard (Menotti-Raymond et al. 1999; Bhagavatula and Singh 2006; Mondol et al. 2009a, b, 2015; Singh et al. 2017; Smith et al. 2018). Given the documented severe population decline and likely genetic impoverishment of this species, it was considered necessary to select markers based on polymorphism; therefore, marker selection was based on the following thresholds: minimum number of alleles (k = 5) and minimum level of heterozygosity (HE > 0.6) per locus. The final set of microsatellite markers comprised the following 16 loci: FCA232, FCA304, FCA126, FCA441, FCA628, FCA310, FCA672, FCA043, FCA090, FCA230, FCA279, FCA229, E7, D15, F41, and ZN (Supplementary Information, Table 2). We designed four multiplexes using Multiplex Manager v1.2, to include the full set of 16 loci. All forward primers were fluorescently labelled with HEX, FAM, or ROX dyes (Supplementary Information, Table 3). Each microsatellite PCR reaction volume (10 µl) contained 5 µl Qiagen Multiplex buffer mix (’Qmix’, Qiagen Inc.), 1 µl primer mix, 2 µl BSA, and 2 µl template DNA. Multiplex PCR cycling conditions comprised an initial hot start of 95 ºC for 1 min; 45 cycles of 94 °C for 30 s; 55 °C for 90 s; 72 °C for 90 s; final incubation period of 10 min at 72 °C. We performed a minimum of three and up to six PCRs for each microsatellite locus for every sample confirmed to have originated from tiger.
Alleles were identified and scored using Geneious Prime v2020.2.4. To validate the true genotype of each sample, we adopted a multi-tube method (Taberlet et al. 1996) followed by a comparative genotyping approach (Frantz et al. 2003; Hansen et al. 2008) whereby genotype profiles were used in the event of too few replicates to obtain consensus genotypes for each sample. Peak calls and motifs were checked using the programme CERVUS v3.0.7 (Kalinowski et al. 2007). To assist with the process of differentiating between samples suspected of coming from the same individual, genotypes were checked visually and against known sample history (sample location and date of collection). Samples with matching but incomplete genotypes were then assessed using the following criteria: (1) collected more than three years apart and (2) collected from different locations that extended beyond the average of an Indochinese tiger’s home range (average between male and female of 168 km2 (7 km radius) (Simcharoen et al. 2014) or separated by more than an average dispersal distance of 49 km (Suttidate et al. 2021). Based on these criteria, samples were considered unlikely to be duplicates from the same individual, though we acknowledge that these criteria could result in a positive bias on the extent of inbreeding. We consider three years to be a minimum duration beyond which it is unlikely that two samples collected in the same area will be from the same individual based on documented knowledge of the high level of recent habitat loss and fragmentation and extreme pressure from illegal poaching, whereby the population has crashed by over 95% (Ten et al. 2021). These specificied conditions were met whereby, following these checks, all samples were retained in the dataset because all individuals were deemed to be unique. The presence of null alleles and allele dropout was checked using MicroChecker (Van Oosterhout et al. 2004).
Assessment of nuclear genetic diversity
We designated samples as belonging to one of two geographically defined putative subpopulations: the Main Range (MR), and Taman Negara National Park and surrounding areas, (hereafter referred to as the Greater Taman Negara region, GTN), based on geographic location of samples, and the location of previous sampling efforts largely focusing on these two tiger strongholds. Allele frequencies, observed (Ho) and expected (He) heterozygosity, null allele frequency, and polymorphic information content (PIC), were calculated using CERVUS v3.0.7, GenAlEx v6.503 (Peakall and Smouse 2012), and Microsatellite toolkit (Park 2001) for subpopulation diversity statistics. We tested for deviation from Hardy-Weinberg Equilibrium and linkage disequilibrium using GenePop v4.7x (Rousset 2008). However, as the overall population was unlikely to meet assumptions of HWE (random mating and large outbred population with minimal genetic drift) and likely to be inbred as a consequence of population decline, we retained all loci in the dataset. We quantified allelic diversity for the global (i.e. peninsular) population and each of the two putative subpopulations using FSTAT v2.9.4 (Goudet 1995), which uses rarefaction to calculate allelic richess. The observed number of alleles in a sample is highly dependent on sample size, whereas allelic richness is a measure of the average number of alleles per locus, independent of sample size, thus allowing a comparison between groups with different sample sizes (Trouvae et al. 2003).
Analyses of structure, gene flow and migration
We performed a Principal Component Analysis (PCA) for an initial assessment of population structure. This approach is sensitive to sample size but enables a visual overview of genotype variation across the sample set. We imported genotype data into R studio for PCA analysis using the R package Adegenet v2.1.0 (Jombart 2008). We next performed Bayesian assignment tests with microsatellite data in STRUCTURE v2.3.4 (Pritchard et al. 2000) to explore fine-scale structure of the tiger population across the sampled landscape of northern Peninsular Malaysia. STRUCTURE infers (a) the most likely number of genetic clusters (K) using Markov Chain Monte Carlo (MCMC) algorithms, and (b) which cluster (population) each individual originated from. The analysis was run with correlated allele frequencies using two ancestry models: Admixture model, whereby each individual has ancestry from one or more of K genetically distinct sources and putative subpopulations are not considered completely isolated and some admixture is assumed; and Population Informed (also with admixture), whereby prior population information is used to test for migrants and assign individuals to clusters. Putative subpopulations are not considered completely isolated and admixture is assumed.
We did not use a model without admixture, as it is unlikely that tiger subpopulations are completely discrete, and therefore some mixing is expected. An accurate detection of the number of genetic clusters can be difficult to estimate if sampling effort or sample sizes are uneven. (Puechmaille 2016). Nevertheless, genotype data were split into two putative subpopulations (the GTN region and MR) given the approximately even distribution of samples amongst these two regions. Population IDs were incorporated as sampling location information using the LOCPRIOR model to improve analytical performance in the event that the data were only weakly informative about population structure (Pritchard et al. 2000). We varied ancestry priors for each subpopulation using Wang’s alternative ancestry priors, which infers a different degree of admixture to account for uneven sampling (Wang 2017). Each analysis was performed with 10 independent runs, each time specifying a value for K between 1 and 3, with each run comprising 500,000 MCMC iterations with a burn-in of 50,000. Results were analysed in Structure Selector (Li and Liu 2018) and Structure Harvester (Earl and von Holdt 2012) using (i) ∆K (Evanno et al. 2005), (ii) K clusters (Puechmaille 2016), and (iii) highest average negative log likelihood (x̄ -LnP), whereby the higher the x̄ -LnP, the more likely that the number of clusters assigned is correct (Frantz et al. 2014). ∆K represents the change in x̄ -LnP with an increase in the number of inferred ancestral groups, with the ∆K peak showing the optimal number of inferred groups. The proportions of individual assignment were plotted at the geographical locations for each sample using ArcMap v10.5.1.
We tested for significance of differences detected between populations (groups) using the hierfstat package in R Studio (Goudet 2005). F-statistics (fixation indices) describe the statistically expected level of heterozygosity in a population; more specifically the extent of (usually) a reduction in heterozygosity compared to what we would expect under HWE. We calculated F-statistics in GenePop v4.7x: allele frequency-based correlation: one-locus estimates following standard ANOVA (Weir and Cockerham 1984), and Fst (global Fst) to describe the level of genetic differentiation across the landscape between subpopulations (MR and GTN).
To investigate contemporary immigration among the two putative subpopulations, we used the programme BayesAss v.3.0 (Wilson and Rannala 2003). BayesAss assumes linkage equilibrium and relaxes the assumption that populations are in HWE. The analysis was run with default settings of seed = 10, 3 × 106 iterations, 1 × 106 burn-in, and samples collected every 2000 iterations (Sharma et al. 2013). Using BayesAss we inferred (posterior mean) migration rates between the two subpopulations, whereby mij is the fraction of individuals in population i that are migrants derived from population j (per generation) (Shriver et al. 1993; Li and Chakraborty 1995; Smith 2012). We tested for the presence of a recent reduction in effective population size (< 4Ne generations in the past), using the programme BOTTLENECK v1.2.02 (Cornuet and Luikart 1996) which assumes that populations that have undergone a dramatic reduction in population size will have lost a large number of alleles, but with limited effect on heterozygosity (e.g. Biswas 2021). Non-bottlenecked populations are assumed to be at mutation-drift equilibrium and so each locus will have an equal probability of heterozygosity excess or deficiency. We used the stepwise mutation model (SMM) which microsatellites are considered to follow more reliably (Shriver et al. 1993; Li and Chakraborty 1995; Smith 2012), whereas the infinite allele model (IAM) tends to predict a lower equilibrium value for heterozygosity and is more likely to indicate that a significant heterozygosity excess exists (Luikart et al. 1998). Significant deviations from the mutation-drift equilibrium are assessed by the probability given by three tests: the Standardised differences test, Wilcoxon test, and Sign test. The one-tailed Wilcoxon text is used to detect excess heterozygosity (HE): a probability of p < 0.05 would allow accepting the hypothesis of a genetic bottleneck within the analysed data. The Standardised differences test requires at least 20 loci to yield reliable results, whilst the Sign test and Wilcoxon test require less (at least four polymorphic loci for the Wilcoxon test) to detect a significant (p < 0.05) bottleneck signal. The standardised differences test and Wilcoxon test are applied to estimate the probability of heterozygosity excess under three mutation models: IAM, SMM, and two-phase mutation model (TPM). Given these different test requirements, we compared results for the heterozygote excess method (Hx) for the IAM, SMM, and TPM mutation models with 10,000 iterations. Significance values for multiple comparisons were adjusted using the Bonferroni correction.
Assessment of mitochondrial genetic diversity
To compare levels of genetic diversity between Malayan tigers and other subspecies, we calculated haplotype and nucleotide diversity measures for the wild populations of five of the six remaining tiger subspecies (Table 1): the Amur tiger (P. t. altaica), Northern Indochinese tiger (P. t. corbetti), South China tiger (P. t. amoyensis), Malayan tiger (P. t. jacksoni), Sumatran tiger (P. t. sumatrae), and Bengal tiger (P. t. tigris). The South China tiger was excluded from the dataset, as it is thought to survive now only in captivity (Seidensticker et al. 2010). Measures of haplotype diversity, nucleotide diversity, and segregating sites for the Malayan tiger were estimated using the DNA Polymorphism function in DnaSP v6.12.03 (Rozas et al. 2017). Sequences for other tiger subspecies were obtained from GenBank for comparison (Rovie-Ryan unpubl. data; Dubach et al. 2013; Cracraft et al. 1998, Supplementary Information, Table 4).
The combined datasets contained 103 unique cytochrome-b sequences. Sequences were initially aligned using Geneious Prime v2020.2.4 (Biomatters, Auckland, New Zealand) and shared sequence fragments then analysed. Malayan tiger samples from this study were analysed separately for subpopulation (geographical clusters) and subpopulation comparisons. We constructed haplotype networks to assess relationships between the Malayan tiger and other tiger subspecies. Networks were constructed using TCS V. 1.21 (Crandall et al. 2000) with a 95% connection limit and gaps treated as a fifth state. TCS networks were illustrated using TCSBU (Múrias dos Santos et al. 2016).
Results
A total of 295 suspected carnivore samples were collected over 12-months, including fresh scat (n = 140), DNA from a fresh pug mark (n = 1), old scat samples between five and 12 years old (n = 138; collected 2009–2016), old tiger spray samples (n = 12; collected 2014–2015), and old blood samples (n = 4; collected 2010–2019, Supplementary Information, Table 5). Fresh scats were found primarily on old logging roads and naturally established animal trails through disturbed and undisturbed forest. Here we define fresh scat as faecal material estimated to have been deposited within one month prior to collection, with estimated age based on appearance. Most fresh scat samples were heavily degraded due to high temperature and humidity, and in some cases only prey remains (hair clump and bone fragments, for example) with little faecal matter remaining. Old scats available for this study were up to 10 years old, the majority of which were also degraded.
DNA amplification was a significant challenge in this study due to low sample detection and subsequent small sample size consisting of mainly poor quality degraded scat samples, leading to a very low DNA amplification success rate (Alam et al. 2021; e.g. Thapa et al. 2018). Nonetheless, amplification success often improved when scat samples were thoroughly air-dried, additional cRNA and ProK added during the extraction process, and positive PCR products purified using ExoProStar 1-Step PCR Clean up (Illustra, USA), though the difficulties associated with DNA extraction from heavily degraded scat samples from humid forest environments meant these patterns were inconsistent. Such issues concerning degraded DNA extraction and downstream genotyping of tiger samples remains a significant challenge and is well-documented in the literature (Alam et al. 2021). 26 samples were authenticated as having originated from tiger. 22 of these samples (84%) yielded enough DNA of sufficient quality for microsatellite analysis and were used in this study. 18 samples (69%) amplified at five or more loci, and 22 samples (84%) yielded sufficient DNA sequence for analysis of mtDNA variation.
Descriptive statistics, inbreeding and bottleneck events
No linkage disequilibrium was found between pairs of microsatellite loci. Significant deviations from HWE after Bonferroni correction were detected at nine loci (441, 232, 090, 279, 043, 229, 628, 310 and 672) (i.e. 56% not in HWE). Following testing of all loci for scoring error, allelic dropout and null alleles, three loci (232, 090, 229) produced significant evidence of null alleles (i.e. 18% with a positive value of ≥ 0.2 and thus a null allele frequency of 20% or more) due to an excess of homozygotes (Supplementary Information, Table 6), though results should be treated with caution due to missing data. To correct for this, adjusted allelic frequencies and genotypes were calculated using the correction algorithm by Van Oosterhout et al. (2004). Corrected genotypes produced comparable results (Table 2) and were used in downstream analysis. Overall, mean observed heterozygosity (Ho) was 0.35 ± SD 0.036 and unbiased expected heterozygosity (uHe) was 0.60 ± SD 0.050 (Table 2). Mean number of alleles per locus was 3.88 ± 1.45. Mean polymorphic information content (PIC) was 0.50.
Unbiased expected heterozygosity (uHe) Nei (1978), observed heterozygosity (Ho), allelic richness, (Ar). Subpopulations: Greater Taman Negara (GTN), Main Range (MR), and Northern Peninsular tiger subpopulation (Global).
Whilst mean He and Ho appeared lower in tigers from the GTN region compared to the MR (GTN – n = 13, uHe 0.45, Ho 0.25, and MR – n = 9, uHe 0.60, Ho 0.40 respectively), the observed difference was not significant (He p = 0.088 and Ho p = 0.139) (Supplementary Information, Fig. S1). Allelic richness was also observed to be higher in the MR tiger subpopulation (3.56) compared to the GTN region (1.81) and the difference was significant (p = 0.000).
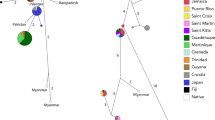
We found expected heterozygosity (He) in Malayan tigers to be comparable with other tiger subspecies but observed heterozygosity (Ho) to be lower - particularly compared to Indochinese and Sumatran tiger subspecies, which have also experienced population decline and isolation (Fig. 2 and Supplementary Information, Table 7). Mean Ho in Malayan tigers is 55% lower than expected. Overall allelic richness was lower in Malayan tigers (3.88 ± SD 1.45), compared to the Bengal tiger subspecies in India (7.30 ± SD 2.25, Luo et al. 2004), Bangladesh (5.5 ± SD 1.65, Aziz et al. 2017) and Indochinese tigers in Northern Indochina (6 ± SD 1.81, Luo et al. 2004), but higher compared to Sumatran tigers in Sumatra (3.60 ± SD 1.48, Luo et al. 2004) and Amur tigers in the Russian Far East (3.47 ± SD 1.22, Luo et al. 2004). To the best of our knowledge, markers used across published studies were directly comparable, however there may be some differences due to ascertainment bias. Tests for significance were also not possible because raw genotype data were not available for all subspecies.

Bar charts illustrating genetic diversity and genetic impoverishment between tiger subspecies with 95% confidence intervals represented by error bars. Circular maps provide visual representations of each subspecies’ range. Southeast Asian subspecies are grouped first (browns; Malayan tiger (this study) in red outline), Bengal tigers from three different geographic locations (Nepal, Bangladesh, India) are grouped second (blues), and Amur tigers are grouped third (pale grey). Tests for significance between subspecies were not possible because raw genotype data were not available for all studies. Diversity metrics were taken from Luo et al. (2004), Aziz et al. (2017), and Thapa et al. (2018)
Estimation of the ‘inbreeding coefficient’ of an individual with respect to its local subpopulation (inbreeding as non-random mating) generated an Fis of 0.463 for the Malayan tiger. This observed level of inbreeding was relatively high in comparison to other tiger subspecies (Table 3). BOTTLENECK detected a significant signal for heterozygosity excess for the entire data set under the IAM mutation model (p = 0.05, Bonferroni-adjusted p = 0.02) with the standardised differences test, however our dataset comprised only 16 loci which is below the minimum recommended (n = 20) (Cornuet and Luikart 1996), and results should thus be considered with caution. However, all other tests for excess heterozygosity found no evidence of a recent bottleneck (p > 0.05). The SMM model of mutation, which provides a more conservative assessment and is considered to reflect microsatellite mutation most closely, did not find evidence of a recent bottleneck.
Population structure, gene flow and migration
We performed principal component analysis (PCA) based on 22 individuals, with samples grouped into either GTN or MR region (Supplementary Information, Fig. S2). Overlap of the centres of the two PCA ellipses indicates similar genetic composition between the two putative subpopulations, though the overlap is not total. The larger-sized ellipse for the MR subpopulation reflects the higher levels of genetic diversity observed there. To deal with missing genotype data, the PCA analysis used mean imputation, whereby missing values were replaced with mean values for the observed allele frequencies of each loci. Whilst this is one approach to dealing with missingness, visualising population structure with mean-imputed missing data may bias the results (Yi and Latch 2022). Although the PCA suggests there is little differentiation in the population and is therefore consistent with other findings in the study, this result should be treated with caution.
STRUCTURE analysis revealed mixed results for each evaluation method. ∆K (Evanno method) consistently inferred structure with two distinct clusters (k = 2). Kplots (Puechmaille method) and x̄-LnP values inferred no structure (k = 1) for the Admixture model and Population Informed model, and STRUCTURE bar plots were consistent with this result (Supplementary Information, Fig. S3). BayesAss analysis indicated a marginally higher level of contemporary migration from MR to GTN (m = 0.2606, 95% CI: − 0.080 ± 0.104), compared to migration in the opposite direction (GTN to MR: m = 0.2365, 95% CI: − 0.138 ± 0.176).
Estimation of average inbreeding relative to the total population (i.e. inbreeding due to population subdivision) generated an Fst of − 0.022 for the Malayan tiger, indicating little or no differentiation between putative subpopulations.
Mitochondrial diversity
In this study, we constructed two hapolotype networks: one specific to the Malayan tiger (174 bp sequence) and a second network to include all tiger supspecies (shared 136 bp sequence), though results should be treated with caution due to limitations of sequence data. We constructed the first haplotype network from a 174 bp cytochrome-b sequence, obtained from 22 tiger samples from Peninsular Malaysia, revealing a total of eight haplotypes (H1-H8, Fig. 3), of which two (H3 and H5) were unique to GTN and two (H4 and H8) were unique to MR. We compared the newly-collected Malayan tiger cytochrome-b sequences in this study to mtDNA haplotypes in Luo et al. (2004) (COR4, COR5, COR6, COR7, COR8) and J. Ryan unpubl. data (EF17935-EF179376 and EU184691-EU184702), and found new sequences from this study overlap with previously discovered haploytypes from Luo et al. (2004) (Supplementary Information, Fig. S4).

Haplotype map for the Malayan tiger containing eight haplotypes (H1 to H8). Coloured circles represent individual tiger samples collected from two geographic areas: the Main Range (orange) and Greater Taman Negara (dark blue). The haplotype network (inset) displays each haplotype as a pie chart, with light/dark blue colour corresponding to the origin of samples belonging to each haplotype (MR or GTN)
We constructed a second haplotype network (Supplementary Information, Fig. S5) from a 136 bp cytochrome-b sequence, available for 103 tiger samples across five subspecies. This network revealed 14 unique haplotypes across all tiger subspecies (H9-H22). One haplotype (H9) represented 39 individuals and was unique (95%) to all subspecies found in Southeast Asia (Malayan, Indochinese, Sumatra) and Amur tigers. Two individual Bengal tigers (5%) shared haplotype H9. Six haplotypes were unique to Malayan tigers: one with six individuals (H10), and the remaining five haplotypes (H11, H12, H13, H14, H15) each with just one individual. Two further haplotypes (H16, H17) were unique to Malayan tigers from a previously unpublished study (Rovie-Ryan unpubl. data), and one (H18) was shared by Sumatran and Bengal tigers.
Haplotype diversity was marginally lower in the MR (h = 0.600) compared to the GTN region (h = 0.709), but nucleotide diversity was marginally higher in the MR compared to the GTN area (π = 0.024 and 0.200, respectively). The Malayan tiger exhibited substantially lower haplotype diversity (upph = 0.260) in comparison to Indochinese, Sumatran and Bengal subspecies (h = 0.691, 0.378 and 0.694, respectively). Malayan tigers have marginally higher nucleotide diversity (π = 0.009) compared to Indochinese tigers (π = 0.007), but notably lower than Sumatran and Bengal tigers (π = 0.018 and 0.024, respectively) (Fig. 4; Table 1).

Bar plots of nucleotide diversity (π) and haplotype diversity (h) between five wild tiger subspecies. The last two bars (pale grey, separated by a dashed line) provide π and h for two putative subpopulations in Peninsular Malaysia, and are given for comparison. Circular maps provide visual representations of each subspecies’ range
Discussion
Here we provide the first insights into the extent and distribution of genetic diversity in the wild tigers of Peninsular Malaysia. Where previous genetic assessments have been conducted on a combination of wild and captive individuals (e.g. Luo et al. 2004), data available for this study was limited by a number of factors, and findings should thus be interpretated appropriately. Nonetheless, we offer fresh information on the contemporary genetic diversity of Malaysia’s remaining wild tigers.
Malayan tigers have declined by 97% and have become increasingly fragmented in the last 70 years (Ten et al. 2021). Although overarching results point to a connected population, the patterns of contemporary nuclear and mitochondrial DNA diversity we have observed in tigers between the two regions allow us to consider that these are very early signals of differentiation - driven by the recent extensive habitat fragmentation and anthropogenic change in land use on the Malay peninsula. This interpretation is consistent with the expectation that at least 10 generations are required to detect changes in levels of genetic diversity and population structure (Mona et al. 2014). For Malayan tigers, nine to ten generations have passed since the onset of population decline in the 1950s (Goodrich et al. 2022; Ten et al. 2021).
Interpretation of population connectivity
Genetic structure develops within a species when random mating ceases and subpopulations form, among which gene exchange via dispersal or mating is impeded (Waples and Gaggiotti 2006; Colonna et al. 2009). STRUCTURE and PCA analysis indicated no evidence of population structure and the pairwise population Fst of − 0.022 also suggested the population is unstructured. However, speculatively, the observed patterns of genetic diversity may be early indications of the impact of genetic drift but tempered with continued dispersal and gene flow. Both Admixture and Population Informed models consistently inferred a cluster of k = 2 under ∆K (Evanno method). This result is consistent with the observation that there are private alleles predominantly found in the GTN region-a possible indication that structure could be emerging as a consequence of loss or fixation of alleles caused by genetic drift, or as a consequence of uneven genotyping success across samples. Indeed, three of the 16 loci (loci 229, 230 and 310) contain private alleles unique to GTN. However, Kplots consistently inferred k = 1. This finding is also consistent with the PCA which found no distinct population structure.
Migration appears to be marginally higher (though not significantly so) from the MR to GTN, which possibly supports the notion that the contiguous highlands that form the backbone of the Main Range serve as a functional corridor that allows tigers to disperse to other areas more easily (Mohamad et al. 2013). We found levels of migration and inferred gene flow between tiger subpopulations in Peninsular Malaysia to be generally high when compared with similar tiger studies in Nepal (Thapa et al. 2018) and Sumatra (Smith et al. 2018). These observed levels of migration are consistent with the STRUCTURE result of observable gene flow between the two putative subpopulations.
Differences in levels of genetic diversity within Peninsular Malaysia
Despite limitations concerning the scale of the study and panel of loci, we observed consistently lower levels of genetic diversity in the GTN region compared to the MR, in terms of He, Ho and Ar, although differences between these two putative subpopulations were not significant. It is possible that statistical power was reduced due to small sample size (n = 22), and the continued collection of samples will help improve this for future assessments. We speculate that the levels of inbreeding observed to be occurring in both subpopulations are consistent with a recent period of population isolation due to contemporary habitat fragmentation, however the exact magnitude of observed inbreeding may be exaggerated by the presence of null alleles or allelic dropout, and proportion of missing data.
Differences in the extent of isolation and presence of natural corridors between these two areas suggests that greater effects of genetic drift on the increasingly fragmented GTN subpopulation-particularly at the peripheries-may have precipitated a more rapid rate of loss of genetic diversity compared to tigers in the MR. Whilst GTN covers a larger area, the region is becoming increasingly fragmented due to forest loss and conversion of surrounding land to monoculture plantations (Taher et al. 2017; Clements et al. 2021), though it may retain a higher level of intactness than the MR and still enable some dispersal. The MR has also suffered fragmentation, but its highland areas continue to provide a corridor of connected habitat from north to south of the central forest spine. Areas in the MR may also remain forested due to the presence of indigenous Orang Asli villages (Sze et al. 2022). The higher observed levels of genetic diversity in the MR compared to the GTN region might also be explained by greater movement of tigers in mountainous areas north to south of the central spine, or the possibility that genetic diversity in the MR has been enhanced via movement of Malayan tigers between Peninsular Malaysia and southern Thailand. This interpretation is supported by the prevalence of mtDNA haplotype H9, which is shared predominantly by Malayan tigers, Indochinese (Thailand and Cambodia), and Sumatran subspecies. However, given the population size in southern Thailand is thought to be small (Suttidate et al. 2021) this scenario is unlikely and further research is needed to validate this hypothesis.
Mitochondrial haplotype analysis inferred a difference in genetic diversity of tigers between GTN and the MR, with four haplotypes corresponding geographically with the two putative subpopulations: H3 and H5 with tigers in GTN, and H4 and H8 with tigers in the MR (Fig. 3). However, these patterns do not completely align with patterns observed for nuclear genetic diversity. Although unique haplotypes were identified in each subpopulation, haplotype diversity was similar in both regions (i.e. number of haplotypes = 4, Table 1). The identification of four unique haplotypes can be interpreted in two ways: (i) a consequence of initial contemporary divergence as a result of recent habitat fragmentation and population decline, with differences in haplotype frequency emerging as haplotypes become lost, perhaps the most likely scenario given the precipitous population decline, or (ii) a consequence of female tiger dispersal behaviour. Female tigers are known to be philopatric and remain close to the maternal home range (Sharma et al. 2011), so haplotype assignment could be the result of maternal clustering due to philopatry. As haplotypes were derived from mtDNA, which is derived from a maternal lineage, this may reflect the patterns of diversity and presence of unique haplotypes. Two additional mtDNA haplotypes unique to Malayan tigers (H16 and H17) were identified from sequences from a previously unpublished analysis (J. Ryan unpubl. data). However, a lack of information on the origin of most of the tigers in the unpublished study limit detailed interpretation. The Malayan tiger subspecies exhibits greater haplotype diversity in comparison to the two other subspecies in Southeast Asia (Indochinese and Sumatran), possibly due to haplotype diversity in Sumatran tigers being constrained due to their geographic range, and loss of haplotypes in Indochinese tigers due to early population collapse (Luo et al. 2004; Ash et al. 2021).
Evidence of genetic impoverishment in the Malayan tiger
The proportional difference in mean Ho and He within Malayan tigers is relatively large (58%), such that the levels of heterozygosity expected in the contemporary population were not subsequently observed—a signal consistent with an inbred population.
Whilst there may be minor differences in marker sets between studies (Aziz et al. 2017; Luo et al. 2004; Thapa et al. 2018) which deserves a degree of caution when comparing marker sets, to the best of our knowledge they are comparable. Comparisons of nuclear genetic diversity between tiger subspecies suggested overall heterozygosity may be lower in Malayan tigers when compared with Sumatran and Amur tigers, but not Indochinese tigers, which have also suffered the effects of habitat fragmentation, population decline and isolation. Indeed, 95% CIs suggest substantially lower Ho in Malayan tigers compared to Amur, Sumatran and Indochinese subspecies, and low He and allelic richness in Malayan tigers compared to Bengal tigers (Bangladesh and India) and Indochinese tigers (Thailand and Cambodia). Higher values for some of the other subspecies may reflect the larger sized populations of tiger (for example, in India n = ~ 3000; (Jhala et al. 2021). Equally, lower values in Sumatran tigers (Smith et al. 2018), Bengal tigers in the Terai Arc, Nepal (Thapa et al. 2018), and Amur tigers in the Russian Far East (Luo et al. 2004) may reflect similar recent effects of extreme population decline and isolation due to habitat fragmentation and poaching. It should also be considered that differences in genotyping or sample quality between studies could account for the recovery or observation of more alleles in other populations, and the limitations of this study in terms of sample size and scale also mean our findings should be treated with caution.
We did not find robust evidence of a population bottleneck in the Malayan tiger population. Whilst it is possible the population has experienced a recent bottleneck, given the widely-documented evidence of extensive habitat loss over several decades (Ten et al. 2021), the low sample size available for this analysis means that failure to detect a bottleneck signature is not surprising. We note that our microsatellite dataset of 22 tiger individuals represents > 10% of the estimated population of ~ 150 tigers remaining in Peninsula Malaysia (Abdullah 2022), though the negative Fst value obtained for the Malayan tiger (Fst = − 0.022) may be a potential indication that sample sizes are too small (Gerlach et al. 2010). Moreover, significant deviations from HWE were detected at ten of the 16 microsatellite loci.
Our findings suggest that there is continuing dispersal and gene flow between tigers in the MR and GTN, in Peninsular Malaysia. Our results did not find consistent evidence of structure in the contemporary population, further indicating a level of geneflow between these two critical tiger areas. We detected relatively small differences in allele frequencies between MR and GTN, consistent with the reasonably recent fragmentation of a once single, large population. However, the continuation of rapid loss and fragmentation of habitat presents a growing threat to gene flow, and we therefore expect the MR and GTN subpopulations to become increasingly genetically differentiated and inbred with time.
Of the two putative tiger subpopulations, the more isolated GTN subpopulation may continue to lose genetic diversity more rapidly than the MR subpopulation because of (i) a greater effect of drift, and (ii) greater movement of tigers from north to south of the mountainous Main Range forest spine, facilitated by highland habitat which can serve as a natural corridor.
The Malayan tiger appears to have low levels of genetic diversity in comparison to most other tiger subspecies. This finding is consistent with a contemporary bottleneck and may reflect the sharp population decline of over 95% that has been documented in the Malayan tiger in the last 70 years (Ten et al. 2021). The levels of genetic impoverishment that we have observed for the Malayan tiger are of potential concern given the role of genetic variation in a population’s resilience and its ability to respond and adapt to future environmental change (Willi et al. 2022).
Given these emerging patterns of genetic diversity, we recommend the following actions for conservation management of tigers in Peninsular Malaysia to enhance functional connectivity between known tiger sites. Actions focus on reducing the net rate of loss of genetic diversity, strengthening gene flow, and slowing the rate of further accumulation of inbreeding.
-
(1)
Preserve existing forest linkages as a critical priority. This should be achieved through the protection and improvement of linkages in the CFS Masterplan (CFS1, Fig. 1) as a matter of urgency. Conservation measures should prioritise:
-
i.
PL1/C-PL1 to facilitate movement between GTN and the MR.
-
ii.
PL3/D-PL1 and PL6/C-PL2 to ensure forest connectivity remains functional from the MR, and specifically PL3/D-PL1 to ensure connectivity on the western side of the MR within Perak State to help tigers from the northern MR reach PL6/C-PL2.
-
iii.
PL2/A-PL1 to facilitate movement between the forest complex containing Hala Bala Wildlife Sanctuary and Bang Lang National Park in Thailand and Royal Belum State Park, with the rest of the MR. This area is the only location where the tiger populations in Malaysia and Thailand are now connected.
-
iv.
PL4/A-PL2, PL5/K-PL1 and PL8/A-PL3 to ensure connectivity to tigers between the MR and Bintang Range. Secondary linkages are also potentially important - particularly when primary linkages are degraded and cannot be relied on for reconnectivity. In this case, SL3/ASL1 is also important due to the short distance between the two forests, as well as its land use, with relatively low human activity.
-
v.
Re-establish and enhance linkages for the tiger population in CFS2 (Fig. 1) to restore genetic connectivity in the Greater Endau Rompin Complex.
-
i.
-
(2)
Restore and reinstate ecological linkages to facilitate tiger movement between landscapes. It is essential to maintain ecological integrity within the forested landscape - particularly where transport infrastructure has been built. More roads are planned across intact forest, including a new major road between Muallim Perak and Kuala Lipis (contruction 21/10/2022-20/10-2026) which will cut across the MR. Infrastructure such as tar roads which provide access deeper into forests to enhance accessibility to interior communities would also make the forest more penetrable and potentially worsen poaching activities. We recommend ensuring there are multiple linkages with good habitat where there are roads to avoid becoming over-reliant on a single small linkage. It is vital to identify further areas as additional linkages to maintain and improve connectivity across roads and other linear infrastructure.
-
(3)
Prohibit development and land-use change around new roads. New transport networks often open up surrounding land to further development, in addition to the degazettement of once-protected forest. We recommend ensuring that forests on both sides of roads (which dissect forested landscape) are not degraded, degazetted, or converted into other land use, including shifting cultivation, forest plantation, or other activities under temporary occupation license, to promote tiger movement and geneflow.
-
(4)
Assess the genetic status of southern tigers in the Endau Rompin Complex, to gain a more comprehensive overview of tigers in Peninsular Malaysia. Tigers in the Endau Rompin Complex are assumed to be largely geographically isolated from both the GTN and MR regions, but their existence there and any additional genetic diversity may yet play a critical role in contributing to the future persistence of tigers in Peninsular Malaysia.
Data availability
Genetic data sets generated from this study can be requested from the Department of Wildlife and National Parks Peninsular Malaysia. All other genetic data used can be found in the supplementary information.
References
Abdullah SM (2022) Malayan Tiger could go extinct in five to 10 years if no drastic action is taken. New Straits Times
Alam MS, Rahaman MA, Begum RA, Shahjahan RM (2021) Non-invasive DNA extraction for molecular identification of royal Bengal tiger Panthera tigris tigris. J Biol Sci 30:325–330
Ash E, Kaszta Ż, Noochdumrong A, Redford T, Chanteap P, Hallam C, Jaroensuk B, Raksat S, Srinoppawan K, Macdonald DW (2021) Opportunity for Thailand’s forgotten tigers: assessment of the indochinese tiger Panthera tigris corbetti and its prey with camera-trap surveys. Oryx 55:204–211
Aziz MA, Tollington S, Barlow A, Greenwood C, Goodrich JM, Smith O, Shamsuddoha M, Islam MA, Groombridge J (2017) Using non-invasively collected genetic data to estimate density and population size of tigers in the Bangladesh Sundarbans. Global Ecol Conserv 12:272–282
Bhagavatula J, Singh L (2006) Genotyping faecal samples of Bengal tiger Panthera tigris tigris for population estimation: a pilot study. BMC Genet 7:48
Biswas S (2021) Metapopulation dynamics of tiger (Panthera tigris tigris) in the Terai-Arc landscape, India. Wildlife Institute of India, Dehradun, India and Saurashtra University: PhD thesis
Biswas S, Bhatt S, Sarkar D, Talukdar G, Pandav B, Mondol S (2020) Assessing tiger corridor functionality with landscape genetics and modelling across Terai-Arc Landscape, India. bioRxiv
Caragiulo A, Kang Y, Rabinowitz S, Dias-Freedman I, Loss S, Zhou XW, Bao WD, Amato G (2015) Presence of the endangered Amur tiger Panthera tigris altaica in Jilin Province, China, detected using non-invasive genetic techniques. Oryx 49:632–635
Clements GR, Rostro-Garcia S, Kamler JF, Liang SH, Hashim AKBA (2021) Conservation status of large mammals in protected and logged forests of the greater Taman Negara Landscape, Peninsular Malaysia. Biodiversitas J Biol Divers 22
Colonna V, Nutile T, Ferrucci RR, Fardella G, Aversano M, Barbujani G, Ciullo M (2009) Comparing population structure as inferred from genealogical versus genetic information. Eur J Hum Genet 17:1635–1641
Cornuet J, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 114:2001–2014
Cracraft J, Feinstein J, Vaughn J, Helm-Bychowski K (1998) Sorting out tigers (Panthera tigris): mitochondrial sequences, nuclear inserts, systematics, and conservation genetics. Anim Conserv Forum 1:139–150
Crandall M, Clement M, Posad D (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
DTCP (2009) Central forest spine (CFS): masterplan for ecological linkages. Kuala Lumpur, Malaysia
Dubach JM, Briggs MB, White PA, Ament BA, Patterson BD (2013) Genetic perspectives on Lion Conservation Units in Eastern and Southern Africa. Conserv Genet 14:741–755
Dutta T, Sharma S, DeFries R (2018) Targeting restoration sites to improve connectivity in a tiger conservation landscape in India. PeerJ 6:5587
Earl DA, von Holdt BM (2012) Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Frankham R, Ballou SEJD, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ, Burke T (2003) Reliable microsatellite genotyping of the eurasian badger (Meles meles) using faecal DNA. Wiley Online Libr 12:1649–1661
Frantz AC, McDevitt AD, Pope LC, Kochan J, Davison J, Clements CF, Elmeros M, Molina-Vacas G, Ruiz-Gonzalez A, Van Den Balestrieri A (2014) Revisiting the phylogeography and demography of European badgers (Meles meles) based on broad sampling, multiple markers and simulations. Heredity 113:443–453
Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P (2010) Calculations of population differentiation based on GST and D: forget GST but not all of statistics! Mol Ecol 19:3845–3852
Goodrich J, Lynam A, Miquelle D, Wibisono H, Kawanishi K, Pattanavibool A, Htun S, Tempa T, Karki J, Jhala Y, Karanth U (2022) Panthera Tigris. The IUCN Red List of Threatened Species
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186
Hansen H, Ben-David ME, Mcdonald DB (2008) Effects of genotyping protocols on success and errors in identifying individual river otters (Lontra canadensis) from their faeces. Mol Ecol Resour 8:282–289
Hansen MC, Wang L, Song XP, Tyukavina A, Turubanova S, Potapov PV, Stehman SV (2020) The fate of tropical forest fragments. Sci Adv. https://doi.org/10.1126/sciadv.aax8574
Hardouin EA, Butler H, Cvitanović M, Ulrich RG, Schulze V, Schilling AK, Lurz PW, Meredith A, Hodder KH (2021) Wildlife conservation in a fragmented landscape: the eurasian red squirrel on the Isle of Wight. Conserv Genet 22:571–583
Hoban S, Bruford M, Jackson JDU, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, Sjögren-Gulve P, Segelbacher G, Vernesi C, Aitken S (2020) Genetic diversity targets and indicators in the CBD post-2020 global biodiversity framework must be improved. Biol Conserv 248:108654
Jhala Y, Gopal R, Mathur V, Ghosh P, Negi HS, Narain S, Yadav SP, Malik A, Garawad R, Qureshi Q (2021) Recovery of tigers in India: critical introspection and potential lessons. People Nat 3:281–293
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Kawanishi K (2015) Panthera tigris Ssp. Jacksoni (Malayan Tiger). The IUCN Red List of Threatened Species
Kitchener AC, Yamaguchi N (2010) What is a tiger? Biogeography, morphology, and taxonomy. In: Tilson R, Nyhus PJ (eds) Tigers of the world. William Andrew Publishing, Norwich
Kleinhans C, Willows-Munro S (2019) Low genetic diversity and shallow population structure in the endangered vulture, Gyps coprotheres. Sci Rep 9:1–11
Kopatz A, Kleven O, Kojola I, Aspi J, Norman AJ, Spong G, Gyllenstrand N, Dalén L, Fløystad I, Hagen SB, Kindberg J (2021) Restoration of transborder connectivity for fennoscandian brown bears (Ursus arctos). Biol Conserv 253:108936
Labisko J, Griffiths RA, Chong-Seng L, Bunbury N, Maddock ST, Bradfield KS, Taylor ML, Groombridge JJ (2019) Endemic, endangered and evolutionarily significant: cryptic lineages in Seychelles’ frogs (Anura: Sooglossidae). Biol J Linn Soc 126:417–435
Li J, Chakraborty R (1995) Population structure, stepwise mutations, heterozygote deficiency and their implications in DNA forensics. Heredity 74:274–285
Li YL, Liu JX (2018) StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Res 18:176–177
Liu YC, Sun X, Driscoll C, Miquelle DG, Xu X, Martelli P, Uphyrkina O, Smith JL, O’Brien SJ, Luo SJ (2018) Genome-wide evolutionary analysis of natural history and adaptation in the world’s tigers. Curr Biol 28:3840–3849
Luikart G, Allendorf F, Cornuet J (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
Luo SJ, Kim JH, Johnson WE, Walt JVD, Martenson J, Yuhki N, Miquelle DG, Uphyrkina O, Goodrich JM, Quigley HB, Tilson R (2004) Phylogeography and genetic ancestry of tigers (Panthera tigris). PLoS Biol 2:442
Mardiastuti A (2018) Lessons learned from the tiger translocation and release in Tambling, Lampung, Indonesia. J Indonesian Nat History 6:42–47
Markert JA, Champlin DM, Gutjahr-Gobell R, Grear JS, Kuhn A, McGreevy TJ, Roth A, Bagley MJ, Nacci DE (2010) Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10:1–13
McManus JS, Dalton DL, Kotzé A, Smuts B, Dickman A, Marshal JP, Keith M (2015) Gene flow and population structure of a solitary top Carnivore in a human-dominated landscape. Ecol Evol 5:335–344
Menotti-Raymond M, David VA, Lyons LA, Schäffer AA, Tomlin JF, Hutton MK, O’Brien SJ (1999) A genetic linkage map of microsatellites in the domestic cat (Felis catus). Genomics 57:9–23
Mohamad SW, Christopher WCT, Sagtia S, Hamirul M, Lau CF, Mohamed A, Rayan DM (2013) Highest recorded elevation of tiger presence in peninsular Malaysia. Cat News 58:39–40
Mona S, Ray N, Arenas M, Excoffier L (2014) Genetic consequences of habitat fragmentation during a range expansion. Heredity 112:291–299
Mondol S, Karanth KU, Ramakrishnan U (2009a) Why the Indian subcontinent holds the key to global tiger recovery. PLoS Genet 5:1000585
Mondol S, Karanth KU, Kumar NS, Gopalaswamy AM, Andheria A, Ramakrishnan U (2009b) Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol Conserv 142:2350–2360
Mondol S, Kumar N, Gopalaswamy A, Sunagar K, Karanth KU, Ramakrishnan U (2015) Identifying species, sex and individual tigers and leopards in the Malenad-Mysore Tiger Landscape, Western Ghats, India. Conserv Genet Resource 7:353–361
Morgan KI, Ewart KM, Nguyen TQ, Sitam FT, Ouitavon K, Lightson AL, Kotze A, McEwing R (2021) Avoiding common numts to provide reliable species identification for tiger parts. Forensic Sci Int Rep 3:100166
Mueller SA, Reiners TE, Middelhoff TL, Anders O, Kasperkiewicz A, Nowak C (2020) The rise of a large Carnivore population in Central Europe: genetic evaluation of lynx reintroduction in the Harz Mountains. Conserv Genet 21:577–587
Múrias dos Santos A, Cabezas MP, Tavares AI, Xavier R, Branco M (2016) tcsBU: a tool to extend TCS network layout and visualization. Bioinformatics 32:627–628
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Oyler-McCance SJ, Fedy BC, Landguth EL (2013) Sample design effects in landscape genetics. Conserv Genet 14:275–285
Park SDE (2001) Trypanotolerance in West African cattle and the population genetic effects of selection. PhD thesis, University of Dublin
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Res 16:608–627
Ratnayeke S, Van Manen FT, Clements GR, Kulaimi NAM, Sharp SP (2018) Carnivore hotspots in Peninsular Malaysia and their landscape attributes. PLoS ONE 13:0194217
Rayan DM, Linkie M (2015) Conserving tigers in Malaysia: a science-driven approach for eliciting conservation policy change. Biol Conserv 184:18–26
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Sanderson EW, Forrest J, Loucks C, Ginsberg J, Dinerstein E, Seidensticker J, Leimgruber P, Songer M, Heydlauff A, O’Brien T, Bryja G (2010) Setting priorities for conservation and recovery of wild tigers. The technical assessment. In: Tilson RL, Nyhus PJ (eds) Tigers of the world: the science, politics, and conservation of Panthera tigris: 2005–2015. Elsevier, New York
Sarkar MS, Ramesh K, Johnson JA, Sen S, Nigam P, Gupta SK, Murthy RS, Saha GK (2016) Movement and home range characteristics of reintroduced tiger (Panthera tigris) population in Panna Tiger reserve, central India. Eur J Wildl Res 62:537–547
Schmidt-Küntzel A, Dalton DL, Menotti-Raymond M, Fabiano E, Charruau P, Johnson WE, Sommer S, Marker L, Kotzé A, O’Brien SJ (2018) Conservation genetics of the cheetah: genetic history and implications for conservation. Biology Conserv. https://doi.org/10.1016/B978-0-12-804088-1.00006-X
Seidensticker J, Dinerstein E, Goyal SP, Gurung B, Harihar A, Johnsingh AJT, Manandhar A, McDougal CW, Pandav B, Shrestha M, Smith JD (2010) Tiger range collapse and recovery at the base of the Himalayas. Biology Conserv Wild Felids 12:305–324
Sharma R, Stuckas H, Bhaskar R, Khan I, Goyal SP, Tiedemann R (2011) Genetically distinct population of Bengal tiger (Panthera tigris tigris) in Terai Arc Landscape (TAL) of India. Mammalian Biology 76:484–490
Sharma S, Dutta T, Maldonado JE, Wood TC, Panwar HS, Seidensticker J (2013) Forest corridors maintain historical gene flow in a tiger metapopulation in the highlands of central India. Biol Sci 280:20131506
Shriver MD, Jin L, Chakraborty R, Boerwinkle E (1993) VNTR allele frequency distributions under the stepwise mutation model: a computer simulation approach. Genetics 134:983–993
Simcharoen A, Savini T, Gale GA, Simcharoen S, Duangchantrasiri S, Pakpien S, Smith JL (2014) Female tiger Panthera tigris home range size and prey abundance: important metrics for management. Oryx 48:370–377
Singh SK, Aspi J, Kvis L, Sharma R, Pandey P, Mishra S, Singh R, Agrawal M, Goyal SP (2017) Fine-scale population genetic structure of the Bengal tiger (Panthera tigris tigris) in a humandominated western Terai Arc Landscape, India. PLoS ONE 12:e0174371
Smith O (2012) Population Genetics and Structure of the Sumatran tiger. London: UCL. PhD thesis
Smith O, Wang J, Carbone C (2018) Evaluating the effect of forest loss and agricultural expansion on Sumatran tigers from scat surveys. Biol Conserv 221:270–278
Suttidate N, Steinmetz R, Lynam AJ, Sukmasuang R, Ngoprasert D, Chutipong W, Bateman BL, Jenks KE, Baker-Whatton M, Kitamura S, Ziółkowska E (2021) Habitat connectivity for endangered indochinese tigers in Thailand. Global Ecol Conserv 29:e01718
Symes WS, Edwards DP, Miettinen J, Rheindt FE, Carrasco LR (2018) Combined impacts of deforestation and wildlife trade on tropical biodiversity are severely underestimated. Nat Commun 9:1–9
Sze JS, Childs DZ, Carrasco LR, Edwards DP (2022) Indigenous lands in protected areas have high forest integrity across the tropics. Curr Biol 32:4949–4956
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194
Taher TM, Abdullah NI, Ariffin NT, Mustapha MA, Lihan T, Patah PA, Razali SHA, Ismail A, Shukor MN (2017) Predicting connectivity between main range forest complex and Taman Negara. J Wildl Parks 32:1–12
Ten DCY, Jani R, Hashim NH, Saaban S, Abu Hashim AK, Abdullah MT (2021) Panthera tigris Jacksoni population crash and impending extinction due to environmental perturbation and human-wildlife conflict. Animals 11:1032
Thapa K, Manandhar S, Bista M, Shakya J, Sah G, Dhakal M, Sharma N, Llewellyn B, Wultsch C, Waits LP, Kelly MJ (2018) Assessment of genetic diversity, population structure, and gene flow of tigers (Panthera tigris tigris) across Nepal’s Terai Arc Landscape. PLoS ONE 13:e0193495
Trouvae S, Degen L, Renaud F, Goudet J (2003) Evolutionary implications of a high selfing rate in the freshwater snail Lymnaea Truncatula. Evolution 57:2303–2314
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wang J (2017) The computer program structure for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17:981–990
Waples R, Gaggiotti O (2006) Invited review: what is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Weir B, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Willi Y, Kristensen TN, Sgrò CM, Weeks AR, Ørsted M, Hoffmann AA (2022) Conservation genetics as a management tool: the five best-supported paradigms to assist the management of threatened species. Proc National Acad Sci 119:e2105076119
Wilson G, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wilting A, Courtiol A, Christiansen P, Niedballa J, Scharf AK, Orlando L, Balkenhol N, Hofer H, Kramer-Schadt S, Fickel J, Kitchener AC (2015) Planning tiger recovery: understanding intraspecific variation for effective conservation. Sci Adv 1:e1400175
Yi X, Latch EK (2022) Nonrandom missing data can bias principal component analysis inference of population genetic structure. Mol Ecol Resour 22:602–661
Acknowledgements
We are very grateful to the Department of Wildlife and National Parks Peninsular Malaysia (permit reference: KATS 600-2/2/21 JLD.9 (7)), Economic Planning Unit (permit reference: UPE 40/200/19//3490 (14)), Forestry Department Peninsular Malaysia (permit reference: JH/ 100 Jld. 22 (29)), and Perak State Parks Corporation (permit reference: PTNPK/T/400/4/32/Bill.(1)dlm/2019) for providing permits for this study. The Orang Asli community played an important role in this research, and we are extremely thankful for their support and expertise in the forest. We are grateful to the teams who collectively contributed samples to this study: WWF-Malaysia, Department of Wildlife and National Parks Peninsular Malaysia-National Wildlife Forensic Laboratory, Panthera Malaysia, Rimba, Perak State Parks Corporation Rangers, and Malayan Rainforest Station. We are especially grateful to Encik Shah Redza, Wai Yee Lam, Dr Rob Pickles, Wong Pui May, Dr Sheema Adbul Aziz and Professor Reuben Clements for guidance, technical advice and support. We thank the team at the National Wildlife Forensic Laboratory for their help and expertise during labwork, especially Frankie Sitam and Millawati Binti Gani. We also thank Dr Md. Abdul Aziz, Dr Simon Tollington, Dr Hazel Jackson, Dr Debbie Fogell, and Dr Claire Raisin, for their advice, training, and assistance with statistical analysis. All samples collected and used in this study were analysed and stored at the National Wildlife Forensic Laboratory in Kuala Lumpur, Malaysia.
Funding
This work was supported by a NERC CASE Studentship (NE/L002582/1) by the EnvEast DTP and a Conservation Research Grant from The Habitat Foundation. The authors declare that no funds, grants or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
K.A. and J.G. conceived the study. K.A., J.G., M.S. and R.O. contributed to the study design. C.W., L.C.F. and K.A.M. assisted with fieldwork design and logistical planning. K.A. performed all statistical analysis. K.A. wrote the first draft of the manuscript, with J.G., M.S., R.O., J.J.R.R., N.A.E., M.D., C.W., L.C.F. and O.S. contributing to manuscript revisions. K.A. and N.A.N.G.A. conducted DNA and genotype analysis. C.W., L.C.F., O.S., M.I.A.M. and M.H.M.Y. contributed samples to the study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by David Hawksworth.
Publisher’s Note
Springer nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Allberry, K., Rovie-Ryan, J.J., Ali, N.A.N.G. et al. Emerging patterns of genetic diversity in the critically endangered Malayan tiger (Panthera tigris jacksoni). Biodivers Conserv 33, 1325–1349 (2024). https://doi.org/10.1007/s10531-024-02799-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-024-02799-9